

Abstract
Hydrogen sulfide (H2S) production in colon cancer cells supports cellular bioenergetics and proliferation. The aim of the present study was to investigate the alterations in H2S homeostasis during the development of resistance to 5-fluorouracil (5-FU), a commonly used chemotherapeutic agent. A 5-FU-resistant HCT116 human colon cancer cell line was established by serial passage in the presence of increasing 5-FU concentrations. The 5-FU-resistant cells also demonstrated a partial resistance to an unrelated chemotherapeutic agent, oxaliplatin. Compared to parental cells, the 5-FU-resistant cells rely more on oxidative phosphorylation than glycolysis for bioenergetic function. There was a significant increase in the expression of the drug-metabolizing cytochrome P450 enzymes CYP1A2 and CYP2A6 in 5-FU-resistant cells. The CYP450 inhibitor phenylpyrrole enhanced 5-FU-induced cytotoxicity in 5-FU-resistant cells. Two major H2S-generating enzymes, cystathionine-β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST) were upregulated in the 5-FU-resistant cells. 5-FU-resistant cells exhibited decreased sensitivity to the CBS inhibitor aminooxyacetate (AOAA) in terms of suppression of cell viability, inhibition of cell proliferation and inhibition of oxidative phosphorylation. However, 5FU-resistant cells remained sensitive to the antiproliferative effect of benserazide (a recently identified, potentially repurposable CBS inhibitor). Taken together, the current data suggest that 5-FU resistance in HCT116 cells is associated with the upregulation of drug-metabolizing enzymes and an enhancement of endogenous H2S production. The anticancer effect of prototypical H2S biosynthesis inhibitor AOAA is impaired in 5-FU-resistant cells, but benserazide remains efficacious. Pharmacological approaches aimed at restoring the sensitivity of 5-FU-resistant cells to chemotherapeutic agents may be useful in the formulation of novel therapeutic strategies against colorectal cancer.
Graphical abstract
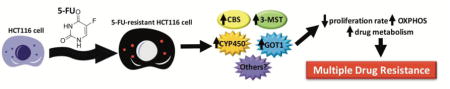
1. Introduction
Colorectal cancer is still the third most common cancer and the second most common cause of cancer-related death worldwide [1,2], with nearly 1.4 million cases a year and ~774,000 deaths worldwide [3]. The chemotherapeutic drug 5-fluorouracil (5-FU) remains widely used in the treatment of colorectal carcinoma.
5-FU is an analog of uracil with a fluorine atom substituted at the carbon-5 position of the pyrimidine ring in place of hydrogen. 5-FU, and other 5-fluorinated pyrimidines have been widely used in the treatment of colorectal cancer [4]. The efficacy of 5-FU is, at least in part, attributed to its ability to induce p53-dependent cell growth arrest and apoptosis. 5-FU is considered an S phase-active chemotherapeutic agent which inhibits cell proliferation and survival [5,6]. While some patients respond initially to chemotherapy, many advanced colorectal cancer patients eventually develop chemotherapy resistance disease, which is a major barrier to achieve effective therapy.
Cystathionine-β-synthase (CBS) is upregulated and hydrogen sulfide (H2S) production is increased in various types of cancer including colon, ovarian, breast and lung cancer [7–12]. The functional consequence of increased cellular H2S production is stimulation of cellular bioenergetics, tumor growth and proliferation (reviewed in [13]). The mechanisms involved in the stimulation of mitochondrial function by H2S are multiple; they involve direct electron donation to Complex II of the mitochondrial electron transport chain [14–16] inhibition of mitochondrial cAMP phosphodiesterases, followed by cAMP-stimulated increases in mitochondrial electron transport [17], mitochondrial antioxidant effects [18,19], stimulation of mitochondrial DNA repair [12,20], direct stimulation of mitochondrial ATP synthase via posttranslational modification (via protein S-sulfhydration) [21] as well as the stimulation of lactate dehydrogenase activity (via protein S-sulfhydration) [22]. All of these effects occur at low-to-intermediate concentrations of H2S, while at higher concentrations, inhibitory effects of H2S on mitochondrial function become apparent – primarily mediated by the inhibition of mitochondrial Complex IV (cytochrome c oxidase) by H2S [23,24]. In 2013, we discovered that CBS is overexpressed in colon cancer and that H2S produced by it serves to support cellular bioenergetics and tumor angiogenesis. Pharmacological inhibition or silencing of CBS reduced tumor bioenergetic function, inhibited tumor angiogenesis and suppressed tumor growth [7]. In contrast, other H2S-producing enzymes such as cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST) were not differentially expressed in colon tumor tissue when compared to normal mucosa [7]. The mitochondrial enzyme 3-MST is a newly identified endogenous source of H2S in various cells and tissues, including vascular endothelial cells [25]. We have recently demonstrated that 3-MST activity is inhibited by oxidative stress in vitro and speculated that this may exert adverse effects on cellular homeostasis [26]. Although we did not observe a significant upregulation of 3-MST in colon cancer [7], in other forms of cancer, an upregulation of 3-MST has been reported [12,27,28].
The aims of the present study were to examine whether H2S homeostasis is altered in 5-FU resistant colon cancer and to characterize the molecular mechanisms that contributed to the development of the resistant phenotype, such as proteins involved in the extrusion and/or metabolism of the chemotherapeutic drug. We also tested how development of 5-FU resistance affected cellular bioenergetic status and sensitivity to CBS inhibition with either aminooxyacetic acid (a prototypical CBS inhibitor) or benserazide. Both of these compounds exert significant inhibitory effects in vitro and in vivo. The results of the present study demonstrate the upregulation of the H2S-generating enzymes CBS and 3-MST during the development of 5-FU resistance in HCT116 cells, positively influencing cellular viability, bioenergetics and proliferation.
2. Materials and Methods
2.1 Cell culture
The parental human colorectal carcinoma cell line, HCT116 (ATCC, Manassas, VA, USA) was cultured in McCoy’s 5A medium supplemented with 10% FBS, 100 IU/mL penicillin and 100 mg/mL streptomycin as described [7]. Cells were grown in a 37°C, 5% CO2 atmosphere.
To established an acquired resistant cell line, HCT116 cells were cultured in medium containing stepwise increased concentrations of 5-FU for 6 months to obtain a 5-FU-resistant HCT116 cell line. Briefly, HCT116 cells were cultured in fresh medium without drugs for 24 h. Subsequently, the medium was changed and 1 μM 5-FU (Cat. # F6627) in complete medium was added. HCT116 cells were exposed to 5-FU for 48–72 h, thereafter the 5-FU-treatment medium was removed and cells were allowed to recover (in normal medium) for about a week. When cells reached 70% confluence, the treatment process was repeated for several times until they were stable. Once stable, cells were subjected 3, 10 and finally 30 μM 5-FU treatment. Thereafter, the 5-FU-resistant cells were maintained in full medium supplemented with 30 μM 5-FU (to maintain 5-FU resistance).
2.2 Western blotting
Cells were lysed in RIPA buffer (Sigma-Aldrich, St. Louis, MO) supplemented with protease inhibitor cocktail (Complete Mini EDTA-free, Roche Applied Science, Indianapolis, IN). Cell homogenates were resolved on 4–12% NuPage Bis-Tris acrylamide gels (Invitrogen), then transferred to nitrocellulose. Membranes were blocked in 10% non-fat dried milk and probed overnight with CBS (Proteintech Group, Inc., Rosemont, IL; Cat. # 14787-1-AP), CSE (Proteintech; Cat. # 12217-1-AP), 3-MST (Abcam; Cat. # ab85377), rhodanese (Proteintech; Cat. # 66018-1), MRP1 (Abcam; Cat. # ab24102), GOT1 (Abcam; Cat. # ab85857), CYP1A2 (Proteintech; Cat. # 19936-1-AP), CYP2A6 (Proteintech; Cat. # 21721-1-AP), or β-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA; Cat. # 47778). After incubation with peroxidase conjugates the blots were detected on a CCD camera based detection system (GBox, Syngene USA, Frederick, MD). ImageJ was used for densitometric analysis.
