

Abstract
Background
Neural precursor cell (NPC) migration toward lesions is key for neurological functional recovery. The neovasculature plays an important role in guiding NPC migration. MicroRNA‐210 (miR‐210) promotes angiogenesis and neurogenesis in the subventricular zone and hippocampus after cerebral ischemia; however, whether miR‐210 regulates NPC migration and the underlying mechanism is still unclear. This study investigated the role of miR‐210 in NPC migration.
Methods and Results
Neovascularization and NPC accumulation was detected around ischemic foci in a mouse model of middle cerebral artery occlusion (MCAO) and reperfusion. Bone marrow–derived endothelial progenitor cells (EPCs) were found to participate in neovascularization. miR‐210 was markedly upregulated after focal cerebral ischemia/reperfusion. Overexpressed miR‐210 enhanced neovascularization and NPC accumulation around the ischemic lesion and vice versa, strongly suggesting that miR‐210 might be involved in neovascularization and NPC accumulation after focal cerebral ischemia/reperfusion. In vitro experiments were conducted to explore the underlying mechanism. The transwell assay showed that EPCs facilitated NPC migration, which was further promoted by miR‐210 overexpression in EPCs. In addition, miR‐210 facilitated VEGF‐C (vascular endothelial growth factor C) expression both in vitro and in vivo. Moreover, the luciferase reporter assay demonstrated that miR‐210 directly targeted the 3′ untranslated region of SOCS1 (suppressor of cytokine signaling 1), and miR‐210 overexpression in HEK293 cells or EPCs decreased SOCS1 and increased STAT3 (signal transducer and activator of transcription 3) and VEGF‐C expression. When EPCs were simultaneously transfected with miR‐210 mimics and SOCS1, the expression of STAT3 and VEGF‐C was reversed.
Conclusions
miR‐210 promoted neovascularization and NPC migration via the SOCS1–STAT3–VEGF‐C pathway.
Keywords: brain ischemia, cell migration, miR‐210, vascular endothelial growth factor
Subject Categories: Ischemic Stroke, Neurogenesis
Clinical Perspective
What Is New?
This study suggests that the mechanism underlying the important role of miR‐210 in promoting neovascularization and neural precursor cell accumulation around ischemic foci after cerebral ischemia is by regulation of the SOCS1 (suppressor of cytokine signaling 1)–STAT3 (signal transducer and activator of transcription 3)–VEGF‐C (vascular endothelial growth factor C) pathway.
What Are the Clinical Implications?
Our findings suggest that increased miR‐210 expression may provide a novel therapeutic target for promoting neovascularization and facilitate nerve repair after cerebral ischemia.
Introduction
Ischemic stroke refers to localized ischemic necrosis or softening of brain tissues caused by cerebral blood supply disorders or hypoxia. This clinical situation accounts for 80% of all stroke cases and is characterized by high morbidity and mortality, as well as high recurrence and disability rates. Ischemic stroke is a heavy burden on families and societies.1, 2 Therefore, the recovery of neurological function following cerebral infarction is an urgent issue that needs specific attention. The migration of neural precursor cells (NPCs) toward lesions is key to the repair process; however, the mechanism underlying NPC migration is still unclear. An increasing number of studies have focused recently on the correlation between neurogenesis and angiogenesis after cerebral ischemia. Neuroblasts in the subependymal zone were observed migrating toward the neovasculature around the ischemic lesion after a stroke, and the NPC migration route was closely related to the orientation of cerebral blood vessels. The findings suggested that the neovasculature in the neurovascular niche acts as a scaffold and plays important roles in guiding NPC migration.3 Based on the in vivo studies, which showed that the injection of endostatin significantly inhibited neuroblast migration while inhibiting angiogenesis and blockade of Angiopoietin receptor TIE2 reduced neuroblast migration. Ohab et al proposed the concept of the neurovascular niche, suggesting that the migration of neuroblasts is closely related to vascular remodeling.4 The findings gave us a clue that the newly formed neurovascular niche around the ischemic lesion may be an important microenvironment that is required for NPC migration and a crucial factor that affects NPC migration.
Adult endothelial progenitor cells (EPCs) reside in the bone marrow and can be mobilized to circulate in the peripheral blood via cytokines released from injured endothelial cells and tissues in pathological conditions.5 The enriched cells in injured areas contribute to the neovascularization. In addition, the increased mobilization of EPCs was also observed after acute ischemic stroke and bone marrow–derived EPCs were shown to be involved in neovascularization around cerebral ischemic lesions.6
MicroRNA‐210 (miR‐210) controls the hypoxia response. Hypoxia induces the upregulation of miR‐210 in cells and tissues.7 Clinical studies demonstrated that miR‐210 levels are significantly higher in stroke patients with good outcomes than in those with poor outcomes. Moreover, miR‐210 can be used as a blood biomarker to predict clinical outcomes.8 In vitro studies have shown that the overexpression of miR‐210 promotes the migration of endothelial cells and the formation of vascular‐like structures, whereas inhibiting miR‐210 expression decreases endothelial cell growth and migration, induces apoptosis, and inhibits the formation of vascular‐like structures.9 The overexpression of miR‐210 in vivo was shown to increase tissue perfusion and capillary density after renal ischemia/reperfusion (I/R) injury and myocardial injury.10, 11, 12 These findings suggested that miR‐210 is related to vascular remodeling after ischemic injuries. It was reported that the in vivo overexpression of miR‐210 increases angiogenesis and neurogenesis in both healthy adult mice and mice with focal cerebral ischemia.13, 14 However, few studies focused on the relationships of miR‐210 with hypoxia, angiogenesis, and NPC migration. In addition, the mechanisms underlying these relationships have not been explored.
Vascular endothelial growth factor (VEGF) is an endothelial cell–specific mitogen and secreted dimer protein. Exposure to hypoxia induces increased VEGF expression and angiogenesis through a variety of pathways.15, 16, 17, 18, 19 VEGF promotes neurogenesis and the survival of new NPCs.20, 21, 22 VEGF and VEGF receptor (VEGFR) expression is upregulated in the ischemic penumbra, which is beneficial for neurovascular remodeling and neurological function recovery after a hypoxic/ischemic cerebral injury.17, 23, 24, 25, 26, 27, 28, 29 Several studies indicated that miR‐210 upregulates VEGF expression.30, 31, 32, 33 Therefore, we hypothesized that EPC homing may be involved in cerebral neovascularization following cerebral ischemia and that miR‐210 in EPCs upregulates VEGF under hypoxic conditions, promoting NPC accumulation and survival around ischemic foci and neurological function recovery.
In this study, the upregulated miR‐210 and VEGF and the increased angiogenesis and distribution of NPCs around ischemic lesions were observed in a focal cerebral ischemia mouse model. In addition, the upregulated miR‐210 and VEGF were observed in EPCs under hypoxic conditions. Moreover, the upregulated miR‐210 in EPCs promoted further NPC migration, whereas downregulated miR‐210 decreased NPC migration in vitro. Furthermore, the expression of SOCS1 (suppressor of cytokine signaling 1), STAT3 (signal transducer and activator of transcription 3), and VEGF was regulated by miR‐210 in vitro.
Materials and Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. All data and supporting materials have been provided with the published article.
Animals
All experiments were performed according to national guidelines and the ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines). The study procedures for the use of laboratory animals were approved by the laboratory animal welfare and ethics committee of the Third Military Medical University, Chongqing, China. C57BL/6 genetic background mice and EGFP (enhanced green fluorescent protein) transgenic mice (Nanjing Biomedical Research Institute of Nanjing University, China) were raised in a temperature‐ and light‐controlled environment. Food and water were provided to the mice ad libitum.
Generation of Chimeric Mice Expressing EGFP in Bone Marrow Cells
Eight‐week‐old C57BL/6 mice were irradiated with cobalt 60 at a dose of 10 Gy to destroy their bone marrow cells. Bone marrow cells were isolated from the EGFP transgenic mice to prepare a cell suspension. After irradiation, the bone marrow cell suspension prepared from the EGFP transgenic mice was injected into the irradiated mice via the jugular vein on the same day. After 8 weeks of bone marrow reconstruction in the mice, bone marrow was withdrawn from the hind limbs to prepare a cell suspension. The percentage of EGFP‐positive cells in the bone marrow was examined using flow cytometry. The mice were used to establish a middle cerebral artery I/R model 8 weeks after bone marrow reconstruction.
Mouse Middle Cerebral Artery Occlusion/Reperfusion Model
The I/R model was prepared using C57BL/6 mice and chimeric mice with bone marrow cells expressing EGFP. The model was established using middle cerebral artery occlusion (MCAO), as in our previous study.34 The animals were divided into 3‐ and 7‐day (after I/R) groups, in addition to the sham‐operated group, with 6 animals in each group. Briefly, the mice were anesthetized with chloral hydrate (300 mg/kg IP). A 2.0‐cm silicone‐coated 8‐0 nylon suture was gently inserted from the external carotid artery stump to the internal carotid artery, stopping at the opening of the middle cerebral artery. The ligation was maintained for 120 minutes before cerebral blood flow was restored. Body temperature of the mice was maintained at 37±0.3°C using a heating pad during surgery. The mice were provided with unlimited access to food and water after awakening. Neurological deficits were graded after the mice recovered using a 4‐point neurologic deficit severity scale: 0=no deficit; 1=forelimb weakness and torso turning to the ipsilateral side when held by the tail; 2=circling to the affected side; 3=unable to bear weight on the affected side; and 4=no spontaneous locomotor activity or barrel rolling.35 Animals with scores of 2 to 3 were used in the subsequent studies, whereas animals with a score of 1 or 4 were excluded from the studies. Sham‐operated mice underwent the same procedure without inserting the suture into the internal carotid artery.
5′‐Bromo‐2′‐Deoxyuridine Labeling In Vivo
On the fourth to sixth days or the first to seventh days after cerebral I/R, BrdU (5′‐Bromo‐2′‐Deoxyuridine) (100 mg/dL in saline) was injected intraperitoneally. The sham‐operated mice were injected with the same dose of BrdU on the same days after the sham operation.
Lentiviral Gene Transfer Into Mouse Brains
Adult C57BL/6 mice were anesthetized with chloral hydrate and then kept immobile on a stereotactic apparatus. Next, 3 µL of a lentivirus solution (LV‐miR‐210 or LV‐miR‐210 inhibitor, 4.0×108 IU, packaged and titered at Shanghai Genechem Co, Ltd, China) were directly injected (0.2 μL/min) into the right striatum. A Stoelting injection system was used to perform the injection at 2 mm lateral and 0.8 mm anterior to the bregma at a depth of 3.0 mm. The needle then was maintained in place for 10 minutes. At 14 days after viral vector injection, the MCAO model was prepared using these mice. Lentivirus‐GFP (green fluorescent protein; LV‐GFP), as a viral marker and a viral control, was delivered to the animals using the same protocol.
Brain Water Content
The brain water content (BWC) of all groups was examined using a previously published method.36 Briefly, mice injected with LV–miR‐210, LV–miR‐210 inhibitor, or LV‐GFP were euthanized using intraperitoneal anesthetization 3 days after successful MCAO model preparation. Cerebral tissues were collected from the ischemic side for examination of the BWC of mice in each group. The BWC was calculated using the following formula: (wet weight−dry weight)/wet weight×100%. The BWC examination was performed in a blinded manner.
