

Abstract
The role of the three gasotransmitter systems - nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) - in cancer cells has not yet been studied simultaneously in the same experimental system. We measured the expression of NO and CO and H2S generating enzymes in primary colon cancer tissues and HCT116 colon cancer cells, and evaluated the effect of their pharmacological inhibition or pharmacological donation on cell proliferation. Increased expression of iNOS, nNOS, HO-1, CBS and 3-MST was detected in colon cancer. Inhibitors of NOS, HO-1/2, CBS/CSE and 3-MST, at lower concentrations, slightly stimulated HCT116 cell proliferation, but inhibited proliferation at higher concentrations. Donors of NO, CO or H2S inhibited HCT116 proliferation in a concentration-dependent manner. Inhibition of the cGMP/VASP pathway, Akt and p44/42 MAPK (Erk1/2) inhibited HCT116 cell proliferation. Endogenous NO and H2S biosynthesis were found to play a role in the maintenance of the activity of the cGMP/VASP pathway in HCT116 cells. We conclude that each of the three gasotransmitters play similar, bell-shaped roles in the control of HCT116 cell proliferation: endogenously produced NO, CO and H2S, at an optimal concentration, support HCT116 proliferation; inhibition of their production (which decreases gasotransmitter levels below optimal concentrations) as well as exogenous delivery of these gasotransmitters (which increases gasotransmitter levels above optimal concentrations) suppresses colon cancer cell proliferation. The current data give a mechanistic explanation for the paradoxical finding that both inhibitors and donors of NO, CO and H2S exert anticancer actions in cancer cells.
Keywords: gasotransmitters, cell proliferation, cancer, apoptosis, bioenergetics, PI3K, signaling, anticancer
Graphical Abstract
1. Introduction
Three gaseous molecules have been identified as gasotransmitters in the last decades: nitric oxide (NO), carbon monoxide (CO) and, most recently, hydrogen sulfide (H2S). Each of these molecules is endogenously produced by different enzymes in various cell types and has multiple roles in normal physiology and in the regulation of various cancer cell functions (e.g. proliferation, invasion, metastasis, and tumor angiogenesis). They also exhibit bimodal or bell-shaped pharmacological character. Modulation of each of these gasotransmitters has previously been reported to exert both anti- or pro-tumor effects, as reviewed recently [1].
In different reports, which employed different tumor cell types and different experimental systems, either pharmacological inhibition or pharmacological donation of NO, CO or H2S has been shown to exert antiproliferative effects and, therefore, has been proposed as a potential future anticancer approach [1]. This paradoxical situation has not yet been explained in focused experiments, in part because the effect of pharmacological modulation (inhibition as well as donation) of all three gasotransmitter systems on cancer cell growth and proliferation have not yet been studied simultaneously in the same experimental/cellular system.
In the present study, we have studied the expression of NO and CO and H2S generating enzymes in primary colon cancer tissues and surrounding normal tissues and in the HCT116 colon cancer cell line, and investigated the effect of pharmacological inhibition and pharmacological donation of each of these transmitters on HCT116 colon cancer cell proliferation. In addition, we evaluated the effect of pharmacological modulators of their putative downstream pathways.
2. Materials and Methods
2.1. Reagents
Fetal bovine serum (FBS) and 0.25% trypsin-EDTA were purchased from Life Technologies (Carlsbad, CA, USA). All other pharmacological agents and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Cell culture
The HCT116 colon cancer cell line (ATCC; Manassas, VA) and the NCM356 cell line (Incell Corporation, LLC; San Antonio, TX) was cultured in McCoy’s 5A medium supplemented with 10% FBS. Cells were grown in a 37 °C, 5% CO2 atmosphere.
2.3. Human colon specimens
Colon cancer specimens (n=15, from 15 different patients), together with their patient-matched surrounding normal tissues (n=15) were purchased from Bioserve (Beltsville, MD, USA). The samples were collected under IRB approved specimen collection protocols and registered sites, IRB approved informed consent forms in such a way that the collection studies did not interfere with patient care. Protection of privacy included assignment of unique de-identified and anonymized patient identifiers – neither BioServe nor the investigators involved in the current study had access to protected health information of donors.
2.4. Preparation of the whole-cell extracts colon specimens for western blots
Total cell extracts were prepared from cells and colon specimens by homogenizing in NP-40 lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% Nonidet P-40) in the presence of protease inhibitor (Roche) followed by clean-up centrifugation at 20,000 × g for 10 min. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The membrane was blocked in Thermo Scientific™ StartingBlock™ Blocking Buffer (Thermo Fisher Scientific, Waltham, MA, USA) followed by exposure to primary antibodies for overnight at 4C°. Secondary antibodies employed: anti-mouse IgG conjugated to horseradish peroxidase and 1:2,000 (Cell Signaling), anti-rabbit IgG conjugated to horseradish peroxidase 1:2,000. After incubating with secondary antibody for 1 hour at room temperature, the membrane was developed with SuperSignal™ West Pico Chemiluminescent Substrate (Pierce) in a GeneBox Detection System (Syngene).
2.5. Cell proliferation monitoring by the XCelligence system
To monitor cellular events in real time we used the XCelligence system to measure electrical impedance across interdigitated micro-electrodes integrated on the bottom of tissue culture E-Plates, as described [2]. The impedance measurement provides quantitative information about the biological status of the cells, including cell number, viability and adherence. Briefly, HCT-116 (6,000 cells/well) were seeded into 200 μl of media in a 96X E-Plate. 24 hours after seeding, cells were treated with different concentration of NO, CO, or H2S donors and biosynthesis inhibitors or various other pharmacological agents as indicated. The proliferation-related readout (Cell Index) was monitored for 48h.
2.6. MTT assay
The MTT method, an assay that detects mitochondrial-dependent conversion of a specific dye 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was performed, as described [3]. Briefly, MTT was added to the cells at a final concentration of 0.5 mg/ml and cells were cultured at 37 °C for 1 h. The cells were washed with PBS and the formazan dye was dissolved in DMSO. The amount of converted formazan dye was measured at 570 nm with a background measurement at 690 nm on a Molecular Devices M2 reader.
2.7. Cell death assay
The detection of apoptosis/necrosis of HCT-116 treated with different compounds was performed with the PE Annexin V Apoptosis Detection Kit (BD Biosciences) by flow cytometry as described [4]. Cells were trypsinized, washed in ice-cold PBS and re-suspended in 1 ml Binding Buffer. Then, 1×105 cells (500 μl) were incubated with 5 μl Annexin V PE and 5 μl 7AAD for 10 min at 25°C in the dark, and analyzed immediately using a Guava EasyCyte Plus Flow Cytometer (Millipore, Billerica, MA). The early and late apoptotic cells, and as well as the necrotic cells, were estimated as the percentage of the total number of cells by CytoSoft 5.3 Software (Millipore, Billerica, MA).