2.3 Enzymatic activity of H2S-generating enzymes
Cells were lysed in ice-cold NP40 lysing buffer (1% NP40; 150mM NaCl; 50mM Tris-Cl, pH 8.0) for 30 min on ice. Cell homogenates (~5–20 μl, 100–200 μg protein) were added to the reaction mixture (50 mM Tris HCL pH 8.0), which was supplemented with 2 mM homocysteine and 2 mM L-cysteine (CBS activity assay), 2 mM L-cysteine (CSE activity assay), or 2 mM 3-mercaptopyruvate (3-MST activity assay). Pyridoxal 5′-phosphate (PLP; 5 μM final concentration) was added to only the CBS and CSE reaction mixtures, followed by the addition of AzMc (7-azido-4-methylcoumarin; 10 μM final concentration) to all the reaction mixtures, bring the total volume of the reaction buffer to 200 μl. The enzyme activity mixture was incubated at 37 °C for 4 h. Immediately thereafter, fluorescence was measured at 450 nm (via Molecular Devices M2 microplate reader). 1% NP40 was used as a vehicle control.
2.3 Extracellular Flux Analysis
The XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies) was used to measure bioenergetic function as described [14,29]. Four key parameters of mitochondrial function (basal respiration, adenosine triphosphate (ATP) turnover, proton leak and maximal respiration) were assessed through the sequential use of 40 μM oligomycin (ATP synthase inhibitor), 13.5 μM FCCP (oxidative phosphorylation un- coupler) and 10 μM rotenone + 1 μM antimycin A (complex I and III inhibitors, respectively). The difference between the maximal and the basal respirations was considered the respiratory reserve capacity (the capacity of a cell to generate ATP via oxidative phosphorylation in response to increased demand for energy). A glycolytic stress test was used to estimate various parameters of cellular glycolysis (glycolysis, glycolytic capacity and glycolytic reserve), which was obtained with the sequential use of 25 mM glucose, 5 μM oligomycin (to block mitochondrial respiration and force the cells to rely on glycolysis for ATP production) and 100 mM 2-deoxyglucose (2-DG, a glucose analog and inhibitor of glycolytic ATP production). Glycolytic reserve was calculated as the difference between the glycolytic capacity and the glycolysis; this parameter is indicative of the cellular ability to increase the glycolytic rate upon increased energy demand. Acidification of carbon dioxide, the end product of the tricarboxylic acid (TCA) cycle, which can be converted to bicarbonate, is considered a major contributor to nonglycolytic acidification. Bioenergetic parameters were normalized to protein content via Lowry reagent (Bio-Rad) using BSA as a standard.
2.4 MTT and LDH assays
The MTT assay and LDH activity measurements were performed as previously described [29]. Briefly, after 48 h of treatment with 1-phenylpyrrole (Cat. # 131474), AOAA (Cat. # C13408) and/or 5-FU, cells were washed and incubated in medium containing 0.5 mg/ml, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Calbiochem, EMD BioSciences, San Diego, CA) for 1 h at 37°C in a 5% CO2 atmosphere. The converted formazan dye was dissolved in DMSO. The plates were read on a Molecular Devices M2 microplate reader at a test wavelength of 570 nm and a reference wavelength of 630 nm. Cell viability was calculated as the ratio of absorbance in treated cultures to absorbance in untreated control cultures.
LDH release was measured by mixing cell culture supernatant (30 μl) with 100 μl LDH assay reagent containing 110 mM lactic acid, 1.35 M nicotinamide adenine dinucleotide (NAD+), 290 mM N-methylphenazonium methyl sulfate (PMS), 685 mM 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (INT) and 200 mM Tris (pH 8.2). The changes in absorbance were read kinetically at 492 nm for 15 min (kinetic LDH assay). LDH activity values are shown as Vmax (mOD/min).
2.5 Cell proliferation assays
For assessment of cell proliferation, the xCELLigence system (Roche Applied Science, Indianapolis, IN) was used as described [9,12]. Briefly, HCT116 cells were cultured until ~70% confluence. After 24 h, cells were detached by trypsin-EDTA and resuspended in fresh culture media at a concentration of 60,000 cells/ml. Cell suspension was added to each well (6,000 cells/well) of an E-plate 96, a specially designed 96-well microtiter plate containing interdigitated microelectrodes to noninvasively monitor the cell proliferation by measuring the relative change in the electrical impedance of the cell monolayer, a unitless parameter named cell index. After 24 h, cells were treated with 5-FU (1–30 μM) with or without AOAA (0.3–1 mM) and proliferation was monitored for 48 h.
2.6 Chemicals and statistical analysis
All chemicals and commercially available enzymes used in this study were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. All data are presented as mean ± SEM and were analyzed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Statistical analyses included Student t test or two-way ANOVA followed by Tukey’s multiple comparisons were used to detect differences between groups. Statistical significance was considered at P < 0.05.
3. Results
3.1 H2S-generating enzymes are upregulated in 5-FU-resistant HCT116 cells
To elucidate the underlying mechanism for 5-FU resistance in CRC, we established an acquired resistant cell line, HCT116, through addition of increasing amount of 5-FU to the HCT116 parental cells for ~6 months. Thereafter, the 5-FU-resistant cells were maintained in medium supplemented with 30 μM 5-FU. MTT assay was used to assess cellular cytotoxicity. Parental HCT116 cells responded to 5-FU challenge (3–100 μM) via decreased MTT reduction, while 5-FU-resistant cells were not responsive to 5-FU treatment, whereby cell viability remained unchanged (Fig. 1A). 5-FU-resistant HCT116 cells proliferated at a markedly slower rate than the parental HCT116 cells. XCELLigence analysis revealed that 30 μM 5-FU treatment significantly inhibited cell proliferation of naive HCT116 cells, but was without antiproliferative effect in 5-FU-resistant HCT116 cells (Fig. 1B). (As a result of the different proliferation rates, XCELLigence data obtained from both cell lines were normalized to their respective, untreated control group.) Surprisingly, low concentrations of 5-FU (3 μM) significantly enhanced cell proliferation in parental HCT116 cells, and this effect became more pronounced in 5-FU-resistant cells (Fig. 1B). No changes were observed in cell necrosis (via LDH release assay) with 5-FU administration in either cell lines (Fig. 1C).
Figure 1. Characterization of 5-FU-resistant HCT116 cells.
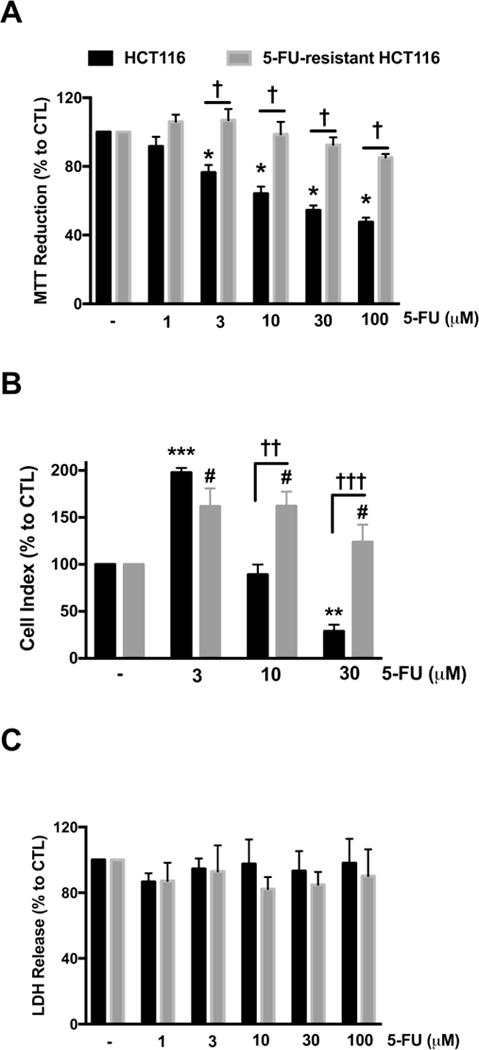
HCT116 and 5-FU-resistant HCT116 cells were treated with 5-FU (3–100 μM) for 48 h. (A) MTT reduction in both HCT116 and 5-FU-resistant HCT116 cells treated with 5-FU. (B) HCT116 and 5-FU-resistant HCT116 cells were seeded in xCELLigence plates and proliferation was monitored for 24 h. Thereafter, cells were treated with or without 5-FU (3–30 μM) and proliferation was monitored for another 48 h. Figure shows statistical analysis of cell proliferation endpoints normalized to cell index and expressed as a percentage of the corresponding control group. (C) Necrotic cell death determined by LDH release in HCT116 and 5-FU-resistant HCT116 cells treated with 5-FU. Data represent mean ± SEM. n = 4 per group. *P < 0.05, **P < 0.01 and ***P < 0.001 vs HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05, ††P < 0.01 and †††P < 0.001 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
Furthermore, we observed partial resistance to the clinically-used anti-cancer drug, oxaliplatin, in the 5-FU-resistant cells with comparison to parental HCT116 cells (Fig. 2). From 1–100 μM, oxaliplatin significantly reduced MTT reduction in both resistant and parental cell lines (Fig. 2A); however, 5-FU-resistant HCT116 cell viability was significantly higher to that of HCT116 cell viability in the presence of 3–100 μM oxaliplatin (Fig. 2A). No significant changes were reported in cell necrosis (via LDH release assay) in either cell line in the presence of oxaliplatin (Fig. 2B). These findings indicate that the current protocol of 5-FU-resistance, in fact, creates multi-drug-resistant cells and predict that the development of 5-FU-resistance is associated with a significant degree of reprogramming of various cellular processes.
