

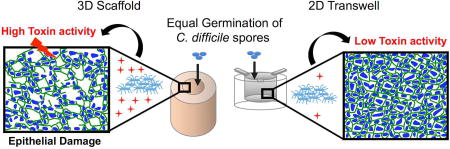
Abstract
Endospore-forming Clostridioides difficile is a causative agent of antibiotic-induced diarrhea, a major nosocomial infection. Studies of its interactions with mammalian tissues have been hampered by the fact that C. difficile requires anaerobic conditions to survive after spore germination. We recently developed a bioengineered 3D human intestinal tissue model and found that low O2 conditions are produced in the lumen of these tissues. Here, we compared the ability of C. difficile spores to germinate, produce toxin and cause tissue damage in our bioengineered 3D tissue model versus in a 2D transwell model in which human cells form a polarized monolayer. 3D tissue models or 2D polarized monolayers on transwell filters were challenged with the non-toxin producing C. difficile CCUG 37787 serotype X (ATCC 43603) and the toxin producing UK1 C. difficile spores in the presence of the germinant, taurocholate. Spores germinated in both the 3D tissue model as well as the 2D transwell system, however toxin activity was significantly higher in the 3D tissue models compared to the 2D transwells. Moreover, the epithelium damage in the 3D tissue model was significantly more severe than in 2D transwells and damage correlated significantly with the level of toxin activity detected but not with the amount of germinated spores. Combined, these results show that the bioengineered 3D tissue model provides a powerful system with which to study early events leading to toxin production and tissue damage of C. difficile with mammalian cells under anaerobic conditions. Furthermore, these systems may be useful for examining the effects of microbiota, novel drugs and other potential therapeutics directed towards C. difficile infections.
Keywords: Clostridioides difficile, Clostridium difficile, 3D intestinal tissue, 2D transwell, germination, toxin activity
Graphical abstract
Introduction
The anaerobic, spore-forming, rod-shaped, Gram-positive Clostridioides difficile, also known as Clostridium difficile, is a causative agent of antibiotic-induced diarrhea (1). In the past two decades, C. difficile has emerged as a major cause of nosocomial infection (1). In the US alone, C. difficile infections (CDI) are responsible for more than 25,000 deaths annually (2). The major risk factor for C. difficile disease is the use of antibiotics, which reduces the normal microbiota in the GI tract (1, 3) that typically suppress germination and growth of C. difficile spores in the colon (3–5). In the absence of the normal microbiota, germinated C. difficile replicates to higher levels in the GI tract, which is a prerequisite for pathogenesis and production of toxins (1, 3). Infections by C. difficile can be very recalcitrant to clearance by antibiotics for several reasons (1, 3). Spores are not sensitive to antibiotics so can remain in the GI tract for long periods. Most antibiotics used to treat C. difficile also continue to disrupt the microbiota that outcompete C. difficile, therefore preventing establishment of normal homeostasis of the gut microflora. As a result, continual C. difficile germination, replication and toxin production occur (1, 3, 4).
Visualizing and studying the interactions between epithelial cells and germinated C. difficile are extremely challenging in animal models (6–8). While recent advances in imaging have enhanced the sensitivity of detecting bacterial-cell interactions in vivo, these assays are still cumbersome and cannot be conducted for extended periods in live animals (9, 10). To define and study early interactions between C. difficile and the polarized intestinal epithelium, cell culture models are required. Even though C. difficile is able to germinate in an aerobic environment, it cannot grow and divide in the presence of oxygen (11). Because of this, studies of its interactions with epithelial cells in culture systems have been challenging since the bacterium and the host have different optimum oxygen requirements. Thus, much work has focused on studying the effects of purified C. difficile glucosyltransferase toxins, TcdA and TcdB on epithelial cells (12, 13). The enterotoxin TcdA and the cytotoxin TcdB are considered critical virulence factors of C. difficile. They function to inactivate Rho family GTPases leading to loss of intestinal barrier function and colonocyte death leading to diarrhea (1, 3). These studies have revealed critical facets of C. difficile toxin biology, but by their nature, such experiments do not permit detailed analysis of bacterial-epithelial interactions (12–14).
We have produced a 3D intestinal tissue model using tubular silk biomaterial scaffolds (15). Our previous work indicated that 3D tissue models developed anaerobic luminal environments when the tissues were placed in a vertical orientation and incubated in Eppendorf tubes (15). Here we show that these 3D tissue models can support C. difficile spore germination, growth, toxin production and epithelium damage over a course of 48 hours and thus provide a valuable model to study early pathogen-cell interactions including toxin production and epithelial damage.