2.8. Statistical analysis
Data are presented as mean ± SEM. Data were analyzed using GraphPad Prism software (GraphPad, San Diego, CA, USA). Statistical analyses included Student’s t test, one- or two-way ANOVA followed by Tukey’s multiple comparisons were used to detect differences between groups. Statistical significance was considered when P<0.05.
3. Results
3.1 Expression analysis of NO-, CO- and H2S-producing enzymes in primary human colon cancer samples
First, we determined the expression of NO-, CO- and H2S-producing enzymes in pairs of primary human colon cancer samples and their surrounding, patient-matched, non-cancerous tissue. The tumor tissue exhibited higher expression of eNOS than the surrounding tissue. In contrast, iNOS and nNOS expression levels were not statistically different (Fig. 1A–C).
Figure 1. Expression of (A) eNOS, (B) iNOS, (C) nNOS, (D) HO-1, (E) HO-2, (F) CBS, (G) CSE and (H) 3-MST in human colon cancer biopsies (C) and matched normal surrounding tissue (N).

For each enzyme, representative Western blots, summary of expression data (mean±SEM) from all samples analyzed (where detectable signal was found) and individual paired sample analysis is shown. Arbitrary relative densitometry units were normalized with β-actin using image analysis software. *P<0.05 T (tumor) vs. N (normal surrounding tissue); n=15.
With respect to the expression of the CO-producing enzymes HO-1 and HO-2, no significant differences were noted between the tumor tissue and the surrounding tissue (Fig. 1D,E), with about half of the individual sample pairs showing increases in the tumors, and the other half showing decreases. HO-2 levels were very low (undetectable) in many of the samples.
With respect to the expression of the H2S-producing enzymes CBS (but not CSE or 3-MST) showed a significant increase between the tumor tissue and the surrounding normal mucosa (Fig. 1F–H). However, the magnitude of the difference within individual patients was highly variable with tumors from three out of 15 analyzed patients showing very high levels of CBS expression compared to normal (Fig. 1F). In most patients, CSE expression levels were very low in both the tumor homogenates and the surrounding tissue (Fig. 1G). Approximately half of the individual sample pairs showed increases in the 3-MST expression level in the tumor tissue compared to the surrounding normal tissue; the other half of the samples showed low 3-MST expression which was comparable between the tumor and its surrounding tissue (Fig. 1H).
3.2 Expression analysis of NO-, CO- and H2S-producing enzymes in HCT116 human colon cancer cell line compared to the adenoma-like colonic epithelial cell line NCM356
Next, we sought to determine the expression of NO-, CO- and H2S-producing enzymes in HCT116 human colon cancer cell line compared to the non-malignant adenoma-like colonic epithelial cell line NCM356. With respect to NO-producing enzymes, HCT116 cells contained lower expression of eNOS than NCM356 cells, while iNOS and nNOS expression levels were not statistically different (Fig. 2A–C). Expression of both the CO-producing enzymes HO-1 and HO-2 was higher in the HCT116 cells compared to NCM356 cells (Fig. 2D–E). Expression of the H2S-producing enzymes CBS and 3-MST was significantly higher in HCT116 cells than in NCM356 cells, while the levels of CSE were comparable between the two cell lines (Fig. 2F–H).
Figure 2. Expression of (A) eNOS, (B) iNOS, (C) nNOS, (D) HO-1, (E) HO-2, (F) CBS, (G) CSE and (H) 3-MST in the human epithelial cell line NCM356 and in the human colon cancer cell line HCT116.

For each enzyme, representative Western blots and summary of expression data (mean±SEM) from all samples analyzed (where detectable signal was found) is shown. Arbitrary relative densitometry units were normalized with β-actin using image analysis software. *P<0.05 HCT116 vs. NCM356; n=9 lanes (3 experimental days, each performed in triplicate).
3.3 Effect of pharmacological inhibition of NO-, CO- and H2S-producing enzymes on HCT116 cell proliferation and mitochondrial activity
Next, we determined the effect of pharmacological inhibition of NO-, CO- and H2S-producing enzymes on HCT116 cell proliferation and mitochondrial function (evaluated as MTT conversion). The non-isoform-selective NOS inhibitor L-NMMA [5] exerted a concentration-dependent inhibitory effect on HCT116 cell proliferation as well as MTT conversion at the highest concentrations used (3 and 10 mM) but it slightly increased proliferation at the lowest concentration used (0.3 mM) (Fig. 3A). A number of additional NOS inhibitors were also tested on HCT116 proliferation. N5-(1-iminoethyl)-L-ornithine (L-NIO), which is a potent nonselective competitive inhibitor of NOS [6] increased HCT116 proliferation at lower concentrations, while it higher concentrations an inhibitory effect was observed (Fig. 4A). Thus, its effects were biphasic, and similar to L-NMMA (another non-isoform-selective inhibitor). L-Nω-propyl-arginine (L-NPLA), or 3-bromo-7-nitroindazole (3B7NI) nNOS-selective inhibitors [6,7] failed to affect HCT116 proliferation (Fig. 4B,C).
Figure 3. Effect of (A) NOS inhibition with L-NMMA, (B) HO inhibition with ZnPPIX, (C) CBS/CSE inhibition with AOAA or (D) 3-MST inhibition with HMPSNE on HCT116 cell proliferation and MTT conversion.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Thereafter, mitochondrial-dependent conversion of MTT was determined. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); middle panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours) and right panels show the summary of 3 MTT conversion experiments conducted on 3 different experimental days for cell proliferation (analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation or MTT conversion compared to the vehicle control; n=9 determinations (3 experimental days, each performed in triplicate).
Figure 4. Effect of (A) NOS inhibition with L-NIO (B) nNOS inhibition with NPLA, (C) nNOS inhibition with 3B7NI or (D) HO inhibition with TINPP on HCT116 cell proliferation.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); right panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation compared to the vehicle control; n=9 determinations (3 experimental days, each performed in triplicate).
To evaluate the effects of pharmacological inhibition of CO-producing enzymes on HCT116 cell proliferation and mitochondrial activity, cells were treated with the zinc-protoporphyrin IX (ZnPPIX) [8]. This compound, at the lowest concentration tested, caused a slight but statistically significant increase in cell proliferation (despite of the fact that it also caused a slight decrease in MTT conversion). At higher concentrations, however, it caused a pronounced, concentration-dependent reduction in the rate of cell proliferation, as well as a decrease in MTT conversion (Fig. 3B). Another HO inhibitor, tin-protoporphyrin IX (TINPP) [8] had similar effects. At low concentrations, TINPP treatment caused a small increase in the rate of cell proliferation and at higher concentrations it inhibited proliferation, but with a lower potency than ZnPPIX (Fig. 4D). The difference between the effects of the two inhibitors, as well as direct cell viability measurements (see below) suggest that the antiproliferative effect of zinc-protoporphyrin IX may be, in part, related to secondary actions unrelated to inhibition of CO production (most likely direct cytotoxicity).