Figure 2. 5-FU-resistant HCT116 cells exhibit partial resistance to oxaliplatin.
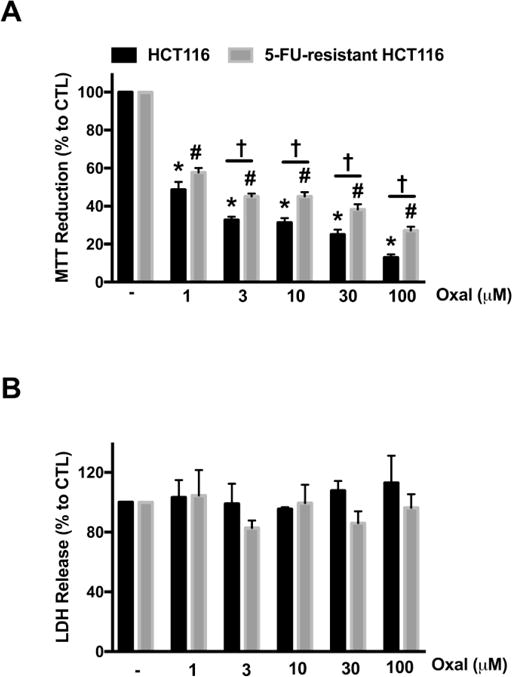
HCT116 and 5-FU-resistant HCT116 cells were treated with oxaliplatin (1–100 μM) for 48 h. (A) MTT reduction in both HCT116 and 5-FU-resistant HCT116 cells treated with oxaliplatin. (B) Necrotic cell death determined by LDH release in HCT116 and 5-FU-resistant HCT116 cells treated with oxaliplatin. Data represent mean ± SEM. n = 5–8 per group. *P < 0.05 vs HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test). Oxal: oxaliplatin.
The physiological production of H2S is due, in large part, to the activity of three enzymes: CBS, CSE, and 3-MST. Colon cancer cells contain high levels of CBS, whereby its product, H2S, promotes the growth and proliferation of colorectal tumor cells [7]. Densitometric analysis of Western blots revealed a 49%, 63% and 107% increase in CBS, 3-MST and the H2S-metabolizing enzyme rhodanese, respectively in 5-FU-resistant cells, compared to parental HCT116 cells (Fig. 3A and Fig. 3B). In agreement with the protein expression data, the enzymatic activities of CBS and 3-MST were enhanced in 5-FU-resistant cells compared to parental HCT116 cells (Fig. 3C). In contrast, CSE protein level and enzymatic activity were similar in both parental and 5-FU-resistant cells (Fig. 3).
Figure 3. Proteins and the enzymatic activities of major H2S-generating enzymes are upregulated in 5-FU-resistant HCT116 cells.
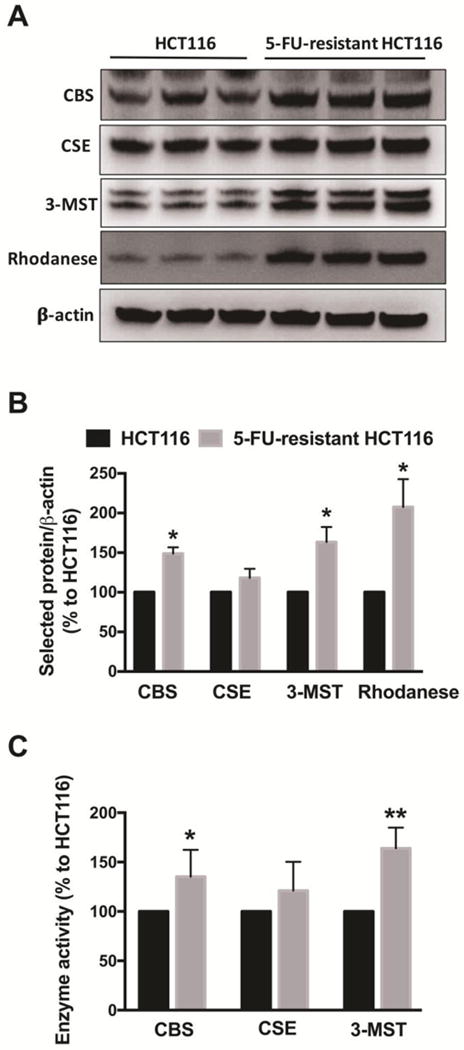
(A) Representative western blot images of CBS, CSE, 3-MST and rhodanese are shown. (B) The density of the H2S-producing enzymes was normalized to that of β-actin and expressed as a percentage of the corresponding HCT116 group. n = 3–6 per group. *P < 0.05 vs. HCT116 CTL. (C) The enzymatic activity of CBS, CSE and 3- MST were analyzed as described in Material and Methods and expressed as a percentage to the corresponding HCT116 group. n = 3–4 per group. Data represent mean ± SEM. *P < 0.05, ** P < 0.01 vs. HCT116 cells (based on unpaired Student’s t test).
3.2 CYP450 and GOT1 proteins, but not MRP1, are upregulated in 5-FU-resistant cells
Members of the ATP-binding cassette (ABC) transporter family proteins are one of the most studied mechanisms of cancer drug resistance, including multidrug resistance-associated protein 1 (MRP1). ABC proteins are involved in reducing drug accumulation by enhancing efflux [30]. We found no change in the protein level of MRP1 in 5-FU-resistant cells compared to parental HCT116 cells (Fig. 4). Glutamate oxaloacetate transaminase 1 (GOT1), also known as aspartate transaminase or AST, is an essential enzyme in the malate/aspartate shuttle [31,32]. Densitometric analysis showed a ~40% increase in GOT1 protein level in 5-FU-resistant cells compared to parental HCT116 cells (Fig. 4).
Figure 4. GOT1 and CYP450 proteins are upregulated in 5-FU-resistant HCT116 cells.
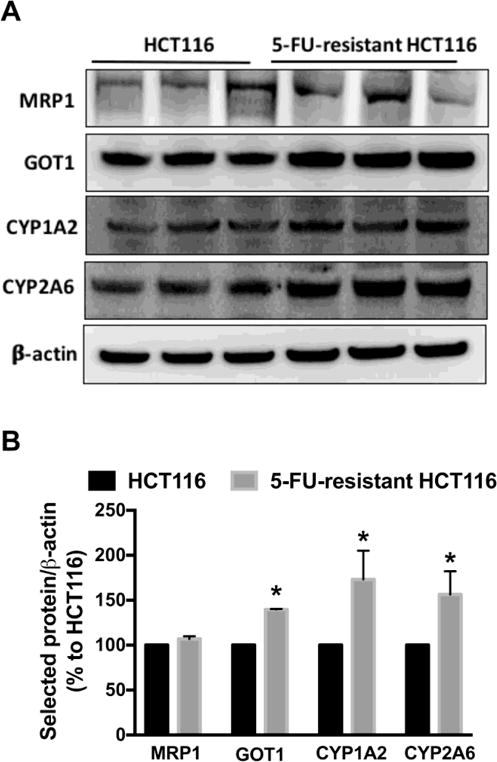
(A) Representative western blot images of MRP1, GOT1, CYP1A2 and CYP2A6 are shown. (B) The density of the selected proteins was normalized to that of β-actin and expressed as a percentage of the corresponding HCT116 group. n = 3–4 per group. Data represent mean ± SEM. *P < 0.05 vs. HCT116 CTL (based on Student’s t test).