Neurological Deficit Scores
Neurological deficits were assessed using a previously published method.37 Briefly, mice injected with LV–miR‐210, LV–miR‐210 inhibitor, or LV‐GFP were evaluated for motor deficits 3 days after occlusion/perfusion using a 4‐point neurologic deficit severity scale. The scoring was performed in a blinded manner by 2 researchers who did not have information on animal grouping. The average score was used as the final score for each mouse.
Isolation, Culture, and Characterization of EPCs From Bone Marrow
Isolation, culture and characterization of EPCs from bone marrow were carried out according to protocols described in the literature.38 Briefly, 6‐week‐old male C57BL/6 mice were euthanized through anesthesia overdose under aseptic conditions, and the tibia and femurs were bluntly isolated. The bone marrow was aspirated with 5 mL of PBS, filtered through 200‐mesh sieves, and centrifuged at 400g for 30 minutes to collect the cloudy cell layer. The cells were suspended in EGM‐2‐MV Bullet Kit medium (Lonza). The medium was changed to remove the suspension cells after 72 hours, followed by medium changes once every 3 days. The cells from day 7 were used in subsequent studies. The expression levels of the EPC surface antigens CD31, CD34, and VEGFR2 were examined on days 1, 4, and 7 using flow cytometry.
NPC Isolation, Culture, and Characterization
NPC isolation, culture, and characterization were carried out according to protocols described in the literature.39 Briefly, pregnant C57BL/6 mice were euthanized at gestational day 12 to 13 by cervical dislocation, and the embryonic telencephalon was isolated and cut into 1‐mm3 pieces using scissors. The tissue was then digested using 0.125% trypsin (containing EDTA) at 37°C for 5 minutes. Medium containing FBS was then added to neutralize trypsin digestion, and the cells were collected through centrifugation at 200g for 5 minutes. The cells were resuspended in NPC medium (DMEM/F12 plus 1% N2 supplement, 2% B27 supplement, 10 ng/mL basic fibroblast growth factor, and 20 ng/mL epidermal growth factor) and inoculated into T‐25 flasks for culture. The NPCs grew into “neuronal spheres,” and the suspension cells were collected after 48 hours for further culture, with medium changes every 2 days. The cells from the third passage were characterized using immunofluorescence. The examined markers included β‐tubulin III, DCX (doublecortin), and nestin. These cells were used in the subsequent studies.
Hypoxic Treatment of EPCs
The EPC culture plates were placed in a mixture of 94% N2, 1% O2, and 5% CO2 for 24 hours. The cells were collected for quantitative real‐time–polymerase chain reaction (qRT‐PCR) to detect the expression of miR‐210 under hypoxic conditions. The expression of VEGF‐C in the supernatant was detected using ELISA. The EPCs that were cultured under normal conditions were used as controls. The samples from each group were assayed in triplicate, in parallel.
Culture of HEK293 Cells
HEK293T cells were obtained from the American Type Culture Collection and cultured in DMEM with 10% FBS.
Constructs
The primers in this study were synthesized by GenePharma. The primers for miR‐210 were forward primer 5′‐GCAGTCTGTGCGTGTGACAGC‐3′ and reverse primer 5′‐GTGCAGGGTCCGAGGT‐3′.
The primers for VEGF‐C were forward primer 5′‐ACTTGCTGTGCTTCTTGT‐3′ and reverse primer 5′‐CTCATCTACGCTGGACAC‐3′.
The miR‐210 mimic and miR‐210 inhibitor were synthesized by GenePharma.
To generate the SOCS1 vector, the full open reading frame cDNA for human SOCS1 was transcribed, and the product was amplified using primers with flanking Spe I and Hind III restriction enzyme sites. The DNA was then inserted into the pcDNA3.1 vector (Invitrogen). SOCS1‐specific small interfering RNA (siRNA; SC‐40997) and control siRNA (SC‐37007) expression vectors were purchased from Santa Cruz Biotechnology.
Cell Transfection
HEK293T cells and EPCs were grown to 60% to 80% confluency and then transfected with miR‐210 mimic, miR‐210 inhibitor, a control siRNA, a siRNA targeting SOCS1, or a SOCS1 overexpression vector (pcDNA3.1‐SOCS1). For other experiments, cells were cotransfected with miR‐210 mimics and pcDNA3.1‐SOCS1. Cell transfection was carried out using Lipofectamine 2000 (Thermo Fisher Scientific), according to the instructions. Cells transfected with the miR‐210 mimic, miR‐210 inhibitor, si_SOCS1, or pcDNA3.1‐SOCS1 were collected 24 and 48 hours after transfection. The miR‐210 and SOCS1 expression levels were examined using qRT‐PCR to determine the optimal time point for successful transfection.
Cell Migration Assay
The cells used in the migration assay were divided into a normal EPC group, a miR‐210–overexpressing EPC group, and a miR‐210–suppressing group. For the blank control, medium (rather than EPCs) was placed in the lower chamber. The EPCs in each group were seeded into a 24‐well plate at a density of 1×104 cells per well in the lower chamber. The NPCs were added to the transwell chamber at a density of 5×103 cells per well in 100‐μL volume. The transwell chamber was removed after 24 hours and fixed with 4% paraformaldehyde, followed by 2 washes with normal saline and staining with 0.1% crystal violet for 3 minutes. A total of 20 fields of view for each group were selected under ×200 magnification for cell counting. The results were then analyzed. The assay was carried out in triplicate for each group. The counting of positive cells and the analysis were performed in a blinded manner.
To observe the effect of VEGFR inhibition on NPC migration, EPCs were placed in the lower chamber, whereas NPCs were placed in the upper chamber. The VEGFR2 antagonist vandetanib and the VEGFR3 antagonist SAR131675 were added to the upper transwell chamber (both at 100‐nmol/L concentrations). This treatment blocked the action of VEGF on NPCs. VEGFR antagonists were not added in the control group.
RNA Extraction and qRT‐PCR
EPCs were collected 24 hours after being cultured under hypoxic conditions for RNA extraction and examination of miR‐210 expression using qRT‐PCR. EPCs cultured under normal conditions were used as controls.
Total RNA from the cultured EPCs was extracted using the TRIzol reagent (Thermo Fisher), and cDNA was synthesized using the iScript cDNA synthesis reagent (Bio‐Rad), according to the manufacturers' instructions. In addition, qRT‐PCR was performed on a Bio‐Rad iCycler with the iQ SYBR Green Supermix (Bio‐Rad) in 96‐well plates, according to the manufacturer's instructions. The 2−ΔΔCT method was adopted for data analysis.
ELISA
To examine VEGF‐C expression by EPCs under hypoxia, supernatant was collected from EPCs that had been cultured under hypoxic conditions. To examine the effect of changes in miR‐210 levels on the secretion of VEGF‐C by EPCs, supernatant from EPCs transfected with miR‐210 mimics or the miR‐210 inhibitor was collected 48 hours after transfection. The ELISA was carried out following the instructions for the VEGF‐C ELISA kit.
Western Blotting
Cultured cells or brain tissues were lysed in RIPA lysis buffer, and the lysates were used in standard Western blotting assays. The anti‐VEGF‐C (1:100), anti‐SOCS1 (1:100), and anti‐STAT3 (1:5000) antibodies and the horseradish peroxidase–conjugated secondary antibody were purchased from Abcam. β‐Actin was used as an internal standard.
Immunofluorescence Staining
The MCAO model was prepared using C57BL/6 mice and chimeric mice with bone marrow cells expressing EGFP. Perfusion fixation was performed 3 and 7 days after cerebral I/R using 4% paraformaldehyde. The brain was collected and dehydrated using 30% sucrose and was cryosectioned. To examine neovascularization and the source of neovasculature cells after cerebral I/R, CD31 immunofluorescence staining was performed on sections from brains that were harvested at 3 and 7 days. To observe NPC proliferation and migration in the subventricular zone (SVZ), brain sections from the BrdU‐labeled I/R mice and sham‐operated mice were collected on day 7 after cerebral I/R and used for anti‐BrdU and anti‐DCX double‐immunofluorescence staining. To observe the accumulation of neuroblasts around the ischemic lesions following cerebral I/R, sections from C57BL/6 mice collected 7 days after cerebral I/R were stained using DCX immunofluorescence. To observe the relationship between neovasculature and the accumulation of neuroblasts around ischemic lesions, brain sections were double‐immunofluorescence stained for CD31 and DCX. Perfusion fixation was performed with mice that were injected with LV–miR‐210 or LV–miR‐210 inhibitor 7 days after cerebral I/R, and the brain was collected and dehydrated using 30% sucrose. These brains were cryosectioned for immunofluorescence staining of CD31 and DCX. These staining experiments were performed to observe the effect of changes in local miR‐210 expression on neovascularization and the accumulation of neuroblasts around ischemic lesions following cerebral I/R. The primary antibodies used in the experiment were rabbit anti–mouse CD31 (1:200), goat anti–mouse DCX (1:100), and mouse anti–mouse BrdU (1:200). The secondary antibodies included Alexa Fluor 647 (1:200; donkey anti‐goat) and Alexa Fluor 594 (1:200; goat anti‐rabbit). The observed results were imaged using confocal microscopy (TCS Sp5; Leica). The numbers of CD31‐positive and DCX‐positive cells were counted in each field using ×400 magnification. The numbers of CD31 and EGFP double‐labeled cells were counted in each field using ×800 magnification. Three fields of view per animal and 6 mice per time point were examined under a confocal microscope for positive cell counting. The positive cell counting was performed in a blinded manner.
Dual‐Luciferase Reporter Assay
To investigate the molecular mechanism of miR‐210 in the regulation of VEGF‐C, we searched for possible downstream target genes of miR‐210 using the PicTar and Target Scan 6.0 websites. We found that miR‐210 may be able to recognize the 3′ untranslated region (UTR) of SOCS1 mRNA. To determine whether SOCS1 is directly regulated by miR‐210, we designed and synthesized the SOCS1 3′‐UTR sequence and a mutated SOCS1 3′‐UTR sequence. We cloned the 2 fragments of the target gene into the pMIR‐REPORT Luciferase dual‐luciferase reporter DNA vector, generating wild‐type and mutant SOCS1 3′‐UTR dual‐luciferase reporter vectors (ie, wild‐type pMIR‐SOCS1‐3′ UTR and mutant pMIR‐SOCS1‐3′ UTR). The recombinant vectors were characterized through PCR, electrophoresis, and sequencing to confirm the successful construction of the vectors. The recombinant vector plasmids were transfected into HEK293T cells together with miR‐210 mimics or a miR‐210 negative control using Lipofectamine 2000, and luciferase activity was examined using the double luciferase assay system.
Statistical Analyses
All data were expressed as mean±SD and analyzed using SPSS 18.0 software (IBM Corp), and graphs were generated using GraphPad Prism 5.0 (GraphPad Software). A t test, 1‐way ANOVA, or nonparametric test was used to examine the differences between groups. A P value <0.05 was considered statistically significant. The statistical analysis was conducted in a blinded manner.