To inhibit CSB and CSE mediated H2S production HCT116 cells were treated with the combined CBS/CSE inhibitor aminooxyacetic acid (AOAA) [9,10,11]. AOAA exerted a concentration-dependent inhibitory effect on HCT116 cell proliferation as well as MTT conversion at the highest concentrations used (100 and 300 μM) but slightly increased proliferation at the lowest concentration used (30 μM) (Fig. 3C). DL-propargylglycine (PAG), which is a compound with low potency, but high selectivity for CSE (over CBS or 3-MST) [10,11] failed to affect HCT116 proliferation (Fig. 4E). The 3-MST inhibitor 2-[(4-hydroxy-6-methylpyrimidin-2-yl)sulfanyl]-1-(naphthalen-1-yl)ethan-1-one [HMPSNE] [11,12] also exerted a concentration-dependent inhibitory effect on HCT116 cell proliferation, with a concentration-response that appears to be biphasic: at the low concentrations (1–30 μM) the compound exerted a partial inhibitory effect on cell proliferation which was comparable at all concentrations tested - and these effects manifested without an effect on MTT conversion, while at higher concentrations (100 μM - 1 mM), a further decrease in proliferation was observed with an associated concentration-dependent decrease in MTT conversion (Fig. 3D).
These data, taken together, suggest that endogenously produced NO, CO and H2S in HCT116 cells stimulates proliferation, but via a bell-shaped concentration-response curve (see: Discussion for further context and implications).
3.4 Effect of pharmacological donors of NO, CO or H2S on HCT116 cell proliferation and mitochondrial activity
Next, we determined the effect of pharmacological donation of NO, CO or H2S- on HCT116 cell proliferation and mitochondrial activity. All donors tested, including the NO donor DETA-NO [13,14] (Fig. 5A), the CO donor CORM3 [15,16] (Fig. 5B), and the mitochondrially targeted H2S donor AP39 [17] (Fig. 5C) induced concentration-dependent inhibitory effects on cell proliferation, and inhibited MTT conversion. At the lower concentrations (100 and 300 μM) the antiproliferative effects of the NO donor were not associated with any inhibition of mitochondrial activity; at the highest concentration tested (1000 μM), the near-complete inhibition of cell proliferation was associated with a slight. but statistically significant inhibition of MTT conversion (Fig. 5A). In contrast, CO and H2S donors exerted comparable effects in terms of % inhibition values for cell proliferation and MTT conversion (Fig. 5B,C).
Figure 5. Effect of (A) NO donation with DETA-NO, (B) CO donation with CORM3 and (C) H2S donation with AP39 on HCT116 cell proliferation and MTT conversion.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Thereafter, mitochondrial-dependent conversion of MTT was determined. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); middle panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours) and right panels show the summary of 3 MTT conversion experiments conducted on 3 different experimental days for cell proliferation (analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation or MTT conversion compared to the vehicle control; n=9 determinations (3 experimental days, each performed in triplicate).
Other gasotransmitter donors, such as the NO donor S-nitroso-N-acetyl-D,L-penicillamine (SNAP) [15] (Fig. 6A), the CO donor CORM A1 (Fig. 6B), and the non-organelle-targeted, slow-releasing H2S donor GYY4137 (GYY) (Fig. 6C) also exerted antiproliferative effects.
Figure 6. Effect of (A) NO donation with SNAP (B) CO donation with CORM-A1, and (C) H2S donation with GYY4137 on HCT116 cell proliferation.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); right panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation compared to the vehicle control; n=9 determinations (3 experimental days, each performed in triplicate).
3.5 Effect of pharmacological modulators of the NO, CO or H2S pathways on HCT116 cell viability
In order to obtain a more detailed insight into the effect of the various pharmacological agents on cell viability, flow cytometry analysis was conducted. Treatment with the NOS inhibitor L-NMMA did not have any marked effects on the distribution of various cell populations, even at the highest concentration (10 mM) tested, where it caused marked antiproliferative effects (Fig. 7A). In contrast, ZnPPIX exerted a marked antiproliferative and inhibition of MTT activity at 10 and 30 μM and was associated with an increase in the population of dead cells (especially late apoptotic and necrotic) (Fig. 7A), most likely due to effects that are unrelated to inhibition of heme oxygenase activity or suppression of CO production. In fact, non-specific, cytotoxic effects of ZnPPIX have previously been reported [18,19]. The antiproliferative effect of AOAA at 100 μM was only associated with small increases in the proportion of dead or dying cells, but at 300 μM its cytotoxicity became apparent, primarily inducing increases in the proportion of early and late apoptotic cells (Fig. 7A). CBS/CSE-unrelated pharmacological effects (for instance, on many other PLP-dependent enzymes) [20] may well contribute to some of the inhibitory effects of AOAA on cell viability.
Figure 7. Effect of (A) the NOS inhibitor L-NMAA, the HO inhibitor ZnPPIX, and the CBS/CSE inhibitor AOAA and (B) the NO donor DETA-NO, the CO donor CORM-3 and the H2S donor AP39 on HCT116 cell viability.

HCT116 cells were treated with various concentrations of the pharmacological agents and at 48 h analyzed for various cell populations as indicated using flow cytometry. Data show mean±SEM of n=9 determinations (3 experimental days, each performed in triplicate).
With respect to the donors, the NO donor DETA did not induce cell death at any of the concentrations tested (Fig. 7B). Likewise, the antiproliferative effects of CORM3 were not associated with significant cell death (Fig. 7B). However, the antiproliferative effect of AP39 (seen at 100 μM) were associated with an increase in the population of early and late apoptotic cells. Even at 30 μM (a concentration that did not induce a significant decrease in HCT116 cell proliferation), some increase in the apoptotic cell populations was detected (Fig. 7B).
3.6 Combined effect of pharmacological modulators of the NO, CO or H2S pathways on HCT116 cell viability
Next, we determined the effect of combined pharmacological inhibition of NO-, CO- and H2S-producing enzymes, or combined pharmacological donation of NO-, CO- and H2S on HCT116 cell proliferation and mitochondrial activity. Whenever possible, we have selected concentrations of the inhibitors or donors that produce approximately 50% antiproliferative effects, and tested their effects on cell proliferation both alone (single agent) and in combination. The results of the inhibitor combination studies (L-NMMA, 10 mM; ZNPPIX, 10 μM; AOAA, 150 μM) are shown in Fig. 8. Addition ZnPPIX to either L-NMMA (Fig. 8A) or to AOAA (Fig. 8C) exerted additional antiproliferative effects, while the combination of AOAA and L-NMMA was not more antiproliferative than either the effect of AOAA alone or L-NMAA alone (Fig. 8B). These data suggest that ZnPPIX enhances the antiproliferative effect of inhibitors of the other two pathways; however, given the fact that ZnPPIX has likely cytotoxic effects on its own, the enhancement may be nonspecific.
