

Abstract
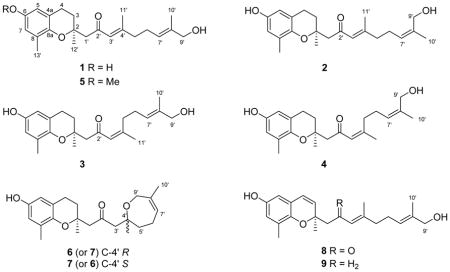
Nine new compounds containing either a chromane or chromene ring moiety were isolated from the monotypic plant Koeberlinia spinosa. Compounds 1 – 4 are chromanes with all possible E and Z isomers of the isoprenoid side chain, with compound 5 a methylated derivative of 1. Compounds 6 and 7 were assigned as diastereomeric cyclized derivatives of 2, and were probably artifacts formed during the extraction or the isolation processes. Compounds 8 and 9 were characterized as new chromenes. Structure elucidation of 1 – 9 was conducted via 1D and 2D NMR spectroscopic data interpretation, and absolute configurations were determined by ECD spectroscopic analysis. Compounds 2, 5, 6, and 7 had weak antiplasmodial activity, while none of the compounds exhibited antiproliferative activity. The isolation, structure elucidation, and biological evaluation of these compounds are presented.
Graphical Abstract
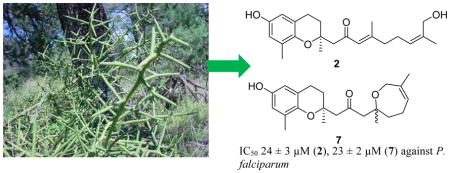
Koeberlinia spinosa Zucc., also known as “crucifixion thorn”, is a monotypic species in the family Koeberliniaceae. The plant is a thorny, leafless, and xerophytic shrub found in North America and located primarily in the southwestern United States and northern Mexico.1 In 2009 Lucero et al. identified 18 known volatile organic compounds from Koeberlinia spinosa via GC-MS analysis of an essential oil extracted from various plant parts by steam distillation, with the purpose of identifying known compounds to aid in biochemical ecology.2 The extraction and analysis methods used by these investigators favored the identification of small volatile molecules, since larger molecules are not extracted or are not detected by GC-MS. In addition, the elevated temperatures of the distillation could have caused thermal decomposition of some larger and more complex compounds, and no phytochemical study of non-volatile compounds was conducted.
Over the past few years our group has been investigating plant extracts that display biological activity against the malaria parasite Plasmodium falciparum in the expectation of finding new antimalarial compounds, since the antimalarial drugs quinine and artemisinin are both plant-derived natural products. We are interested in this vector-borne disease due to its disproportionate effect in poor and underdeveloped countries without access to western medicine, and due to the appearance of artemisinin-resistant parasites in five countries and the high failure rates with four different artemisinin-based combination therapies in Cambodia.3 This growing resistance to artemisinin means that there is an urgent need for new and affordable antimalarial medicines. Therefore, when an extract of K. spinosa from the Natural Products Discovery Institute (NPDI) was found to have moderate activity (IC50 value 5 μg/mL) against the Dd2 drug-resistant strain of Plasmodium falciparum, it was selected for bioassay-guided isolation. This is the first phytochemical investigation on a whole plant extract from this genus and species.
RESULTS AND DISCUSSION
Isolation and Structure Elucidation
A 901 g sample of the whole plant of Koeberlinia spinosa was collected in 1997 and extracted with ethanol to yield 82 g of extract, of which 5.2 g were provided for this study. Liquid-liquid partition of 3.7 g of this extract between aqueous ethanol and dichloromethane afforded 2.3 g of a moderately active dichloromethane fraction with an IC50 value of 5 μg/mL against P. falciparum. This fraction was loaded onto a polyamide column and eluted with methanol to produce 1.9 g of a detanninized fraction with an IC50 value between 2.5 and 5 μg/mL. Further bioassay-guided fractionation utilizing reversed-phase (RP) solid-phase extraction (SPE), RP-HPLC, diol HPLC, and open silica gel columns afforded the seven new chromanes 1 – 7 and the two new chromenes 8 and 9.
Compound 1 was isolated as a clear oil. Its chemical formula was determined to be C22H30O4 by HRESIMS ([M+Na]+ m/z 381.2017, calcd for C22H30NaO4+ m/z 381.2036) and 13C NMR spectroscopy. Summation of all integrals from the 1H NMR spectrum in CD3OD indicated the presence of 28 protons, consistent with the presence of two hydroxy groups (Table 1 and Table S.1, Supporting Information).
Table 1.
NMR Spectra of Compounds 1 – 5 in CD3OD (J in Hz).a
| position | 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH | δC, type | δH | δC, type | δH | δC, type | δH | δC, type | δH | δC, type | |
| 2 | 76.2, C | 76.2, C | 76.2, C | 76.2, C | 76.4, C | |||||
| 3 | 1.90, dt (13.6, 6.9) 1.79, dt (13.6, 6.9) |
33.1, CH2 | 1.90, dt (13.6, 6.8) 1.79, dt (13.6, 6.8) |
33.1, CH2 | 1.90c, m 1.79, dt (13.6, 6.9) |
33.2, CH2 | 1.91, dt (13.8, 6.9) 1.79, dt (13.8, 6.9) |
33.2, CH2 | 1.92, dt (13.6, 6.7) 1.81, dt (13.6, 6.7) |
33.0, CH2 |
| 4 | 2.71, t (6.9) | 23.3, CH2 | 2.71, t (6.8) | 23.3, CH2 | 2.7, t (6.9) | 23.3, CH2 | 2.71, t (6.9) | 23.3, CH2 | 2.76, t (6.7) | 23.4, CH2 |
| 4a | 122.0, C | 122.0, C | 122.0, C | 122.0, C | 121.9, C | |||||
| 5 | 6.35, d (2.9) | 113.7, CH | 6.35, d (2.9) | 113.7, CH | 6.34, d (2.9) | 113.7, CH | 6.34, d (2.9) | 113.7, CH | 6.47, d (2.9) | 112.1, CH |