Various cytochromes P450 (CYP) - including isoforms 1A2 and 2A6 - are involved in drug metabolism in cancer cells [33–35]. We observed a significant increase in the protein levels of CYP1A2 and CYP2A6 in 5-FU-resistant HCT116 cells compared to parental HCT116 cells (Fig. 4). Densitometric analysis of Western blots revealed a 73% and 56% increase in CYP1A2 and CYP2A6, respectively in 5-FU-resistant cells (Fig. 4A and Fig. 4B).
We next tested whether or not treatment with the CYP450 inhibitor, phenylpyrrole, would sensitize 5-FU-resistant cells to 5-FU treatment. Both resistant and parent cell lines were treated with 100 μM phenylpyrrole (or its vehicle control), followed by increasing concentrations of 5-FU (i.e. 1–100 μM). There was a steady decline in parental HCT116 cell viability with increasing 5-FU concentration (Fig. 5A). Parental HCT116 cell viability did not further decline with the co-administration of 100 μM phenylpyrrole and 5-FU (Fig. 5A). In contrast, a significant enhancement of 5-FU cytotoxicity (at 100 μM) was noted in 5-FU-resistant cells in the presence of phenylpyrrole (Fig. 5B). These data suggest that increased metabolism (inactivation) of 5-FU (but not increased extrusion of 5-FU from the resistant cells) may underlie, at least in part, the drug resistance observed in the 5-FU-resistant cells in the current study.
Figure 5. Inhibition of CYP450 enzymes sensitizes 5-FU-resistant HCT116 cells to 5-FU treatment.
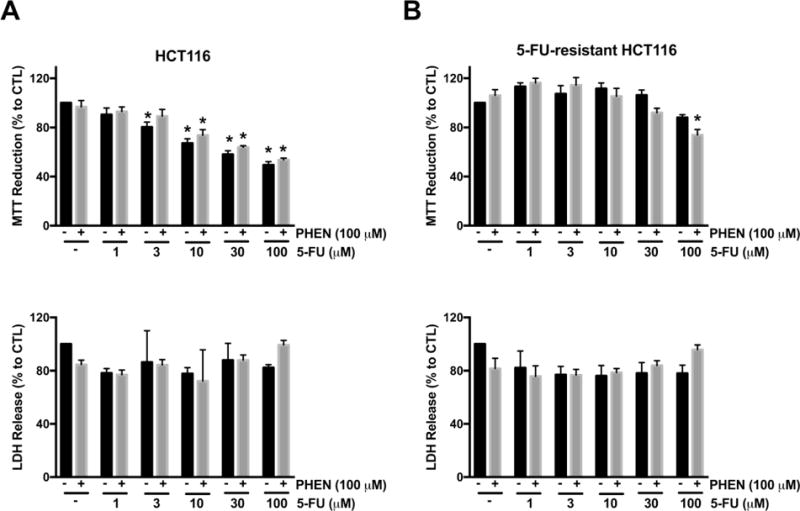
HCT116 (A) and 5-FU-resistant HCT116 cells (B) were treated with 5-FU (1–100 μM) and/or 100 μM phenylpyrrole, a potent CYP450 inhibitor, for 48 h. Top panels in both (A) and (B) shows cell viability via MTT reduction under various treatment conditions. n = 3–5, for each group. Bottom panels in both (A) and (B) show necrotic cell death determined by LDH release under various treatment conditions. n = 4 per group. Data represent mean ± SEM. *P < 0.05 vs. HCT116 CTL; #P < 0.05 vs. 5-FU-resisant CTL (based on two-way ANOVA corrected with Tukey’s post-hoc test). PHEN: phenylpyrrole.
3.3 The efficacy of AOAA is impaired in 5-FU-resistant cells
We previously showed that the prototypical CBS inhibitor aminooxyacetic acid (AOAA) suppresses the proliferation of colon cancer cells in vitro and reduces tumor growth in vivo [7,9]. AOAA is a potent pharmacological inhibitor of both CBS and CSE [36]. To assess whether the excessive generation of H2S contributed to 5-FU resistance in the 5-FU-resistant HCT116 cells, cells were treated with a combination of AOAA and 5-FU. 5-FU-resistant cells developed a resistance against AOAA treatment; in 5-FU-resistant cells AOAA only decreased viability at millimolar concentrations (i.e. 1–3 mM), and even then, it only produced a partial effect (Fig. 6A, top panel). When co-treatment of AOAA and 5-FU was tested, the 5-FU-resistant cells remained insensitive to the combination of drugs and cell viability maintained (Fig. 6B, top panel). Fig. 6B (bottom panel) shows that a significant increase in cell necrosis (via LDH release assay) was only noted at the highest concentration of AOAA and 5-FU combination; in these conditions both resistant and parental cell lines responded with a comparable degree of LDH release (Fig. 6B, bottom panel).
Figure 6. 5-FU-resistant HCT116 cells are resistant against AOAA treatment.
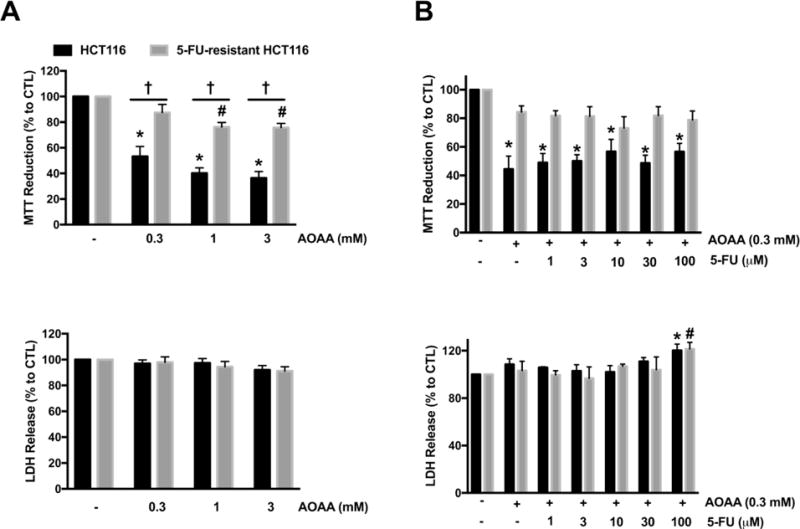
(A) HCT116 and 5-FU-resistant HCT116 cells were treated with AOAA (0.3–1 mM) for 48 h. Top panel: MTT reduction in both HCT116 and 5-FU-resistant HCT116 cells treated with 5-FU. Bottom panel: Necrotic cell death determined by LDH release in HCT116 and 5-FU-resistant HCT116 cells treated with AOAA. n = 5–8 per group. (B) HCT116 and 5-FU-resistant HCT116 cells were treated with 5-FU (1–100 μM) with or without 0.3 mM AOAA for 48 h. Top panel: MTT reduction in both HCT116 and 5-FU-resistant HCT116 cells; bottom panel: necrotic cell death determined by LDH release in HCT116 and 5-FU-resistant HCT116 cells. n = 3–5 per group. Data represent mean ± SEM. *P < 0.05 vs HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
In agreement with our previous findings [7,9], AOAA treatment significantly inhibited HCT116 cell proliferation (Fig. 7). 5-FU-resistant cells showed resistance to AOAA (Fig. 7). At 300 μM AOAA produced a stimulatory effect on cell proliferation in the 5-FU-resistant cells, while at 1 mM AOAA, only a minor (statistically not significant) inhibitory effect was observed on the proliferation of 5-FU-resistant HCT116 cells (Fig. 7).
Figure 7. The antiproliferative effects of AOAA is absent in 5-FU-resistant HCT116 cells.
