

Abstract
Background
For many indications, the negative chronotropic effect of β‐blockers is important to their efficacy, yet the heart rate (HR) response to β‐blockers varies. Herein, we sought to use a genome‐wide association approach to identify novel single nucleotide polymorphisms (SNPs) associated with HR response to β‐blockers.
Methods and Results
We first performed 4 genome‐wide association analyses for HR response to atenolol (a β1‐adrenergic receptor blocker) as: (1) monotherapy or (2) add‐on therapy, in 426 whites and 273 blacks separately from the PEAR (Pharmacogenomic Evaluation of Antihypertensive Responses) study. A meta‐analysis was then performed between the genome‐wide association analysis performed in PEAR atenolol monotherapy and add‐on therapy, in each race separately, using the inverse variance method assuming fixed effects. From this analysis, SNPs associated with HR response to atenolol at a P<1E‐05 were tested for replication in whites (n=200) and blacks (n=168) treated with metoprolol (a β1‐adrenergic receptor blocker). From the genome‐wide association meta‐analyses, SNP rs17117817 near olfactory receptor family10 subfamily‐p‐member1 (OR10P1), and SNP rs2364349 in sorting nexin‐9 (SNX9) replicated in blacks. The combined studies meta‐analysis P values for the rs17117817 and rs2364349 reached genome‐wide significance (rs17117817G‐allele; Meta‐β=5.53 beats per minute, Meta‐P=2E‐09 and rs2364349 A‐allele; Meta‐β=3.5 beats per minute, Meta‐P=1E‐08). Additionally, SNPs in the OR10P1 and SNX9 gene regions were also associated with HR response in whites.
Conclusions
This study highlights OR10P1 and SNX9 as novel genes associated with changes in HR in response to β‐blockers.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00246519.
Keywords: atenolol, heart rate, metoprolol, pharmacogenomics, β‐blockers
Subject Categories: Genetic, Association Studies; Cardiovascular Disease; Hypertension
Clinical Perspective
What Is New?
Herein, we used a genome‐wide association approach to identify novel genetic polymorphisms associated with heart rate (HR) response to β‐blockers.
From the genome‐wide analyses, we identified and replicated 2 genetic signals, rs17117817 in OR10P1 genetic region and rs2364349 in SNX9, with clinically relevant effects on HR response to β‐blockers.
Results from the meta‐analysis revealed that participants with rs17117817 T/T and G/T genotypes had ≈11 and 5.5 beats per minute reductions in their HR response to β‐blockers, respectively, compared with noncarriers.
Similarly, rs2364349G/G and A/G genotype carriers had ≈7 and 3.5 beats per minute reductions in their HR, respectively, compared with noncarriers.
What Are the Clinical Implications?
The results of this study highlight OR10P1 and SNX9 as novel genes associated with changes in HR in response to β‐blockers.
Replication of the findings from this study in large, well‐designed independent studies is still needed, which may help guide the selection of antihypertensive therapy in the future.
Future investigation of the association between OR10P1 and SNX9 and HR response to β‐blockers may provide novel insights into the mechanism underlying HR response.
Over the past 5 decades, β‐adrenergic receptor blockers (β‐blockers) have been a cornerstone therapy for heart failure, post–myocardial infarction, symptomatic angina, and other cardiovascular diseases.1, 2 Additionally, they remain one of the most commonly prescribed classes of drugs in the United States.3 β‐Blockers work by inhibiting the β‐adrenergic receptors in the heart, which prevent the binding of epinephrine and norepinephrine to these receptors and eventually reduce cardiac contractility and heart rate (HR).4 HR lowering in response to β‐blockers has been associated with lower risk of incident heart failure and cardiovascular diseases.5, 6 Additionally, results from clinical trials and meta‐analyses have shown a significant association between β‐blockers’ HR‐lowering effect and improvement in clinical outcomes.7, 8, 9, 10 Altogether, these data highlight that the HR‐modulating effects of β‐blockers are a critical efficacy component for this class of drugs and contributes to their beneficial therapeutic effects.
Despite the widespread use of β‐blockers, interindividual differences in HR‐lowering response to β‐blockers have been observed.11, 12 These differences might be attributed to several factors, including genetics.13 Over the past 2 decades, considerable resources have been allocated to elucidate the genetic contributors of variability in drug response to β‐blockers. Most of this research has focused on explaining differences in their effects on blood pressure, heart failure phenotypes, and effects on cardiovascular outcomes.14, 15, 16, 17, 18 However, only a few candidate gene pharmacogenetic studies have focused on evaluating the role of genetics in explaining the differences in β‐blockers’ negative chronotropic response. Not surprisingly, ADRB1, the gene encoding the β1 receptor, has been the focus of most studies evaluating variability in negative chronotropic response to β‐blockers.12, 19, 20, 21, 22, 23, 24 However, there is a paucity of information about other potential genes that may be associated with changes in β‐blockers’ negative chronotropic response.
Since autonomic regulation of HR is dependent on a vast array of proteins, change in HR in response to a β‐blocker may be controlled via multiple genes. Therefore, a thorough understanding of genetic determinants associated with changes in HR in response to a β‐blocker is needed. Accordingly, in this study, we sought to conduct a genome‐wide association analysis to identify novel genetic predictors associated with changes in HR in response to β‐blockers therapy in European‐Americans (whites) and African‐Americans (blacks).
Methods
Study Design and Participants
PEAR study
A description of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study (clinicaltrials.gov #NCT00246519), including the study rationale, design, protocol, and safety procedures, has been previously published.25 PEAR genotype and phenotype data used in this study have been made publicly available at the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000649.v1.p1; dbgap study accession: phs000649.v1.p1). Briefly, PEAR was a randomized, open‐label, multicenter, prospective study designed to evaluate the role of genetic variation on the blood pressure response to the β‐blocker atenolol and the thiazide diuretic hydrochlorothiazide (HCTZ). In total, 768 individuals with mild‐to‐moderate essential hypertension, of any race or ethnicity, and between the ages of 17 and 65 years completed the PEAR study. Following a washout period and collection of baseline data, study participants were randomized to either atenolol 50 mg or HCTZ 12.5 mg, both administered once daily, with the dose doubled (ie, atenolol 100 mg or HCTZ 25 mg) in patients with a systolic blood pressure >120 mm Hg and/or diastolic blood pressure >70 mm Hg. Responses to treatment were assessed after 6 to 9 weeks of monotherapy (response assessment #1; Figure 1), after which those subjects who were still not at goal had the alternate drug added with a similar dose titration scheme and a response assessment following 6 to 9 weeks on combination therapy (response assessment #2). In PEAR, >85% of the participants treated with atenolol as a mono‐ or add‐on therapy had atenolol dose titrated to 100 mg per day. Data collected included home, office, and 24‐hour ambulatory systolic blood pressure, diastolic blood pressure, and HR. Both HR measurement and blood pressure response were used to guide dosage adjustments, as those individuals with HR <55 beats per minute were precluded from receiving a higher dose of atenolol. Institutional Review Boards at each clinical research center approved the study protocol (University of Florida, Gainesville, FL; Emory University, Atlanta, GA; Mayo Clinic, Rochester, MN) and written informed consent was obtained from all participants.
Figure 1.
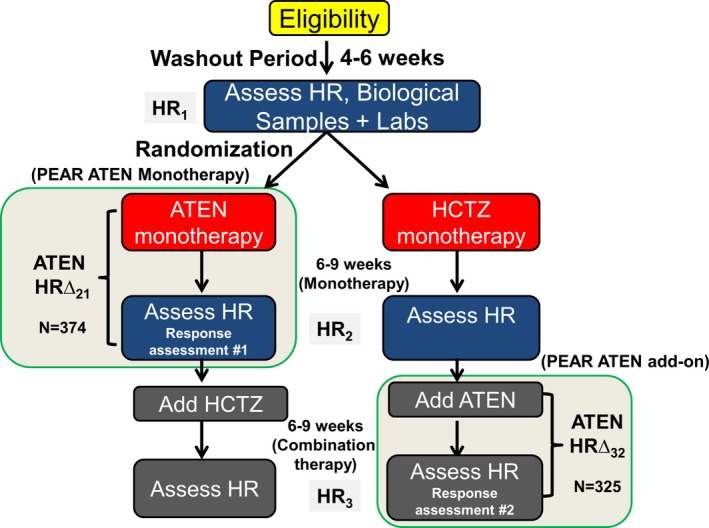
PEAR study design. ATEN indicates atenolol; HCTZ, hydrochlorothiazide; HR, heart rate; PEAR, Pharmacogenomics Evaluation of Antihypertensive Response.