Material and methods
3D intestinal tissue construction
3D intestinal tissues were constructed and maintained as previously described (15). Briefly, 3D porous silk scaffolds with hollow channels were prepared using a sequential six step process involving silk regeneration, cylindrical PDMS mold casting, insertion of Teflon-coated wires across the cylinder, application of the silk solution, lyophilization and β-sheet induction (15). The fabrication method resulted in a scaffold consisting of a hollow channel space (2 mm in diameter) and a bulk space around the channel that contained interconnected pores. Primary human intestinal myofibroblasts (H-InMyoFibs) (Cat # CC-2902, Lonza) were delivered into the spongy silk bulk using type I rat tail collagen 2.01 mg/ml (First Link, UK) and then human intestinal epithelial cell lines, Caco-2 (ATCC CRL-2102) and HT29-MTX cells (Public Health England Culture Collections, Salisbury, Great Britain) were seeded in the hollow channel of the 3D scaffolds at a ratio of 3:1. The 3D scaffolds seeded with intestinal cells were maintained routinely in co-culture media DMEM (Cat# MT10-013CV) and SMGM (Cat# CC-3182) at a ratio of 1:1. Media were changed every other day. The scaffolds were incubated horizontally for 3 weeks until robust intestinal tissue formed (15).
2D infection model
Transwell inserts of 0.4 µm pore size were coated overnight at room temperature with 50 µg/ml collagen dissolved in 60% ethanol. They were seeded with 1×105 cells/cm2 composed of a mixture of Caco-2 and HT29-MTX cells at a ratio of 3:1. Inserts were placed in 24-well plates seeded with 1×104 cells/well of human intestinal myofibroblasts. Media was replaced in the upper and lower chambers every other day and human intestinal myofibroblasts were replaced every 4 days. Transwells were maintained at 37°C in a CO2 incubator for 14 days in co-culture media DMEM:SMGM (1:1). Transwells were monitored for transepithelial electrical resistance (TEER) and were infected with C. difficile spores when the TEER reached 1000 Ohms.
Spore preparation
Spores of C. difficile non-toxigenic strain CCUG 37787 serotype X (ATCC 43603) (16) and the toxin producing UK1, a NAP1/027/BI-type hypervirulent strain (17), were prepared by culturing on SMC medium (1.5 g Tris, 1 g (NH4)2SO4, 5 g proteose peptone, 90 g peptone supplemented with 0.1% L-cysteine) for 7 days anaerobically. Spores were harvested from the plates and incubated in water overnight at 4°C. Spores were washed twice with 0.1% Tween 80 in PBS and then with PBS. Spores were diluted with 1% bovine serum albumin (BSA) in PBS, heated at 60°C for 10 minutes and dispersed by shaking in a Qialyzer for 90 seconds, prior to inoculation of scaffolds.
To determine the germination efficiency of C. difficile spores in the absence of taurocholate, spores were diluted with pre-reduced PBS containing 0.1% cysteine and plated on BHIC plates with and without 0.1% sodium taurocholate (Na-TA) (Sigma-Aldrich).
Infecting and collecting samples for C. difficile CFU and toxin activity levels
Scaffolds and transwells were infected with a total of 5.0×106 spores in co-culture media containing 2 mM Na-TA. To inoculate the scaffolds with spores, the medium was removed from the hollow channel. The horizontally positioned scaffolds were inoculated with 50 µl containing 2.5×106 spores that were directly pipetted into the lumen and incubated at 37°C in a CO2 incubator for 15 min. To ensure an even distribution of the spores in the hollow channel, scaffolds were then flipped 180° and infected with an additional 2.5×106 spores in 50 µl followed by incubation at 37°C in a CO2 incubator for an additional 15 minutes. At the end of the infection period, scaffolds were placed vertically in 2 ml sterile Eppendorf tubes and submerged in 1.5 ml co-culture medium containing 2 mM Na-TA. The scaffolds were incubated at 37°C in CO2 incubator for 4, 24 or 48 hours. At the designated time points, a subset of scaffolds was moved to an anaerobic chamber and 100 µl was removed from the lumen; 10 µl was used for plating and 90 µl was stored at −80°C for later analysis of toxin activity as described below. To quantify vegetative cells recovered from scaffolds, samples were serially diluted with pre-reduced PBS containing 0.1% cysteine and plated on BHIC plates without taurocholate.