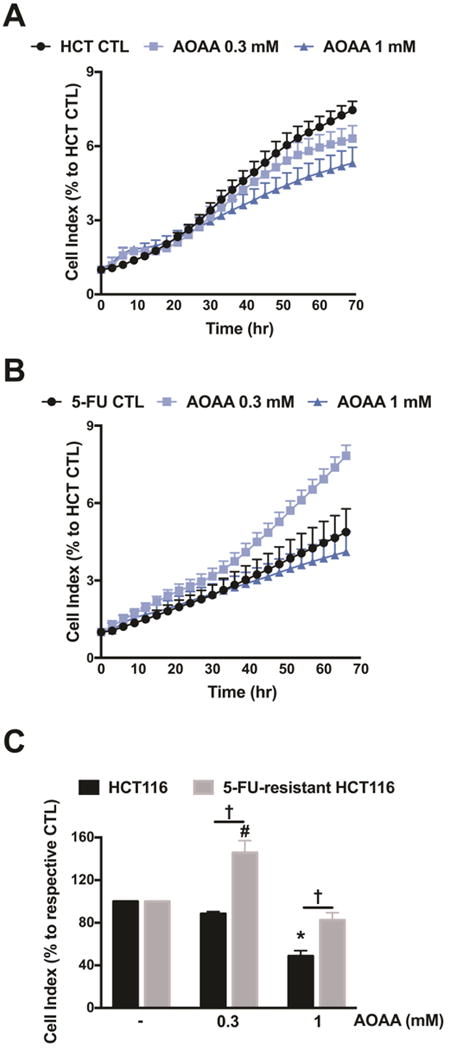
Both cell lines were seeded in xCELLigence plates and proliferation was monitored for 24 h. Thereafter, cells were treated with or without AOAA (0.3–1 mM), and proliferation was monitored for another 48 h. Cell proliferation is shown over time for HCT116 cells (A) and 5-FU-resistant HCT116 cells (B) under various treatments. (C) Statistical analysis of cell proliferation endpoints normalized to cell index and expressed as a percentage of the corresponding control group. Data represent mean ± SEM. n = 3–4 per group. *P < 0.05 vs. HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
When analyzing bioenergetic responses, we noted that 5-FU-resistant cells had a comparable basal respiration rate as parental control cells, but an increased respiratory spare capacity (in response to the uncoupling agent FCCP) (Fig. 8). AOAA markedly suppressed respiratory spare capacity in control cells, but only had a slight inhibitory effect in 5-FU-resistant cells (Fig. 8D). 5-FU-resistant cells exhibited a lower basal glycolytic rate compared to the parental HCT116 cells and AOAA inhibited glycolysis to a comparable degree in both wild-type and resistant cells (Fig. 9). Taken together, the bioenergetic data indicate that the 5-FU resistant cells, compared to the parental controls, tend to rely more on oxidative phosphorylation than on glycolysis. Also, the 5-FU-resistant cells become more resistant to the oxidative-phosphorylation-inhibitory effects of AOAA, than against its glycolysis-inhibitory effects. However, the functional data (cell viability and cell proliferation) suggest that whatever small suppressive effect AOAA exerts on bioenergetic parameters in the 5-FU-resistant cells, it is insufficient to produce any significant suppression of cell viability or cell proliferation.
Figure 8. Comparison of aerobic respiration in control and 5-FU-resistant HCT116 cells; effect of AOAA.
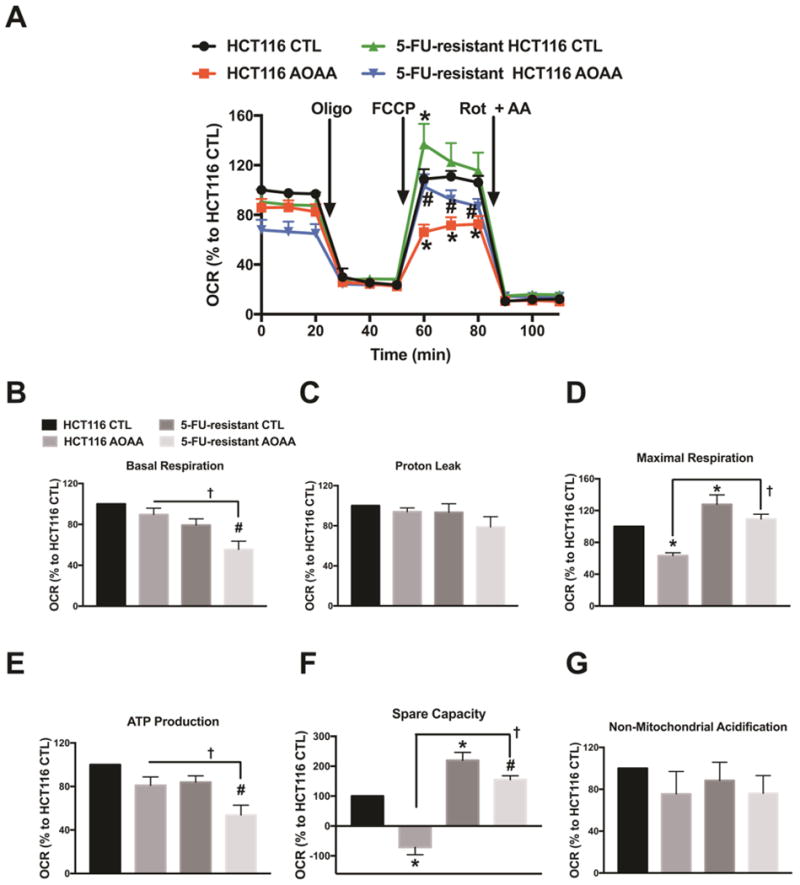
(A) Oxygen consumption rate (OCR) in HCT116 and 5-FU-resistant HCT116 cells (FCCP: carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, a mitochondrial oxidative phosphorylation uncoupler; Rot: rotenone; and AA: antimycin A; inhibitors of complex I and III, respectively). (B–E) Comparison of cellular bioenergetics parameters based on OCR. Data represent mean ± SEM. n = 4–6 per group. *P < 0.05 vs. HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
Figure 9. Comparison of glycolysis in control and 5-FU-resistant HCT116 cells; effect of AOAA.
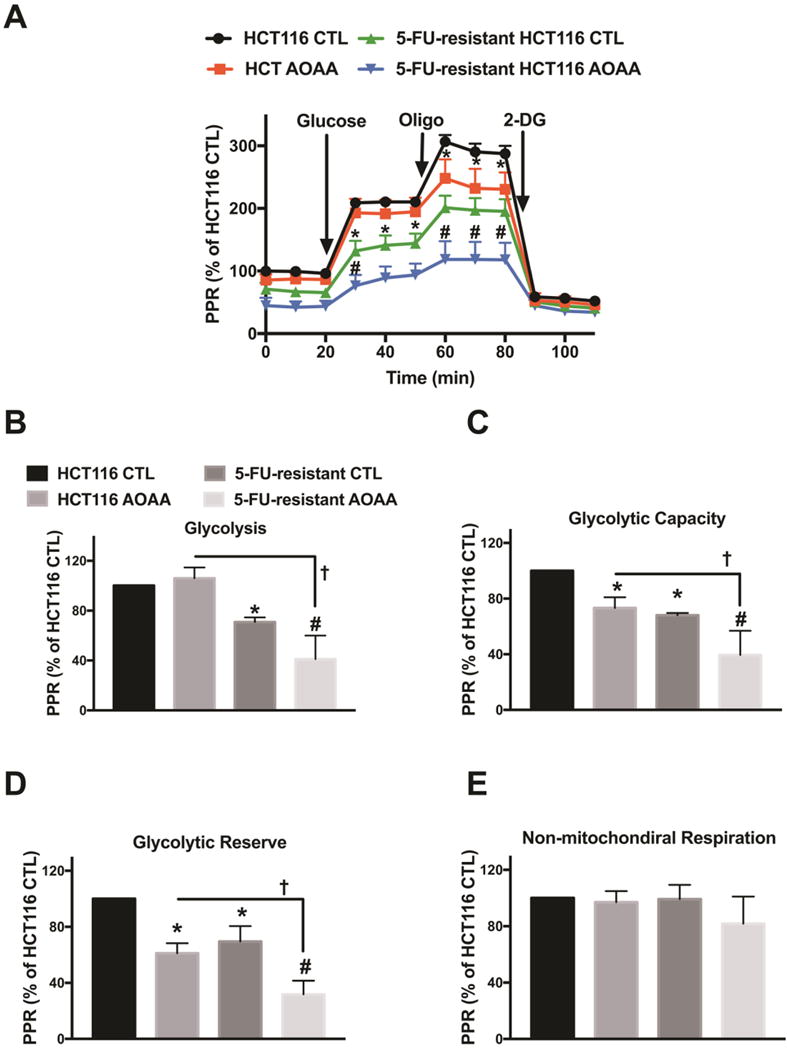
HCT116 and 5-FU-resistant HCT116 cells were treated with or without 0.3 mM AOAA for 1 h. Extracellular Flux Analysis was then performed. (A) Changes in the prolife of proton production rate (PPR) in HCT116 or 5-FU-resistant HCT116 cells (oligo: oligomycin, an inhibitor of ATP synthase; 2-DG: 2-deoxy-D-glucose; an inhibitor of glycolysis). (B–E) Comparison of cellular bioenergetics parameters based on PPR. Data represent mean ± SEM. n = 4–6 per group. *P < 0.05 vs. HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
3.4 The efficacy of benserazide is largely maintained in 5-FU-resistant HCT116 cells
To further assess the effect of CBS inhibition in 5-FU-resistant HCT116 cells, cells were treated with benserazide. We have recently reported that benserazide, a compound that is clinically used as a DOPA decarboxylase inhibitor, inhibits CBS activity in vitro and colon tumor growth in vivo [37]. Parental HCT116 cells treated with benserazide at concentrations of 30 μM and above showed a significant decrease in cell viability and an increase in LDH (Fig. 10). By comparison, although 5-FU-resistant HCT116 cells showed resistance to benserazide at the low concentration of 30 μM, the drug maintained efficacy at higher concentrations; reducing cell viability and increasing cell death (LDH release) (Fig. 10).