Figure 8. Effect of (A) L-NMAA (10 mM), ZnPPIX (20 μM) or their combination (B) L-NMAA (10 mM), AOAA (150 μM) or their combination or (C) ZnPPIX (20 μM), AOAA (150 μM) or their combination on HCT116 cell proliferation.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); right panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation compared to the vehicle control; #p<0.05 shows significant difference between the two groups as indicated; n=9 determinations (3 experimental days, each performed in triplicate).
With respect to the combination effect of various gasotransmitter donors that induce approximately 50% antiproliferative effects on their own (DETA, 100 μM; CORM3, 1 mM; AP39, 60 μM), we were unable to detect any additive or synergistic effects. The combination of DETA and CORM3 was, in fact, less antiproliferative than either agents alone (Fig. 10A). The combination of DETA and AP39 was comparable to the effect of either of the two individual agents alone (Fig. 10B), and a similar relationship pertains to the combination of CORM3 and AP39 (Fig. 10C).
Figure 10. Effect of (A) DETA-NO (100 μM), CORM3 (1 mM) or their combination (B) DETA-NO (100 μM), AP39 (60 μM) or their combination or (C) CORM3 (1 mM), AP39 (60 μM) or their combination on HCT116 cell proliferation.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); right panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation compared to the vehicle control; # P <0.05 shows significant difference between the two groups as indicated; n=9 determinations (3 experimental days, each performed in triplicate).
3.7 Potential signaling pathways downstream from NO, CO or H2S in colon cancer biopsies or HCT116 cells
Next, we determined the role of potential putative proliferative signaling pathways that are downstream from NO, CO or H2S. We focused on the PI3K/Akt pathway, the p44/42 MAPK (Erk1/2) pathway and the cGMP/VASP pathway [1,20–26].
In the tumor tissues, we detected lower phosphorylation of Akt, of p44/42 MAPK and of VASP than in the surrounding normal tissues (Fig. 11). In the HCT116 cells, we also detected lower phosphorylation of Akt and of p44/42 MAPK than in NCM356 cells (Fig. 12A,B). In contrast, HCT116 cells exhibited higher VASP phosphorylation than NCM356 cells (Fig. 12C).
Figure 11. Expression of (A) pAkt and Akt, (B) p-p44/42 MAPK and p44/42 MAPK, and (C) pVASP and VASP in normal surrounding tissue (Normal) and human colon cancer biopsies (Tumor).

For each enzyme, representative Western blots, summary of expression data (mean±SEM; phosphorylated forms normalized to non-phosphorylated) from all samples analyzed (where detectable signal was found) and individual paired sample analysis is shown. *P<0.05 Tumor vs. Normal; n=8.
Figure 12. Expression of (A) pAkt and AktT, (B) p-p44/42 MAPK and p44/42 MAPK, and (C) pVASP and VASP in HCT116 cells and NCM356 cells.

For each enzyme, representative Western blots and summary of expression data (mean±SEM; phosphorylated forms normalized to non-phosphorylated) from all samples analyzed. *P<0.05 NCM356 vs. HCT116; n=6.
Next, we have tested the effect of the respective pharmacological inhibitors of the PI3K/Akt pathway, the p44/42 MAPK (Erk1/2) pathway and the cGMP/VASP pathway on HCT116 cell proliferation. LY294002 was used as an inhibitor of PI3K [27], GSK690593 was used as an Akt pathway inhibitor [25], U126 was used as an ERK1/2 pathway inhibitor [28] and the soluble guanylate cyclase inhibitor ODQ (1H-[1,2,4]oxadiazolo[4,3-a] quinoxalin-1-one) [29] was used to inhibit guanylate cyclase activation (and to prevent the activation of the downstream phosphorylation of VASP). All of these compounds inhibited HCT116 cell proliferation, but they also exerted inhibitory effects on MTT conversion (Fig. 13).
Figure 13. Effect of (A) the PI3K inhibitor LY294002, (B) the Akt pathway inhibitor GSK 690593, (C) the MEK1/2 inhibitor U0126 or (D) the guanylate cyclase inhibitor ODQ on HCT116 cell proliferation and MTT conversion.

HCT116 cells were seeded at the density of 6,000 cells per well in xCELLigence plates, treated with various concentrations of the pharmacological inhibitors and proliferation was monitored for 48 h. Thereafter, mitochondrial-dependent conversion of MTT was determined. Left panels show individual xCELLigence experiments run in triplicate wells (mean±SEM); middle panels show the summary of 3 experiments conducted on 3 different experimental days for cell proliferation (normalized Cell Index [CI]: analyzed at 48 hours) and right panels show the summary of 3 MTT conversion experiments conducted on 3 different experimental days for cell proliferation (analyzed at 48 hours). *P<0.05 shows significant differences in cell proliferation or MTT conversion compared to the vehicle control; n=9 determinations (3 experimental days, each performed in triplicate).
If endogenous NO, CO and/or H2S production play constitutive roles in the maintenance of the activation of the Akt, p44/42 MAPK (Erk1/2) and/or cGMP/VASP pathways, then one would expect that pharmacological inhibition of NO, CO and/or H2S production would suppress the phosphorylation of AKT, p44/42 MAPK or VASP. Contrary to our hypothesis, we found that L-NMMA did not decrease Akt phosphorylation, but, rather, increased it, while it did not affect p44/42 MAPK phosphorylation. However, L-NMAA decreased VASP phosphorylation. ZnPPIX increased both AKT and p44/42 MAPK and did not affect VASP phosphorylation. AOAA did not affect Akt or p44/42 MAPK phosphorylation. However, AOAA decreased VASP phosphorylation (Fig. 14).
Figure 14. Effect of AOAA, L-NMMA or ZnPPIX on the (A) expression of pAkt and AKT, (B) p-p44/42 MAPK and p44/42 MAPK, and (C) pVASP and VASP in HCT116 cells.

For each enzyme, representative Western blots and summary of expression data (mean±SEM; phosphorylated forms normalized to non-phosphorylated) are shown. *P<0.05 or **P<0.01 cells treated with test compounds vs. vehicle control; n=6.