For inoculation of transwells, medium was removed from the transwell and 5.0×106 spores in 100 µl of co-culture media containing 2 mM Na-TA were added to the transwell surface adjacent to the differentiated monolayers of Caco-2:HT29 cells formed on the transwell filter, then covered and incubated at 37°C in a CO2 incubator for 30 minutes. An additional 400 µl of co-culture medium containing 2 mM Na-TA was added to the apical side of the transwells. Transwells were sampled for germinated C. difficile and toxin activity after 4, 24 and 48 hours by collecting 100 µl from the surface of the transwell insert; 10 µl was used for plating and 90 µl was stored at −80°C for later analysis of toxin activity described below.
After sampling for germinated C. difficile and toxin activity, scaffolds and transwells were washed 3 times with PBS then fixed in 4% paraformaldehyde (PFA) for 30 minutes at room temperature followed by washing three times with PBS and then stored in PBS at 4°C until they were processed for imaging.
Toxin Cell Rounding Assay
Toxin activity was measured by detecting mammalian cell rounding, as described (18), with the following modifications. Mouse Embryonic Fibroblast cells (MEF) (ATCC SCRC-1040) were seeded in 96-well flat-bottom microtiter plates at a density of 1×104 cells/well in 100 µl of DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin and incubated overnight at 37°C in a CO2 incubator. Medium collected from each scaffold lumen or transwell at the indicated time points was diluted 3-fold and serial 3-fold dilutions were added to MEF cell monolayers. Plates were incubated at 37°C in 5% CO2 incubator overnight. Samples were scored for toxin activity by monitoring the degree of cell rounding visually using a 10× objective 24 hours after incubation. Toxin activity is expressed as the log of the reciprocal of the highest dilution necessary to result in 100% rounding of the MEF cells.
Immunofluorescence and confocal imaging
Silk scaffolds and transwells were permeabilized using 0.1% Triton X-100 in phosphate-buffered saline (PBS, Gibco), then blocked with 5% BSA (Sigma) for 1 hour. The specimens were incubated overnight at 4°C with anti-human Zona Occludens-1 (ZO-1) (Invitrogen Cat# 339100), then immersed in Alexa Fluor 488 donkey anti-mouse (Invitrogen, Cat# A-21202) at a dilution of 1:200. Scaffolds and transwells were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Invitrogen) before being mounted using Vectashield mounting medium (Vector Laboratories) for scaffolds and Pro-Long Gold (Invitrogen, Cat# P36930) for transwells. The 3D scaffolds and 2D transwell inserts were scanned using a confocal Nikon A1R (Nikon Instruments Inc.) with Z-series capability using a filter set for DAPI (Ex/Em: 350/470nm) and GFP/FITC (Ex/Em: 488/514 nm). Confocal 3D maximum projection images were assembled with a NIS-Elements AR software package (ver 4.20.01, Nikon) and ImageJ.
Pictures from scaffolds and transwells were taken and graded blindly for tight junction (TJ) disruption and monolayer integrity (MLI) by three investigators using a scale from 0–3. For tight junction integrity as assessed by ZO-1 staining, a score of 0 indicated TJ were observed between all cells in the field; 1, a few cells lacking TJ; 2, 25–50% of cells lacking TJ; 3, greater than 50% lacking TJ. For estimation of MLI, as assessed by DAPI staining, a score of 0 indicated no gaps between cells; 1, a few gaps; 2, several gaps making up 25–50% of the monolayer; 3, >50% gaps in visualized tissue.
Scanning Electron Microscopy (SEM)
3D intestinal tissues were cross-linked with 2.5% glutaraldehyde (19), followed by progressive dehydration with a graded series of ethanol solutions (30%, 50%, 75%, 95% and twice in 100%, 30 minutes at each concentration). The samples were subsequently dried by critical point drying with a liquid CO2 dryer (AutoSamdri-815, Tousimis Research Corp.). Prior to imaging using a scanning electron microscope (Zeiss UltraPlus SEM or Zeiss Supra 55 VP SEM, Carl Zeiss SMT Inc.) at a voltage of 2 ~ 3 kV, the samples were coated with a thin layer (10 nm thick) of Pt/Pd using a sputter coater (208HR, Cressington Scientific Instruments Inc., Cranberry Twp).
Statistical Analysis
Statistical analyses were performed by GraphPad Prism version 7 (GraphPad Software, San Diego, CA).