Results
Neovascularization Around Ischemic Lesions in Mice Following Cerebral I/R
A focal cerebral ischemia model was established in C57BL/6 mice and confirmed using TTC (2,3,5‐triphenyltetrazolium chloride) staining (Figure 1A). The mortality rate of the C57BL/6 mice after I/R was ≈25% on the third day and ≈40% on the seventh day. The mice were euthanized at 3 and 7 days after I/R, and perfusion fixation was performed. Sham‐operated mice were used as the control. The brain was collected for cryosectioning and immunofluorescence staining. CD31 (red) was used as a vascular endothelial cell marker. CD31‐positive vascular endothelial cells were found in the infarct border zone (IBZ) 3 and 7 days after MCAO (Figure 1B). The number of vascular endothelial cells increased over time, suggesting gradually increasing neovascularization with prolonged time after I/R (Figure 1B).
Figure 1.
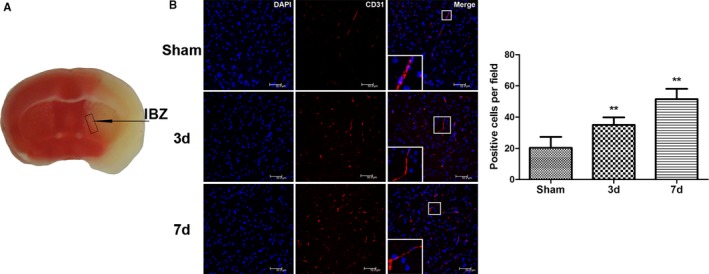
Neovascularization around ischemic foci after cerebral ischemia/reperfusion in mice. A, Schematic drawing of ischemic foci and the infarction border zone (IBZ) after cerebral ischemia/reperfusion in mice. B, Compared with the sham‐operated control group, the number of CD31‐stained vascular endothelial cells around the ischemic foci increased significantly 3 and 7 days after middle cerebral artery occlusion/reperfusion in the treated mouse group; this number gradually increased with time after ischemia/reperfusion (n=6, **P<0.01). d indicates days; DAPI, 4′,6‐diamidino‐2‐phenylindole.
Bone Marrow–Derived EPC Homing to Ischemic Foci and Neovascularization After Cerebral I/R
Eight‐week old C57BL/6 mice were irradiated with cobalt 60 to destroy their bone marrow. A suspension of bone marrow cells from the EGFP transgenic mice was injected into the irradiated mice through the jugular vein on the same day as the irradiation. The bone marrow was collected from the hind limb of these mice after 8 weeks to prepare bone marrow cell suspensions. Flow cytometry showed that 74.5% of the bone marrow cells were EGFP positive, demonstrating that most of the bone marrow cells in these mice had been replaced by EGFP‐positive cells. This result indicated the successful generation of chimeric mice with EGFP‐expressing bone marrow cells (Figure 2A).
Figure 2.
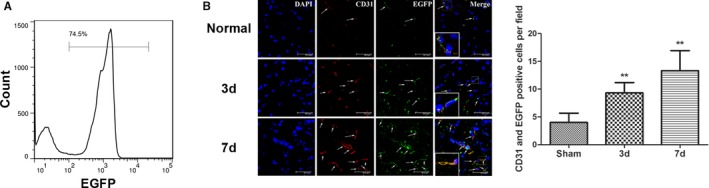
Successful construction of chimeric mice with bone marrow cells expressing enhanced green fluorescent protein (EGFP) and the mobilization and migration of bone marrow cells to the ischemic foci after cerebral ischemia/reperfusion (I/R) in the chimeric mice. A, The ratio of EGFP‐positive cells in the bone marrow cells of chimeric mice at 8 weeks after construction as determined using flow cytometry. B, The number of EGFP‐expressing (green) bone marrow–derived cells was significantly increased 3 and 7 days after middle cerebral artery occlusion in the treated group compared with the control group; this number gradually increased with time after I/R. EGFP‐expressing bone marrow–derived cells were colocalized with CD31‐positive vascular endothelial cells (n=6, **P<0.01). The arrows indicate the EGFP and CD31 double‐labeled cells. d indicates days.
A focal cerebral I/R model was established next using chimeric mice with EGFP‐expressing bone marrow cells. CD31‐labeled vascular endothelial cells (red) were present around the ischemic foci at 3 and 7 days after I/R, and the number of vascular endothelial cells increased with prolonged time after I/R. The EGFP‐positive cells were bone marrow derived, and the coexpression of the vascular endothelium‐specific marker CD31 and EGFP indicated that bone marrow‐derived EPCs participated in neovascularization (Figure 2B).
Increased NPCs Around Ischemic Foci After Cerebral I/R in Mice
NPCs were labeled with DCX. The number of NPCs in the SVZ and IBZ was significantly increased 7 days after MCAO in the mice (Figure 3C). We also observed the NPC proliferation in the SVZ of the ischemic side. When BrdU was injected intraperitoneally on fourth to sixth days after I/R, some BrdU/DCX double‐labeled cells were found in the SVZ and along the corpus callosum on day 7 after cerebral I/R as well as in sham operated control. More DCX‐positive‐only cells were observed in the SVZ and striatum on day 7 after cerebral I/R than after sham operation (Figure 3A). When BrdU was injected intraperitoneally on the first to seventh days after I/R, the number of BrdU/DCX double‐labeled cells were found in the SVZ and striatum increased markedly on day 7 after I/R, which was more than that were found after sham operation (Figure 3B).
Figure 3.
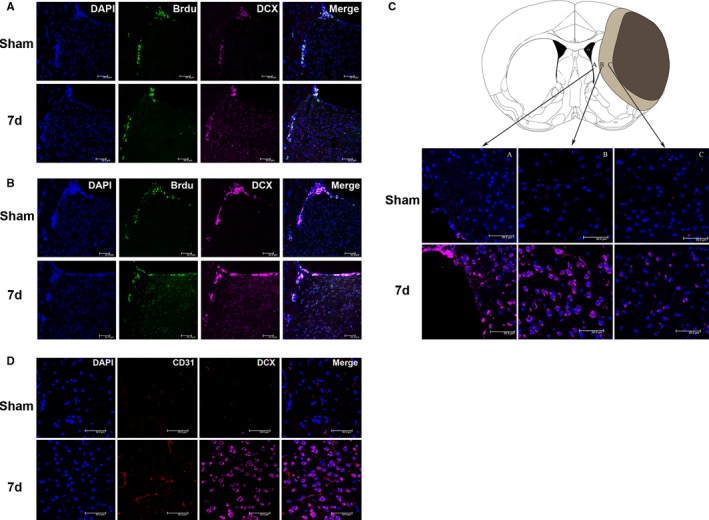
Neural precursor cells (NPCs) accumulated around the ischemic foci and were distributed close to the neovasculature after cerebral ischemia/reperfusion (I/R) in mice. A, BrdU (5′‐bromo‐2′‐deoxyuridine) was injected intraperitoneally on the fourth to sixth days. In subventricular zone (SVZ) and along corpus callosum, BrdU and DCX (doublecortin) double‐labeled cells were found in cerebral I/R group and sham operated group. Whereas compare to the sham‐operated control, there are more DCX‐positive‐only cells in the SVZ and striatum on the 7th day after cerebral I/R. B, BrdU was injected intraperitoneally on the first to seventh days. BrdU/DCX double‐labeled cells increased in SVZ and striatum on day 7 after cerebral I/R compared with those in sham‐operated control mice. C, Compared with the sham‐operated control group, the number of DCX‐labeled NPCs was significantly increased in SVZ and IBZ at 7 days after middle cerebral artery occlusion (MCAO). D, Compared with the control group, CD31 and DCX double immunofluorescence staining showed that vascular endothelial cell proliferation was enhanced, and the number of neuroblasts was increased around ischemic foci 7 days after MCAO. d indicates days; DAPI, 4′,6‐diamidino‐2‐phenylindole.
Double immunofluorescence staining of CD31 and DCX showed remarkably increased numbers of CD31‐positive vascular endothelial cells and DCX‐positive neuroblasts around ischemic foci at 7 days after cerebral I/R. This result suggests that the neovasculature around ischemic foci may be involved in the homing of NPCs toward ischemic foci (Figure 3D).
Significantly Increased miR‐210 Levels After Cerebral I/R in Mice
Changes in miR‐210 expression were examined in mice after cerebral I/R. Expression levels of miR‐210 in the cortex on the ischemic side were examined using qRT‐PCR at 1, 3, and 7 days after MCAO. The results showed that miR‐210 expression was significantly increased at each time point after cerebral I/R compared with sham operation, with the highest expression level observed on day 3. The miR‐210 expression level on day 7 was lower than that on day 3 but was still markedly higher than that of the control group (Figure 4A), suggesting that miR‐210 may participate in the pathophysiological process of cerebral ischemia.
Figure 4.