| 6 | 150.8, C | 150.8, C | 150.8, C | 150.8, C | 154.1, C | |||||
| 7 | 6.43, d (2.9) | 116.8, CH | 6.43, d (2.9) | 116.8, CH | 6.43, d (2.9) | 116.8, CH | 6.43, d (2.9) | 116.8, CH | 6.55, d (2.9) | 116.1, CH |
| 8 | 127.8, C | 127.9, C | 127.9, C | 127.89, C | 128.0, C | |||||
| 8a | 145.7, C | 145.8, C | 145.7, C | 145.7, C | 146.6, C | |||||
| 1′ | 2.82, d (13.1) | 53.7, CH2 | 2.81, d (13.1) | 53.7, CH2 | 2.81, d (13.0) | 53.8, CH2 | 2.81, d (13.1) | 53.8, CH2 | 2.82, d (13.3) | 53.7, CH2 |
| 2.55, d (13.1) | 2.56, d (13.1) | 2.52, d (13.0) | 2.52, d (13.1) | 2.57, d (13.3) | ||||||
| 2′ | 201.7, C | 201.7, C | 201.0, C | 201.1, C | 201.5, C | |||||
| 3′ | 6.19, brs | 126.2, CH | 6.18, brs | 126.3, CH | 6.19, brs | 126.7, CH | 6.19, brs | 126.8, CH | 6.18, brs | 126.2, CH |
| 4′ | 159.5, C | 159.4, C | 160.4, C | 160.3, C | 159.6, C | |||||
| 5′ | 2.19b, m | 41.9, CH2 | 2.18, m | 42.4, CH2 | 2.62, m | 34.6, CH2 | 2.59, m | 35.1, CH2 | 2.19d, m | 41.9, CH2 |
| 6′ | 2.20b, m | 26.7, CH2 | 2.24, m | 26.6, CH2 | 2.21, q (7.4) | 27.4, CH2 | 2.22, q (7.8) | 27.3, CH2 | 2.20d, m | 26.7, CH2 |
| 7′ | 5.36, brt (6.7) | 125.1, CH | 5.22, brt (7.2) | 127.2, CH | 5.44, brt (7.4) | 125.9, CH | 5.31, t (7.5) | 127.86, CH | 5.35, m | 125.1, CH |
| 8′ | 137.0, C | 136.9, C | 136.7, C | 136.6, C | 137.0, C | |||||
| 9′ | 3.89, s | 68.7, CH2 | 4.04, s | 61.3, CH2 | 3.9, s | 68.9, CH2 | 4.09, s | 61.3, CH2 | 3.89, s | 68.7, CH2 |
| 10′ | 1.63, d (1.4) | 13.7, CH3 | 1.74, d (1.3) | 21.5, CH3 | 1.66, d (1.4) | 13.7, CH3 | 1.75, d (1.4) | 21.5, CH3 | 1.63, d (1.3) | 13.7, CH3 |
| 11′ | 2.12, d (1.2) | 19.6, CH3 | 2.11, d (1.2) | 19.6, CH3 | 1.88, d (1.4) | 25.9, CH3 | 1.87, d (1.4) | 25.8, CH3 | 2.12, d (1.4) | 19.6, CH3 |
| 12′ | 1.34, s | 25.3, CH3 | 1.33, s | 25.3, CH3 | 1.33, s | 25.2, CH3 | 1.33, s | 25.1, CH3 | 1.35, s | 25.3, CH3 |
| 13′ | 2.06, s | 16.5, CH3 | 2.06, s | 16.5, CH3 | 2.06, s | 16.4, CH3 | 2.06, s | 16.4, CH3 | 2.09, s | 16.6, CH3 |
| OCH3 | 3.70, s | 55.9, CH3 | ||||||||
1H NMR data obtained at 500 MHz; 13C NMR data acquired in CD3OD at 125 MHz.
Overlapping signals. Chemical shifts determined from the HSQC spectrum.
Obscured signal.
Further examination of the spectrum revealed two meta-coupled aromatic protons at δH 6.43 (1H, d, J = 2.9 Hz, H-7) and δH 6.35 (1H, d, J = 2.9 Hz, H-5), as well as an aromatic methyl group at δH 2.06 (3H, s, H-13′), indicating the presence of a highly substituted aromatic ring. A deshielded one-proton signal at δH 6.19 (1H, br s, H-3′) was consistent with the presence of a vinyl proton α to a carbonyl group, and a methyl group with long-range allylic coupling at δH 2.12 (3H, d, J = 1.2 Hz, H-11′) suggested the presence of a methyl group at the β position of an α,β-unsaturated carbonyl group. A broad vinyl triplet was observed at δH 5.36 (1H, br t, J = 6.7 Hz, H-7′) and the presence of another methyl with long-range allylic coupling at δH 1.63 (3H, d, J = 1.4 Hz, H-10′) indicated the presence of an isoprene unit. A singlet for deshielded methylene protons at δH 3.89 (2H, s, H-9′) indicated that one of the hydroxy groups is attached to an isolated methylene unit. Two isolated and geminally coupled doublets at δH 2.82 (1H, d, J = 13.1 Hz, H-1′a) and δH 2.55 (1H, d, J = 13.1 Hz, H-1′b) indicated the presence of diastereotopic protons of a methylene unit next to a stereocenter and α to a carbonyl group. A benzylic methylene triplet was observed at δH 2.71 (2H, t, J = 6.9 Hz, H-4) and was coupled to two geminally coupled diastereotopic protons at δH 1.90 (1H, dt, J = 13.6, 6.9 Hz, H-3a) and δH 1.79 (1H, dt, J = 13.6, 6.9 Hz, H-3b). The remaining proton signals belonged to two overlapping methylene units at δH 2.20 (4H, m, H-5′ and H-6′) and a methyl group at δH 1.34 (3H, s, H-12′).
The 13C NMR spectrum of 1 (Table 1) indicated the presence of 22 carbons, corresponding to the calculated molecular formula. The 13C NMR and HSQC spectra showed signals for an α,β-unsaturated ketone, aromatic carbons, two double bonds, five methylenes, one oxygen-bearing methylene, one oxygen-bearing quaternary carbon, and four methyl carbons.
The HMBC spectrum of 1 (Figure 1) exhibited correlations from the aromatic methyl at δH 2.06 (CH3-13′) to carbons at δC 127.8 (C-8), δC 145.7 (C-8a), and δC 116.8 (CH-7) and from the aromatic proton at δH 6.43 (1H, d, J = 2.9 Hz, H-7) to carbons at δC 127.8 (C-8), δC 150.8 (C-6), and δC 113.7 (CH-5). Correlations from the aromatic proton at δH 6.35 (1H, d, J = 2.9 Hz, H-5) to carbons at δC 150.8 (C-6), δC 145.7 (C-8a), δC 122.0 (C-4a), and δC 23.3 (CH2-4) and from the benzylic-methylene protons at δH 2.71 (2H, t, J = 6.9 Hz, H-4) to carbons at δC 33.1 (CH2-3) and δC 76.2 (C-2) were also observed, and confirm the presence of a 6,8-disubstituted chromane ring system. HMBC correlations from the methyl at δH 1.34 (CH3-12′) to δC 76.2 (C-2), δC 33.1 (CH2-3), and δC 53.7 (CH2-1′) confirmed the attachment of this methyl group and the terpenoid side chain at C-2 on the chromane ring. These observations are consistent with the spectroscopic data of other reported chromanes.4,5
Figure 1.