PEAR‐2 study
PEAR‐2 was a multicenter, prospective, open‐label, sequential monotherapy clinical trial (clinicaltrials.gov #NCT01203852).26 The PEAR‐2 genotype and phenotype data used in this study are currently in the process of being uploaded to dbGaP and will soon be available to other researchers under dbGaP accession phs000649.v2.p1. The Institutional Review Boards of each enrolling institution (University of Florida, Gainesville, FL; Emory University, Atlanta, GA; Mayo Clinic, Rochester, MN) approved the study and all subjects provided written informed consent. Eligible study participants received metoprolol tartrate 50 mg twice daily, and after 2 weeks if an inadequate response was observed (>120/70 mm Hg and HR >55 bpm), the dose was increased to 100 mg twice daily for an additional 6 weeks. Of note, >95% of the participants treated with metoprolol had their dose titrated to 100 mg twice day. Metoprolol and atenolol are both β1‐adrenergic receptor blockers, and they have the same mechanism of reducing HR via their inhibitory effect on the β1‐adrenergic receptor. Thus, we used data from PEAR‐2 metoprolol‐treated participants for replicating the results discovered from PEAR atenolol‐treated participants.
Heart Rate Assessment
In both the PEAR and PEAR‐2 studies, HR data were generated from measurements collected using a home blood pressure monitor, the Microlife model 3AC1‐PC home blood pressure monitor (Minneapolis, MN), which has been validated for accuracy.27 Blood pressure and HR were measured in triplicate mode in the morning and evening, averaged by the blood pressure, and recorded with a date and time stamp. At each assessment period, study participants were required to have 5 morning and evening recordings in the previous 7 days. Thus, HR values used in the analysis described herein were based on a minimum of 30 HR recordings. Baseline HR was measured at the end of an average 4‐week washout of antihypertensive drugs (HR1). HR response following monotherapy with atenolol or HCTZ was measured after 6 to 9 weeks (HR2), and HR response following addition of atenolol to HCTZ was measured after 6 to 9 weeks of combination therapy (HR3). In PEAR‐2, the HR response to metoprolol was noted as HR2 and the baseline HR was HR1.
Genetic Analyses and Genotyping
Genome‐wide association study approach
Details of the genome‐wide genotyping, quality control, and imputation performed on PEAR samples were previously described.28 In brief, PEAR DNA samples underwent genotyping using the Illumina Human Omni‐1Million Quad BeadChip (Illumina, San Diego CA). Genotypes were called using GenTrain2 Illumina clustering algorithm in the software package GenomeStudio (Illumina, San Diego, CA). MaCH software (version 1.0.16) was used to impute single nucleotide polymorphisms (SNPs) based on HapMapIII haplotypes. SNPs with minor allele frequency <5% or imputation r 2 <0.3 were excluded from the analysis. After quality control, we had ≈1.1 million SNPs that were included in the genome‐wide association discovery analyses in white participants in PEAR monotherapy and PEAR add‐on therapy and ≈1.2 million SNPs in blacks. For PEAR‐2, genotyping was conducted using Human Omni2.5 S BeadChip (Illumina), and imputation based on 1000 Genomes Phase I reference panels was performed using Minimac.29 Participants from PEAR or PEAR‐2 were excluded if sample genotype call rates were below 95%. Sample contamination was tested by checking sex mismatches using X chromosome genotype data, and those who were discordant were excluded. Additionally, cryptic relatedness was estimated by pairwise identity‐by‐descent analysis implemented using PLINK.30 We also ran principal component analysis on all samples, using the EIGENSTRAT method,30 to assess the ancestral background. Heterozygosity was also assessed using PLINK, by estimating the inbreeding coefficient. After imputing PEAR2 genotyping data and running quality control procedures, as explained above, we had ≈6.5 million SNPs in whites and ≈8.9 million SNPs in blacks treated with metoprolol therapy, which were used for the replication efforts in this study.
The analysis approach used in this study is summarized in Figure 2. First, we aimed to identify new genetic variants that contribute to the observed interindividual variability in HR‐lowering effect in response to atenolol by conducting a genome‐wide association study (GWAS). A meta‐analysis was then performed, in each race separately, between PEAR atenolol monotherapy and PEAR atenolol add‐on therapy. To replicate our findings, SNPs with P<1E‐05 from the PEAR meta‐analysis were evaluated for association with changes in HR in response to metoprolol in PEAR‐2. Given the fact that patterns of linkage disequilibrium often differ across different ancestral groups, further validation of SNPs of interest was performed by looking up regions of the SNPs of interest in the alternate ancestral group.
Figure 2.
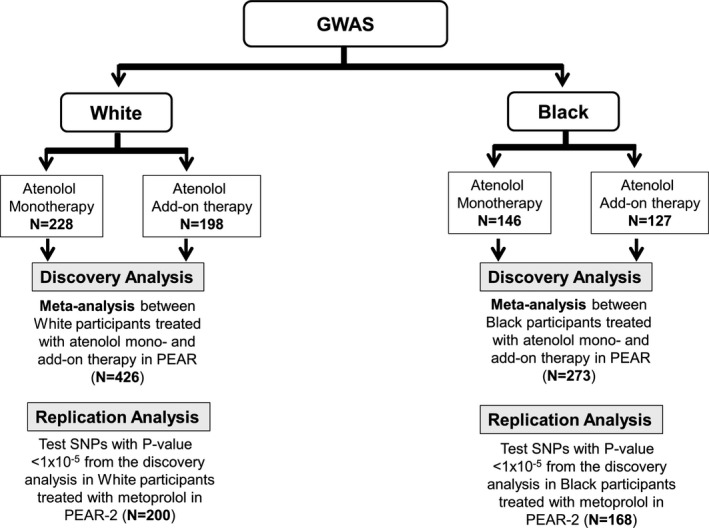
The overall analysis framework of the study. GWAS indicates genome‐wide association study; PEAR, Pharmacogenomics Evaluation of Antihypertensive Response; SNP, single nucleotide polymorphism.
Statistical Analysis
In PEAR, the change in HR in response to atenolol was determined as follows (Figure 1). For the monotherapy, HR1 was subtracted from HR2 (HRΔ21); for the add‐on therapy, HR2 was subtracted from HR3 (HRΔ32). Hence, HR1 and HR2 values were considered baseline HR for monotherapy and add‐on therapy, respectively. In PEAR‐2, the change in HR in response to metoprolol was obtained from subtracting HR2 from HR1 (HRΔ21).
For the GWAS discovery analyses, 4 separate genome‐wide association analyses were conducted for association with β‐blockers’ negative chronotropic response; analyses of response to atenolol as: (1) monotherapy, and (2) add‐on therapy, in whites and blacks. Association analyses between SNPs and HR responses were performed using a linear regression analysis in PLINK.30 All analyses were conducted under an additive model of inheritance and adjusted for age, sex, baseline HR, and principal components 1 and 2. Although >85% of the atenolol‐treated participants in PEAR monotherapy and add‐on therapy were taking the same dose of therapy (atenolol 100 mg per day), we adjusted for differences in dose in the analyses to prevent any confounding results. A meta‐analysis was then performed between PEAR atenolol monotherapy and PEAR atenolol add‐on therapy, in each race separately, assuming fixed effects and using inverse‐variance weighting as implemented in the METAL software.31 For the GWAS analysis, a typical genome‐wide P value of <5E‐08 was used to judge the significance of SNPs in both black and white participants. SNPs were considered suggestive with meta‐analysis P<1E‐05. Power analysis was performed using Quanto (http://biostats.usc.edu/software) to identify the power and effect sizes that can be detected using our study sample size. We found that our GWAS discovery analysis in whites (n=426) has >80% power to detect a 5 bpm difference in β‐blocker HR response by genotype, assuming a SD of 10 bpm and SNPs of minor allele frequency ≥15%, with 2‐sided α level=1E‐05 (suggestive α level used in the study) (Table S1). This shows that this study is only powered to detect common SNPs with large effect size. Similarly, the power calculation in blacks shows that we only have power to detect common SNPs with large effect sizes (Table S1). This reveals that the sample size used in this study is not powered to detect SNPs with small effect sizes.