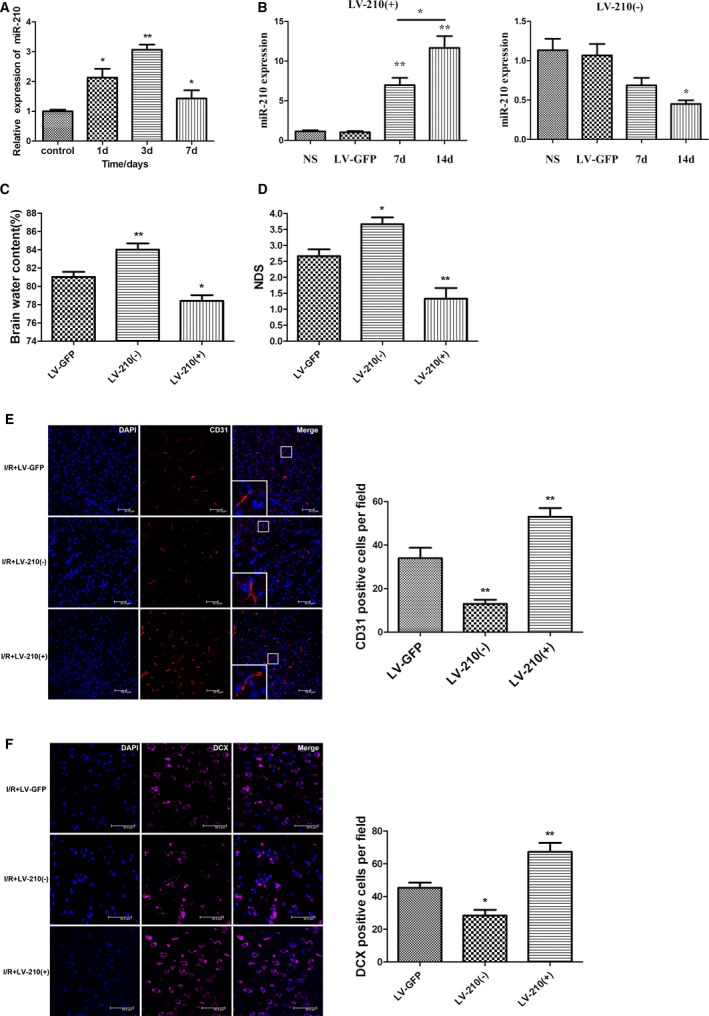
The expression of miR‐210 was significantly increased after the cerebral ischemia/reperfusion (I/R) in mice, and miR‐210 overexpression promoted neovascularization and neural precursor cell migration toward ischemic foci to alleviate neural function damage. A, The expression of miR‐210 in the ischemic brain tissue of the middle cerebral artery occlusion (MCAO) group was quantified using quantitative real‐time–polymerase chain reaction (qRT‐PCR) at 1, 3 and 7 days after I/R. Compared with the control group, miR‐210 expression levels were significantly higher in the MCAO group, with the highest level being observed at day 3 (*P<0.05, **P<0.01). B, Lentivirus (3 μL) with the miR‐210 overexpression construct (LV‐210[+]) or a miR‐210 inhibitor construct (LV‐210[−]) was injected into the right striatum of the mice. An empty vector (LV‐GFP) or normal saline (NS) injection was used as a control. The expression of miR‐210 in the right striatum was detected by qRT‐PCR on days 7 and 14 after the injection. The expression of miR‐210 was increased significantly on days 7 and 14 after LV‐miR‐210 injection (**P<0.01). The expression of miR‐210 was significantly higher on day 14 than on day 7 (*P<0.05). The expression on miR‐210 was significantly decreased on day 14 after injection of the LV‐miR‐210 inhibitor (*P<0.05). C and D, The right striatum of the mice was injected using a stereotactic apparatus with 3 μL of lentivirus with LV‐210(+), LV‐210(−), or an empty vector (LV‐GFP). These mice were used to establish the I/R model 14 days after virus injection. The neurological deficit score (NDS) was obtained 3 days after I/R, and the brain water content (BWC) on the ischemic side of the brain was calculated. Compared with the control group injected with LV‐GFP, the overexpression of miR‐210 attenuated I/R‐induced cerebral edema, whereas the BWC was higher in the group with miR‐210 expression inhibition. The NDS was significantly decreased in the miR‐210 overexpression group and increased in the miR‐210 inhibition group (n=6, *P<0.05, **P<0.01). E and F, The right striatum of the mice was injected using a stereotactic apparatus with 3 μL of LV‐210(+) or LV‐210(−), or LV‐GFP, and the mice were used to establish the I/R model at 14 days after virus injection. At 7 days after I/R, the vascular endothelial cell‐ and DCX (doublecortin)–positive neuroblast numbers around the ischemic foci increased significantly in the miR‐210 overexpression group compared with the control group injected with LV‐GFP, whereas these numbers decreased significantly in the miR‐210 inhibition group (n=6, *P<0.05, **P<0.01). d indicates days; DAPI, 4′,6‐diamidino‐2‐phenylindole.
To investigate the role of miR‐210 in cerebral I/R, a lentivirus was injected into the mouse striatum to locally overexpress or suppress miR‐210. The effect of altered miR‐210 levels on cerebral edema, neurological deficits, angiogenesis, and NPC migration after cerebral ischemia was then observed. Perfusion fixation of the mice was performed at 7 and 14 days after unilateral injection of the lentivirus into the striatum. The brains were collected and sectioned. The transduction of the lentivirus in local brain cells was observed under a confocal microscope, with green fluorescence in cells around the injection site indicating that the lentivirus had been successfully transfected. More green fluorescence–positive cells were found at day 14. Mouse striata were collected on days 7 and 14 to quantify miR‐210 expression using qRT‐PCR. The results showed that miR‐210 expression increased significantly on days 7 and 14 after LV–miR‐210 injection (P<0.01); expression of miR‐210 was increased more significantly on day 14 than on day 7 (P<0.05). Expression of miR‐210 was decreased significantly on day 14 after LV–miR‐210 inhibitor injection (P<0.05; Figure 4B). Consequently, the MCAO model was established 14 days after lentivirus injection.
Neurological deficit scores were compared at 3 days after I/R among the mouse groups. The mice were euthanized, and the BWC was measured and compared. Compared with the LV‐GFP group, miR‐210 inhibition increased the BWC 72 hours after I/R (P<0.01, n=6; Figure 4C), whereas miR‐210 overexpression reduced the BWC (P<0.05, n=6; Figure 4C). Similarly, the neurological deficit score of the miR‐210 inhibition group was significantly higher than that of the LV‐GFP group (P<0.05, n=6; Figure 4D), whereas the neurological deficit score of the miR‐210 overexpression group was significantly lower than that of the LV‐GFP group (P<0.01, n=6; Figure 4D). These results indicate that miR‐210 overexpression can reduce cerebral edema around the ischemic lesion and alleviate neurological deficits.
Next, the effect of miR‐210 on angiogenesis and NPC accumulation around ischemic foci was examined. The MCAO focal cerebral I/R model was established 14 days after lentiviral injection. Perfusion fixation of the mice was performed at 7 days after cerebral I/R, and the brains were collected and sectioned for immunofluorescence staining. The virus was still expressed on the 21st day after lentiviral injection. Compared with the LV‐GFP group, the miR‐210 overexpression group exhibited a significantly increased number of vascular endothelial cells in the cerebral tissue around the ischemic foci (P<0.01; Figure 4E), whereas the miR‐210 inhibition group showed a remarkably decreased number of vascular endothelial cells (P<0.01; Figure 4E). Similarly, the number of NPCs in the cerebral tissues around the ischemic lesions was significantly increased in the miR‐210 overexpression group (P<0.01; Figure 4F), whereas the number of NPCs was clearly decreased in the miR‐210 inhibition group (P<0.05; Figure 4F). This result suggests that miR‐210 overexpression may promote neovascularization and NPC accumulation around ischemic lesions in vivo.
Significantly Increased miR‐210 Expression Under Hypoxic Conditions in EPCs and Enhanced NPC Migration by miR‐210–Overexpressing EPCs In Vitro
To examine the effect of hypoxia on miR‐210 expression in EPCs, bone marrow EPCs were cultured in vitro, and the EPCs exhibited cobblestone‐like growth when observed under a microscope (Figure 5A). The cultured cells were examined for CD31, CD34, and VEGFR2 expression using flow cytometry at days 1, 4, and 7 to confirm successful isolation of EPCs (Figure 5B). The EPCs were then cultured in a hypoxic incubator with a gas mixture of 94% N2, 1% O2, and 5% CO2 (by volume) for 24 hours. The effect of hypoxia on miR‐210 expression in EPCs was examined using qRT‐PCR. The results showed that miR‐210 expression was significantly increased in EPCs cultured under hypoxic conditions for 24 hours compared with that in EPCs cultured under normal conditions (P<0.01; Figure 5C). Next, the EPCs were transfected with miR‐210 mimics or a miR‐210 inhibitor, and miR‐210 expression was examined by qRT‐PCR. The increase in miR‐210 expression was most evident 48 hours after the transfection of EPCs with miR‐210 mimics, whereas the inhibition of miR‐210 expression was most obvious 48 hours after the transfection of EPCs with miR‐210 inhibitor. Consequently, 48 hours was selected as the time point of successful transfection (Figure 5D).
Figure 5.
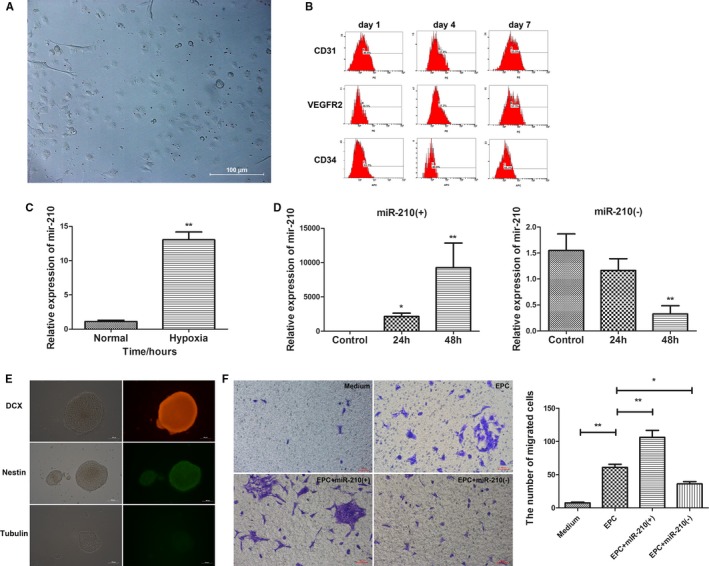
Endothelial progenitor cells (EPCs) promoted neural precursor cell (NPC) migration, which is further enhanced by the overexpression of miR‐210 in EPCs. A, Cobblestone‐like growth of mouse bone marrow EPCs under an inverted microscope (×200). B, The successful isolation of EPCs confirmed by examination of CD31, CD34, and VEGFR2 (vascular endothelial growth factor receptor 2) expression at 1, 4 and 7 days after isolation via flow cytometry. C, Significantly increased miR‐210 expression was observed in EPCs cultured under hypoxic conditions for 24 hours compared with EPCs cultured under normal conditions (P<0.01). D, EPCs were transfected with the miR‐210 mimic (miR‐210[+]) or miR‐210 inhibitor (miR‐210[−]), and the expression of miR‐210 was detected by quantitative real‐time–polymerase chain reaction. The manipulation of miR‐210 expression by miR‐210(+) (**P<0.01) and miR‐210(−) (**P<0.01) was most significant 48 hours after transfection. E, The isolation, culture, and characterization of mouse NPCs. The NPCs exhibited spheroid growth under an inverted microscope (×200). The cells were DCX (doublecortin) and nestin positive and β‐tubulin III negative, confirming the successful isolation and culture of NPCs. F, A transwell assay indicated that NPC migration was significantly enhanced by EPCs compared with the medium‐alone condition (n=3, **P<0.01). The effect of EPCs was further boosted by miR‐210 overexpression (n=3, **P<0.01), whereas the effect was decreased by the inhibition of miR‐210 (n=3, *P<0.05). h indicates hours.
To further investigate the role of miR‐210 in NPC migration, NPCs were isolated and cultured, exhibiting spheroid growth under the microscope. These cells were shown to be positive for nestin and DCX and negative for β‐tubulin III in immunofluorescence staining, confirming the success of NPC culture (Figure 5E). To observe the effects of altered miR‐210 expression in EPCs on NPC migration, EPCs that were successfully transfected with miR‐210 mimics or the miR‐210 inhibitor were placed in the lower chamber of the transwell apparatus, and NPCs were placed in the upper chamber. The cells were cultured for 24 hours to observe the impact of miR‐210 overexpression or inhibition in EPCs on NPC migration. Only medium was placed in the lower chamber of the transwell apparatus in the blank control, whereas normally cultured EPCs were put in the lower chamber as an additional control. After coculture of EPCs and NPCs for 24 hours, crystal violet staining was used to visualize the migrated NPCs on the membrane, and the number of migrated cells was counted. Compared with the blank control, normal EPCs apparently promoted NPC migration (P<0.01), and the number of migrated cells in the miR‐210–overexpressing EPC group was significantly higher than that in the normal EPC group (P<0.01). In contrast, compared with the normally cultured EPCs, the number of migrated NPCs was remarkably reduced in the group of EPCs with inhibited miR‐210 expression (P<0.05; Figure 5F). These findings suggest that EPCs promote NPC migration and that miR‐210 overexpression in EPCs enhances NPC migration, whereas the inhibition of miR‐210 expression in EPCs inhibits NPC migration.