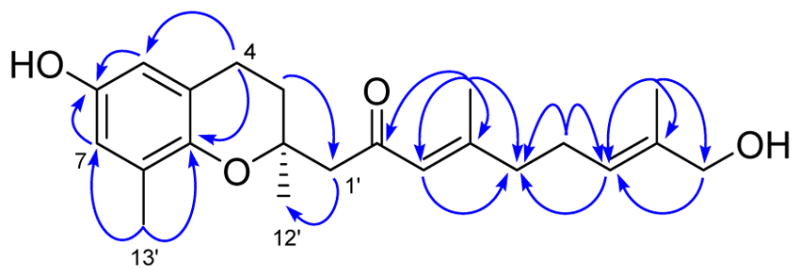
Selected HMBC correlations of 1.
The position of attachment of the terpenoid side chain was also confirmed by HMBC correlations from the diastereotopic methylene protons at δH 2.82 (H-1′a) and δH 2.55 (H-1′b) to the quaternary oxygen-bearing carbon at δC 76.2 (C-2), as well as to the carbonyl carbon at δC 201.7 (C-2′), the methyl carbon at δC 25.3 (C-12′), and the α-carbon at δC 126.2 (CH-3′). HMBC correlations from the methyl at δH 2.12 (H-11′) to the quaternary carbon at δC 159.5 (C-4′), the α-carbon at δC 126.2 (CH-3′), and the methylene unit at δC 41.9 (CH2-5′) confirmed the locations of the β-carbon and the end of this isoprene unit. HMBC correlations from the vinyl proton at δH 5.36 (H-7′) to the methylene carbons at δC 26.7 (CH2-6′) and δC 41.9 (CH2-5′) supported the attachment of the final isoprene unit. HMBC correlations from the oxygen-bearing methylene proton at δH 3.89 (H-9′) with carbons at δC 137.0 (C-8′), δC 125.1 (CH-7′), and δC 13.7 (CH3-10′) confirmed that the isoprenoid chain has a terminal hydroxy group.
Selective NOE experiments were conducted on the protons of the C-3′/4′ and C-7′/8′ double bonds on the terpenoid side chain to determine their configurations (Figure 2). Saturation of the α-proton at δH 6.19 (H-3′) enhanced the methylene signal at δH 2.19 (H-5′), and saturation of the vinyl proton at δH 5.36 (H-7′) enhanced the methylene unit at δH 3.89 (H-9′). These observations confirm that the double bonds at C-3′/4′ and C-7′/8′ both have the E configuration.
Figure 2.
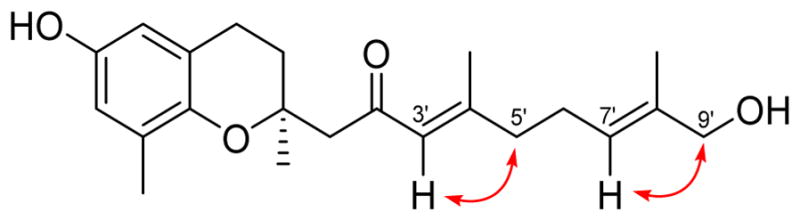
NOE correlations for 1.
The absolute configuration at the C-2 asymmetric center in 1 was determined to be S on the basis of an observed negative Cotton effect at 275 – 315 nm ([Δε]296 nm −3.96) in the ECD spectrum, in agreement with ECD data on tocopherols and tocotrienols (Figure 3).5,6 Thus, 1 was determined to be the new compound (3E,7E)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Figure 3.

(A) ECD spectra of 1 – 7 in MeOH. (B) UV of 1 – 7 in MeOH. (C) ECD spectra of 8 and 9 in CHCl3 at 1.0 mM. (D) UV spectra of 8 and 9 in CHCl3. All compounds except 1 were measured at 1.0 mM while 1 was at 0.5 mM.
Compound 2 was isolated as a colorless oil, and its molecular formula was determined to be C22H30O4 by HRESIMS and 13C NMR spectroscopy. A side-by-side comparison of the NMR spectroscopic data with those of 1 showed them to be nearly identical except for the final isoprene unit on the side chain (Table 1). Examination of the HMBC spectrum for the vinyl proton at δH 5.22 (1H, br t, J = 7.2 Hz, H-7′), the methyl group at δH 1.74 (3H, d, J = 1.4 Hz, H-10′), and the methylene unit at δH 4.04 (2H, s, H-9′) all confirmed the same connectivity as in 1. The largest chemical shift difference was observed in the 13C NMR spectrum for the methyl at δC 21.5 (CH3-10′) compared to the C-10′ methyl in 1 at δC 13.7.
Selective NOE experiments (Figures S2.6 and S2.7, Supporting Information) confirmed the configuration of the double bonds at C-3′/4′ and C-7′/8′ as E and Z, respectively. Thus, saturation of the α-proton at δH 6.18 (1H, d, J = 1.2 Hz, H-3′) caused enhancement of the methylene protons at δH 2.18 (2H, m, H-5′) confirming that the double bond at C-3′/4′ is E, while saturation of the vinyl proton at δH 5.22 (H-7′) caused enhancement of the methyl protons at δH 1.74 (H-10′) confirming that the double bond at C-7′/8′ is in the Z configuration.
The absolute configuration of the chiral center at C-2 was assigned as S from examination of the ECD spectroscopic data (Figure 3). Thus, 2 was determined to be the new compound (3E,7Z)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Compound 3 was assigned the composition, C22H30O4, by HRESIMS and 13C NMR spectroscopy. Comparison of the NMR spectroscopic data of 3 with those of 1 indicated a few significant differences in the 1H NMR data (Table 1), with the C-11′ methyl group resonating at δH 1.88 (1H, d, J = 1.4 Hz) instead of δH 2.1 in 1 and 2. The H-5′ methylene protons resonated at δH 2.62 (2H, m), as compared with about δH 2.2 in 1 and 2. Differences were also observed in their 13C NMR data (Table 2), with chemical shifts at δC 25.2 (CH3-11′) and δC 34.6 (CH2-5′) as compared with δC 19.6 and 41.9 for 1. These observations indicated that the compounds differ in the configuration of the double bond at C-3′/4′. A selective NOE experiment with saturation of H-3′ at δH 6.19 (Figure S3.6, Supporting Information) displayed enhancement of the methyl group at δH 1.88 (H-11′), confirming the C-3′/4′ double bond configuration to be Z. Thus, saturation of the vinyl proton at δH 5.44 (Figure S3.7, Supporting Information) displayed enhancement of the methylene at δH 3.90 (2H, s, H-9′) confirming the double bond configuration at C-7′/8′ to be E.