PLINK30 software was used to explore the linkage disequilibrium between SNPs with P<1E‐05 from PEAR atenolol mono‐ and add‐on therapy GWAS meta‐analysis. SNPs were then pruned based on linkage disequilibrium by removing any SNP with an r 2 >0.5 with any other SNP in a 50 SNP window. SNPs that remained after pruning were considered independent SNPs and were moved forward for replication in PEAR‐2 participants treated with metoprolol (Figure 2). A Bonferroni correction was used to define a significant threshold for this analysis (0.05/number of independent SNPs tested). Deviation from Hardy–Weinberg equilibrium was tested by χ2 or Fisher exact test in each race separately, and SNPs with Hardy‐Weinberg P<1E‐06 were excluded from the analysis.
To evaluate the effect of multiple response alleles on the HR response to β‐blockers and to investigate the relative contribution of our genetic findings toward the phenotype, we constructed a genetic response score based on replicated SNPs. Points were given for the genotypes of the replicated SNPs in which the homozygous genotype of each SNP with the greatest HR‐lowering effect had 2 points, while heterozygous genotype had 1 point, and homozygous genotype associated with the worst HR‐lowering effect had 0. Alleles with HR‐lowering effect were then summed up for inclusion in a linear regression model to test the association between the response score and changes in HR in response to β‐blockers in PEAR and PEAR‐2 participants. The latter analyses were adjusted for age, sex, baseline HR, differences in dose, and PC1 and 2.
Network and In Silico Analyses
To further investigate the potential physiological roles of the replicated SNPs identified in this study and the possible interactions between them, we used STRING database (version 10.5) (https://string-db.org/).32 Replicated SNPs, from the GWAS meta‐analysis, were used to build networks showing the potential interactions between the identified SNPs/genes and other potential interacting genes that might be involved in the mechanism underlying β‐blocker HR‐lowering effects.
Results
Baseline demographic characteristics for the PEAR and PEAR‐2 study participants treated with β‐blockers are summarized in Table 1. Age, sex, body mass index, baseline HR, and HR response were similar between white atenolol mono‐ and add‐on therapy and metoprolol monotherapy–treated participants. Similarly, age, sex, body mass index, baseline HR, and HR response were similar between black atenolol mono‐ and add‐on therapy and metoprolol monotherapy‐treated participants. The mean and 95% confidence interval for atenolol HR‐lowering response following mono‐ and add‐on therapy were similar, as shown in Table S2, suggesting minimal chronotropic response following HCTZ treatment, which provides the justification for combining these in the meta‐analysis. We did not conduct any comparisons between whites and blacks because analyses in these groups were all done separately and the purpose of this study is not to evaluate differences between whites and blacks.
Table 1.
Characteristics of β‐Blocker‐Treated Participants in PEAR and PEAR‐2
| Characteristics | PEAR Atenolol Monotherapy (N=374) | PEAR Atenolol Add‐on (N=325) | PEAR‐2 Metoprolol Monotherapy (N=368) | |||
|---|---|---|---|---|---|---|
| White (n=228) | Black (n=146) | White (n=198) | Black (n=127) | White (n=200) | Black (n=168) | |
| Age, mean (SD) y | 49.5±9.5 | 47.2±8.5 | 49.9±9.5 | 47.4±8.8 | 51.0±9.0 | 50.0±9.2 |
| Women, N (%) | 109 (47.3) | 107 (73.2) | 91 (45.9) | 92 (77.3) | 110 (55.0) | 89 (53.0) |
| BMI, mean (SD) kg×m−2 | 30.3±5.6 | 31.6±6.3 | 30.3±4.9 | 31.5±5.4 | 30.8±5.1 | 30.8±5.2 |
| Pretreatment HR, mean (SD) bpm | 76.5±9.2 | 79.9±9.2 | 77.8±9.7 | 81.2±9.6 | 77.7±9.6 | 79.8±9.5 |
| Change in HR, mean (SD) bpm | −12.8±5.5 | −11.0±6.8 | −13.4±6.0 | −11.1±7.4 | −12.3±7.2 | −11.2±7.0 |
BMI indicates body mass index; bpm, beats per minute; HR, heart rate; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses.
Genome‐Wide Association Analyses
Four separate genome‐wide association analyses were conducted for association with β‐blockers’ negative chronotropic response; these included analyses of response to atenolol as monotherapy and add‐on therapy in whites and blacks, followed by meta‐analysis of the 2 data sets within race. SNPs with a P<1E‐05 for atenolol monotherapy associations in whites and blacks are reported in Tables S3 and S4, respectively, and those for associations with response to atenolol add‐on therapy are in Tables S5 and S6, respectively. Manhattan plots and q‐q plots for each GWAS analysis are presented in Figures S1 through S4.
In blacks, the meta‐analysis of atenolol monotherapy and add‐on therapy revealed 35 SNPs achieving the suggestive P value level, representing 20 independent genetic signals (Table 2); these were tested for replication in PEAR‐2 blacks treated with metoprolol. Replicated SNPs were defined as those with a Bonferroni corrected P<0.0025 (0.05/20 independent genetic signals), and with HR‐lowering effects in the same direction observed in the discovery analysis. From this analysis, we found 2 SNPs—SNP rs17117817 located 7 kb away from 5′ of olfactory receptor family10 subfamily p‐member1 (OR10P1) gene, and SNP rs2364349 within the sorting nexin‐9 gene (SNX9)—to be significantly associated with metoprolol's HR‐lowering effect in black participants (Table 2). Using an additive genetic model, blacks carrying the rs17117817 G‐allele or rs2364349 A‐allele had a smaller HR‐lowering response to β‐blockers (Figure 3). The combined 3‐cohort meta‐analysis P value for the rs17117817 and rs2364349 reached genome‐wide significance (rs17117817G‐allele; β=5.53 bpm, P=2E‐09 and rs2364349 A‐allele; β=3.5 bpm, P=1E‐08, respectively). Additionally, we validated the association between the OR10P1 and SNX9 genetic regions and changes in HR in response to β‐blockers in whites where we found significant SNPs in the OR10P1 and SNX9 genetic regions associated with HR changes in response to β‐blockers (rs79860936 in OR10P1 gene region; P=7.6E‐04, rs7772983 in the SNX9 gene region; P=1.7E‐03, Figure 4).
Table 2.