4. Discussion
4.1. Expression patterns of NO, CO and H2S-producing enzymes in primary colon cancer tissues and in HCT116 colon cancer cells
The first aim of the current study was to determine the expression of NO-, CO- and H2S-producing enzymes in colon cancer cells. We have employed two approaches: First, we studied the expression of eNOS, iNOS, nNOS, HO-1, HO-2, CBS, CSE and 3-MST in pairs of primary human colon cancer samples and their surrounding, patient-matched, non-cancerous tissue. Next, we have measured the expression of the same enzymes in HCT116 cells, a human colon cancer cell line, and compared it to the expression of the same enzymes in NCM356 cells, a non-transformed epithelial cell line (which we used as it approximates a normal epithelial cell line, as far as any cell line grown in culture can approximate a ‘normal’ cell). As far as we know, prior to our study a simultaneous, comprehensive analysis of all gasotransmitter-producing enzymes has not yet been conducted, neither in primary cancer tissues, nor in cell lines. However, separate prior studies (reviewed in [1]) have studied the expression patterns of either the NO-producing enzymes, or the CO-producing enzymes, or the H2S-producing enzymes, in colon cancer cells, as well as in many other forms of cancer. With respect to NO-producing enzymes, previous studies have established that in many tumors, including colon cancer [33,34] and breast cancer [35,36] iNOS expression is increased; in other tumors (e.g. melanoma, myeloma) iNOS and nNOS are upregulated [37,38], while in renal cell carcinoma and sarcoma iNOS and eNOS levels were elevated [39,40]. In the colon cancer samples analyzed in the current study, we found that tumor contained higher expression of eNOS, while iNOS and nNOS expression levels were not statistically different. In contrast, HCT116 cells contained lower expression of eNOS than the control comparator NCM356 epithelial cells cells, while iNOS and nNOS expression levels were not statistically different between the two cell lines.
With respect to CO-producing enzymes, previous studies are fairly uniform in demonstrating that HO-1 is upregulated in many forms of tumors, including colon cancer [41,42], breast cancer [43,44], melanoma, sarcoma and others [1,45,46]. In the primary colon cancer tissues studied in the present report, about half of the individual sample pairs showed increases in HO-1 in the tumors, and the other half showing decreases. These data indicate that HO-1 expression levels are likely heterogeneous in colon cancer, possibly depending on the tumor subtype, stage, perhaps the concomitant therapies and other factors - all of which remain to be studied in future experiments. As far as the NCM/HCT comparison is concerned, expression of HO-1 was markedly higher in the HCT116 cells than in NCM356 cells, but the expression of HO-2 was also higher in HCT116 cells than in NCM356 cells.
With respect to the expression of the H2S-producing enzymes, previous studies have demonstrated the upregulation of CBS, CSE and/or 3-MST in many forms of tumors, including colon cancer [9], lung cancer [2], ovarian cancer [47], liver cancer [48] and others [1]. In the current report – in line with our previous results in a smaller, different set of patient specimens [9] – CBS (but not CSE or 3-MST) showed a significant increase between the tumor tissue and the surrounding normal mucosa, but there was also a trend for elevation of 3-MST in about 50% of the samples analyzed. In line with the findings in primary tissues, expression of CBS and 3-MST was also significantly higher in HCT116 cells when compared to NCM356 cells.
Taken together, these data show that some (but not all) NO, CO and H2S-producing enzymes are elevated in colon cancer tissues from patients. It is likely that the expression of these enzymes is not only dependent on cancer type, but it is also likely to be dependent on the patient population (geographical origin of the patients, stage of the disease, concomitant therapies etc.).
The HCT/NCM comparison did not mirror the patterns of the expression of the various enzymes studied in the primary human samples, although there were some notable overlaps (HO-1 expression, CBS expression, and perhaps 3-MST expression). It is a limitation of the current study that we have only studied one single human colon cancer cell line. Further work, in additional colon cancer cell lines, is likely to provide a more comprehensive picture of the expression patterns, even though it is our opinion that studies using primary human tumor samples are more likely to be clinically more relevant than additional expression analysis in further cell lines.
In the case of NO-producing enzymes, the HCT/NCM comparison was highly discordant from the analysis of the primary human samples (i.e. in the primary tumors, eNOS was increased, while in the cell line comparison, eNOS was decreased). We speculate that this particular difference in eNOS expression between primary tumors vs. HCT116 cells may be due to the fact that the primary human tissue likely contains various cell types (including microvessels, due to tumor cell angiogenesis, which likely increases the amount of eNOS the homogenate contains). In fact, it is a limitation of all studies using primary tumor homogenates, that the material consists of multiple cell types (tumor cells, tumor-associated fibroblasts, tumor-infiltrating immune cells, vascular cells, etc).
4.2 Pharmacological modulation of NO-, CO- and H2S-producing enzymes in HCT116 cells
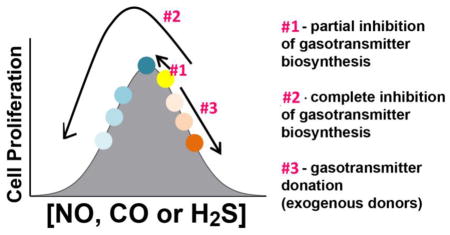
Next, we studied the effect of pharmacological inhibitors of NO-, CO- and H2S-producing enzymes, as well as NO-, CO- and H2S donors on HCT116 cell proliferation and MTT conversion. The results obtained, for each gasotransmitter, were consistent with our working hypothesis [1] and suggest that all three gasotransmitters, at the optimal concentration - i.e. when produced endogenously by the tumor cells - contribute to the maintenance of cellular energetics and/or cell proliferation pathways. Inhibitors of each gasotransmitter (e.g. the NOS inhibitor L-NMMA or the HO inhibitor ZnPPIX, or the CBS/CSE inhibitor AOAA) exerted concentration-dependent inhibitory effect on HCT116 cell proliferation as well as MTT conversion at the highest concentrations used. Interestingly (and consistently for each gasotransmitter pathway), at the lowest concentration used, most of the inhibitors studied slightly increased proliferation. From these findings, we conclude that endogenously produced NO (most likely from eNOS and/or iNOS), CO (most likely from HO-1) and H2S (most likely from CBS) regulate cell proliferation via a bell-shaped concentration-response curve, where in resting cells, the cells are “to the right” from the peak of the bell-curve, and therefore a small (partial) inhibition of the respective enzymatic activity, is pro-proliferative, while additional (more complete) inhibition of the activity of the same enzyme exerts antiproliferative effects (Fig. 15).
Figure 15. Working hypothesis: consequences of the bell-shaped pharmacological profile of NO, CO and H2S on the proliferation of HCT116 cells.

Optimal concentrations of nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S), produced endogenously by NO synthases (eNOS, iNOS, nNOS), heme oxygenases (HO-1 and HO-2) and the H2S-producing enzymes (primarily cystathionine-β-synthase [CBS] and 3-mercaptopyruvate sulfurtransferase, 3-MST), respectively, support tumor growth and proliferation. Each of the three gasotransmitters regulates tumor cell proliferation according to a bell-shaped concentration-response curve. The data obtained in the current study can be best interpreted if we hypothesize that the resting point of the tumor cells, under control conditions, is to the right from the top of the bell-shaped curve. Partial inhibition of NO, CO or H2S production moves the cells towards the top of the curve, thereby explaining the slight enhancement of cell proliferation seen with low inhibitor concentrations (Arrow #1). More complete inhibition of NO, CO or H2S production moves the cells down on the left shoulder of the curve thereby explaining the dose-dependent inhibition of cell proliferation seen at higher inhibitor concentrations. (Arrow #2) (At the highest inhibitor concentrations, the inhibitors may also affect other cellular processes, best exemplified by the effects of ZnPPIX in the current study). Donation of excess NO, CO or H2S (in addition to what the cells already produce endogenously) production moves the cells down on the right shoulder of the curve (Arrow #3), thereby explaining the concentration-dependent inhibition of cell proliferation seen with donors, and also explaining why low concentrations of the donors - as opposed to the inhibitors - do not increase cell proliferation.