Figure 10. 5-FU-resistant HCT116 cells respond to benserazide treatment.
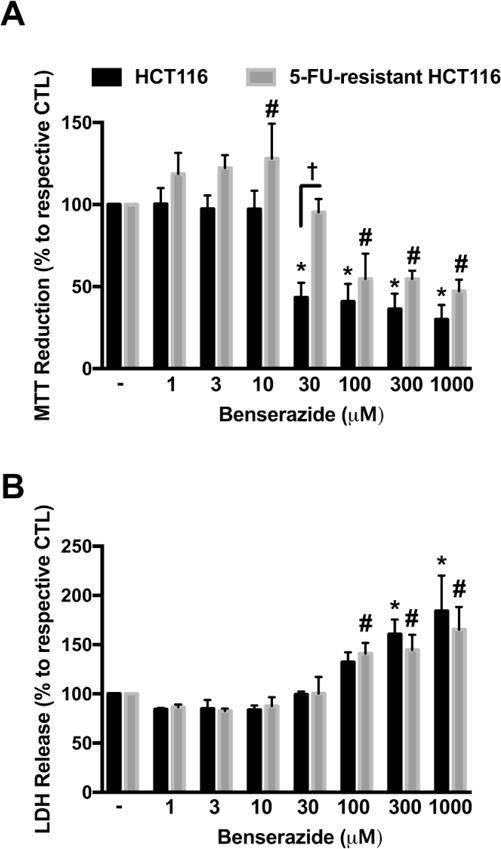
HCT116 and 5-FU-resistant HCT116 cells were treated with benserazide (1–1000 μM) for 48 h. (A) MTT reduction in both HCT116 and 5-FU-resistant HCT116 cells treated with benserazide. (B) Necrotic cell death determined by LDH release in HCT116 and 5-FU-resistant HCT116 cells treated with benserazide. n = 4 per group. *P < 0.05 vs HCT116 CTL; #P < 0.05 vs. 5-FU-resistant HCT116 CTL; †P < 0.05 (based on two-way ANOVA corrected with Tukey’s post-hoc test).
4. Discussion
The major findings of the present study are the following: 1) protein levels and enzymatic activities of the H2S-generating enzymes, CBS and 3-MST are significantly upregulated in 5-FU-resistant HCT116 cells. Rhodanese, an enzyme involved in the oxidation and thus removal of endogenous H2S, was also upregulated in 5-FU-resistant cells; 2) 5-FU-resistant cells express elevated CYP1A2 and CYP2A6 proteins and pharmacological data suggest that increased drug metabolism may contribute to the development of drug resistance; 3) 5-FU-resistant HCT116 cells are resistant (in terms of cell proliferation and MTT/LDH parameters) to the anticancer effect of the prototypical CBS inhibitor AOAA, even though partial effects of AOAA on bioenergetic parameters remain detectable; 4) 5-FU-resistant cells remain largely responsive to the antiproliferative effect of the “non-professional” (repurposable) CBS inhibitor benserazide.
Many different molecular mechanisms have been implicated in the resistance to cytotoxic chemotherapy or targeted therapy, particularly for the acquired resistance [30,33–35,38,39]. Although upregulation of drug transporters has been previously implicated in drug resistance [30], we did not detect an upregulation of ABC proteins. We did, however, detect an upregulation of CYP450 enzymes, which have also been previously implicated in oncological studies [33–35]. CYP450 enzymes are key players in the phase I-dependent metabolism of drugs and other xenobiotics and these enzymes mediate the metabolic activation of numerous precarcinogens and participate in the inactivation and activation of anticancer drugs. The presence of functionally active CYP450 enzymes in tumor cells can inhibit the efficacy of chemotherapy-mediated tumor cell death through the deactivation of various anti-cancer drugs [33]. In the current study, CYP1A2 and 2A6 protein levels were significantly upregulated in 5-FU-resistant cells. Furthermore, we observed a partial restoration of 5-FU cytotoxicity in 5-FU-resistant cells upon co-treatment of phenylpyrrole (a CYP450 inhibitor). Therefore, these results suggest that CYP450-mediated metabolism may contribute to HCT116 cell resistance to 5-FU, and subsequently the CBS inhibitor AOAA.
In addition to the upregulation of CYP450 enzymes, we have also observed a significant upregulation of GOT1 protein. GOT1 is an essential component of the malate/aspartate shuttle [31]. In cancer cells, this mechanism has been shown to link glycolysis to the transfer of electron donors into the mitochondria [40]. Moreover, the function of GOT1 is linked to glutaminolysis, another partially cancer-selective metabolic pathway and anti-cancer target [31]. It is possible that the elevated level of GOT1 seen in 5-FU-resistant cells may have affected some of the cells’ metabolic parameters and may have affected the balance of oxidative phosphorylation and glycolysis. However, the role of GOT1 and its possible relationship with the H2S pathway has not been fully explored in the current study and this remains to be a subject of future investigations.
Importantly, in our studies, we detected a significant upregulation of CBS and 3-MST in 5-FU-resistant HCT116 cells. CBS is selectively upregulated in various human colon cancer lines (HCT116, HT-29, LoVo) as well as in patient-derived colon tumor specimens [7,8]. Although 3-MST was not previously found to be upregulated in colon tumor tissue, we observed a significantly upregulation in the 5-FU-resistant cells. Pharmacological inhibition of CBS (via AOAA treatment) was markedly less effective in suppressing cell viability in 5-FU-resistant HCT116 cells than in parental cells. The mechanism responsible for the loss of AOAA’s efficacy may be due to the fact that: (a) other H2S-producing enzymes, such as the 3-MST system are also contributing to H2S production in these cells and therefore inhibition of CBS results in a lesser suppression of cellular H2S levels; (b) AOAA may be metabolized and inactivated by the upregulated cytochrome P450 enzymes and/or; (c) in 5-FU-resistant cells other pathways are mobilized to maintain cell viability and cell proliferation and these pathways are not affected by AOAA. The fact that AOAA remained partially effective in suppressing various bioenergetic parameters in 5-FU-resistant cells, and yet it failed to affect functional parameters (viability, proliferation) indirectly supports the last hypothesis.