Replication of the Linkage‐Disequilibrium Pruned SNPs With P Value <0.00001 From the GWAS Meta‐Analysis of β‐Blockers HR Response in PEAR‐2 Participants Treated With Metoprolol
| Race | SNP | A1 | FRQ | Rsqa | Meta Effect | Meta SE | Meta P Value | Dir | PEAR‐2 Effect | PEAR‐2 SE | PEAR‐2 P Value | Rsqb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blackc | rs17117817c | Tc | 0.93c | 1.00 to 0.82c | −5.73c | 1.11c | 2E‐07c | −−c | −5.05c | 1.65c | 2.2E‐03c | 1.00c |
| Black | rs10809249 | T | 0.82 | 0.95 to 0.99 | −3.52 | 0.74 | 2E‐06 | −− | −0.57 | 0.98 | 0.56 | 0.99 |
| Black | rs12890215 | T | 0.82 | 1.01 to 1.03 | −3.4 | 0.72 | 2E‐06 | −− | −0.48 | 0.92 | 0.602 | 1.00 |
| Black | rs1045580 | T | 0.85 | 0.95 to 1.02 | 3.56 | 0.76 | 2E‐06 | ++ | 0.62 | 1.02 | 0.53 | 1.00 |
| Black | rs2806495 | A | 0.10 | 0.96 to 0.82 | 4.67 | 0.99 | 2E‐06 | ++ | −0.53 | 1.29 | 0.67 | 1.00 |
| Black | rs7738600 | A | 0.51 | 1.05 to 1.03 | −2.6 | 0.56 | 4E‐06 | −− | −0.24 | 0.71 | 0.73 | 1.00 |
| Black | rs11757000 | T | 0.77 | 0.99 to 0.91 | −3.05 | 0.67 | 5E‐06 | −− | 0.35 | 0.91 | 0.69 | 1.00 |
| Black | rs7042878 | C | 0.09 | 1.05 to 1.10 | 4.31 | 0.95 | 5E‐06 | ++ | 1.65 | 1.35 | 0.22 | 1.00 |
| Blackc | rs2364349c | Ac | 0.18c | 0.87 to 0.92c | 3.43c | 0.76c | 7E‐06c | ++c | 3.64c | 1.03c | 4E‐04c | 0.98c |
| Black | rs4733278 | T | 0.46 | 0.82 to 0.99 | −2.62 | 0.58 | 7E‐06 | −− | 0.01 | 0.75 | 0.98 | 0.99 |
| Black | rs11069252 | T | 0.92 | 0.92 to 1.00 | −4.76 | 1.06 | 7E‐06 | −− | 1.21 | 1.37 | 0.37 | 1.00 |
| Black | rs12417208 | A | 0.84 | 0.89 to 0.82 | −3.76 | 0.84 | 8E‐06 | −− | −1.43 | 1.09 | 0.19 | 0.93 |
| Black | rs3759422 | T | 0.46 | 0.91 to 1.06 | 2.5 | 0.56 | 8E‐06 | ++ | 0.77 | 0.69 | 0.26 | 1.00 |
| Black | rs1018353 | T | 0.9 | 0.95 to 0.85 | −4.29 | 0.96 | 8E‐06 | −− | 0.94 | 1.37 | 0.49 | 1.00 |
| Black | rs4554901 | T | 0.07 | 0.93 to 1.03 | 4.85 | 1.09 | 8E‐06 | ++ | −0.09 | 1.41 | 0.94 | 0.99 |
| Black | rs10145648 | T | 0.74 | 0.98 to 1.00 | −2.85 | 0.64 | 8E‐06 | −− | −0.34 | 0.77 | 0.65 | 1.00 |
| Black | rs10809367 | A | 0.14 | 1.18 to 1.18 | 3.41 | 0.77 | 9E‐06 | ++ | 0.95 | 1.18 | 0.42 | 1.00 |
| Black | rs10499 | A | 0.95 | 0.94 to 1.10 | −5.67 | 1.28 | 9E‐06 | −− | −2.01 | 2.00 | 0.31 | 1.00 |
| Blackc | rs6455914c | Tc | 0.22c | 0.97 to 0.93c | 3.08c | 0.7c | 1E‐05c | ++c | 1.73c | 0.83c | 0.03c | 0.99c |
| Black | rs6470259 | A | 0.80 | 0.94 to 0.88 | −3.2 | 0.72 | 1E‐05 | −− | −0.76 | 1.01 | 0.44 | 0.94 |
| White | rs955395 | A | 0.36 | 0.99 to 0.96 | −1.7 | 0.35 | 9E‐07 | −− | 0.21 | 0.59 | 0.69 | 0.99 |
| White | rs11727192 | T | 0.21 | 0.94 to 0.97 | −1.92 | 0.42 | 5E‐06 | −− | −0.80 | 0.77 | 0.29 | 1.00 |
| White | rs10516175 | A | 0.12 | 1.09 to 1.02 | 2.17 | 0.48 | 6E‐06 | ++ | 0.46 | 0.79 | 0.64 | 0.99 |
| White | rs13160161 | A | 0.37 | 1.07 to 0.97 | −1.5 | 0.33 | 7E‐06 | −− | −0.34 | 0.67 | 0.71 | 1.00 |
| White | rs11641210 | T | 0.89 | 0.99 to 1.00 | 2.32 | 0.52 | 8E‐06 | ++ | −0.50 | 1.00 | 0.61 | 0.99 |
A1 indicates coded allele; Dir, the direction of effect; FRQ, coded allele frequency; GWAS, genome‐wide association study; HR, heart rate; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses; POS, position; SNP, single nucleotide polymorphism.
Rsq, r‐squared imputation quality metric in the discovery analysis in which the first number represents the imputation quality for the presented SNP in PEAR monotherapy and the second number represents the imputation quality for the presented SNP in PEAR add‐on therapy.
Rsq, r‐squared imputation quality metric in the replication analysis (PEAR2).
Represent polymorphisms with a P value <0.05 in the replication cohort and the effect of the polymorphism on HR in response to β‐blockers is in the same direction in the discovery (Meta Effect) and replication (PEAR‐2 Effect) analysis. Meta represents meta‐analysis between PEAR β‐blocker mono‐ and add‐on therapy.
Figure 3.
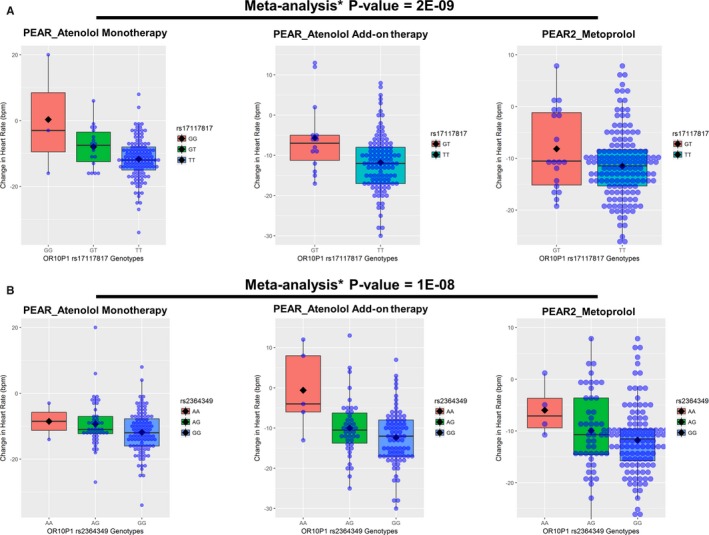
Effect of rs17117817 and rs2364349 polymorphisms on the β‐blocker HR response in whites within PEAR and PEAR‐2 studies. A, rs17117817. B, rs2364349. The box represents the values from the 25% to 75% percentile. The horizontal line represents the median. The black diamond represents the mean. The vertical line extends from the minimum to the maximum value. Each blue dot represents an individual. HR response was adjusted for age, sex, baseline HR, differences in dose, and principal components 1 and 2. Two‐sided P values represented are for the contrast of adjusted means between different genotype groups. *Meta‐analysis was performed assuming fixed effects and using inverse‐variance weighting. bpm indicates beats per minute; HR, heart rate; OR10P1, olfactory receptor family 10 subfamily p member 1; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses; SNX9, sorting nexin 9.
Figure 4.