The results of the follow-on studies - with pharmacological donors of NO, CO or H2S - on HCT116 cell proliferation and mitochondrial activity - produced additional findings that further support our working hypothesis. All donors tested induced a concentration-dependent suppression of cell proliferation and MTT conversion, which we interpret as follows: these three gasotransmitters, at the optimal concentration contribute to the maintenance of cellular energetics and/or cell proliferation pathways; elevation of their concentrations beyond this (endogenously maintained) optimum level moves the gasotransmitter balance to the “right shoulder” of the concentration-response curve and may become anti-proliferative. The bell-shaped concentration-response concept and the effects of the various pharmacological modulators is summarized in Fig. 15. To our knowledge, this is the first study where all three gasotransmitters have been studied simultaneously in the same cell line, and where both donors and inhibitors have been tested; the scheme in Fig. 15 puts all of these findings into a coherent concept, and explains the seemingly paradoxical findings that both inhibitors and donors of the same pathway can have, ultimately, the same functional effect on the proliferation of cancer cells.
4.3. Analysis of the effector pathways downstream from gasotransmitters in HCT116 cells
In order to investigate the pathways responsible for the maintenance of HCT116 cell proliferation, we have conducted two types of experiments: (a) gasotransmitter combination studies and (b) specific proliferation pathway analysis. In the combination studies, we have determined the effect of combined pharmacological inhibition of NO-, CO- and H2S-producing enzymes, or the effect of combined pharmacological donation of NO-, CO- and H2S on HCT116 cell proliferation and mitochondrial activity. Part of the reason for these studies was practical (combinations that may be most efficacious in terms of antiproliferative effects may have future translational potential) and part mechanistic, the working hypothesis being that if the various inhibitors act on pathways that are identical or converge on common effectors, then combination of these agents will not have any marked synergistic effects. If, however, each of these agents mobilizes their own distinct downstream pathways, then additive or synergistic combination effects may be more likely. The conclusion of the combination studies was, that, for most part, the combination of two donors or two inhibitors was not more potent in terms of antiproliferative action than the effect of either of the single agent. These findings suggest that the downstream processes of the three gasotransmitters may converge on common effectors. It should be noted, nevertheless, that combination studies of this type are inherently difficult to interpret, because there is a great deal of interactions between the various gasotransmitters and the enzyme systems that produce them. For example, CO, as well as NO, can inhibit CBS, and thereby, can suppress endogenous H2S biosynthesis [49,50]; H2S can inhibit iNOS expression [51] and eNOS activity [52,53] and thereby H2S donation may suppress endogenous NO production.
As far as specific proliferation pathway effectors, we have selected pathways that are commonly known to be involved in cancer cell proliferation - e.g. PI3K/Akt pathway, the p44/42 MAPK (Erk1/2) pathway - as well as a pathway that are common for gasotransmitter effects, the cGMP/VASP pathway [1,21–30]. Unexpectedly, in the tumor tissues and HCT116 cells, we detected lower phosphorylation of Akt and of p44/42 MAPK than in their corresponding controls. The only pathway that showed elevation was the activation of VASP (evidenced by VASP phosphorylation) in the HCT116 cells. Thus, perhaps surprisingly, the activity of most of the canonical cancer proliferation pathways was not found to be higher in tumors than in normal control tissues - at least in the current experimental conditions. This finding, nevertheless, does not exclude the possibility that these pathways play significant roles in the stimulation of colon cancer cell proliferation, probably in synergy with various other pathways. Therefore, we have tested the effect of the respective pharmacological inhibitors of these pathways on HCT116 cell proliferation. Inhibition of PI3K, Akt and of ERK1/2 all inhibited HCT116 cell proliferation, and so did the pharmacological inhibitor of the soluble guanylate cyclase activation.
The question was whether endogenous NO, CO and/or H2S production play a constitutive roles in the maintenance of the activation of the above-mentioned proliferation pathways. If this was the case, pharmacological inhibition of NO, CO and/or H2S production would be expected to inhibit the constitutive activity of these pathways. However, this was not the case for most of these pathways; the only pathway where an effect was noted was the VASP pathway, where two inhibitors, the NOS inhibitor L-NMMA and the CBS/CSE inhibitor AOAA suppressed VASP phosphorylation. These observations are consistent with the fact that the NO and the H2S pathways converge at the level of cGMP and VASP through multiple mechanisms including (a) stimulation of eNOS phosphorylation and elevation of eNOS activity by H2S and (b) activation of the guanylate cyclase system (by NO) and simultaneous stabilization of intracellular cGMP levels by H2S (via inhibition of cGMP phosphodiesterase) [30,31,54].
4.4. Conclusions and implications
In conclusion, the current study shows that some - but not all - of the enzymes responsible for NO, CO and/or H2S production are upregulated in colon cancer cells. The data also show that this upregulation depends on the experimental system used (i.e. human colon cancer biopsies vs. a human colon cancer cell line). The findings with the pharmacological donors and inhibitors are consistent with the hypothesis that the three gasotransmitters plays bell-shaped roles in the control of HCT116 cell proliferation: endogenously produced low-to-mid concentrations of H2S, NO or CO support HCT116 proliferation (and, consequently, inhibition of either of them suppresses this response), while exogenous delivery of either of the gasotransmitters H2S, NO or CO (using their respective pharmacological donors) can also suppress the proliferation of the HCT116 colon cancer cells (Fig. 15).
Based on the results with their respective pharmacological inhibitors, we also conclude that the Akt, p44/42 MAPK (Erk1/2) and/or cGMP/VASP pathways - obviously, in addition to a wide variety of additional pathways and mechanisms not studied here - appear to be functionally relevant in the maintenance of HCT116 cell proliferation (although they do not appear to be activated compared to control/non-transformed cells). In this respect, however, it should be pointed out that the NCM356 cells have an adenoma-type phenotype, and therefore these cells may also have some signaling pathways activated (compared to healthy normal epithelial cells). In fact, engineering a single gasotransmitter-producing enzyme, CBS, into NCM356 cells is sufficient to induce an invasive phenotype [32].
Endogenous production of NO or H2S appears to support the constitutive activity of the cGMP/VASP pathway in HCT116 cells. Pharmacological inhibition of this pathway should be further explored in the experimental therapy of colon cancer.