Table 2.
NMR Spectra of Compounds 6 – 9 in CD3OD (J in Hz).a
| position | 6 | 7 | 8 | 9 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC, type | δH | δC, type | δH | δC, type | δH | δC, type | |
| 2 | 76.1, C | 76.0, C | 78.3, C | 78.8, C | ||||
| 3 | 1.94, dt (13.6, 6.0) 1.80b, m |
33.2, CH2 | 1.93d, m 1.80d, m |
32.5, CH2 | 5.71, d (9.8) | 130.7, CH | 5.61, d (9.7) | 131.5, CH |
| 4 | 2.69, m | 23.3, CH2 | 2.69e, m | 23.3, CH2 | 6.28f, d (9.8) | 124.3, CH | 6.27h, d (9.7) | 124.2, CH |
| 4a | 122.3, C | 122.2, C | 122.5, C | 122.7, C | ||||
| 5 | 6.36, d (2.9) | 113.8, CH | 6.34, d (2.9) | 113.7, CH | 6.29f, s | 111.4, CH | 6.28h, s | 111.3, CH |
| 6 | 151.1, C | 150.9, C | 151.8, C | 151.3, C | ||||
| 7 | 6.45, d (2.9) | 116.9, CH | 6.43, d (2.9) | 116.8, CH | 6.44, d (2.9) | 118.3, CH | 6.43, d (2.9) | 117.9, CH |
| 8 | 127.8, C | 127.9, C | 127.1, C | 126.8, C | ||||
| 8a | 145.7, C | 145.7, C | 144.7, C | 145.3, C | ||||
| 1′ | 3.00c, d (14.1) 2.50, d (14.1) |
52.6, CH2 | 2.84, d (14.8) 2.74, d (14.8) |
53.9, CH2 | 2.79, d (13.4) 2.75, d (13.4) |
55.2, CH2 | 1.65, m | 41.9, CH2 |
| 2′ | 211.0, | 210.2, | 200.6, C | 2.12i, m | 23.7, CH2 | |||
| 3′ | 3.03c, d (13.7) 2.37, d (13.7) |
52.9, CH2 | 2.94, d (15.0) 2.64, d (15.0) |
53.6, CH2 | 6.18, brs | 125.8, CH | 5.14, brt (7.1) | 125.7, CH |
| 4′ | 78.7, C | 78.4, C | 159.8, CH | 135.8, CH | ||||
| 5′ | 1.85b, m 1.76b, m |
40.2, CH2 | 1.89d, m 1.85d, m |
39.6, CH2 | 2.15g, m | 41.8, CH2 | 2.12i, m | 27.3, CH2 |
| 6′ | 2.18, m 2.03, m |
23.96, CH2 | 2.11, m | 24.1, CH2 | 2.17g, m | 26.7, CH2 | 2.00, t (7.6) | 40.5, CH2 |
| 7′ | 5.36, m | 125.8, CH | 5.41, m | 126.0, CH | 5.35, brt (6.9) | 125.1, CH | 5.37, brt (7.2) | 126.5, CH2 |
| 8′ | 137.1, C | 137.2, C | 136.9, C | 135.9, | ||||
| 9′ | 3.83, d, (16.6) 3.68, d (16.6) |
65.6, CH2 | 4.08, d (16.7) 3.98, d (16.7) |
65.8, CH2 | 3.89, s | 68.7, CH2 | 3.89, s | 69.0, CH2 |
| 10′ | 1.51, d (2.1) | 21.4, CH3 | 1.58, s | 21.4, CH3 | 1.63, d (1.4) | 13.7, CH3 | 1.62, s | 13.7, CH3 |
| 11′ | 1.20, s | 24.04, CH3 | 1.26, s | 24.1, CH3 | 2.04, d (1.3) | 19.6, CH3 | 1.58, d (1.4) | 15.9, CH3 |
| 12′ | 1.35, s | 25.7, CH3 | 1.34, s | 25.0, CH3 | 1.47, s | 27.0, CH3 | 1.33, s | 26.3, CH3 |
| 13′ | 2.09, s | 16.5, CH3 | 2.09, s | 16.5, CH3 | 2.08, s | 15.8, CH3 | 2.10, s | 15.7, CH3 |
1H NMR data obtained at 500 MHz; 13C NMR data acquired in CD3OD at 125 MHz.
Overlapping signals. Chemical shifts determined from HSQC.
Obscured signal.
The absolute configuration of the asymmetric center at C-2 was confirmed as S from examination of the ECD spectroscopic data (Figure 3). Thus, 3 was determined to be the new compound (3Z,7E)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Compound 4 was assigned the composition C22H30O4 by HRESIMS and 13C NMR spectroscopy. A side-by-side comparison of its NMR spectroscopic data with those of compounds 1 – 3 (Table 2) indicated that 4 is the C-7′/8′ Z isomer of 3. This observation was confirmed by selective NOE experiments (Figures S4.6 and S4.7, Supporting Information) that confirmed that the double bonds at both the C-3′/4′ and C-7′/8′ positions are in the Z configuration. The absolute configuration of the asymmetric center at C-2 was confirmed as S from examination of its ECD spectroscopic data (Figure 3). Thus, 4 was determined to be the new compound (3Z,7Z)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Compound 5 was determined to have the composition C23H32O4 from HRESIMS and carbon NMR spectroscopy. The 1H and 13C NMR spectra of 5 were extremely similar to those of 1 (Table 1), with the only major difference being signals for a new methoxy methyl group at δH 3.70 (3H, s, OCH3-6). HMBC correlations from the methoxy methyl at δH 3.70 to the aromatic ring at δC 154.1 (C-6) showed that the methyl group is attached to the ring by an ether linkage. Selective NOE experiments (Figures S5.6 and S5.7, Supporting Information) confirmed that the double bonds are both in the E configuration. The absolute configuration for the asymmetric center at C-2 was assigned as S from examination of ECD spectroscopic data (Figure 3). Thus, 5 was determined to be the new compound (3E,7E)-9-hydroxy-1-[(S)-6-methoxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Figure 3.

Selected HMBC correlations for 6 and 7. Green correlations were obtained after the spectrum was optimized for a coupling of 12 Hz.