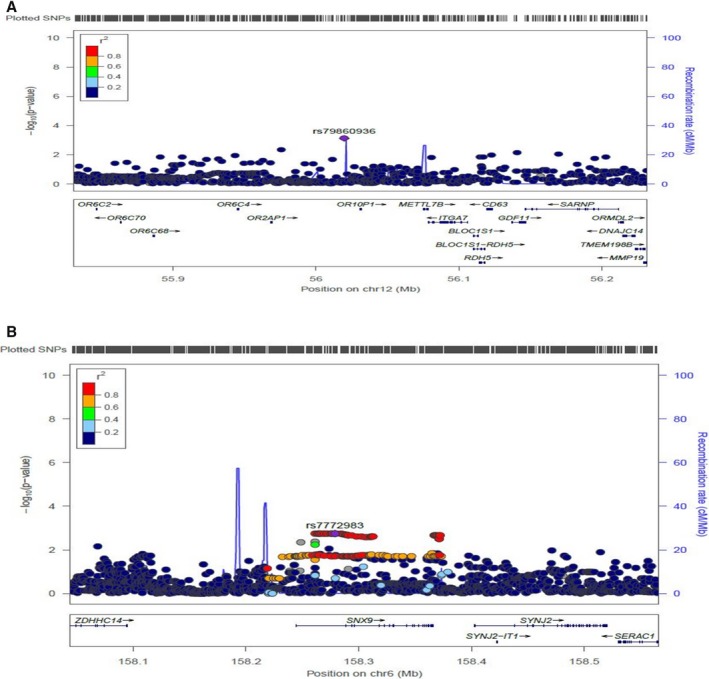
Regional plot showing the significance of the associations of SNPs with changes in HR in response to β‐blockers used in whites. A, In olfactory receptor family 10 subfamily p member 1 (OR10P1) genetic region. B, In sorting nexin‐9 (SNX9) genetic region. HR indicates heart rate; SNP, single nucleotide polymorphism.
To make sure that the 2 replicated signals were not previously associated with HR and that they are pharmacogenomics independent signals, we searched NHGRI GWAS Catalog, (https://www.ebi.ac.uk/gwas/search?query=SNX9) and the dbGap phenotype‐genotype integrator (https://www.ncbi.nlm.nih.gov/gap/phegeni) for any GWAS associations that have been previously reported between OR10P1 or SNX9 and HR. The results from this search showed no previously reported association between SNX9 or OR10P1 and HR. Although we adjusted for baseline HR in our analysis, we also tested the association between SNP rs2364349 and SNP rs17117817 and baseline HR in black participants treated with β‐blockers in PEAR and PEAR‐2 participants included in this study. We found no association between SNP rs2364349 and baseline HR in PEAR monotherapy (P=0.23), PEAR add‐on therapy (P=0.37), and PEAR‐2 (P=0.70). Similarly, we found no association between SNP rs17117817 and baseline HR in PEAR monotherapy (P=0.18), PEAR add‐on therapy (P=0.79), and PEAR‐2 (P=0.30).
To assess the relative contribution of the replicated SNPs toward our changes in HR in response to β‐blockers, we created a response score as previously discussed in the Methods section. As expected, in black participants treated with atenolol mono‐ and add‐on therapy in PEAR and metoprolol in PEAR2, individuals with a higher score had a greater HR reduction in response to β‐blockers compared with individuals with lower score (Figure 5). The meta‐analysis of the 3 data sets reveal that, on average, for each HR‐lowering allele carried by an individual, there is ≈3.4 bpm reduction in HR in response to β‐blockers (P=2.4E‐12). Such an approach requires validation in an independent cohort but highlights the potential approach to clinical use of such data.
Figure 5.
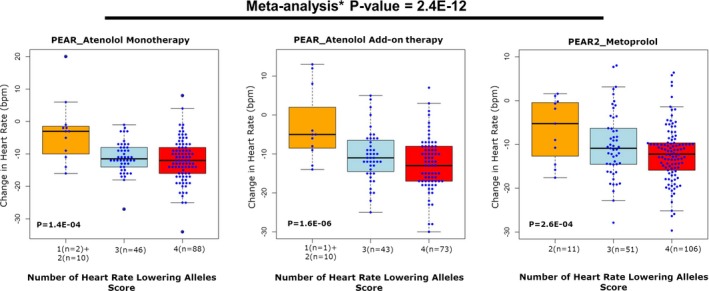
β‐Blocker's HR response score in PEAR and PEAR‐2. HR responses were adjusted for age, sex, baseline HR, differences in dose, and principal components 1 and 2. Genetic variants were coded as follows: (A) rs17117817 (OR10P1 genetic region) T/T=2, G/T=1, G/G=zero, and (B) rs2364349 (SNX9 genetic region) G/G=2, A/G=1, A/A=zero. The box represents the values from the 25% to 75% percentile. The horizontal line represents the median. The vertical line extends from the minimum to the maximum value. Each blue dot represents an individual. bpm indicates beats per minute HR, heart rate; PEAR, Pharmacogenomic Evaluation of Antihypertensive Responses. *Meta‐analysis was performed between PEAR atenolol monotherapy, PEAR atenolol add‐on therapy, and PEAR‐2 metoprolol groups, assuming fixed effects and using inverse‐variance weighting.
In whites, the meta‐analysis between PEAR atenolol mono‐ and add‐on therapy revealed 13 SNPs with a P<1E‐05, which represent 5 independent genetic signals (Table 2). However, none of these SNPs were replicated when tested in white participants treated with metoprolol in PEAR‐2 (Table 2).
Network Analysis
Lastly, we conducted a network analysis using the genes discovered in this study (OR10P1 and SNX9) and the β‐adrenergic receptor genes (ADRB1 and ADRB2), which encode the protein targets for β‐blockers, to identify potential interactions between the replicated genetic signals that might be involved in β‐blockers’ HR‐lowering effects mechanism. From this analysis, using STRING database, we found a direct interaction between SNX9 and ADRB2, and an indirect interaction between OR10P1 and ADRB1 and ADRB2 via the effect of OR10P1 on either ADRBK1 or GNB1 (Figure 6). These results further support the association we found between OR10P1 and SNX9 and changes in HR in response to β‐blockers and suggest that OR10P1 and SNX9 may be playing a potential role in the HR‐lowering mechanism underlying β‐blocker therapy.
Figure 6.
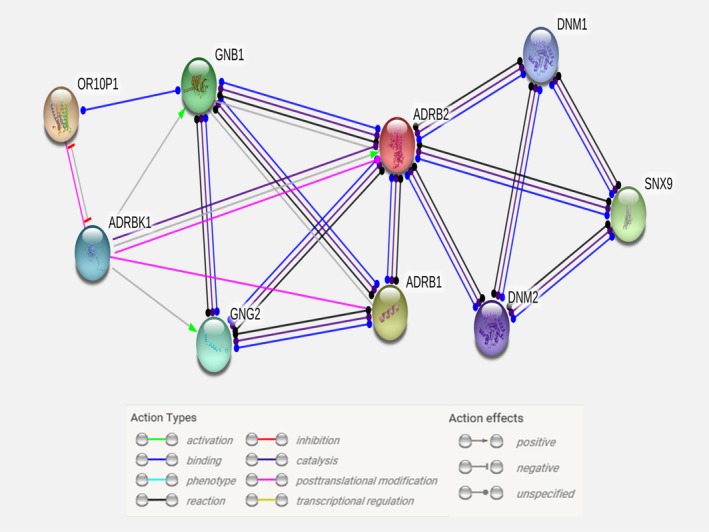
Interaction network of replicated genetic signals and protein targets for β‐blockers. Network was created using the STRING database (https://string-db.org/).
Discussion
β‐Blockers are one of the most commonly prescribed classes of drugs and have been used for decades to treat various cardiovascular conditions, including myocardial infarction, heart failure, hypertension, and angina.1, 3 The HR‐lowering effect of β‐blockers is a critical efficacy component for this class of drugs and contributes substantially to their beneficial effects in acute‐ and post–myocardial infarction, heart failure, angina, and arrhythmias.5, 6, 33 However, considerable interpatient variability in HR response to β‐blockers has been observed,11, 12 indicating that a substantial proportion of β‐blocker‐treated patients fail to achieve cardioprotection with β‐blockers. This reveals that the current approach for β‐blockers selection and achieving target HR is suboptimal. Thus, identifying factors that are associated with the observed changes in HR in response to β‐blockers is important and holds the promise to optimize the use of this class of drugs. Given the growing evidence in the literature highlighting the significant influence of genetics on HR,34, 35, 36 we sought herein to identify genetic factors associated with the observed interindividual variability in the negative chronotropic response to atenolol and metoprolol using a genome‐wide association approach. To our knowledge, the present study is the first GWAS to investigate genetic variations associated with changes in HR in response to β‐blocker therapy in whites and blacks.