Consistent with our findings, an earlier study that exposed CT26 mouse colon carcinoma cancer cells to cytotoxic concentrations of hydrogen peroxide to yield a potentially lethal damage-recovered cells population, showed that the recovered cells increased expression of CBS, CSE and 3-MST [41]. Furthermore, the cells with upregulated H2S-producing enzyme expression underwent a significant degree of dedifferentiation, which was, at least in part, due to elevation of intracellular NAD+ levels in the damage-recovered cells [41]. However, in the CT26 study, the effect of H2S biosynthesis inhibitors on the proliferation or viability of parental or damage-recovered cells was not evaluated.
Endogenous H2S is cleared at least in part, through the mitochondrial sulfide oxidation pathway. The major oxidation products of H2S are thiosulfate and sulfate whose ratio and production rate vary in a tissue-specific manner. The sulfide oxidation pathway begins with sulfide quinone oxidoreductase (SQR) and includes a sulfur dioxygenase, sulfite oxidase and rhodanese. Rhodanese has a 60% homology to 3-MST [42]. Rhodanese catalyzes the formation of thiosulfate primarily via transferring sulfhydryl groups to cyanide, forming thiocyanate and sulfate [42]. However, rhodanese also has the ability to produce H2S from thiosulfate [43,44]. Furthermore, Kimura et al. showed that rhodanese utilizes H2S to generate polysulfides (i.e. H2S3) [45]. There are some reports indicating that rhodanese expression is altered in cancer cells compared to non-transformed cells [46–48]; however, changes in rhodanese expression during drug resistance, prior the current study, have not been investigated. The current results indicate a substantial upregulation of rhodanese in 5-FU-resistant cells; however, the functional importance of this change remains to be explored. It appears that the increased H2S degradation that would be expected from increased rhodanese is not able to counterbalance the increased H2S biosynthesis, as evidenced by the direct measurement of H2S production in tissue homogenates (Fig. 3C). One would expect that if upregulation of rhodanese (or other H2S-degrading pathways) were a major factor contributing to the phenotype of the 5-FU-resistant cells, then pharmacological inhibition of H2S production (e.g. with AOAA) would maintain most of its efficacy in 5-FU-resistant cell line; however, this is not the case, according to our data. Future experiments remain to be conducted to investigate the functional role of rhodanese upregulation in drug resistance.
One of the observations of the current study is that 5-FU-resistant HCT116 cells proliferated at a slower basal rate than parental HCT116 cells. It is possible that the excessively elevated endogenous H2S level in 5-FU-resistant cells produced an anti-mitogenic effect in resistant cells. Indeed, others have demonstrated an antiproliferative effect of H2S (at millimolar concentrations) in various cell lines via the induction of the cyclin-dependent kinase inhibitor p21Cip1, a protein associated with cell cycle arrest [49,50]. Wu and colleagues [49] observed that 1 mM NaHS (an H2S concentration similar to those found in the human colon) induced a substantial accumulation of HT-29 cells (a colon epithelial cell line) at the G0/G1-phase and an upregulation of the cyclin-dependent kinase inhibitor p21Cip1 in a time-dependent manner. The AMPK/mTOR cascade plays a central role in mediating the antiproliferative effect of H2S. Wu et al. [49] also demonstrated that H2S increased the phosphorylation of AMPK at Thr-172, whereby the pharmacological inhibition of AMPK by compound C prevented the anti-mitogenic effect of H2S in both HT-29 and HCT116 cells. Additionally, another group showed that H2S inhibited cell proliferation and induced cell cycle arrest via the induction of p21Cip1 in Ca9-22 cells (a human gingival epithelial cell line) [50]. Further work is needed to delineate the pathways and mechanisms responsible for the slower proliferation rate observed in the 5-FU-resistant HCT116 cells.
We can only speculate on the nature of the genes and pathways that are altered in the current 5-FU-resistant cell population and further work remains to be conducted to evaluate the possible connections between these alterations and the upregulation of various H2S pathways. For example, it has been previously suggested that 5-FU leads to the activation of ataxia-telangiectasia mutated serine/threonine protein kinase (ATM), AMP kinase (AMPK) and PGC-1α, which could promote mitochondrial biogenesis and thus stimulate OXPHOS [51–53]. In fact, H2S was shown to enhance the expression and activity of PGC-1α leading to increased mitochondrial biogenesis in primary hepatocytes [54]. The potential connection between the H2S pathway and the PGC-1α pathway in the current experimental paradigm remains to be further investigated.
We previously showed that pharmacological inhibition of CBS with AOAA significantly reduced the growth rate of patient-derived colon cancer xenografts and reduced tumor blood flow to the tumor xenografts in nude mice [7]. We also showed that AOAA significantly decreased tumor growth and size in nude mice bearing subcutaneous HCT116 cancer cell xenografts [9]. Based on the current results, we anticipate that the efficacy of AOAA would diminish in mice bearing relapsing/multi-drug resistant colon cancer cell xenografts. However, the current data showing that the anticancer efficacy of another CBS inhibitor, benserazide may be encouraging in this respect. Benserazide has been identified by a screening campaign that was seeking to identify clinically used, and potentially repurposable drugs for CBS inhibition [37]. The mode of benserazide’s inhibition of CBS is likely related to its metabolic conversion to an active metabolite, which interferes with the PLP prosthetic group in the active center of CBS [37]. Since benserazide likely affects several enzymes (its “original” mode of action is inhibition of DOPA decarboxylase, another PLP-dependent enzyme) it is possible that some of its antitumor effects rely on the combination of these additional targets. Benserazide is not known to be metabolized by those cytochrome enzymes that we found upregulated in the 5-FU-resistant HCT116 cells. Our working hypothesis, therefore, is that the 5-FU-resistant colon cancer cells metabolize benserazide to a lesser degree than other compounds (e.g. 5-FU or AOAA), and this metabolic stability may account for its maintained antiproliferative efficacy in 5-FU-resistant cells. Further studies are needed to test whether the efficacy of benserazide in vivo is maintained in animals bearing drug-resistant colon cancer cell lines.
The prognosis of advanced colorectal cancer remains poor [55]. Chemotherapy with 5-FU has been the first line treatment option for advanced colorectal cancer, but many patients develop cancer relapse after initial treatment. Therefore, investigating cellular molecular mechanisms leading to anti-cancer drug resistance is of paramount importance. By further understanding the molecular mechanisms of drug resistance - including the mechanism and functional consequences of enhanced H2S generation in 5-FU-resistant colon cancer - may be useful to formulate rational strategies to counteract multidrug resistance.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R21TR001734 and R01GMCA175803).
Abbreviations
- 2-DG
2-deoxyglucose
- 3-MST
3-mercaptopyruvate sulfurtransferase
- 5-FU
5-Fluorouracil
- AA
antimycin A
- ABC
ATP-binding cassette
- AMPK
AMP kinase
- AOAA
aminooxyacetic acid
- ATM
ataxia-telangiectasia mutated serine/threonine protein kinase
- ATP
adenosine triphosphate
- AzMc
7-azido-4-methylcoumarin
- CBS
cystathionine-β-synthase
- CSE
cystathionine-γ-lyase
- CTL
control
- CYP450
cytochromes P450
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- GOT1
glutamate oxaloacetate transaminase 1
- H2S
hydrogen sulfide
- INT
2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride
- MRP1
multidrug resistance-associated protein 1
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- NAD+
nicotinamide adenine dinucleotide
- OCR
oxygen consumption rate
- oligo
oligomycin
- PGC-1α
peroxisome proliferator-activated receptor-γ- coactivator-1α
- PMS
N-methylphenazonium methyl sulfate
- PPR
proton production rate
- Rot
rotenone
- SQR
sulfide quinone oxidoreductase
- TCA
tricarboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author disclosure statement
C.S. and M.R.H. are founders, officers and shareholders of CBS Therapeutics Inc., an UTMB spin-off company focusing on therapeutic approaches around H2S biosynthesis inhibition in cancer cells. A.P. is a shareholder and officer of the same company.
For the other authors, no competing financial interests exist.
Author contributions
AAU – carried out molecular biology and pharmacological assays, contributed to the writing of the paper. TP – contributed to the production of the 5-FU-resistant HCT116 cells. ND – performed enzymatic activity analyses. AP – contributed to the design of the project and to data interpretation. MRH and CS – conceived and supervised the project and contributed to the writing of the paper.