Compound 6 was isolated as a clear oil with a chemical composition of C22H30O4 as determined by HRESIMS and 13C NMR data. Compound 6 has the same chemical composition as compounds 1 – 4 and the same chromane ring system, as confirmed by comparison of its 1H and 13C NMR spectra with those of 1 – 4 (Tables 1 and 2). Major differences were, however, observed for the terpenoid side chain. In the first place, the 1H NMR spectra obtained in CD3OD for compounds 1 – 4 indicated the presence of two exchangeable protons, but the 1H NMR spectrum of 6 indicated the presence of only one exchangeable proton. Another significant difference was the loss of the resonance at about δH 6.2 for C-3′ and its replacement by a pair of one-proton doublets at δH 3.03 and 2.37. The 13C NMR spectrum of 6 had resonances at δC 52.9 and 78.7 assigned to C-3′ and C-4′, respectively, consistent with alkoxylation of the 3′,4′ double bond. HMBC correlations were observed from the geminally coupled diastereotopic protons at δH 3.00 (1H, d, J = 14.1 Hz, H-1′a) and δH 2.50 (1H, d, J = 14.1 Hz, H-1′b) to C-2′, CH2-3, CH3-12′, C-2 and also from the diastereotopic protons at δH 3.03 (1H, d, J = 13.7 Hz, H-3′a) and δH 2.37 (1H, d, J = 13.7 Hz, H-3′b) to C-2′, C-4′, CH2-5′, and CH3-11′ (Figure 3). These observations confirmed that the carbonyl carbon is still attached at the C-2′ position on the side chain.
The fact that the diastereotopic protons at CH2-3′ have very different chemical shifts suggested that they are close to a chiral center, consistent with C-4′ being the center involved. Since protons H-3′a, H-3′b, the methyl at δH 1.20 (3H, s, H-11′), and the diastereotopic methylene protons at δH 1.85 (1H, m, H-5′-a) and δH 1.76 (1H, m, H-5′-b) all gave HMBC correlations with the oxygen-bearing quaternary carbon at δC 78.7, this was assigned as the asymmetric quaternary carbon at C-4′. HSQC and HMBC correlations established the signals for the methylene units at C-5′ and C-6′.
The diastereotopic protons at δH 1.85 (m, 1H, H-5′a) and δH 1.76 (m, 1H, H-5′b) are attached to the carbon at δC 40.2 (C-5′) and gave HMBC correlations with C-3′, C-4′, C-11′, another methylene at δC 23.96 (C-6′) and a vinyl carbon at δC 125.8 (C-7′); these correlations were used to establish the carbon at δC 40.2 as C-5′. The diastereotopic protons at δH 2.18 (1H, m, H-6a′) and δH 2.03 (1H, m, H-6b′) are attached to the carbon at δC 23.96 and gave HMBC correlations with C-4′, C-5′, the vinyl carbon at δC 125.8 (C-7′), and a quaternary carbon at δC 137.1 (C-8′), which established the carbon at δC 23.96 as C-6′. The methyl group at δH 2.03 (3H, d, J = 2.1 Hz, H-10′) is attached to the carbon at δC 21.4 (C-10′) and showed HMBC correlations with the vinyl carbon at δC 125.8 (C-7′), the quaternary carbon at δC 137.1 (C-8′), and the oxygen-bearing carbon at δC 65.6 (C-9′). The vinyl proton at δH 5.36 (1H, m, H-7′) had HMBC correlations with C-5′, C-6′, the methyl at δC 21.4 (C-10′), and the oxygen-bearing carbon at δC 65.6 (C-9′), when the experiment was optimized for a coupling of 12 Hz. The geminally coupled protons at δH 3.83 (1H, d, J = 16.6 Hz, H-9a′) and δH 3.68 (1H, d, J = 16.6 Hz, H-9b′) had HMBC correlations with the vinyl carbon at δC 125.8 (C-7′), the quaternary carbon at δC 137.1 (C-8′), and the methyl at δC 21.4 (C-10′). These correlations taken together were used to determine the end unit of the side chain. Finally, the geminally coupled methylene protons at CH2-9′, which occurred as singlets in compounds 1 – 5, had one additional HMBC correlation with the new oxygen-bearing quaternary carbon C-4′. This correlation confirmed that the α,β-unsaturated ketone of 1 underwent an internal cyclization with the hydroxy group at the end of the side chain to generate a cyclized seven-membered ring. A selective NOE experiment with saturation of the vinyl proton confirmed that the C-7′/8′ double bond is in the Z configuration. The configuration of the chiral center at C-2 was confirmed as S from examination of the ECD spectroscopic data (Figure 3), but the configuration of the new chiral center at C-4′ could not be determined. Compound 6 was thus assigned as either the new compound 1-[(R)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one or 1-[(S)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one. The difficulty of assigning the configuration of the tetrahydrooxepine ring has been noted by other investigators.7,8
Compound 7 was isolated as a clear oil and was found to have the elemental composition C22H30O4 from its HRESIMS, 13C NMR, and the HSQC spectroscopic data. Twenty-nine of the 30 protons were observed in its 1H NMR spectrum obtained in CD3OD, indicating the presence of only one hydroxy group, as in 6. A side-by-side comparison of the 13C NMR spectra of 6 and 7 (Table 2) revealed them to be extremely similar. A similar comparison of the 1H NMR spectra of 6 and 7 (Table 2) indicated that the major differences were for the protons in close proximity to the chiral center at C-4′, and its HMBC correlations confirmed the same connectivity for the cyclized side chain as were observed for 6 (Figure 6). Although 6 and 7 are very similar in almost every respect, they differ in HPLC retention times, with a tR of 22.03 min for 6 and 23.67 min for 7 under standard conditions (Figures S6.9 and S7.12, Supporting Information). These findings indicated that 6 and 7 are diastereomers, and since the chiral center at C-2 was again confirmed as S from examination of its ECD spectroscopic data (Figure 3), 7 was concluded as being diastereomeric with 6 at C-4′. The ECD spectroscopic data and specific rotations of 6 and 7 were very similar, and could not be used to assign the configurations at C-4′. Thus, 7 was determined to be either the new compound 1-[(S)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one or 1-[(R)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one. The observation that compounds 6 and 7 are diastereomeric at C-4′ suggests that the two compounds might be artifacts formed during extraction or isolation. A trace of acid could protonate the carbonyl group of 2 and activate the α,β-unsaturated ketone to Michael attack by the C-10′ hydroxy group to generate the seven-membered ring seen in 6 and 7 (Scheme 1). Deprotonation and tautomerization would afford diastereomers 6 and 7.
Scheme 1.