The results from the GWAS analyses have revealed 2 novel genetic signals, SNP rs17117817 in the OR10P1 genetic region, and SNP rs2364349 in the SNX9 gene, with clinically relevant effects on HR in response to β‐blocker therapy used in blacks. We also validated these findings in whites, by identifying significant signals, in the OR10P1 and SNX9 genetic regions, associated with changes in HR of whites treated with β‐blockers. According to the meta‐analysis results, rs17117817 T/T and G/T genotype carriers have ≈11 and 5.5 bpm reductions in their HR in response to β‐blockers, respectively, compared with noncarriers. Similarly, rs2364349 G/G and A/G genotype carriers have ≈7 and 3.5 bpm reductions in their HR in response to β‐blockers, respectively, compared with noncarriers. Moreover, we created a response score to assess the relative contribution of the replicated SNPs and their effect on changes in HR in response to β‐blockers. From this analysis, we found that for each HR‐lowering allele carried by an individual, there is ≈3.4 bpm reduction in HR in response to β‐blockers (P=2.4E‐12). These observed reductions in HR in response to β‐blockers represent clinically significant differences that have been previously reported to be significantly associated with cardiovascular morbidity and mortality in the general population and in patients with heart disease.37, 38, 39, 40 Several studies have reported the pivotal role of β‐blockers HR reduction effect in reducing cardiovascular mortality in heart failure.41, 42 For instance, a meta‐analysis of 35 studies has shown that β‐blocker HR reduction of 5 bpm could provide 14% risk reduction in mortality.10 These data reveal the clinical significance of our findings on HR response to β‐blockers and their clinical implications on cardiovascular mortality in patients treated with β‐blockers. However, further validation of the response score and the genetic variants, identified in this study, in large well‐designed independent cohorts are still needed.
OR10P1 is a member of the olfactory receptor gene family, which represents a large family of genes that encode G‐protein coupled receptors. OR10P1 has been shown to be highly expressed in the heart,43 yet little is known about the role of OR10P1 in the regulation of HR. Herein, using network analyses, we have seen an indirect interaction between OR10P1 and ADRB2. Consistent with this finding, previous studies have demonstrated the interaction between protein members of the olfactory receptor family and β2‐adrenergic receptors.44 Additionally, several studies have shown that inactivation of β‐adrenergic receptor kinase‐2 and β‐arrestin both have a well‐known regulatory role on β‐adrenergic receptors, results in increased olfactory receptor stimulation of cAMP formation,45, 46; the latter mediates the catecholaminergic control on HR and contractility.47 Moreover, a recent study by Jovancevic et al48 has shown that the activation of olfactory receptors (ie, OR51E1) induces a negative chronotropic effect in human stem cell–derived cardiomyocytes in a dose‐dependent manner. This literature evidence, along with the results of this study, suggests that olfactory receptors, including OR10P1, may be important regulators of HR in response to β‐blockers.
SNX9, a member of the sorting nexin gene family, has been shown to be highly expressed in the heart and placenta.49 It was first described in 1999 as a Src homology 3‐domain and phox homology‐domain protein that regulates metalloproteinases ADAM9 and ADAM15.49 SNX9 has also been shown to play a substantial role in endocytosis via clathrin‐coated pits, which is well identified in the downregulation of transmembrane signal transduction but can also promote sustained signal transduction.50 Interestingly, studies have reported that SNX9 interacts and stimulates the basal GTPase activity of dynamin, a GTPase that plays a central role in clathrin‐induced endocytosis and agonist‐mediated sequestration of ADRB2.51, 52, 53 Studies have also shown that the rapid and efficient recycling of G‐protein coupled receptors, like ADRB2, to the plasma membrane after ligand‐induced endocytosis is essential for the functional resensitization of receptors‐mediated signaling.54, 55 This highlights the importance of clathrin‐mediated endocytosis, which is regulated by SNX9, in controlling the activity of G‐protein coupled receptors like ADRB2.
In addition, studies have also revealed an interaction between SNX9 and adaptor protein 2,56, 57 an important multimeric protein that binds to cargo proteins and regulates the assembly of the clathrin‐coated vesicle and their endocytosis.58 Furthermore, adaptor protein 2 has been shown to interact with β‐arrestin,59 an interaction that is required for the recycling of the ADRB2. Since β‐arrestin is known to block the activation of adenylyl cyclase, it eventually reduces ADRB responsiveness to catecholamines.60 Thus, it may be possible that SNX9 mediates its effect on HR response to β‐blockers via β‐arrestin. Moreover, the results of the network analysis that we performed revealed that SNX9 might be interacting with ADRB2 in an indirect way via its effect on dynamin 1 (DNM1) or dynamin 2 (DNM2) proteins. Although all these possible hypotheses are intriguing, follow‐up studies are needed to test these hypotheses.
The present study has several strengths. To our knowledge, this is the first GWAS conducted to identify genetic variants associated with changes in HR in response to β‐blockers in whites and blacks. Using this genome‐wide approach, we are the first to uncover the novel association between OR10P1 and SNX9 and changes in HR in response to β‐blockers. Additionally, the replication and the validation of the OR10P1 and SNX9 signals in blacks and whites, respectively, highlight the novelty of these findings. Moreover, the highly consistent effect of the OR10P1 and SNX9 genetic signals on changes in HR, across 3 independent cohorts, with 2 β‐blockers, emphasizes the importance of these findings and suggests the involvement of the OR10P1 and SNX9 in the mechanism underlying β‐blockers’ HR‐lowering effect.
The present study also has a few noteworthy limitations. First, the small sample sizes used for the genetic analyses may have limited our power to identify additional novel markers and to replicate some of our genetic signals. Second, we have used resting instead of exercise HR as an end point. However, in clinical practice, dose titration of β‐blockers is performed based on resting HR and blood pressure response. Thus, this is the more clinically relevant phenotype.
In summary, the present study sheds light on OR10P1 and SNX9 and their association with changes in HR in response to β‐blockers. Future studies are still needed to investigate the role of OR10P1 and SNX9 in the mechanism underlying HR response to β‐blockers. Such studies may provide new insights into the HR‐lowering mechanism, and help in the identification of novel drug targets based on a deeper understanding of the determinants of HR response to β‐blockers. Perhaps as we move forward towards a personalized medicine approach, future use of the genetic signals identified in this study might help in guiding the selection of β‐blockers and optimize their use.
Sources of Funding
PEAR was supported by the National Institute of Health Pharmacogenetics Research Network grant U01 GM074492 and the National Center for Advancing Translational Sciences under the award number UL1 TR000064 (University of Florida); UL1 TR000454 (Emory University); and UL1 TR000135 (Mayo Clinic). PEAR was also supported by funds from the Mayo Foundation. Gonzalez receives support for research from the National Institute of Child Health and Human Development (K23HD083465) and the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org).
Disclosures
None.
Supporting information
Table S1. Power Calculation for Whites and Blacks Involved in the Discovery Analysis
Table S2. Heart Rate Response to Atenolol and Hydrochlorothiazide in the PEAR Study Participants
Table S3. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Whites Treated With Atenolol Monotherapy in the PEAR Study
Table S4. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Blacks Treated With Atenolol Monotherapy in the PEAR Study
Table S5. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Whites Treated With Atenolol Add‐on Therapy in the PEAR Study
Table S6. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Blacks Treated With Atenolol Add‐on Therapy in the PEAR Study
Figure S1. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR white participants treated with atenolol monotherapy using HR∆21 as the phenotype.
Figure S2. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR black participants treated with atenolol monotherapy using HR∆21 as the phenotype.
Figure S3. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR white participants treated with atenolol add‐on therapy using HR∆32 as the phenotype.
Figure S4. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR black participants treated with atenolol add‐on therapy using HR∆32 as the phenotype.
Acknowledgments
We appreciate the valuable contributions of the study participants of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) and PEAR‐2. We also would like to thank PEAR and PEAR‐2 support staff and study physicians.