The current study not only puts all three gasotransmitters into a logical mechanistic framework with respect to the regulation of colon cancer cell proliferation, but also underlines the translational potential of pharmacological modulation of gasotransmitters (whereby either NO, CO or H2S donors or NOS, HO-1 or CBS or 3-MST inhibitors) may have the potential to suppress colon cancer cell proliferation. Obviously - and as discussed recently [1] - each of these approaches has advantages and potential drawbacks including selectivity issues, delivery issues and potential issues with respect to their feasibility for human therapeutic translation. Nevertheless, it is hoped that the current report helps to clarify some of the controversies and conceptual paradoxes in the field of gasotransmitters and cancer and serves as a useful point to stimulate further work in this field.
Figure 9. Effect of AOAA (150 μM), ZnPPIX (20 μM) or their combination on HCT116 cell viability.

HCT116 cells were treated with the pharmacological agents and at 48 h analyzed for various cell populations using flow cytometry. Top panels show individual cell analysis, bottom bars show summary data.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 CA175803) and the Cancer Prevention Research Institute of Texas (DP150074). AP39 and GYY4137 were kindly provided by Dr. Matt Whiteman (University of Exeter).
Abbreviations
- 3-MST
3-mercaptopyruvate sulfurtransferase
- 3B7NI
3-bromo-7-nitroindazole
- AOAA
aminooxyacetic acid
- CBS
cystathionine-β-synthase
- CO
carbon monoxide
- CSE
cystathionine gamma lyase
- eNOS
endothelial nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- GYY
GYY4137 (P-(4-methoxyphenyl)-P-4-morpholinyl-phosphinodithioic acid)
- H2S
hydrogen sulfide
- HMPSNE
2-(4-hydroxy-6-methylpyrimidin-2-yl)sulfanyl]-1-(naphthalen-1-yl)ethan-1-one
- DETA-NO
diethylenetriamine nitric oxide adduct
- HO
heme oxygenase
- iNOS
inducible nitric oxide synthase
- INT
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride
- L-NIO
N5-(1-iminoethyl)-L-ornithine
- L-NMMA
NG-methyl-L-arginine
- L-NPLA
L-Nω-propyl-arginine
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- NAD+
nicotinamide adenine dinucleotide
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a] quinoxalin-1-one
- PAG
DL-propargylglycine
- PI3K
phosphoinositide 3-kinase
- PLP
pyridoxal phosphate
- SNAP
S-nitroso-N-acetyl-D,L-penicillamine
- TINPP
tin-protoporphyrin IX
- ZnPPIX
zinc-protoporphyrin IX
Footnotes
Author Disclosure Statement
C.S. and M.R.H. are founders and shareholders of CBS Therapeutics Inc., an UTMB spin-off company focusing on therapeutic approaches around H2S biosynthesis inhibition in cancer cells. For the other authors, no competing financial interests exist.
Author Contributions
GO – carried out molecular biology and pharmacological assays, prepared the figures and contributed to writing the paper. KM - contributed to experimental design, Western analysis and data interpretation. GT- contributed to experiments, data interpretation, data presentation, and manuscript writing. MRH – contributed to experimental design and data interpretation and manuscript writing. BS- contributed to experimental design, data interpretation and manuscript writing. CS – was responsible for the conceptual design of the study, planned and supervised the experiments and wrote the results/discussion section of the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov. 2016;15:185–203. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P, et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci Rep. 2016;6:36125. doi: 10.1038/srep36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 4.Chao C, Zatarain JR, Ding Y, Coletta C, Mrazek AA, Druzhyna N, et al. Cystathionine-β-synthase inhibition for colon cancer: enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol Med. 2016;22:361–79. doi: 10.2119/molmed.2016.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Southan GJ, Szabo C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol. 1996;51:383–94. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 6.Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;2012:318087. doi: 10.1155/2012/318087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bland-Ward PA, Moore PK. 7-Nitro indazole derivatives are potent inhibitors of brain, endothelium and inducible isoforms of nitric oxide synthase. Life Sci. 1995;57:PL131–5. doi: 10.1016/0024-3205(95)02046-l. [DOI] [PubMed] [Google Scholar]
- 8.Pittalà V, Salerno L, Romeo G, Modica MN, Siracusa MA. A focus on heme oxygenase-1 (HO-1) inhibitors. Curr Med Chem. 2013;20:3711–32. doi: 10.2174/0929867311320300003. [DOI] [PubMed] [Google Scholar]
- 9.Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. 2013;110:12474–9. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–32. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabo C, Papapetropoulos A International Union of Basic and Clinical Pharmacology. CII. Pharmacological modulation of hydrogen sulfide (H2S) levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol Rev. 2017 doi: 10.1124/pr.117.014050. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanaoka K, Sasakura K, Suwanai Y, Toma-Fukai S, Shimamoto K, Takano Y, et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci Rep. 2017;7:40227. doi: 10.1038/srep40227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25:674–8. [PubMed] [Google Scholar]
- 14.Keefer LK. Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annu Rev Pharmacol Toxicol. 2003;43:585–607. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- 15.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 16.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–43. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 17.Szczesny B, Módis K, Yanagi K, Coletta C, Le Trionnaire S, Perry A, et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–30. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowis D, Bugajski M, Winiarska M, Bil J, Szokalska A, Salwa P, et al. Zinc protoporphyrin IX, a heme oxygenase-1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer. 2008;8:197. doi: 10.1186/1471-2407-8-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Avery JE, Hannafon BN, Lind SE, Ding WQ. Zinc protoporphyrin suppresses cancer cell viability through a heme oxygenase-1-independent mechanism: the involvement of the Wnt/β-catenin signaling pathway. Biochem Pharmacol. 2013;85:1611–8. doi: 10.1016/j.bcp.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 20.Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine β-synthetase/hydrogen sulfide inhibition in cancer. Antioxid Redox Signal. 2015;22:424–48. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YL, Jiang SX, Yang YM, Xu H, Liu JL, Wang XS. USP22 acts as an oncogene by the activation of BMI-1-mediated INK4a/ARF pathway and Akt pathway. Cell Biochem Biophys. 2012;62:229–35. doi: 10.1007/s12013-011-9287-0. [DOI] [PubMed] [Google Scholar]