Proposed mechanism for the formation of 6 and 7 from 2.
Compound 8 was isolated as a pale yellow oil and was found to have the molecular composition of C22H28O4 as determined by its HRESIMS and 13C NMR data. Its 1H NMR spectrum was similar to that of 1 (Tables 1 and 2), but it contained signals for two new deshielded doublets at δH 6.28 (1H, d, J = 9.0 Hz, H-4) and δH 5.71 (1H, d, J = 9.0 Hz, H-3). Examination of the aliphatic region revealed that the benzylic methylene protons and the diastereotopic protons at position C-3 were missing (Table 2). Integration of the 1H NMR spectrum in CD3OD revealed 26 protons, indicating the presence of two hydroxy groups. Comparison of the 13C NMR spectrum of 8 with that of 1 revealed that the chromane ring moiety is modified as a chromene ring moiety, and this conclusion was confirmed by HMBC correlations from the methyl at δH 1.47 (3H, s, H-12′) to δC 78.3 (C-2), δC 130.7 (C-3), and δC 55.2 (C-1′) and from one of the new signals at δH 6.28 (1H, d, J = 9.0 Hz, H-4) to δC 122.5 (C-4a) and δC 144.7 (C-8a). These assignments agreed with those of other reported chromenes.9,10 Comparison of the 1H and 13C NMR data for the side chain with those of 1 led to the same assignments, and the side-chain double bond configurations were determined as E by selective NOE experiments. The stereochemistry of the chiral center at C-2 was determined to be R by observation of a negative Cotton effect at 290 – 370 nm ([Δε]321 nm −6.00) in the ECD spectrum (Figure 3). Thus, 8 was assigned as the new compound (3E,7E)-9-hydroxy-1-[(R)-6-hydroxy-2,8-dimethyl-2H-chromen-2-yl]-4,8-dimethylnona-3,7-dien-2-one.
Compound 9 was also isolated as a pale yellow oil, with the elemental composition of C22H30O3, as deduced from its HRESIMS and 13C NMR data. Summation of the integration in CD3OD only accounted for only 28 of the 30 protons, indicating the presence of two hydroxy groups. Comparison of the 1H and 13C NMR data of 8 with 9 (Table 2) revealed the presence of the same chromene ring moiety as well as the loss of the carbonyl oxygen at C-2′. Signals for a vinyl proton coupled to a new methylene unit were observed in the 1H NMR spectrum at δH 5.14 (1H, m, H-3′) and δH 2.12 (2H, m, H-2′), indicating the loss of the C-2′ carbonyl group. This was confirmed by HMBC correlations from the methylene protons at δH 1.65 (2H, m, H-1′) to δC 131.5 (C-3), δC 78.8 (C-2), δC 23.7 (C-2′), and δC 125.7 (C-3′). The rest of the side-chain chemical shifts matched those of 8. The double bond configuration was determined by utilizing selective NOE experiments to confirm that both double bonds on the side chain are E. The configuration of the asymmetric center at C-2 was assigned as R by a negative Cotton effect in the ECD spectrum between 290 – 370 nm ([Δε]321 nm −5.27). Thus, compound 9 was assigned as the new chromene (R)-2-[(3E,7E)-9-hydroxy-4,8-dimethylnona-3,7-dien-1-yl]-2,8-dimethyl-2H-chromen-6-ol.
Biological Activities
Chromane and chromene derivatives have been reported to possess potent antioxidant properties.11 All compounds were evaluated against the chloroquine-resistant Dd2 strain of P. falciparum and the A2780 ovarian cancer cell line (Table 3).
Table 3.
IC50 Values of Compounds 1 – 9 against P. falciparum Dd2 and A2780 Ovarian Cancer Cells
| compound |
P. falciparum Dd2 (μM) |
A2780 (μM) |
|---|---|---|
| 1 | > 112 | > 10 |
| 2 | 24 ± 3 | > 10 |
| 3 | > 112 | > 10 |
| 4 | > 112 | > 10 |
| 5 | 24 ± 2 | > 10 |
| 6 | 109 ± 8 | > 10 |
| 7 | 23 ± 2 | > 10 |
| 8 | > 112 | > 10 |
| 9 | > 117 | > 10 |
| artemisinin | 0.009 ± 0.003 | NTa |
| paclitaxel | NTa | 0.013 ± 0.001 |
NT = not tested.
Comparison of the antiplasmodial activities of the E and Z isomers 1 – 4 demonstrates that the configuration of both double bonds is crucial for this activity. Thus compound 2, with an E C-3′/4′ double bond and a Z C-7′/8′ double bond, showed the most potent biological activity among these compounds. In addition 1 was inactive against P. falciparum, but its methylated derivative 5 proved to be moderately active, suggesting that the methoxy group is also important to the observed biological activity. The antiplasmodial activities of the cyclized compounds 6 and 7 also differ by a factor of 4.
A good antimalarial drug needs to be nontoxic to the individual taking the drug, so the isolated compounds were also tested for antiproliferative biological activity against the drug-sensitive A2780 ovarian cancer cell line as an approximate indication of toxicity to human cells. None of the compounds tested were cytotoxic (IC50 > 10 μM) against the A2780 cell line under the conditions used.
Conclusions
Nine new compounds (1 – 9) were isolated from K. spinosa. All compounds were determined to contain either a chromane or chromene ring moiety with an isoprenoid side chain. Compounds 6 and 7 were determined to be diastereomers, but the absolute configuration at the new stereocenter could not be determined; the compounds are most likely artifacts generated during the extraction or isolation process. Bioassay results from testing against the Dd2 strain of P. falciparum indicated that both stereospecific cyclization of the side chain and an aromatic methoxy group modestly increased antiplasmodial activity.
EXPERIMENTAL SECTION
General Experimental Procedures
All chemicals used were purchased from either Sigma-Aldrich or the Fisher Scientific Company and were used without further purification. Optical rotations were measured on a JASCO P-2000 polarimeter. UV spectra and ECD analysis were obtained on a JASCO J-810 spectropolarimeter with a 1 cm cell in methanol or chloroform at room temperature with constant nitrogen flushing under the following conditions: speed 100 nm/min, time constant 1 s, bandwidth 1.0 nm. 1H and 13C NMR spectra were obtained on a Bruker Avance 500 spectrometer. Mass spectra were obtained on an Agilent 6220 LC-TOF-MS spectrometer. Semipreparative HPLC was performed using Shimadzu LC-10AT pumps coupled with a Shimadzu SPD-M10A diode array detector, a SCL-10A system controller, and a Phenomenex 5 μm, 100 Å, Luna C18(2) (250 × 10 mm) column or a Kromasil 5 μm, 60 Å, Diol (250 × 10 mm) column. Solid-phase extractions were conducted with Thermo Scientific Hypersep C18 500 mg, 3 mL SPE cartridges, and all reverse phase TLC was conducted with Sorbtech C18-W silica TLC plates, w/UV254 on an aluminum backing with a thickness of 150 μm.