EPCs Modulate VEGF Activation Through miR‐210 Expression
Previous studies have shown that VEGF can promote the differentiation of EPCs and angiogenesis.40 Consequently, the effect of changes in miR‐210 expression on VEGF expression was investigated in this study. Lentivirus was injected into mouse striatum using stereotactic positioning to induce local miR‐210 overexpression or inhibition. These mice were used to establish the MCAO I/R model 14 days after virus injection. Cerebral tissues were collected from the ischemic side 7 days after model construction, and VEGF‐C expression was examined using Western blotting. VEGF‐C secretion was significantly increased in the miR‐210 overexpression group compared with the group infected with the LV‐GFP control virus, whereas VEGF‐C expression markedly declined when miR‐210 expression was suppressed (Figure 6A).
Figure 6.
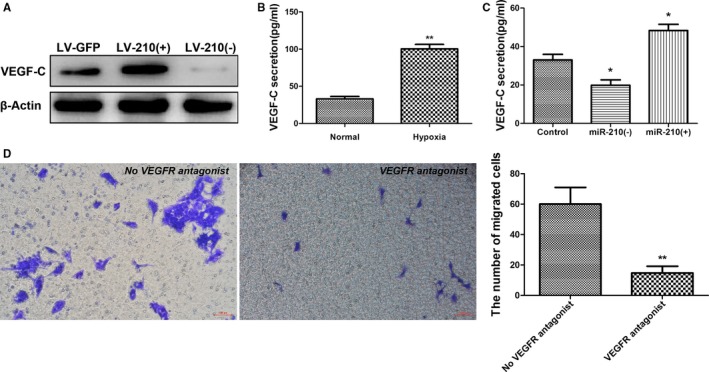
miR‐210 promotes VEGF‐C (vascular endothelial growth factor C) expression. A, The right striatum of the mice was injected with 3 μL of LV‐210(+) or LV‐210(−) or LV‐GFP, and the mice were used to establish the middle cerebral artery occlusion ischemia/reperfusion (I/R) model 14 days after injection. Brain tissues were collected from the ischemic side 7 days after I/R for examination of VEGF‐C expression using Western blotting. Compared with the control group, VEGF‐C protein expression was significantly increased in the miR‐210 overexpression group and decreased in the miR‐210 inhibition group. B, The level of VEGF‐C in the supernatant of endothelial progenitor cell (EPC) culture was determined via ELISA after 24 hours of culture under hypoxia. The secretion of VEGF‐C was significantly increased in the EPCs cultured under hypoxic conditions compared with the EPCs cultured under normal conditions (n=3, **P<0.01). C, The level of VEGF‐C in the supernatant of EPCs transfected with miR‐210 mimics or a miR‐210 inhibitor was determined by ELISA 48 hours after transfection. The secretion of VEGF‐C was significantly increased in the miR‐210 overexpression group and significantly decreased in the miR‐210 inhibition group compared with the control group (n=3, *P<0.05). D, The impact of VEGFR (vascular endothelial growth factor receptor) antagonists on the promotion of neural precursor cell (NPC) migration by EPCs was evaluated using a transwell assay. Compared with the untreated EPC group, the blockade of VEGFR significantly decreased the EPCs' effect in promoting NPC migration (n=3, **P<0.01).
As mentioned, after being cultured under hypoxic conditions for 24 hours, EPCs exhibited significantly increased miR‐210 expression compared with EPCs that were cultured under normal conditions; therefore, the effect of EPCs cultured under hypoxic conditions on VEGF‐C secretion was examined using ELISA. It was found that VEGF‐C expression by EPCs cultured under hypoxic conditions for 24 hours was significantly elevated compared with that by EPCs cultured under normal conditions (P<0.01; Figure 6B). To further confirm the regulation of VEGF‐C by miR‐210, EPCs were transfected with miR‐210 mimics or a miR‐210 inhibitor, and cell culture supernatant was collected after 48 hours for ELISA. Compared with the blank control, VEGF‐C secretion by EPCs was significantly increased in the miR‐210 overexpression group (P<0.05; Figure 6C), whereas VEGF‐C secretion by EPCs significantly dropped in the miR‐210 inhibition group (P<0.05; Figure 6C).
Next, to elucidate the role of VEGF‐C in NPC migration, we examined the effect of blocking the VEGF–VEGFR interaction on NPC migration. EPCs were seeded in the lower chamber of a transwell apparatus, and NPCs were placed in the upper chamber. The VEGFR2 antagonist vandetanib and the VEGFR3 antagonist SAR131675 were also added to the upper chamber to block the VEGF–VEGFR interaction. We then observed the effect of this manipulation on NPC migration. The transwell apparatus was removed after 24 hours of culture, fixed with paraformaldehyde, and stained with crystal violet. The results showed that the number of migrated NPCs in the group treated with both antagonists was significantly lower than that in the group not treated with the antagonists (P<0.01; Figure 6D). This result indicates that blocking the VEGF–VEGFR interaction can significantly attenuate the effect of EPCs in promoting NPC migration.
STAT3–VEGF Activation Is Modulated by miR‐210 Through SOCS1 Targeting
MicroRNA acts through posttranscriptional regulation of target genes, leading to the degradation of target gene mRNAs or translational inhibition. The results from this study and previous studies showed increased VEGF and STAT3 expression when miR‐210 was overexpressed.30, 31, 33 Consequently, we hypothesized that VEGF and STAT3 may not be direct targets of miR‐210. Through bioinformatics analysis, we found that miR‐210 can bind to the 3′‐UTR of SOCS1 mRNA, and SOCS1 may be the downstream target gene of miR‐210. It has been reported that SOCS1 can negatively regulate the STAT3–VEGF signaling pathway.41, 42, 43, 44 To determine whether SOCS1 is a direct target gene of miR‐210, the wild‐type 3′‐UTR and a mutant lacking miR‐210 binding sites were cloned downstream of the firefly luciferase coding region in a luciferase reporter vector (Figure 7A). These constructs were cotransfected into HEK293T cells with pRL‐TK and the miR‐210 mimic or the normal control mimic. The relative luciferase activity was reduced by 30% in cells transfected with pGL‐3 vectors containing the wild‐type SOCS1 3′‐UTR, but this reduction was not observed in cells expressing the vector with the mutant 3′‐UTR (Figure 7B). To further determine whether miR‐210 can inhibit the expression of SOCS1, miR‐210 mimics were transfected into HEK293T cells and EPCs, and changes in SOCS1 expression were examined using Western blotting after 48 hours. The results showed that miR‐210 overexpression decreased SOCS1 protein expression in the HEK293T cells and EPCs compared with the normal control mimic group (Figure 7C). Previous studies have shown that SOCS1 negatively regulates STAT3 expression and that STAT3 is a positive upstream regulator of VEGF.41, 42, 43, 44 Consequently, we examined the effect of miR‐210 overexpression on STAT3–VEGF‐C expression, in addition to its effect on SOCS1 expression. Interestingly, HEK293 cells and EPCs exhibited significant upregulation of STAT3 and VEGF‐C expression compared with the miR‐NC group after treatment with the miR‐210 mimic (Figure 7C). To determine whether the regulation of STAT3–VEGF‐C by miR‐210 was SOCS1‐dependent, we constructed a siRNA targeting SOCS1 and a SOCS1 overexpression vector (pcDNA3.1‐SOCS1), which were transfected into EPCs. Changes in the expression of SOCS1, STAT3, and VEGF‐C were examined using Western blotting. EPCs treated with the siRNA targeting SOCS1 exhibited significantly reduced SOCS1 protein expression, along with remarkably increased STAT3 and VEGF‐C protein expression. Meanwhile, overexpressing SOCS1 through pcDNA3.1‐SOCS1 clearly elevated SOCS1 protein expression levels, whereas the expression levels of STAT3 and VEGF‐C significantly declined (Figure 7D). These results demonstrated that SOCS1 is a signaling molecule that is upstream of STAT3–VEGF‐C.
Figure 7.
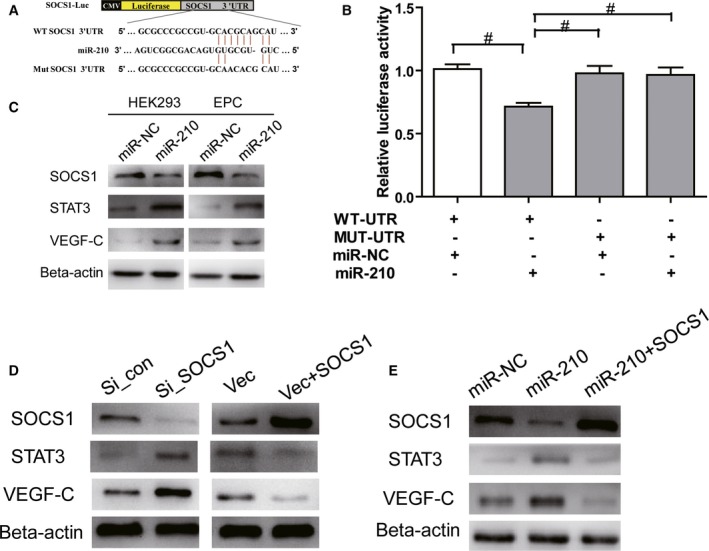
miR‐210 modulates VEGF (vascular endothelial growth factor) activation and secretion through STAT3 (signal transducer and activator of transcription 3) by targeting SOCS1 (suppressors of cytokine signaling 1). A, Schematic representation of miR‐210 recognition site in wild‐type SOCS1 3′ untranslated region (WT SOCS1 3′ UTR; WT‐UTR) and mutant SOCS1 3′UTR (MUT SOCS1 3′ UTR; MUT‐UTR). B, Luciferase activity assays of luciferase vectors containing WT‐UTR or MUT‐UTR were performed after transfection with normal control mimic (miR‐NC) or miR‐210 mimics in HEK293 cells (n=3, # P<0.05). C, Western blot showing SOCS1, STAT3, and VEGF‐C expression in the sham and miR‐210 mimic transfection groups at 48 hours in HEK293T cells and EPCs (n=3). D, Western blot assays showing the protein expression of SOCS1, STAT3, and VEGF‐C in endothelial progenitor cells (EPCs) transfected with a control small interfering RNA (si_con), a siRNA targeting SOCS1 (si_SOCS1), pcDNA3.1, or pcDNA3.1‐SOCS1. β‐Actin served as an internal reference (n=3). E, Western blot assays showing the protein expression of SOCS1, STAT3, and VEGF‐C in EPCs 48 hours after transfection with control mimics or miR‐210 mimics or cotransfection with miR‐210 mimics and the pcDNA3.1‐SOCS1 vector (n=3).