- 22.Wu DM, Zhang P, Liu RY, Sang YX, Zhou C, Xu GC, et al. Phosphorylation and changes in the distribution of nucleolin promote tumor metastasis via the PI3K/Akt pathway in colorectal carcinoma. FEBS Lett. 2014;588:1921–9. doi: 10.1016/j.febslet.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 23.Krech T, Thiede M, Hilgenberg E, Schäfer R, Jürchott K. Characterization of Akt independent effects of the synthetic Akt inhibitors SH-5 and SH-6 using an integrated approach combining transcriptomic profiling and signaling pathway perturbations. BMC Cancer. 2010;10:287. doi: 10.1186/1471-2407-10-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YS, Kim SY, Song SJ, Hong HK, Lee Y, Oh BY, et al. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget. 2016;7:36842–36853. doi: 10.18632/oncotarget.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Bian H, Li X, Wu H, Bi Q, Yan Y, Wang Y. Hydrogen sulfide promotes cell proliferation of oral cancer through activation of the COX2/AKT/ERK1/2 axis. Oncol Rep. 2016;35:2825–32. doi: 10.3892/or.2016.4691. [DOI] [PubMed] [Google Scholar]
- 26.Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3β/β-catenin pathways. Mol Cancer. 2017;16:70. doi: 10.1186/s12943-017-0629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berrie CP. Phosphoinositide 3-kinase inhibition in cancer treatment. Expert Opin Investig Drugs. 2001;10:1085–98. doi: 10.1517/13543784.10.6.1085. [DOI] [PubMed] [Google Scholar]
- 28.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 29.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–8. [PubMed] [Google Scholar]
- 30.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;109:9161–6. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu D, Hu Q, Ma F, Zhu YZ. Vasorelaxant effect of a new hydrogen sulfide-nitric oxide conjugated donor in isolated rat aortic rings through cGMP pathway. Oxid Med Cell Longev. 2016;2016:7075682. doi: 10.1155/2016/7075682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips CM, Zatarain JR, Nicholls ME, Porter C, Widen SG, Thanki K, et al. Cystathionine-β-synthase upregulation induces metabolic reprograming and promotes colon carcinogenesis. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-3480. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yagihashi N, Kasajima H, Sugai S, Matsumoto K, Ebina Y, Morita T, Murakami T, Yagihashi S. Increased in situ expression of nitric oxide synthase in human colorectal cancer. Virchows Arch. 2000;436:109–14. doi: 10.1007/pl00008208. [DOI] [PubMed] [Google Scholar]
- 34.Gochman E, Mahajna J, Shenzer P, Dahan A, Blatt A, Elyakim R, Reznick AZ. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem. 2012;114:827–35. doi: 10.1016/j.acthis.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Granados-Principal S, Liu Y, Guevara ML, Blanco E, Choi DS, Qian W, et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015;17:25. doi: 10.1186/s13058-015-0527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas DD, Wink DA. NOS2 as an Emergent Player in Progression of Cancer. Antioxid Redox Signal. 2017;26:963–965. doi: 10.1089/ars.2016.6835. [DOI] [PubMed] [Google Scholar]
- 37.Grimm EA, Ellerhorst J, Tang CH, Ekmekcioglu S. Constitutive intracellular production of iNOS and NO in human melanoma: possible role in regulation of growth and resistance to apoptosis. Nitric Oxide. 2008;19:133–7. doi: 10.1016/j.niox.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendes RV, Martins AR, de Nucci G, Murad F, Soares FA. Expression of nitric oxide synthase isoforms and nitrotyrosine immunoreactivity by B-cell non-Hodgkin’s lymphomas and multiple myeloma. Histopathology. 2001;39:172–8. doi: 10.1046/j.1365-2559.2001.01189.x. [DOI] [PubMed] [Google Scholar]
- 39.Cássio Zequi Sd, Fregnani JH, Favaretto RL, Costa WH, Madeira Campos RS, Fonseca FP, et al. The impact of immunohistochemical expression of nitric oxide synthases on clinical and pathological features of renal cell carcinoma. World J Urol. 2013;31:1197–203. doi: 10.1007/s00345-012-0878-1. [DOI] [PubMed] [Google Scholar]
- 40.Weninger W, Rendl M, Pammer J, Mildner M, Tschugguel W, Schneeberger C, et al. Nitric oxide synthases in Kaposi’s sarcoma are expressed predominantly by vessels and tissue macrophages. Lab Invest. 1998;78:949–55. [PubMed] [Google Scholar]
- 41.Becker JC, Fukui H, Imai Y, Sekikawa A, Kimura T, Yamagishi H, et al. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand J Gastroenterol. 2007;42:852–8. doi: 10.1080/00365520701192383. [DOI] [PubMed] [Google Scholar]
- 42.Kang KA, Maeng YH, Zhang R, Yang YR, Piao MJ, Kim KC, et al. Involvement of heme oxygenase-1 in Korean colon cancer. Tumour Biol. 2012;33:1031–8. doi: 10.1007/s13277-012-0336-0. [DOI] [PubMed] [Google Scholar]
- 43.Bahmani P, Hassanshahi G, Halabian R, Roushandeh AM, Jahanian-Najafabadi A, Roudkenar MH. The expression of heme oxygenase-1 in human-derived cancer cell lines. Iran J Med Sci. 2011;36:260–5. [PMC free article] [PubMed] [Google Scholar]
- 44.Duechler M, Peczek L, Zuk K, Zalesna I, Jeziorski A, Czyz M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology. 2014;219:158–65. doi: 10.1016/j.imbio.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Was H, Dulak J, Jozkowicz A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets. 2010;11:1551–70. doi: 10.2174/1389450111009011551. [DOI] [PubMed] [Google Scholar]
- 46.Hjortsø MD, Andersen MH. The expression, function and targeting of haem oxygenase-1 in cancer. Curr Cancer Drug Targets. 2014;14:337–47. doi: 10.2174/1568009614666140320111306. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia H, Ye J, You J, Shi X, Kang W, Wang T. Role of the cystathionine β-synthase/H2S system in liver cancer cells and the inhibitory effect of quinolone-indolone conjugate QIC2 on the system. Oncol Rep. 2017;37:3001–3009. doi: 10.3892/or.2017.5513. [DOI] [PubMed] [Google Scholar]
- 49.Vicente JB, Colaço HG, Mendes MI, Sarti P, Leandro P, Giuffrè A. NO binds human cystathionine β-synthase quickly and tightly. J Biol Chem. 2014;289:8579–87. doi: 10.1074/jbc.M113.507533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carballal S, Cuevasanta E, Marmisolle I, Kabil O, Gherasim C, Ballou DP, et al. Kinetics of reversible reductive carbonylation of heme in human cystathionine β-synthase. Biochemistry. 2013;52:4553–62. doi: 10.1021/bi4004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, et al. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med. 2006;41:106–19. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 52.Kubo S, Kurokawa Y, Doe I, Masuko T, Sekiguchi F, Kawabata A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology. 2007;241:92–7. doi: 10.1016/j.tox.2007.08.087. [DOI] [PubMed] [Google Scholar]
- 53.Geng B, Cui Y, Zhao J, Yu F, Zhu Y, Xu G, et al. Hydrogen sulfide downregulates the aortic l-arginine/nitric oxide pathway in rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1608–18. doi: 10.1152/ajpregu.00207.2006. [DOI] [PubMed] [Google Scholar]
- 54.Szabo C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications. Am J Physiol Cell Physiol. 2017;312:C3–C15. doi: 10.1152/ajpcell.00282.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]