Results
C. difficile spores germinate in 3D tissue scaffolds
3D tissue models designed to mimic aspects of the human intestine topology were described in our previous communication (15) and are illustrated in Figure 1. Briefly, the hollow chamber of silk scaffolds was seeded with Caco-2 and HT29 cells while the outer surface was seeded with human myofibroblasts (Fig. 1). Building on the observation that these 3D tissue scaffolds generate an anaerobic lumen when incubated vertically (15), we inoculated the 3D scaffolds with 5×106 spores of either the non-toxigenic (NT) C. difficile strain CCUG 37787 serotype X (ATCC 43603) (16) or the NAP1/027/BI-type hypervirulent toxin-producing strain UK1 (17). The inoculated 3D scaffolds were incubated vertically in tissue culture medium containing the spore germinant, sodium taurocholate. In the absence of taurocholate, less than 0.01% of the NT and UK1 spores formed colonies (i.e., germinated and multiplied) on BHIC plates. At 4, 24 and 48 hours post-inoculation, medium was withdrawn from the lumen of the scaffolds under anaerobic conditions, diluted and plated on BHIC medium lacking taurocholate. For comparison, we also infected polarized monolayers, composed of Caco-2 and HT29 cells on transwell filters and human myofibroblast cells seeded in the lower compartment, with NT and UK1 spores (Fig. 1).
Figure 1. Schematic of C. difficile infection in the 3D scaffold tissue model and the 2D transwell system.

3D scaffolds were seeded with a mixture of Caco-2 and HT29 cells in the hollow channel, while the porous exterior bulk was seeded with human myofibroblasts. For comparison, the commonly used transwell inserts were seeded with the same mixture of Caco-2 and HT29 on the filters, and human myofibroblasts were added in the bottom of the well. Spores (5×106) of the non-toxigenic C. difficile strain ATCC 43603 (NT) or the toxin-producing strain UK1 were used to inoculate the channels of scaffolds or the surfaces of transwell filters. After 4, 24 and 48 hours, samples from each system were collected for analysis of bacterial growth, toxin activity and tissue damage.
After 4 hours of incubation in the 3D scaffolds, the mean vegetative CFU was 2.9×106 CFU/scaffold and 2.7×106 CFU/scaffold for the NT and UK1 strains respectively (Fig. 2A). C. difficile vegetative cells persisted in the lumens of 3D scaffolds throughout the next 44 hours for both strains, with the NT and UK1 strains reaching an average of 1.8×107 CFU/scaffold and 7.3×106 CFU/scaffold, respectively at 48 hours (Fig. 2A). Spores seeded in the 2D transwell system germinated after 4 hours with a mean of 9.3×106 CFU/transwell for the NT strain and 3.5×106 CFU/transwell for the UK1 strain, however, no further increase was detected for the next 44 hours (Fig. 2B). Importantly, by 48 hours, viable vegetative C. difficile cells were recovered at similar levels both in the 3D scaffolds and 2D transwells with no significant difference when comparing either strains between the two systems. (CFU of NT at 48 hours in 3D Scaffold versus the 2D transwell p =0.07; and UK1 at 48 hours in 3D Scaffold versus the 2D transwell p =0.5, using Mann-Whitney test).
Figure 2. C. difficile spores germinate in 3D scaffolds and 2D transwells.

Spores (5×106) of the non-toxigenic strain ATCC 43603 (NT) or the toxin-producing strain UK1 were used to inoculate the channels of 3D scaffolds or the surfaces of 2D transwells. Samples were collected from (A) 3D scaffolds and (B) 2D transwells after 4, 24 and 48 hrs, under anaerobic conditions and plated on BHIC plates in the absence of taurochlorate to measure viable vegetative C. difficile cells. Each dot represents the result from an individual scaffold or transwell; the horizontal bars indicate the geometric mean. Statistical significance between the time points for each strain in either system was determined by One Way ANOVA analysis with Tukey’s multiple comparisons post-test (*p=0.0412, **p=0.0064, *** p<0.0001). Only statistically significant comparisons are shown. Composite results are shown for at least 2 independent experiments for the NT strain and at least 3 independent experiments for the UK1 strain using 2–3 replicates per experiment.