References
- 1.Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal carcinoma: a general overview and future perspectives in colorectal cancer. Int J Mol Sci. 2017;18:E197. doi: 10.3390/ijms18010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karpinski P, Sasiadek MM, Blin N. Aberrant epigenetic patterns in the etiology of gastrointestinal cancers. J Appl Gen. 2008;49:1–10. doi: 10.1007/BF03195243. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 4.Papanastasopoulos P, Stebbing J. Molecular basis of 5-fluorouracil-related toxicity: lessons from clinical practice. Anticancer Res. 2014;34:1531–1535. [PubMed] [Google Scholar]
- 5.Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res. 2001;7:2168–2181. [PubMed] [Google Scholar]
- 6.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 7.Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Módis K, Coletta C, Asimakopoulou A, Szczesny B, Chao C, Papapetropoulos A, et al. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide. 2014;41:146–156. doi: 10.1016/j.niox.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao C, Zatarain JR, Ding Y, Coletta C, Mrazek AA, Druzhyna N, et al. Cystathionine-β-synthase inhibition for colon cancer: Enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol Med. 2016;22:361–379. doi: 10.2119/molmed.2016.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PloS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S, et al. Role of cystathionine β-synthase in human breast Cancer. Free Rad Biol Med. 2015;86:228–238. doi: 10.1016/j.freeradbiomed.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 12.Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P, et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci Rep. 2016;6:36125. doi: 10.1038/srep36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov. 2016;15:185–203. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 15.Abou-Hamdan A, Guedouari-Bounihi H, Lenoir V, Andriamihaja M, Blachier F, Bouillaud F. Oxidation of H2S in mammalian cells and mitochondria. Methods Enzymol. 2015;554:201–228. doi: 10.1016/bs.mie.2014.11.042. [DOI] [PubMed] [Google Scholar]
- 16.Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 17.Modis K, Panopoulos P, Coletta C, Papapetropoulos A, Szabo C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem Pharmacol. 2013;86:1311–1319. doi: 10.1016/j.bcp.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci USA. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad A, Olah G, Szczesny B, Wood ME, Whiteman M, Szabo C. AP39, a mitochondrially targeted hydrogen sulfide donor, exerts protective effects in renal epithelial cells subjected to oxidative stress in vitro and in acute renal injury in vivo. Shock. 2016;45:88–97. doi: 10.1097/SHK.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczesny B, Modis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A, et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–130. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Modis K, Ju Y, Ahmad A, Untereiner AA, Altaany Z, Wu L, et al. S-sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol Res. 2016;113:116–124. doi: 10.1016/j.phrs.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Untereiner AA, Oláh G, Módis K, Hellmich MR, Szabo C. H2S-induced S- sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem Pharmacol. 2017;136:86–98. doi: 10.1016/j.bcp.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabo C, Ransy C, Modis K, Andriamihaja M, Murghes B, Coletta C, et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171:2099–2122. doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modis K, Bos EM, Calzia E, van Goor H, Coletta C, Papapetropoulos A, et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br J Pharmacol. 2014;171:2123–2146. doi: 10.1111/bph.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009;146:623–626. doi: 10.1093/jb/mvp111. [DOI] [PubMed] [Google Scholar]
- 26.Modis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013;433:401–407. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- 27.Bronowicka-Adamska P, Bentke A, Wróbel M. Hydrogen sulfide generation from l-cysteine in the human glioblastoma-astrocytoma U-87 MG and neuroblastoma SHSY5Y cell lines. Acta Biochim Pol. 2017;64:171–176. doi: 10.18388/abp.2016_1394. [DOI] [PubMed] [Google Scholar]
- 28.Gai JW, Qin W, Liu M, Wang HF, Zhang M, Li M, et al. Expression profile of hydrogen sulfide and its synthases correlates with tumor stage and grade in urothelial cell carcinoma of bladder. Urol Oncol. 2016;34:166.e15–20. doi: 10.1016/j.urolonc.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 29.Módis K, Gerő D, Erdélyi K, Szoleczky P, DeWitt D, Szabo C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol. 2012;83:633–643. doi: 10.1016/j.bcp.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szakács G, Váradi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) Drug Discov Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: An overview. Cancers. 2014;6:1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda K, Yoshisue K, Matsushima E, Nagayama S, Kobayashi K, Tyson CA, et al. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome P-450 2A6 in human liver microsomes in vitro. Clin Cancer Res. 2000;6:4409–4415. [PubMed] [Google Scholar]
- 35.Shen H, He MM, Liu H, Wrighton SA, Wang L, Guo B, et al. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug metab dispos. 2007;35:1292–1300. doi: 10.1124/dmd.107.015354. [DOI] [PubMed] [Google Scholar]
- 36.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Druzhyna N, Szczesny B, Olah G, Modis K, Asimakopoulou A, Pavlidou A, et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine beta-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol Res. 2016;113:18–37. doi: 10.1016/j.phrs.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373–388. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denise C, Paoli P, Calvani M, Taddei ML, Giannoni E, Kopetz S, et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. 2015;6:41706–41721. doi: 10.18632/oncotarget.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal. 2015;22:424–448. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, Tabibzadeh S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S-Nampt. Exp Cell Res. 2015;330:135–150. doi: 10.1016/j.yexcr.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 42.Picton R, Eggo MC, Merrill GA, Langman MJ, Singh S. Mucosal protection against sulphide: importance of the enzyme rhodanese. Gut. 2002;50:201–205. doi: 10.1136/gut.50.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikami Y, Shibuya N, Kimura Y, Nagahara N, Ogasawara Y, Kimura H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem J. 2011;439:479–485. doi: 10.1042/BJ20110841. [DOI] [PubMed] [Google Scholar]
- 44.Ubuka T, Ohta J, Yao WB, Abe T, Teraoka T, Kurozumi Y. L-Cysteine metabolism via 3-mercaptopyruvate pathway and sulfate formation in rat liver mitochondria. Amino Acids. 1992;2:143–155. doi: 10.1007/BF00806085. [DOI] [PubMed] [Google Scholar]
- 45.Kimura Y, Toyofuku Y, Koike S, Shibuya N, Nagahara N, Lefer D, et al. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci Rep. 2015;5:14774. doi: 10.1038/srep14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenthal O. Rhodanese activity of resting, regenerating, and neoplastic liver tissue of the rat. J Natl Cancer Inst. 1955;15:S1611–1614. [PubMed] [Google Scholar]
- 47.Navone NM, Vázquez ES, Polo CF, Batlle AM. Rhodanese and ALA-S in mammary tumor and liver from normal and tumor-bearing mice. Comp Biochem Physiol B. 1992;102:83–85. doi: 10.1016/0305-0491(92)90276-w. [DOI] [PubMed] [Google Scholar]
- 48.Ramasamy S, Singh S, Taniere P, Langman MJ, Eggo MC. Sulfide-detoxifying enzymes in the human colon are decreased in cancer and upregulated in differentiation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G288–296. doi: 10.1152/ajpgi.00324.2005. [DOI] [PubMed] [Google Scholar]
- 49.Wu YC, Wang XJ, Yu L, Chan FK, Cheng AS, Yu J, et al. Hydrogen sulfide lowers proliferation and induces protective autophagy in colon epithelial cells. PLoS One. 2012;7:e37572. doi: 10.1371/journal.pone.0037572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takeuchi H, Setoguchi T, Machigashira M, Kanbara K, Izumi Y. Hydrogen sulfide inhibits cell proliferation and induces cell cycle arrest via an elevated p21 Cip1 level in Ca9-22 cells. J Periodontal Res. 2008;43:90–95. doi: 10.1111/j.1600-0765.2007.00999.x. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011;2:393–397. doi: 10.3892/etm.2011.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo L, Huang W, Tao R, Hu N, Xiao Z-X, Luo Z. ATM and LKB1 dependent activation of AMPK sensitizes cancer cells to etoposide-induced apoptosis. Cancer Letts. 2013;328:114–119. doi: 10.1016/j.canlet.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279:53272–53281. doi: 10.1074/jbc.M406879200. [DOI] [PubMed] [Google Scholar]
- 54.Untereiner AA, Fu M, Modis K, Wang R, Ju Y, Wu L. Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide. 2016;58:67–76. doi: 10.1016/j.niox.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Siegel RL, Miller KD, Jemal A. Cancer Statistics. Cancer. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]