To further demonstrate that the activity of miR‐210 to regulate STAT3–VEGF‐C is dependent on SOCS1, we performed a rescue experiment. As expected, EPCs transfected with miR‐210 mimics exhibited significantly reduced SOCS1 expression 48 hours after transfection, whereas the downstream molecules STAT3 and VEGF‐C showed significantly elevated expression. However, the expression of STAT3 and VEGF‐C returned to normal when the EPCs were cotransfected with pcDNA3.1‐SOCS1 and miR‐210 mimics (Figure 7E). These results demonstrated that miR‐210 modulates STAT3–VEGF activation by targeting SOCS1.
Discussion
The relationship between angiogenesis and neurogenesis after cerebral ischemia has been widely studied. Consistent with previous reports,45, 46an increased number of CD31‐positive cells around an induced brain infarct were detected in the study. In addition, an increased density of vessels was observed 3 days after MCAO reperfusion, and the neovasculature further increased on day 7.
It was reported that vascular endothelial cells around infarcts proliferate and differentiate 12 to 24 hours after cerebral ischemia; immature capillaries begin to appear around infarcts after 3 days; and vascular density increases to a high level on day 7 and remains at a high level for several weeks. The neovascularization around infarcts can improve local blood supply to provide an appropriate microenvironment for neuronal remodeling.47, 48, 49, 50, 51, 52, 53, 54, 55 Previous studies suggested that the neovasculature originates from the expansion and migration of vascular endothelial cells, as well as the migration and differentiation of EPCs adjacent to infarcts.56 In this study, bone marrow cells from EGFP transgenic mice were transplanted into wild‐type C57BL/6 mice that had been irradiated with cobalt 60 to kill endogenous bone marrow cells. Flow cytometry analysis of bone marrow cell suspensions from these mice after 8 weeks of bone marrow reconstruction showed that 74.5% of the cells were EGFP positive, indicating that most bone marrow cells in these mice were replaced by donor cells. After construction of the focal cerebral I/R model in these mice, angiogenesis near the ischemic lesion increased 3 and 7 days after reperfusion, and the new blood vessels were both CD31 and EGFP positive, suggesting that donor bone marrow–derived cells were involved in neovascularization around ischemic lesions after cerebral ischemia. A significant increase in the mobilization of bone marrow EPCs following acute ischemic stroke is a rapid response to ischemia and hypoxia.57, 58, 59, 60, 61, 62, 63, 64, 65, 66 Our findings provide evidence that bone marrow–derived endogenous EPCs are a source of endothelial cells for the neovasculature and participate in neovascularization and vascular repair.
The repair of ischemic brain injury includes (1) neurogenesis and angiogenesis in the SVZ, (2) angiogenesis around the ischemic foci, and (3) migration of newborn SVZ neurons to the ischemic foci to form the neurovascular nest. In addition, nutrients produced by neovasculature provide an appropriate microenvironment and nutritional support for the survival of the original and newborn neurons and provide scaffolds for migration of newborn neurons.67, 68, 69 BrdU is used to label the proliferated cells because BrdU is a thymidine analogue that can be incorporated into DNA when proliferating cells enter the S cell cycle. DCX is a 43‐ to 53‐kDa intermediate filament protein expressed on both dividing NPCs and immature neurons. Both BrdU and DCX have been widely used to detect neurogenesis and neuronal migration.70, 71, 72 Consistent with the previous studies,20, 44, 73 the number of DCX‐positive cells was found to be increased in the SVZ and IBZ at 7 days after I/R in this study. In addition, similar to the prior studies showing that SVZ neurogenesis increased and neuroblasts migrated into the adjacent striatum after cerebral ischemia,74, 75, 76 many BrdU/DCX double‐labeled cells were observed in the SVZ and striatum on day 7 after cerebral I/R compared with sham operation in this study. The findings mentioned strongly suggest that NPCs in the SVZ proliferated after cerebral ischemia and that the increased NPCs in the IBZ might migrate from the SVZ. When BrdU was injected intraperitoneally on the fourth to sixth days, more DCX‐positive‐only cells were observed in the SVZ and striatum on day 7 after cerebral I/R than in those of the sham‐operated control mice. In contrast, when BrdU was injected intraperitoneally on first to seventh days, more BrdU/DCX double‐labeled cells were observed in the SVZ and striatum on day 7 after cerebral I/R than after sham operation. The long cell cycle of the NPCs may explain the phenomena. Neurogenesis was reported to be linked mechanically with angiogenesis by several studies.27, 77 In our study, the observation of a relatively higher number of microvessels and NPCs around the ischemic area indicated that the local microenvironment provided by new blood vessels facilitates NPC accumulation around ischemic foci. Our in vivo results discovered that bone marrow–derived EPCs are involved in the formation of new blood vessels; therefore, we examined the in vitro effects of EPCs on NPC migration and found that more NPCs migrated when cultured with normal EPCs in the lower chamber of the transwell apparatus than with medium alone. The findings revealed that EPCs promote NPC migration and confirmed in vitro that the local microenvironment produced by EPCs may affect NPC migration.
After cerebral ischemia, the tissues at the center of the ischemic foci undergo ischemic hypoxic degeneration and necrosis. The blood supply to the tissue at the periphery of ischemic foci decreases, leading to a hypoxic state, and VEGF expression increases as a result.16 VEGF‐C expression was detected to significantly increase when EPCs were cultured for 24 hours under hypoxic conditions compared with EPCs cultured under normal conditions. VEGF is an endothelial cell–specific mitogen and a secretory protein dimer. The exogenous administration or overexpression of VEGF can promote injury‐induced angiogenesis, induce the expansion of NPCs in the SVZ, and promote the migration and survival of newborn neurons.77, 78, 79 In this study, angiogenesis and the number of neuroblasts around ischemic foci were found to increase in vivo. In vitro analysis showed that EPCs cultured under normal conditions secreted VEGF‐C, the levels of which were higher when EPCs were cultured under hypoxic conditions. EPCs promoted the migration of NPCs. The overexpression of miR‐210 increased the expression of VEGF‐C by EPCs, further enhancing the positive effect of EPCs on NPC migration. These data suggested that VEGF produced by new blood vessels in the local microenvironment could promote NPC accumulation around ischemic foci and acted as an important link between angiogenesis and neurogenesis. Taken together, these findings further confirmed that VEGF plays an important role in neurovascular remodeling during hypoxic–ischemic brain damage.25, 26
As a highly conserved hypoxia‐regulated noncoding miRNA, miR‐210 is expressed in a variety of hypoxic tissues and plays an important role in cell survival, migration, and differentiation.29, 80, 81 Consistent with previous reports,6, 82 the expression of miR‐210 in the ischemic side of the cerebral cortex rose 1 day after focal cerebral I/R, peaked after 3 days, and began to decline at 7 days in the in vivo study. In the in vitro experiments, miR‐210 expression was upregulated in EPCs after 24 hours of culture under hypoxia. The findings confirmed that miR‐210 expression increased in both hypoxic brain tissue and EPCs. The overexpression of miR‐210 in endothelial cells in vitro promoted the formation of capillary‐like structures,7, 43, 83 and the local injection of miR‐210 promoted vascular proliferation in injured joints, the Achilles tendon, and the myocardium.10, 31 The overexpression of miR‐210 in the striatum by local injection of lentivirus was previously shown to enhance angiogenesis in the basal ganglia and neurogenesis in the SVZ and hippocampus of both normal and cerebral ischemia mice.11, 12 The present detection revealed increased expression of miR‐210 in the ischemic side of the cerebral cortex in a model of cerebral ischemia and enhanced neovascularization around the ischemic foci, suggesting that increased miR‐210 expression in the tissues around ischemic foci after cerebral ischemia was an early response of the brain to hypoxia to promote angiogenesis. This study showed that miR‐210 expression in the cerebral cortex on the ischemic side increased after focal brain ischemia. VEGF expression was also reported to increase after cerebral ischemia.84, 85, 86, 87, 88, 89, 90 To further clarify the relationship between these 2 phenomena, we injected a lentivirus to alter the local expression of miR‐210 in the striatum. A focal cerebral I/R model was established 14 days after lentivirus injection. Seven days after I/R model establishment, the expression of VEGF‐C in the cerebral cortex on the ischemic side was found to be significantly higher in the group injected with the miR‐210 lentivirus than in the group injected with the LV‐GFP lentivirus. In addition, the numbers of microvessels and DCX‐positive cells around the ischemic foci increased in the miR‐210 overexpression group, consistent with the prior reports by Zeng et al.13 EPCs were shown to be involved in angiogenesis around ischemic foci. To elucidate the relationship between angiogenesis around ischemic foci and NPC migration, miR‐210 mimics were used to overexpress miR‐210 in EPCs in vitro, and the data revealed that both VEGF‐C expression and the number of migrated NPCs significantly increased in the miR‐210 overexpression group compared with the untreated EPC group. In contrast, inhibition of miR‐210 expression using a miR‐210 inhibitor significantly decreased VEGF‐C expression and the number of migrated NPCs, suggesting that miR‐210 promoted angiogenesis and upregulated VEGF expression in EPCs to enhance NPC migration further. The findings were consistent with the previous studies in vitro and in vivo that showed miR‐210 upregulates VEGF expression.28, 29, 31 Unfortunately, no study has explored the mechanism underlying this effect so far. To elucidate the regulatory mechanism between miR‐210 and VEGF, we conducted a bioinformatics analysis using PicTar and Target Scan 6.0 and found that miR‐210 could bind to the 3′‐UTR of SOCS1 mRNA, which was confirmed using a luciferase reporter system. Moreover, our data showing that miR‐210 overexpression decreased SOCS1 protein expression in HEK293 cells and EPCs further verified that miR‐210 can reduce SOCS1 protein levels by inhibiting the transcription of SOCS1 mRNA. Many studies have demonstrated that SOCS1 is a negative modulator of the JAK/STAT pathway.91, 92, 93 The overexpression of SOCS1 inhibits STAT3 expression, reduces VEGF‐A production, and decreases myocardial vessel density.94 STAT3 can bind to the VEGF promoter and activating STAT3 upregulates VEGF expression. The interference of STAT3 signaling via the expression of a dominant‐negative STAT3 protein, a STAT3 inhibitor, or STAT3 antisense RNA was previously shown to downregulate VEGF expression.28, 94 The transfection of normal endometrial stromal cells with miR‐210 induced cell proliferation and VEGF production and inhibited apoptosis by activating STAT3.28 In the present study we found that SOCS1 expression decreased and STAT3 and VEGF expression increased in miR‐210–overexpressing HEK293 cells and EPCs. The expression of STAT3 and VEGF‐C significantly increased after following treatment with SOCS1 siRNA, whereas their expression significantly decreased when SOCS1 was overexpressed using pcDNA3.1‐SOCS1. These results suggested that SOCS1 is a link in the regulation of the STAT3–VEGF axis by miR‐210. When EPCs were cotransfected with pcDNA3.1‐SOCS1 and miR‐210 mimics, no significant differences in the levels of SOCS1, STAT3, and VEGF‐C were observed compared with control. These findings revealed that miR‐210 upregulates STAT3 to increase VEGF expression via the downregulation of SOCS1, thereby regulating VEGF.