A significantly higher toxin activity detected in the 3D tissue scaffolds compared to 2D polarized epithelial cells
To determine whether vegetative C. difficile cells produced toxin in these model systems, medium from both systems was collected from within the luminal space of the 3D scaffolds or the surface of the insert of the 2D transwell system, diluted, and applied to Mouse Embryo Fibroblast (MEF) cells. Toxin activity was defined as the log of the reciprocal of the highest dilution that resulted in 100% cell rounding of the MEF cells (Fig. 3). At 4 hours post-inoculation of 3D scaffolds and 2D transwells, cell-rounding activity was not detected in any samples (data not shown), although C. difficile spores had germinated (Fig 2). At 24 hours post-infection, cell-rounding activity was detected in 43% of supernatants from the UK1-infected 3D scaffolds but not in any of the supernatants from 2D transwell samples infected with UK1 (Fig. 3A). At 48 hours, cell-rounding activity was detected in 100% of the UK1 inoculated 3D scaffolds and in 33% of the 2D transwell samples (Fig. 3B). Noteworthy, the toxin activity was significantly higher in the 3D scaffold samples than in the 2D transwells. Culture medium collected from either system infected with the NT strain showed no MEF cell rounding, suggesting that cytotoxicity was due to toxin production from UK1. These results indicate that although UK1 germinated and persisted in both systems (Fig. 2), the 3D scaffold environment supported a higher toxin activity compared to the 2D transwell system (Fig. 3). Combined, these data are consistent with the hypothesis that the low oxygen tension produced in the lumen of 3D tissue scaffolds allows C. difficile spores to germinate and produce toxins.
Figure 3. A significantly higher toxin activity detected in 3D scaffolds compared to 2D transwells.

Toxin activity was measured by a MEF cell rounding assay. Serial three-fold dilutions of supernatants from 3D scaffolds (SC) or 2D transwells (TW) infected with NT or UK1 C. difficile spores for (A) 24 hrs or (B) 48 hrs were applied to MEF cells for 24 hrs and scored visually at 10× magnification for 100% cell rounding. The log of the inverse of the highest dilution that produced 100% cell rounding of the MEF cells is plotted. Each dot represents the result from an individual scaffold or transwell; the horizontal bars indicate the mean; the horizontal dashed line indicates the limit of detection. Statistical significance between the supernatants from the SC and TW for each strain at each time point was determined by the Mann-Whitney t-test (*p=0.02, and **p<0.0001).
After incubation with C. difficile spores, more damage to the intestinal epithelium occurs in the 3D tissue scaffolds than in 2D polarized epithelial cells
Damage to the epithelium is the hallmark of C. difficile infection in the intestine. Several studies have shown that C. difficile toxins cause disruption of tight junction proteins leading to cell barrier disruption (20–22). We next investigated whether the germinated C. difficile cells caused damage to the intercellular tight junctions (TJ) of the intestinal epithelium cells in these model systems. Tight junction protein ZO-1 was visualized in uninfected controls and spore-exposed 3D scaffolds and 2D transwells by immunofluorescence using confocal microscopy at 4 and 48 hours post-inoculation (Fig. 4). In uninfected 3D scaffolds and 2D transwells, ZO-1 protein was observed at the margins of individual cells in a typical chicken wire-like pattern (Fig. 4 A, D, G, J), indicating intact tight junction complexes. Four hours following inoculation with NT or UK1 spores, minimal changes in ZO-1 distribution were observed in both systems compared to the uninfected controls (Fig. 4 A–C, G–I and M). At 48 hours, 3D scaffolds and 2D transwells inoculated with NT spores displayed similar ZO-1 staining pattern as the corresponding uninfected controls in each system (Fig. 4 D, E, J, K and N). However, the level of damage to TJ with the toxin-producing UK1 strain at 48 hours in the scaffold system was significantly higher compared to uninfected scaffolds, scaffolds infected with the NT strain, and transwells infected with the UK1 or NT strains (Fig. 4 D–F, K–L, and N). Assessment of monolayer integrity (MLI) by DAPI staining for the presence of epithelium mirrored the results of TJ disruption (Fig. 4). At 48 hours, significantly more monolayer disruption was observed in the 3D scaffolds inoculated with UK1 spores than in the 2D transwells (Fig. 4 D–F, J–L and P). Importantly, in the 3D scaffolds, the level of damage as assessed by both loss of tight junction and monolayer disruption significantly correlated with the toxin activity levels (TJ; r =0.97, p =0.03, MLI; r =0.95, p =0.04) but not with the CFU (TJ; r =0.5, p =0.45, MLI; r =0.3, p =0.7). Likewise, the few 2D transwells that had detectable toxin activity also displayed a higher damage score, and, similar to the 3D scaffolds, correlated significantly with toxin activity levels (TJ; r =0.97, p=0.0002, MLI; r =0.96, p =0.0006) but not with the CFU (TJ; r =0.67, p =0.096, MLI; r =0.6, p =0.1).
Figure 4. C. difficile infection causes more damage in 3D scaffolds compared to 2D transwells.