(J Am Heart Assoc. 2018;7:e006463 DOI: 10.1161/JAHA.117.006463.)29478026
References
- 1. Hollenberg NK. The role of beta‐blockers as a cornerstone of cardiovascular therapy. Am J Hypertens. 2005;18:165S–168S. [DOI] [PubMed] [Google Scholar]
- 2. Shin J, Johnson JA. Beta‐blocker pharmacogenetics in heart failure. Heart Fail Rev. 2010;15:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. IMS Institute . Medicines use and spending in the U.S. A review of 2015 and outlook to 2020. Available at: https://structurecms-staging-psyclone.netdna-ssl.com/client_assets/dwonk/media/attachments/590c/6aa0/6970/2d2d/4182/0000/590c6aa069702d2d41820000.pdf?1493985952. Accessed February 7, 2017.
- 4. Ladage D, Schwinger RH, Brixius K. Cardio‐selective beta‐blocker: pharmacological evidence and their influence on exercise capacity. Cardiovasc Ther. 2013;31:76–83. [DOI] [PubMed] [Google Scholar]
- 5. Okin PM, Kjeldsen SE, Julius S, Hille DA, Dahlof B, Devereux RB. Effect of changing heart rate during treatment of hypertension on incidence of heart failure. Am J Cardiol. 2012;109:699–704. [DOI] [PubMed] [Google Scholar]
- 6. Kaplan JR, Manuck SB, Adams MR, Weingand KW, Clarkson TB. Inhibition of coronary atherosclerosis by propranolol in behaviorally predisposed monkeys fed an atherogenic diet. Circulation. 1987;76:1364–1372. [DOI] [PubMed] [Google Scholar]
- 7. Cucherat M. Quantitative relationship between resting heart rate reduction and magnitude of clinical benefits in post‐myocardial infarction: a meta‐regression of randomized clinical trials. Eur Heart J. 2007;28:3012–3019. [DOI] [PubMed] [Google Scholar]
- 8. Flannery G, Gehrig‐Mills R, Billah B, Krum H. Analysis of randomized controlled trials on the effect of magnitude of heart rate reduction on clinical outcomes in patients with systolic chronic heart failure receiving beta‐blockers. Am J Cardiol. 2008;101:865–869. [DOI] [PubMed] [Google Scholar]
- 9. Kolloch R, Legler UF, Champion A, Cooper‐Dehoff RM, Handberg E, Zhou Q, Pepine CJ. Impact of resting heart rate on outcomes in hypertensive patients with coronary artery disease: findings from the INternational VErapamil‐SR/trandolapril STudy (INVEST). Eur Heart J. 2008;29:1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW. Meta‐analysis: beta‐blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784–794. [DOI] [PubMed] [Google Scholar]
- 11. Materson BJ, Reda DJ, Cushman WC, Massie BM, Freis ED, Kochar MS, Hamburger RJ, Fye C, Lakshman R, Gottdiener J. Single‐drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med. 1993;328:914–921. [DOI] [PubMed] [Google Scholar]
- 12. Cotarlan V, Brofferio A, Gerhard GS, Chu X, Shirani J. Impact of beta(1)‐ and beta(2)‐adrenergic receptor gene single nucleotide polymorphisms on heart rate response to metoprolol prior to coronary computed tomographic angiography. Am J Cardiol. 2013;111:661–666. [DOI] [PubMed] [Google Scholar]
- 13. Petrashevskaya NN, Koch SE, Bodi I, Schwartz A. Calcium cycling, historic overview and perspectives. Role for autonomic nervous system regulation. J Mol Cell Cardiol. 2002;34:885–896. [DOI] [PubMed] [Google Scholar]
- 14. Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. Beta 1‐adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther. 2003;74:44–52. [DOI] [PubMed] [Google Scholar]
- 15. Liu J, Liu ZQ, Yu BN, Xu FH, Mo W, Zhou G, Liu YZ, Li Q, Zhou HH. beta1‐Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin Pharmacol Ther. 2006;80:23–32. [DOI] [PubMed] [Google Scholar]
- 16. Mialet Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW, Liggett SB. Beta 1‐adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003;9:1300–1305. [DOI] [PubMed] [Google Scholar]
- 17. Terra SG, Hamilton KK, Pauly DF, Lee CR, Patterson JH, Adams KF, Schofield RS, Belgado BS, Hill JA, Aranda JM, Yarandi HN, Johnson JA. Beta1‐adrenergic receptor polymorphisms and left ventricular remodeling changes in response to beta‐blocker therapy. Pharmacogenet Genomics. 2005;15:227–234. [DOI] [PubMed] [Google Scholar]
- 18. Liggett SB, Mialet‐Perez J, Thaneemit‐Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause‐Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conserved beta(1)‐adrenergic receptor motif alters cardiac function and beta‐blocker response in human heart failure. Proc Natl Acad Sci USA. 2006;103:11288–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beitelshees AL, Zineh I, Yarandi HN, Pauly DF, Johnson JA. Influence of phenotype and pharmacokinetics on beta‐blocker drug target pharmacogenetics. Pharmacogenomics J. 2006;6:174–178. [DOI] [PubMed] [Google Scholar]
- 20. de Groote P, Helbecque N, Lamblin N, Hermant X, Mc Fadden E, Foucher‐Hossein C, Amouyel P, Dallongeville J, Bauters C. Association between beta‐1 and beta‐2 adrenergic receptor gene polymorphisms and the response to beta‐blockade in patients with stable congestive heart failure. Pharmacogenet Genomics. 2005;15:137–142. [DOI] [PubMed] [Google Scholar]
- 21. Karlsson J, Lind L, Hallberg P, Michaelsson K, Kurland L, Kahan T, Malmqvist K, Ohman KP, Nystrom E, Melhus H. Beta(1)‐adrenergic receptor gene polymorphisms and response to beta(1)‐adrenergic receptor blockade in patients with essential hypertension. Clin Cardiol. 2004;27:347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kurnik D, Li C, Sofowora GG, Friedman EA, Muszkat M, Xie HG, Harris PA, Williams SM, Nair UB, Wood AJ, Stein CM. Beta‐1‐adrenoceptor genetic variants and ethnicity independently affect response to beta‐blockade. Pharmacogenet Genomics. 2008;18:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu J, Liu ZQ, Tan ZR, Chen XP, Wang LS, Zhou G, Zhou HH. Gly389Arg polymorphism of beta(1)‐adrenergic receptor is associated with the cardiovascular response to metoprolol. Clin Pharmacol Ther. 2003;74:372–379. [DOI] [PubMed] [Google Scholar]
- 24. Sehrt D, Meineke I, Tzvetkov M, Gultepe S, Brockmoller J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics. 2011;12:783–795. [DOI] [PubMed] [Google Scholar]