Antimalarial Bioassay
Each fraction and isolated compound was tested against the chloroquine/mefloquine-resistant Dd2 strain of Plasmodium falciparum in a 72-hour treatment assay using the malaria SYBR green I-based fluorescence assay as described previously.12,13 The antimalarial drug artemisinin was used as a positive control, with IC50 values of 9.0 ± 3 nM.
Antiproliferative Bioassay
The A2780 ovarian cancer cell line assay was performed at Virginia Tech as previously reported.14,15 The A2780 cell line in a drug-sensitive ovarian cancer cell line.16
Plant Material
A sample of Koeberlinia spinosa was collected on April 25th, 1997 by D. E. Atha and Favio González of the New York Botanical Garden. Collection was in Pecos County, Texas between Sanderson and Fort Stockton on U.S. 285, about 35 miles south of East Dickinson Blvd. at an altitude of 1067 m. The plant was a shrub, 1.3 to 2.0 m. tall, growing in a roadside habitat. Its NYBG barcode is 01159061: http://sweetgum.nybg.org/science/vh/specimen_details.php?irn=762431.
Extraction and Isolation
A dried and ground sample of stems, twigs, and root of Koeberlinia spinosa (901 g) was extracted with ethanol (ca. 5 L) to yield 82 g of extract, of which 5.2 g was supplied to Virginia Tech as 0038278-09B (X-4535) for isolation studies.
The crude plant extract (3.7 g) was dissolved in 60% aqueous ethanol (1 L) and extracted with 3 × 600 mL of dichloromethane to afford a dichloromethane fraction (2.3 g) with an IC50 of about 5 μg/mL against the Dd2 strain of P. falciparum and an inactive aqueous EtOH fraction. The active dichloromethane fraction was then loaded on a polyamide column, which was eluted with MeOH to remove tannins to give 1.9 g of a fraction with an IC50 value between 2.5 and 5 μg/mL. The active detanninized fraction was then separated into the six fractions C1 – C6 by C18 solid-phase extraction (SPE) eluted with 50:50, 25:75, 10:90, 0:100 aqueous acetonitrile mixtures followed by a chloroform flush, which afforded the two active fractions C2 (939.9 mg) and C3 (331.2 mg) with IC50 values of between 2.5 and 5 μg/mL and ≪ 1.25 μg/mL, respectively. SPE fraction C2 was separated further into five fractions (D1 – D5) by reversed-phase HPLC on a Phenomenex C18(2), 5 μm, 250 × 10 mm column with a flow rate of 3 mL/min and an aqueous acetonitrile mobile phase starting at 55:45 to 50.8:49.2 over 25 min then a 7 min continuous flow at 0:100 H2O-CH3CN. Fraction D3 (tR 18 to 24 min, 353.3 mg) was weakly active with an IC50 value between 5 and 10 μg/mL. Fraction D5 was the most active fraction, and its constituents were available in a much greater quantity in fraction C3. Fraction D3 was further separated into eight fractions (E1 – E8) by C18 HPLC with a gradient from 30:70 to 22.5:77.5 H2O-MeOH over 35 min. Four fractions (E2 (17.0 mg, tR 16.83 min), E4 (82.0 mg, tR 18.37 min), and E7 (10.6 mg, tR 21.31 min) each contained one major component with minor impurities, and final purification was achieved on silica gel columns using 1:1 hexanes-EtOAc to afford compounds 8 (20.0 mg), 1 (78.3 mg), and 4 (9.5 mg) from E2, E4, and E7 respectively. Fraction E6 (50.5 mg, tR 19.47 min) was found to be an isomeric mixture of two compounds. Final isolation was achieved on E6 with diol HPLC using isopropanol-hexanes (15:85) at 3 mL/min for seven minutes followed by gradient to isopropanol/hexanes (17:83) over 20 min to afford the two pure compounds 2 (5.2 mg, tR 21.61 min) and 3 (13.5 mg, tR 24.43 min). SPE fraction C3 (331.2 mg, IC50 value ≪ 1.25 μg/mL) was separated into four subsequent fractions (F1 – F4) via C18 HPLC starting with a gradient from 55:45 to 50.8:49.2 H2O/MeCN over 25 min, then CH3CN alone for 7 min. Fraction F4 (224.6 mg, IC50 value ≪< 1.25 μg/mL) was then separated further into twelve fractions (G1 – G12) via C18 HPLC with a gradient starting at 30:70 to 16.5:83.5 H2O-MeOH for 30 min at 3 mL/min, then MeOH alone for 7.5 min. Four semi-pure compounds G4 (8.4 mg, tR 21.69 min), G6 (8.8 mg, tR 23.20 min), G8 (10.1 mg, tR 27.10 min), and G10 (16.2 mg, tR 28.63 min) were obtained and were purified on silica gel open columns with 1:1 hexanes-EtOAc to afford compounds 7 (7.2 mg), 5 (5.7 mg), and 9 (6.5 mg) from fractions G6, G8, and G10, respectively. Fraction G4-1 (10.1 mg) was not homogeneous, and was finally purified via diol HPLC with a flow rate of 3 mL/min using an isocratic flow at 8:92 isopropanol-hexane to afford compound 6 (4.1 mg, tR 18.68 min). Fractions G7 (12.1 mg, tR 24 to 27 min) and G12 (68.4 mg, tR 31 to 42.5 min) were both complex mixtures and found to be highly active (IC50 value ≪< 1.25 μg/mL). Fraction G7 was dropped after each subsequent fractionation (open silica gel column, then another round of C18 HPLC, and finally diol HPLC) led to a decrease in activity and insufficient material to continue isolating. Fraction G12 suffered from the same problems as G7 and was also dropped.
(3E,7E)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one (1): clear oil; 13.8 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 206 (3.25), 236 (2.97), 293 (2.57) nm; ECD (CH3OH) [Δε]296 nm −3.96; 1H and 13C NMR spectra, see Table 1; HRESIMS m/z 381.2017 [M+Na]+ (calcd for C22H30NaO4+, 381.2036).
(3E,7Z)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one (2): clear oil; 7.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.26), 238 (4.10) 293 (3.51) nm; ECD (CH3OH) [Δε]290 nm −8.86; 1H and 13C NMR spectra, see Table 1; HRESIMS m/z 381.2037 [M+Na]+ (calcd for C22H30NaO4+, 381.2036).