Uninfected 3D scaffolds (A and D) or 2D transwells (G and J), NT C. difficile infected 3D scaffolds (B and E) or 2D transwells (H and K) and UK1 C. difficile infected 3D scaffolds (C and F) or 2D transwells (I and L) were fixed at 4hrs (SC: A–C and TW: G–I), and 48 hrs (SC: D–F and TW: J–L) and immunostained with antibody against ZO-1 (Green) and with DAPI (Blue). Samples were visualized by confocal microscopy. Representative images of 3D scaffolds and 2D transwells are shown at 20× magnification and 40× magnfication, respectively. White arrows indicate intact tight junctions. The extent of tight junction loss (M–N) and monolayer disruption (O–P) was quantified by three individuals who scored blinded samples. The scores were averaged for each sample and scores for each infection condition at each time point were averaged. The average and SD for tight junction loss (M–N) and monolayer disruption (O–P) are presented. Statistical significance was calculated using the Kruskal-Wallis test followed by Dunn’s post-test. Only statistically significant comparisons are shown (* p= 0.03, **p= 0.01, ***p=0.003,****p= 0.0009).
Morphological changes to intestinal cells and bacteria in the 3D tissue scaffolds were examined in 3D scaffolds infected with UK1 spores at 4, 24 and 48 hours after inoculation using scanning electron microscopy (Fig. 5). Four hours after inoculation, C. difficile spores of UK1 adhered to the intestinal epithelium and rod-shaped bacteria were detected (Fig. 5D). At 24 hours post inoculation, both small clumps of rod-shaped bacteria and some rounded epithelial cells were detected (Fig. 5E). At 48 hours, the epithelial cells were largely absent from the infected tissues (Fig. 5F) indicating extensive tissue damage and consistent with DAPI staining (Fig. 4F). Collectively, these data demonstrated that despite the fact that the CFU of UK1 in both systems were similar at 48 hours, only the UK1 spores inoculated into 3D scaffolds produced high toxin activity levels and these toxin activity levels significantly correlated with damage.
Figure 5. Scanning electron microscopy of 3D scaffolds infected with UK1 C. difficile for different times.

Scaffolds were visualized by scanning electron microscopy at different times after infection with 5×106 C. difficile UK1 spores. (A–C) uninfected controls; (D–F) scaffolds infected with UK1 spores. Samples were collected for SEM at 4 hrs (A and D), 24 hrs (B and E) or 48 hrs (C and F). White arrows indicate germinated spores. Samples shown are representative of at least 4 scaffolds examined at each time point from at least two independent experiments.
DISCUSSION
C. difficile infection of and damage to the intestinal epithelium is a multi-faceted process and many of the steps remain poorly understood at a molecular mechanistic level. While previous work has shown that C. difficile cells bind to polarized Caco-2 and HT29 cells (19), investigation into subsequent events has been hampered because C. difficile requires anaerobic conditions to grow whereas intestinal epithelium requires oxygen. Here we demonstrate that infection with C. difficile spores of 3D bioengineered tissues based on silk scaffolds seeded with colonic intestinal-like cells (15) led to germination of the bacteria, toxin production and epithelial tissue damage. C. difficile spores germinated in both the 3D scaffold and 2D transwell systems in the first 4 hours (Fig. 2A–B) with no significant difference in their overall levels at 48 hours between the two systems for each strain. Strikingly, despite similar CFU at 48 hours, a significantly higher toxin activity was detected in the UK1-infected 3D scaffolds than in the UK1-infected 2D transwells (Fig. 2A–B, Fig. 3). Likewise, epithelium damage reflected by the loss of TJ and monolayer disruption in the UK1-infected 3D scaffolds was significantly higher compared to the UK1-infected 2D transwells. Importantly, no significant damage was detected when these models were inoculated with a non-toxigenic strain even though this strain germinated at levels comparable to or higher than those for the UK1 strain. Since the NT strain is not an isogenic mutant of the UK1 toxin producing strain, the observed epithelium damage by the UK1 strain could be driven via toxin-dependent and/or toxin- independent mechanisms. However, because a significant correlation was found between tissue damage and the level of toxin activity detected, but no correlation was observed with CFU recovered, we favor the hypothesis that the toxin(s) contributed to cell damage. Supporting this, damage was only observed in the few UK1-infected 2D transwells that had low but detectable levels of toxin activity. In summary, the 3D scaffold system supports an environment that not only allows C. difficile to germinate, but also to produce toxin and cause damage.