This study has some limitations. Although we found that NPCs proliferated in the SVZ and some NPCs accumulated in the IBZ after cerebral I/R, we could not demonstrate accurately that the NPCs accumulated in the IBZ migrated from the SVZ in vivo because it is difficult to label NPCs in the brain.
In summary, this study showed that miR‐210 expression and the number of DCX‐positive neuroblasts increased around ischemic foci after cerebral ischemia in vivo. Moreover, bone marrow–derived EPCs were involved in neovascularization around ischemic foci, and miR‐210 overexpression promoted neovascularization around ischemic foci and NPC accumulation around these foci. The in vitro study results demonstrated that miR‐210 expression was upregulated after hypoxia, which promoted NPC migration by enhancing VEGF‐C expression in EPCs. This study also investigated the regulatory effect of miR‐210 on VEGF‐C in vitro, and the results suggested that miR‐210 decreased SOCS1 expression by inhibiting SOCS1 transcription, thereby upregulating STAT3–VEGF signaling. As an important regulatory pathway in the reconstruction of the neurovascular microenvironment after cerebral ischemia, the miR‐210–SOCS1–STAT3–VEGF axis promotes neovascularization and NPC accumulation around ischemic foci after cerebral ischemia and facilitates nerve repair.
Sources of Funding
The work was supported by grants from the National Natural Science Foundation of China (81100927 and 81571164) and Chongqing Science and Technology Commission (cstc2011jjA10079).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e005052 DOI: 10.1161/JAHA.116.005052.)29478968
References
- 1. O'Donnell MJ, Chin SL, Rangarajan S, Xavier D, Liu L, Zhang H, Rao‐Melacini P, Zhang X, Pais P, Agapay S, Lopez‐Jaramillo P, Damasceno A, Langhorne P, McQueen MJ, Rosengren A, Dehghan M, Hankey GJ, Dans AL, Elsayed A, Avezum A, Mondo C, Diener HC, Ryglewicz D, Czlonkowska A, Pogosova N, Weimar C, Iqbal R, Diaz R, Yusoff K, Yusufali A, Oguz A, Wang X, Penaherrera E, Lanas F, Ogah OS, Ogunniyi A, Iversen HK, Malaga G, Rumboldt Z, Oveisgharan S, Al Hussain F, Magazi D, Nilanont Y, Ferguson J, Pare G, Yusuf S. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case‐control study. Lancet. 2016;388:761–775. [DOI] [PubMed] [Google Scholar]
- 2. Zhou M, Wang H, Zhu J, Chen W, Wang L, Liu S, Li Y, Liu Y, Yin P, Liu J, Yu S, Tan F, Barber RM, Coates MM, Dicker D, Fraser M, Gonzalez‐Medina D, Hamavid H, Hao Y, Hu G, Jiang G, Kan H, Lopez AD, Phillips MR, She J, Vos T, Wan X, Xu G, Yan LL, Yu C, Zhao Y, Zheng Y, Zou X, Naghavi M, Wang Y, Murray CJ, Yang G, Liang X. Cause‐specific mortality for 240 causes in China during 1990–2013: a systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet. 2016;387:251–272. [DOI] [PubMed] [Google Scholar]
- 3. Kojima T, Hirota Y, Ema M, Takahashi S, Miyoshi I, Okano H, Sawamoto K. Subventricular zone‐derived neural progenitor cells migrate along a blood vessel scaffold toward the post‐stroke striatum. Stem Cells. 2010;28:545–554. [DOI] [PubMed] [Google Scholar]
- 4. Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou WJ, Zhu DL, Yang GY, Zhang Y, Wang HY, Ji KD, Lu YM, Gao PJ. Circulating endothelial progenitor cells in Chinese patients with acute stroke. Hypertens Res. 2009;32:306–310. [DOI] [PubMed] [Google Scholar]
- 6. Bai YY, Peng XG, Wang LS, Li ZH, Wang YC, Lu CQ, Ding J, Li PC, Zhao Z, Ju SH. Bone marrow endothelial progenitor cell transplantation after ischemic stroke: an investigation into its possible mechanism. CNS Neurosci Ther. 2015;21:877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F. MicroRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther. 2010;125:92–104. [DOI] [PubMed] [Google Scholar]
- 8. Zeng L, Liu J, Wang Y, Wang L, Weng S, Tang Y, Zheng C, Cheng Q, Chen S, Yang GY. MicroRNA‐210 as a novel blood biomarker in acute cerebral ischemia. Front Biosci (Elite Ed). 2011;3:1265–1272. [DOI] [PubMed] [Google Scholar]
- 9. Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA‐210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin‐A3. J Biol Chem. 2008;283:15878–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alaiti MA, Ishikawa M, Masuda H, Simon DI, Jain MK, Asahara T, Costa MA. Up‐regulation of miR‐210 by vascular endothelial growth factor in ex vivo expanded CD34+ cells enhances cell‐mediated angiogenesis. J Cell Mol Med. 2012;16:2413–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu F, Lou YL, Wu J, Ruan QF, Xie A, Guo F, Cui SP, Deng ZF, Wang Y. Upregulation of microRNA‐210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney Blood Press Res. 2012;35:182–191. [DOI] [PubMed] [Google Scholar]
- 12. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC. MicroRNA‐210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010;122:S124–S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeng LL, He XS, Liu JR, Zheng CB, Wang YT, Yang GY. Lentivirus‐mediated overexpression of microRNA‐210 improves long‐term outcomes after focal cerebral ischemia in mice. CNS Neurosci Ther. 2016;22:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeng L, He X, Wang Y, Tang Y, Zheng C, Cai H, Liu J, Fu Y, Yang GY. MicroRNA‐210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. 2014;21:37–43. [DOI] [PubMed] [Google Scholar]
- 15. Yu D, Chen W, Ren J, Zhang T, Yang K, Wu G, Liu H. VEGF‐PKD1‐HDAC7 signaling promotes endothelial progenitor cell migration and tube formation. Microvasc Res. 2014;91:66–72. [DOI] [PubMed] [Google Scholar]
- 16. Holzer LA, Cor A, Pfandlsteiner G, Holzer G. Expression of VEGF, its receptors, and HIF‐1α in Dupuytren's disease. Acta Orthop. 2013;84:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dzietko M, Derugin N, Wendland MF, Vexler ZS, Ferriero DM. Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl Stroke Res. 2013;4:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schoch HJ, Fischer S, Marti HH. Hypoxia‐induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–2557. [DOI] [PubMed] [Google Scholar]
- 19. Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA. Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res. 2007;29:375–381. [DOI] [PubMed] [Google Scholar]
- 20. Moriyama Y, Takagi N, Hashimura K, Itokawa C, Tanonaka K. Intravenous injection of neural progenitor cells facilitates angiogenesis after cerebral ischemia. Brain Behav. 2013;3:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Rooijen E, Voest EE, Logister I, Bussmann J, Korving J, van Eeden FJ, Giles RH, Schulte‐Merker S. von Hippel‐Lindau tumor suppressor mutants faithfully model pathological hypoxia‐driven angiogenesis and vascular retinopathies in zebrafish. Dis Model Mech. 2010;3:343–353. [DOI] [PubMed] [Google Scholar]
- 22. Rosell A, Morancho A, Navarro‐Sobrino M, Martinez‐Saez E, Hernandez‐Guillamon M, Lope‐Piedrafita S, Barcelo V, Borras F, Penalba A, Garcia‐Bonilla L, Montaner J. Factors secreted by endothelial progenitor cells enhance neurorepair responses after cerebral ischemia in mice. PLoS One. 2013;8:e73244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W. Hypoxia‐induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beck H, Acker T, Wiessner C, Allegrini PR, Plate KH. Expression of angiopoietin‐1, angiopoietin‐2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol. 2000;157:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Y, Lee C, Shen F, Du R, Young WL, Yang GY. Angiopoietin‐2 facilitates vascular endothelial growth factor‐induced angiogenesis in the mature mouse brain. Stroke. 2005;36:1533–1537. [DOI] [PubMed] [Google Scholar]
- 26. Zhang ZG, Zhang L, Tsang W, Soltanian‐Zadeh H, Morris D, Zhang R, Goussev A, Powers C, Yeich T, Chopp M. Correlation of VEGF and angiopoietin expression with disruption of blood‐brain barrier and angiogenesis after focal cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:379–392. [DOI] [PubMed] [Google Scholar]
- 27. Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM. Vascular endothelial growth factor receptor‐2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao H, Bao XJ, Wang RZ, Li GL, Gao J, Ma SH, Wei JJ, Feng M, Zhao YJ, Ma WB, Yang Y, Li YN, Kong YG. Postacute ischemia vascular endothelial growth factor transfer by transferrin‐targeted liposomes attenuates ischemic brain injury after experimental stroke in rats. Hum Gene Ther. 2011;22:207–215. [DOI] [PubMed] [Google Scholar]
- 29. Ruan L, Wang B, ZhuGe Q, Jin K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015;1623:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Okamoto M, Nasu K, Abe W, Aoyagi Y, Kawano Y, Kai K, Moriyama M, Narahara H. Enhanced miR‐210 expression promotes the pathogenesis of endometriosis through activation of signal transducer and activator of transcription 3. Hum Reprod. 2015;30:632–641. [DOI] [PubMed] [Google Scholar]
- 31. Kawanishi Y, Nakasa T, Shoji T, Hamanishi M, Shimizu R, Kamei N, Usman MA, Ochi M. Intra‐articular injection of synthetic microRNA‐210 accelerates avascular meniscal healing in rat medial meniscal injured model. Arthritis Res Ther. 2014;16:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA‐directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Usman MA, Nakasa T, Shoji T, Kato T, Kawanishi Y, Hamanishi M, Kamei N, Ochi M. The effect of administration of double stranded microRNA‐210 on acceleration of Achilles tendon healing in a rat model. J Orthop Sci. 2015;20:538–546. [DOI] [PubMed] [Google Scholar]
- 34. Gui L, Duan W, Tian H, Li C, Zhu J, Chen JF, Zheng J. Adenosine A 2A receptor deficiency reduces striatal glutamate outflow and attenuates brain injury induced by transient focal cerebral ischemia in mice. Brain Res. 2009;1297:185–193. [DOI] [PubMed] [Google Scholar]
- 35. Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD. Chronic behavioral testing after focal ischemia in the mouse: functional recovery and the effects of gender. Exp Neurol. 2004;187:94–104. [DOI] [PubMed] [Google Scholar]
- 36. Zeynalov E, Chen CH, Froehner SC, Adams ME, Ottersen OP, Amiry‐Moghaddam M, Bhardwaj A. The perivascular pool of aquaporin‐4 mediates the effect of osmotherapy in postischemic cerebral edema. Crit Care Med. 2008;36:2634–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S. The flavanol (‐)‐epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab. 2010;30:1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo Y, Peng R, Liu Q, Xu D. Exercise training‐induced different improvement profile of endothelial progenitor cells function in mice with or without myocardial infarction. Int J Cardiol. 2016;221:335–341. [DOI] [PubMed] [Google Scholar]