(3Z,7E)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one (3): clear oil;. 7.5 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.28), 224 (4.06), 292 (3.52) nm; ECD (CH3OH) [Δε]294 nm −8.36; 1H and 13C NMR spectra, see Table 1; HRESIMS m/z 381.2041 [M+Na]+ (calcd for C22H30NaO4+, 381.2036).
(3Z,7Z)-9-hydroxy-1-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one (4): clear oil; 6.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.27), 224 (4.06), 293 (3.51) nm; ECD (CH3OH) [Δε]292 nm −8.94; 1H and 13C NMR spectra, see Table 1; HRESIMS m/z 381.2020 [M+Na]+ (calcd for C22H30NaO4+, 381.2036).
(3E,7E)-9-hydroxy-1-[(S)-6-methoxy-2,8-dimethylchroman-2-yl]-4,8-dimethylnona-3,7-dien-2-one (5): clear oil; 10.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.26), 231 (4.06), 290 (3.45) nm; ECD (CH3OH) [Δε]292 nm −7.78; 1H and 13C NMR spectra, see Table 1; HRESIMS m/z 395.2179 [M+Na]+ (calcd for C23H32NaO4+, 395.2193).
1-[(S or R)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one (6): clear oil; 4.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.31), 293 (3.49) nm; ECD (CH3OH) [Δε]287 nm −8.39; 1H and 13C NMR spectra, see Table 2; HRESIMS m/z 381.2049 [M+Na]+ (calcd for C22H30NaO4+ 381.2036).
1-[(R or S)-2,6-dimethyl-2,3,4,7-tetrahydrooxepin-2-yl]-3-[(S)-6-hydroxy-2,8-dimethylchroman-2-yl]propan-2-one (7): clear oil; 7.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 207 (4.30), 291 (3.47) nm; ECD (CH3OH) [Δε]289 nm −8.67; 1H and 13C NMR spectra, see Table 2; HRESIMS m/z 381.2033 [M+Na]+ (calcd for C22H30NaO4+ 381.2036).
(3E,7E)-9-hydroxy-1-[(R)-6-hydroxy-2,8-dimethyl-2H-chromen-2-yl]-4,8-dimethylnona-3,7-dien-2-one (8): light yellow oil; 4.0 (c 0.1, CHCl3); UV (CHCl3) λmax (log ε) 248 (3.28), 328 (3.47) nm; ECD (CHCl3) [Δε]321 nm −6.00; 1H and 13C NMR spectra, see Table 2; HRESIMS m/z 379.1861 [M+Na]+ (calcd for C22H28NaO4+ 379.1880).
(R)-2-[(3E,7E)-9-hydroxy-4,8-dimethylnona-3,7-dien-1-yl]-2,8-dimethyl-2H-chromen-6-ol (9): light yellow oil; 10.0 (c 0.1, CHCl3); UV (CHCl3) λmax (log ε) 244 (4.14), 330 (3.57) nm. ECD (CHCl3) [Δε]325 nm −5.27; 1H and 13C NMR spectra, see Table 2; HRESIMS m/z 365.2052 [M+Na]+ (calcd for C22H30NaO3+ 365.2087).
Supplementary Material
Acknowledgments
This project was supported by the National Center for Complementary and Integrative Health under award 1 R01 AT008088, and this support is gratefully acknowledged. This work was also supported by the National Science Foundation under Grant No. CHE- 0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant No. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank D. E. Atha and Favio González for the plant collection, Mr. B. Bebout and Dr. M. Ashraf-Khorassani for obtaining the mass spectra, and Dr. T. Grove for the use of the JASCO J-815 spectrometer. The image used in the graphical abstract was reproduced with permission from Russ Kleinman http://wnmu.edu/academic/nspages/gilaflora/koeberlinia_spinosa_small.jpg.
Footnotes
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.005799.
1H, 13C, gHMBC, gHSQC, selective NOESY, and UV and ECD spectra for compounds 1 – 9 (PDF)
References
- 1.Tobe H, Raven PH. Am J Bot. 2008;95:1475–1486. doi: 10.3732/ajb.0800218. [DOI] [PubMed] [Google Scholar]
- 2.Lucero M, Estell R, Tellez M, Fredrickson E. Phytochem Anal. 2009;20:378–384. doi: 10.1002/pca.1137. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. World Malaria Report 2016. Geneva: 2016. http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ [Google Scholar]
- 4.Iwashima M, Tako N, Hayakawa T, Matsunaga T, Mori J, Saito H. Chem Pharm Bull. 2008;56:124–128. doi: 10.1248/cpb.56.124. [DOI] [PubMed] [Google Scholar]
- 5.Jang KH, Lee BH, Choi BW, Lee HS, Shin J. J Nat Prod. 2005;68:716–723. doi: 10.1021/np058003i. [DOI] [PubMed] [Google Scholar]
- 6.Mazzini F, Pescitelli G, Bari LD, Netscher T, Salvadori P. Chirality. 2009;21:35–43. doi: 10.1002/chir.20591. [DOI] [PubMed] [Google Scholar]
- 7.Kreipl AT, König WA. Phytochemistry. 2004;65:2045–2049. doi: 10.1016/j.phytochem.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Kaneko I, Hoshino T. J Org Chem. 2016;81:6657–6671. doi: 10.1021/acs.joc.6b01313. [DOI] [PubMed] [Google Scholar]
- 9.Muckensturm B, Diyani F, Reduron JP, Hildenbrand M. Phytochemistry. 1997;45:549–550. [Google Scholar]
- 10.Kil YS, Park J, Han AR, Woo HA, Seo EK. Molecules. 2015;20:5965–5974. doi: 10.3390/molecules20045965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terashima K, Takaya Y, Niwa M. Bioorg Med Chem. 2002;10:1619–1625. doi: 10.1016/s0968-0896(01)00428-x. [DOI] [PubMed] [Google Scholar]
- 12.Bennett TN, Paguio M, Gligorijevic B, Seudieu C, Kosar AD, Davidson E, Roepe PD. Antimicrob Agents Chemother. 2004;48:1807–1810. doi: 10.1128/AAC.48.5.1807-1810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan E, Harinantenaina L, Brodie PJ, Miller JS, Callmander MW, Rakotonandrasana S, Rakotobe E, Rasamison VE, Kingston DGI. J Nat Prod. 2010;73:1792–1795. doi: 10.1021/np100411d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao S, Brodie PJ, Miller JS, Randrianaivo R, Ratovoson F, Birkinshaw C, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.