Our 3D-intestinal tissue model joins several other recently developed systems that use Ussing chambers or organoids to study C. difficile infection in intestinal tissue models (23–26). Each system has advantages and limitations. Recent advances using an Ussing chamber system to create an apical anaerobic environment and an oxygen- containing basolateral environment with polarized epithelial cells showed that these epithelial cells had much stronger responses to C. difficile infection than when polarized cells were infected in an aerobic environment (25). The study by Jafari et al., using polarized T84 cells grown in this chamber highlights the importance of studying C. difficile under anaerobic conditions by showing that vegetative C. difficile elicits significantly higher cytokine responses, tight junction disruption and greater loss of TEER after an 8 hour incubation under anaerobic conditions versus a standard aerobic transwell system (25). Furthermore, several studies have reported infections of C. difficile in organoid and enteroid systems (23, 24, 26). Infection of human organoids (derived from embryonic stem cells) and colonoids (derived from adult pluripotent stem cells of the colon) provides the opportunity to study the interaction of C. difficile with a greater variety of cells found in the colon (23, 24, 26–28). Importantly, one can study the interactions of C. difficile from organoids from donors of different genetic backgrounds and donors with diseased colons and measure a wider variety of host responses (23, 24). A recent study employing organoids to study C. difficile reported disruption of the epithelial cell barrier of these organoids in a toxin-dependent manner, despite the fact that the bacteria lose viability during the 12 hour course of infection (26). Other studies employing organoids reported reduction in MUC2 mucin, and inhibition of NHE3 (Na+/H+ exchanger isoform 3) production during C. difficile infection consistent with effects observed on human intestinal biopsy samples (23, 24). One caveat to organoid systems, however, is that, to date, it is difficult to manipulate environmental conditions, such as O2 levels or intestinal flow. While these systems are promising, the urgent requirement for new and more refined treatments for CDI combined with the need to better understand C. difficile pathogenesis supports the development of additional new experimental models.
These studies demonstrate the importance and power of using tissue culture model systems that more closely mimic in vivo conditions to study bacterial physiology and the impact of infection on host cells. In addition, they provide exciting avenues with which to explore unknown facets of C. difficile development and impact on intestinal tissues. Importantly, the 3D tissue scaffold provides us with a means to add additional cellular complexity by seeding with different types of cells as well as to manipulate the system with various engineering controls. Such new systems serve as valuable alternatives to animal infections to study C. difficile physiological and pathological processes and to visualize the effect of C. difficile infection, germination and toxin production on intestinal epithelial cells. Future studies on our 3D tissue model will focus on developing a more complex intestinal model with the introduction of critical immune cells and studying the interactions of C. difficile in these complex tissues.
Highlights.
-
-
A bioengineered 3D tissue model provides an anaerobic environment to support studying C. difficile in vitro.
-
-
A significantly higher toxin activity is detected in the 3D tissue model infected with toxigenic C. difficile compared to the 2D transwell system.
-
-
More severe damage to the epithelial tissue is observed in the 3D tissue model infected with toxigenic C. difficile compared to the 2D transwells.
-
-
Epithelial damage correlated significantly with the level of toxin activity detected in both models, but did not correlate with the CFU.
Acknowledgments
We thank Linc Sonenshein for advice, reagents, useful discussions and critical reading of the manuscript and Aimee Shen for useful discussions. We thank Giang Nguyen, Rebecca Silver, Anne McCabe, Charmaine Nganje and Ruby Pina for critical reading of the manuscript and useful discussions. The confocal imaging was performed at the Tufts Center for Neuroscience Research, P30 NS047243. The SEM studies were performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN). CNS is part of the Faculty of Arts and Sciences at Harvard University. This work was supported by NIH P41EB002520 to DLK, and U19 AI131126 to DLK and Ralph Isberg (Tufts University School of Medicine); NIH R01 AI118898 to CAK; NIH RO1 AI113166 and R21AI128093 to JM. LS was supported by NIH 4T32 AI007422; ACF was supported by NIH T32 AI07077. The sources of funding had no involvement in study design; collection, analysis, interpretation of data; in the writing of this manuscript or the decision to submit this article for publication.