- 39. Eom TY, Roth KA, Jope RS. Neural precursor cells are protected from apoptosis induced by trophic factor withdrawal or genotoxic stress by inhibitors of glycogen synthase kinase 3. J Biol Chem. 2007;282:22856–22864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. VEGF contributes to postnatal neovascularization by mobilizing bone marrow‐derived endothelial progenitor cells. EMBO J. 1999;18:3964–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou J, Zhang J, Jin Z, Yi D. The effects of curcumin post‐treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol. 2012;107:263. [DOI] [PubMed] [Google Scholar]
- 42. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. Constitutive Stat3 activity up‐regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. [DOI] [PubMed] [Google Scholar]
- 43. Doti N, Scognamiglio PL, Madonna S, Scarponi C, Ruvo M, Perretta G, Albanesi C, Marasco D. New mimetic peptides of the kinase‐inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem J. 2012;443:231–240. [DOI] [PubMed] [Google Scholar]
- 44. Xiong H, Du W, Zhang YJ, Hong J, Su WY, Tang JT, Wang YC, Lu R, Fang JY. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter‐associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol Carcinog. 2012;51:174–184. [DOI] [PubMed] [Google Scholar]
- 45. Lou YL, Guo F, Liu F, Gao FL, Zhang PQ, Niu X, Guo SC, Yin JH, Wang Y, Deng ZF. MiR‐210 activates notch signaling pathway in angiogenesis induced by cerebral ischemia. Mol Cell Biochem. 2012;370:45–51. [DOI] [PubMed] [Google Scholar]
- 46. Chu M, Hu X, Lu S, Gan Y, Li P, Guo Y, Zhang J, Chen J, Gao Y. Focal cerebral ischemia activates neurovascular restorative dynamics in mouse brain. Front Biosci (Elite Ed). 2012;4:1926–1936. [DOI] [PubMed] [Google Scholar]
- 47. Thored P, Wood J, Arvidsson A, Cammenga J, Kokaia Z, Lindvall O. Long‐term neuroblast migration along blood vessels in an area with transient angiogenesis and increased vascularization after stroke. Stroke. 2007;38:3032–3039. [DOI] [PubMed] [Google Scholar]
- 48. Sawada M, Matsumoto M, Sawamoto K. Vascular regulation of adult neurogenesis under physiological and pathological conditions. Front Neurosci. 2014;8:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li WL, Fraser JL, Yu SP, Zhu J, Jiang YJ, Wei L. The role of VEGF/VEGFR2 signaling in peripheral stimulation‐induced cerebral neurovascular regeneration after ischemic stroke in mice. Exp Brain Res. 2011;214:503–513. [DOI] [PubMed] [Google Scholar]
- 50. Coyle P, Jokelainen PT. Dorsal cerebral arterial collaterals of the rat. Anat Rec. 1982;203:397–404. [DOI] [PubMed] [Google Scholar]
- 51. Hedhli N, Dobrucki LW, Kalinowski A, Zhuang ZW, Wu X, Russell RR 3rd, Sinusas AJ, Russell KS. Endothelial‐derived neuregulin is an important mediator of ischaemia‐induced angiogenesis and arteriogenesis. Cardiovasc Res. 2012;93:516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hamada Y, Gonda K, Takeda M, Sato A, Watanabe M, Yambe T, Satomi S, Ohuchi N. In vivo imaging of the molecular distribution of the VEGF receptor during angiogenesis in a mouse model of ischemia. Blood. 2011;118:e93–e100. [DOI] [PubMed] [Google Scholar]
- 53. Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–1798. [DOI] [PubMed] [Google Scholar]
- 54. Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–180. [DOI] [PubMed] [Google Scholar]
- 55. Marti HH, Bernaudin M, Petit E, Bauer C. Neuroprotection and angiogenesis: dual role of erythropoietin in brain ischemia. News Physiol Sci. 2000;15:225–229. [DOI] [PubMed] [Google Scholar]
- 56. Janic B, Arbab AS. The role and therapeutic potential of endothelial progenitor cells in tumor neovascularization. ScientificWorldJournal. 2010;10:1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iskander A, Knight RA, Zhang ZG, Ewing JR, Shankar A, Varma NR, Bagher‐Ebadian H, Ali MM, Arbab AS, Janic B. Intravenous administration of human umbilical cord blood‐derived AC133+ endothelial progenitor cells in rat stroke model reduces infarct volume: magnetic resonance imaging and histological findings. Stem Cells Transl Med. 2013;2:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beck H, Voswinckel R, Wagner S, Ziegelhoeffer T, Heil M, Helisch A, Schaper W, Acker T, Hatzopoulos AK, Plate KH. Participation of bone marrow‐derived cells in long‐term repair processes after experimental stroke. J Cereb Blood Flow Metab. 2003;23:709–717. [DOI] [PubMed] [Google Scholar]
- 59. Hess DC, Hill WD, Martin‐Studdard A, Carroll J, Brailer J, Carothers J. Bone marrow as a source of endothelial cells and NeuN‐expressing cells after stroke. Stroke. 2002;33:1362–1368. [DOI] [PubMed] [Google Scholar]
- 60. Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow‐derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90:284–288. [DOI] [PubMed] [Google Scholar]
- 61. Gao D, Nolan D, McDonnell K, Vahdat L, Benezra R, Altorki N, Mittal V. Bone marrow‐derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta. 2009;1796:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002;109:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hu Y, Davison F, Zhang Z, Xu Q. Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation. 2003;108:3122–3127. [DOI] [PubMed] [Google Scholar]
- 64. Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. [DOI] [PubMed] [Google Scholar]
- 65. Taguchi A, Matsuyama T, Moriwaki H, Hayashi T, Hayashida K, Nagatsuka K, Todo K, Mori K, Stern DM, Soma T, Naritomi H. Circulating CD34‐positive cells provide an index of cerebrovascular function. Circulation. 2004;109:2972–2975. [DOI] [PubMed] [Google Scholar]
- 66. Leone AM, Rutella S, Bonanno G, Abbate A, Rebuzzi AG, Giovannini S, Lombardi M, Galiuto L, Liuzzo G, Andreotti F, Lanza GA, Contemi AM, Leone G, Crea F. Mobilization of bone marrow‐derived stem cells after myocardial infarction and left ventricular function. Eur Heart J. 2005;26:1196–1204. [DOI] [PubMed] [Google Scholar]
- 67. Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–464. [DOI] [PubMed] [Google Scholar]
- 68. Guo H, Zhou H, Lu J, Qu Y, Yu D, Tong Y. Vascular endothelial growth factor: an attractive target in the treatment of hypoxic/ischemic brain injury. Neural Regen Res. 2016;11:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia‐Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone‐derived neuroblasts migrate and differentiate into mature neurons in the post‐stroke adult striatum. J Neurosci. 2006;26:6627–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Couillard‐Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005;21:1–14. [DOI] [PubMed] [Google Scholar]
- 71. Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule‐associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. [DOI] [PubMed] [Google Scholar]
- 72. Brown JP, Couillard‐Després S, Cooper‐Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. [DOI] [PubMed] [Google Scholar]
- 73. Cheng X, Wang Z, Yang J, Ma M, Lu T, Xu G, Liu X. Acidic fibroblast growth factor delivered intranasally induces neurogenesis and angiogenesis in rats after ischemic stroke. Neurol Res. 2011;33:675–680. [DOI] [PubMed] [Google Scholar]
- 74. Ruan L, Lau BW, Wang J, Huang L, Zhuge Q, Wang B, Jin K, So KF. Neurogenesis in neurological and psychiatric diseases and brain injury: from bench to bedside. Prog Neurobiol. 2014;115:116–137. [DOI] [PubMed] [Google Scholar]
- 75. Yang Z, You Y, Levison SW. Neonatal hypoxic/ischemic brain injury induces production of calretinin‐expressing interneurons in the striatum. J Comp Neurol. 2008;511:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003;24:171–189. [DOI] [PubMed] [Google Scholar]
- 77. Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF‐induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111:1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang Y, Jin K, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA. VEGF‐overexpressing transgenic mice show enhanced post‐ischemic neurogenesis and neuromigration. J Neurosci Res. 2007;85:740–747. [DOI] [PubMed] [Google Scholar]
- 79. Thau‐Zuchman O, Shohami E, Alexandrovich AG, Leker RR. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J Cereb Blood Flow Metab. 2010;30:1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Agrawal R, Pandey P, Jha P, Dwivedi V, Sarkar C, Kulshreshtha R. Hypoxic signature of microRNAs in glioblastoma: insights from small RNA deep sequencing. BMC Genomics. 2014;15:686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jiang Y, Li L, Tan X, Liu B, Zhang Y, Li C. MiR‐210 mediates vagus nerve stimulation‐induced antioxidant stress and anti‐apoptosis reactions following cerebral ischemia/reperfusion injury in rats. J Neurochem. 2015;134:173–181. [DOI] [PubMed] [Google Scholar]
- 82. Qiu J, Zhou XY, Zhou XG, Cheng R, Liu HY, Li Y. Neuroprotective effects of microRNA‐210 on hypoxic‐ischemic encephalopathy. Biomed Res Int. 2013;2013:350419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pulkkinen K, Malm T, Turunen M, Koistinaho J, Yla‐Herttuala S. Hypoxia induces microRNA miR‐210 in vitro and in vivo ephrin‐A3 and neuronal pentraxin 1 are potentially regulated by miR‐210. FEBS Lett. 2008;582:2397–2401. [DOI] [PubMed] [Google Scholar]
- 84. Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in an MCA‐occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999;58:654–666. [DOI] [PubMed] [Google Scholar]
- 85. Croll SD, Wiegand SJ. Vascular growth factors in cerebral ischemia. Mol Neurobiol. 2001;23:121–135. [DOI] [PubMed] [Google Scholar]
- 86. Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. [DOI] [PubMed] [Google Scholar]
- 87. Coyle P, Heistad DD. Blood flow through cerebral collateral vessels one month after middle cerebral artery occlusion. Stroke. 1987;18:407–411. [DOI] [PubMed] [Google Scholar]
- 88. Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt. Expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–1872; discussion 1872‐1873. [DOI] [PubMed] [Google Scholar]
- 89. Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood‐brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. [DOI] [PubMed] [Google Scholar]
- 91. Cittadini A, Monti MG, Iaccarino G, Castiello MC, Baldi A, Bossone E, Longobardi S, Marra AM, Petrillo V, Saldamarco L, During MJ, Sacca L, Condorelli G. SOCS1 gene transfer accelerates the transition to heart failure through the inhibition of the gp130/JAK/STAT pathway. Cardiovasc Res. 2012;96:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113(Pt 16):2813–2819. [DOI] [PubMed] [Google Scholar]
- 93. Shi J, Wei L. Regulation of JAK/STAT signalling by SOCS in the myocardium. Cardiovasc Res. 2012;96:345–347. [DOI] [PubMed] [Google Scholar]
- 94. Funamoto M, Fujio Y, Kunisada K, Negoro S, Tone E, Osugi T, Hirota H, Izumi M, Yoshizaki K, Walsh K, Kishimoto T, Yamauchi‐Takihara K. Signal transducer and activator of transcription 3 is required for glycoprotein 130‐mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem. 2000;275:10561–10566. [DOI] [PubMed] [Google Scholar]