Abbreviations
- NT
non-toxigenic C. difficile
- SC
3D scaffold system
- TW
2D transwell system
- TJ
tight junctions
- MLI
monolayer integrity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leffler DA, Lamont JT. Clostridium difficile Infection. N Engl J Med. 2015;373:287–288. doi: 10.1056/NEJMc1506004. [DOI] [PubMed] [Google Scholar]
- 2.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 4.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- 6.Hutton ML, Mackin KE, Chakravorty A, Lyras D. Small animal models for the study of Clostridium difficile disease pathogenesis. FEMS Microbiol Lett. 2014;352:140–149. doi: 10.1111/1574-6968.12367. [DOI] [PubMed] [Google Scholar]
- 7.Lawley TD, Young VB. Murine models to study Clostridium difficile infection and transmission. Anaerobe. 2013;24:94–97. doi: 10.1016/j.anaerobe.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 9.Geva-Zatorsky N, Alvarez D, Hudak JE, Reading NC, Erturk-Hasdemir D, Dasgupta S, von Andrian UH, Kasper DL. In vivo imaging and tracking of host-microbiota interactions via metabolic labeling of gut anaerobic bacteria. Nat Med. 2015;21:1091–1100. doi: 10.1038/nm.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Progatzky F, Dallman MJ, Lo Celso C. From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus. 2013;3:20130001. doi: 10.1098/rsfs.2013.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol. 2010;192:4983–4990. doi: 10.1128/JB.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Lacy DB. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 2012;8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Lacy DB. Clostridium difficile Toxins TcdA and TcdB Cause Colonic Tissue Damage by Distinct Mechanisms. Infect Immun. 2016;84:2871–2877. doi: 10.1128/IAI.00583-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasendra M, Barrile R, Leuzzi R, Soriani M. Clostridium difficile toxins facilitate bacterial colonization by modulating the fence and gate function of colonic epithelium. J Infect Dis. 2014;209:1095–1104. doi: 10.1093/infdis/jit617. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Lin Y, Davis KM, Wang Q, Rnjak-Kovacina J, Li C, Isberg RR, Kumamoto CA, Mecsas J, Kaplan DL. Robust bioengineered 3D functional human intestinal epithelium. Sci Rep. 2015;5:13708. doi: 10.1038/srep13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tasteyre A, Karjalainen T, Avesani V, Delmee M, Collignon A, Bourlioux P, Barc MC. Phenotypic and genotypic diversity of the flagellin gene (fliC) among Clostridium difficile isolates from different serogroups. J Clin Microbiol. 2000;38:3179–3186. doi: 10.1128/jcm.38.9.3179-3186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald LC, Killgore GE, Thompson A, Owens RC, Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 18.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol. 2008;8:192. doi: 10.1186/1471-2180-8-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eveillard M, Fourel V, Barc MC, Kerneis S, Coconnier MH, Karjalainen T, Bourlioux P, Servin AL. Identification and characterization of adhesive factors of Clostridium difficile involved in adhesion to human colonic enterocyte-like Caco-2 and mucus-secreting HT29 cells in culture. Mol Microbiol. 1993;7:371–381. doi: 10.1111/j.1365-2958.1993.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 20.Nusrat A, von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. 2001;69:1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125:413–420. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 22.Sutton PA, Li S, Webb J, Solomon K, Brazier J, Mahida YR. Essential role of toxin A in C. difficile 027 and reference strain supernatant-mediated disruption of Caco-2 intestinal epithelial barrier function. Clin Exp Immunol. 2008;153:439–447. doi: 10.1111/j.1365-2249.2008.03690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engevik MA, Engevik KA, Yacyshyn MB, Wang J, Hassett DJ, Darien B, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: inhibition of NHE3 and microbiota profile. Am J Physiol Gastrointest Liver Physiol. 2015;308:G497–509. doi: 10.1152/ajpgi.00090.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol. 2015;308:G510–524. doi: 10.1152/ajpgi.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jafari NV, Kuehne SA, Minton NP, Allan E, Bajaj-Elliott M. Clostridium difficile-mediated effects on human intestinal epithelia: Modelling host-pathogen interactions in a vertical diffusion chamber. Anaerobe. 2016;37:96–102. doi: 10.1016/j.anaerobe.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB, Spence JR. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun. 2015;83:138–145. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leslie JL, Young VB. A whole new ball game: Stem cell-derived epithelia in the study of host–microbe interactions. Anaerobe. 2016;37:25–28. doi: 10.1016/j.anaerobe.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Yamamoto Y, Wilson LH, Zhang T, Howitt BE, Farrow MA, Kern F, Ning G, Hong Y, Khor CC, Chevalier B, Bertrand D, Wu L, Nagarajan N, Sylvester FA, Hyams JS, Devers T, Bronson R, Lacy DB, Ho KY, Crum CP, McKeon F, Xian W. Cloning and variation of ground state intestinal stem cells. Nature. 2015;522:173–178. doi: 10.1038/nature14484. [DOI] [PMC free article] [PubMed] [Google Scholar]