- 25. Johnson JA, Boerwinkle E, Zineh I, Chapman AB, Bailey K, Cooper‐DeHoff RM, Gums J, Curry RW, Gong Y, Beitelshees AL, Schwartz G, Turner ST. Pharmacogenomics of antihypertensive drugs: rationale and design of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study. Am Heart J. 2009;157:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamadeh IS, Langaee TY, Dwivedi R, Garcia S, Burkley BM, Skaar TC, Chapman AB, Gums JG, Turner ST, Gong Y, Cooper‐DeHoff RM, Johnson JA. Impact of CYP2D6 polymorphisms on clinical efficacy and tolerability of metoprolol tartrate. Clin Pharmacol Ther. 2014;96:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Brien E, Waeber B, Parati G, Staessen J, Myers MG. Blood pressure measuring devices: recommendations of the European Society of Hypertension. BMJ. 2001;322:531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Turner ST, Boerwinkle E, O'Connell JR, Bailey KR, Gong Y, Chapman AB, McDonough CW, Beitelshees AL, Schwartz GL, Gums JG, Padmanabhan S, Hiltunen TP, Citterio L, Donner KM, Hedner T, Lanzani C, Melander O, Saarela J, Ripatti S, Wahlstrand B, Manunta P, Kontula K, Dominiczak AF, Cooper‐DeHoff RM, Johnson JA. Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension. 2013;62:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome‐wide association studies through pre‐phasing. Nat Genet. 2012;44:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Freemantle N, Cleland J, Young P, Mason J, Harrison J. beta Blockade after myocardial infarction: systematic review and meta regression analysis. BMJ. 1999;318:1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eppinga RN, Hagemeijer Y, Burgess S, Hinds DA, Stefansson K, Gudbjartsson DF, van Veldhuisen DJ, Munroe PB, Verweij N, van der Harst P. Identification of genomic loci associated with resting heart rate and shared genetic predictors with all‐cause mortality. Nat Genet. 2016;48:1557–1563. [DOI] [PubMed] [Google Scholar]
- 35. den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, Nolte IM, Segre AV, Holm H, Handsaker RE, Westra HJ, Johnson T, Isaacs A, Yang J, Lundby A, Zhao JH, Kim YJ, Go MJ, Almgren P, Bochud M, Boucher G, Cornelis MC, Gudbjartsson D, Hadley D, van der Harst P, Hayward C, den Heijer M, Igl W, Jackson AU, Kutalik Z, Luan J, Kemp JP, Kristiansson K, Ladenvall C, Lorentzon M, Montasser ME, Njajou OT, O'Reilly PF, Padmanabhan S, St Pourcain B, Rankinen T, Salo P, Tanaka T, Timpson NJ, Vitart V, Waite L, Wheeler W, Zhang W, Draisma HH, Feitosa MF, Kerr KF, Lind PA, Mihailov E, Onland‐Moret NC, Song C, Weedon MN, Xie W, Yengo L, Absher D, Albert CM, Alonso A, Arking DE, de Bakker PI, Balkau B, Barlassina C, Benaglio P, Bis JC, Bouatia‐Naji N, Brage S, Chanock SJ, Chines PS, Chung M, Darbar D, Dina C, Dorr M, Elliott P, Felix SB, Fischer K, Fuchsberger C, de Geus EJ, Goyette P, Gudnason V, Harris TB, Hartikainen AL, Havulinna AS, Heckbert SR, Hicks AA, Hofman A, Holewijn S, Hoogstra‐Berends F, Hottenga JJ, Jensen MK, Johansson A, Junttila J, Kaab S, Kanon B, Ketkar S, Khaw KT, Knowles JW, Kooner AS, Kors JA, Kumari M, Milani L, Laiho P, Lakatta EG, Langenberg C, Leusink M, Liu Y, Luben RN, Lunetta KL, Lynch SN, Markus MR, Marques‐Vidal P, Mateo Leach I, McArdle WL, McCarroll SA, Medland SE, Miller KA, Montgomery GW, Morrison AC, Muller‐Nurasyid M, Navarro P, Nelis M, O'Connell JR, O'Donnell CJ, Ong KK, Newman AB, Peters A, Polasek O, Pouta A, Pramstaller PP, Psaty BM, Rao DC, Ring SM, Rossin EJ, Rudan D, Sanna S, Scott RA, Sehmi JS, Sharp S, Shin JT, Singleton AB, Smith AV, Soranzo N, Spector TD, Stewart C, Stringham HM, Tarasov KV, Uitterlinden AG, Vandenput L, Hwang SJ, Whitfield JB, Wijmenga C, Wild SH, Willemsen G, Wilson JF, Witteman JC, Wong A, Wong Q, Jamshidi Y, Zitting P, Boer JM, Boomsma DI, Borecki IB, van Duijn CM, Ekelund U, Forouhi NG, Froguel P, Hingorani A, Ingelsson E, Kivimaki M, Kronmal RA, Kuh D, Lind L, Martin NG, Oostra BA, Pedersen NL, Quertermous T, Rotter JI, van der Schouw YT, Verschuren WM, Walker M, Albanes D, Arnar DO, Assimes TL, Bandinelli S, Boehnke M, de Boer RA, Bouchard C, Caulfield WL, Chambers JC, Curhan G, Cusi D, Eriksson J, Ferrucci L, van Gilst WH, Glorioso N, de Graaf J, Groop L, Gyllensten U, Hsueh WC, Hu FB, Huikuri HV, Hunter DJ, Iribarren C, Isomaa B, Jarvelin MR, Jula A, Kahonen M, Kiemeney LA, van der Klauw MM, Kooner JS, Kraft P, Iacoviello L, Lehtimaki T, Lokki ML, Mitchell BD, Navis G, Nieminen MS, Ohlsson C, Poulter NR, Qi L, Raitakari OT, Rimm EB, Rioux JD, Rizzi F, Rudan I, Salomaa V, Sever PS, Shields DC, Shuldiner AR, Sinisalo J, Stanton AV, Stolk RP, Strachan DP, Tardif JC, Thorsteinsdottir U, Tuomilehto J, van Veldhuisen DJ, Virtamo J, Viikari J, Vollenweider P, Waeber G, Widen E, Cho YS, Olsen JV, Visscher PM, Willer C, Franke L, Erdmann J, Thompson JR, Pfeufer A, Sotoodehnia N, Newton‐Cheh C, Ellinor PT, Stricker BH, Metspalu A, Perola M, Beckmann JS, Smith GD, Stefansson K, Wareham NJ, Munroe PB, Sibon OC, Milan DJ, Snieder H, Samani NJ, Loos RJ. Identification of heart rate‐associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45:621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van den Berg ME, Warren HR, Cabrera CP, Verweij N, Mifsud B, Haessler J, Bihlmeyer NA, Fu YP, Weiss S, Lin HJ, Grarup N, Li‐Gao R, Pistis G, Shah N, Brody JA, Muller‐Nurasyid M, Lin H, Mei H, Smith AV, Lyytikainen LP, Hall LM, van Setten J, Trompet S, Prins BP, Isaacs A, Radmanesh F, Marten J, Entwistle A, Kors JA, Silva CT, Alonso A, Bis JC, de Boer R, de Haan HG, de Mutsert R, Dedoussis G, Dominiczak AF, Doney ASF, Ellinor PT, Eppinga RN, Felix SB, Guo X, Hagemeijer Y, Hansen T, Harris TB, Heckbert SR, Huang PL, Hwang SJ, Kahonen M, Kanters JK, Kolcic I, Launer LJ, Li M, Yao J, Linneberg A, Liu S, Macfarlane PW, Mangino M, Morris AD, Mulas A, Murray AD, Nelson CP, Orru M, Padmanabhan S, Peters A, Porteous DJ, Poulter N, Psaty BM, Qi L, Raitakari OT, Rivadeneira F, Roselli C, Rudan I, Sattar N, Sever P, Sinner MF, Soliman EZ, Spector TD, Stanton AV, Stirrups KE, Taylor KD, Tobin MD, Uitterlinden A, Vaartjes I, Hoes AW, van der Meer P, Volker U, Waldenberger M, Xie Z, Zoledziewska M, Tinker A, Polasek O, Rosand J, Jamshidi Y, van Duijn CM, Zeggini E, Jukema JW, Asselbergs FW, Samani NJ, Lehtimaki T, Gudnason V, Wilson J, Lubitz SA, Kaab S, Sotoodehnia N, Caulfield MJ, Palmer CNA, Sanna S, Mook‐Kanamori DO, Deloukas P, Pedersen O, Rotter JI, Dorr M, O'Donnell CJ, Hayward C, Arking DE, Kooperberg C, van der Harst P, Eijgelsheim M, Stricker BH, Munroe PB. Discovery of novel heart rate‐associated loci using the Exome Chip. Hum Mol Genet. 2017;26:2346–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palatini P, Julius S. Elevated heart rate: a major risk factor for cardiovascular disease. Clin Exp Hypertens. 2004;26:637–644. [DOI] [PubMed] [Google Scholar]
- 38. Benetos A, Thomas F, Bean K, Albaladejo P, Palatini P, Guize L. Resting heart rate in older people: a predictor of survival to age 85. J Am Geriatr Soc. 2003;51:284–285. [DOI] [PubMed] [Google Scholar]
- 39. Kannel WB. Risk stratification in hypertension: new insights from the Framingham Study. Am J Hypertens. 2000;13:3S–10S. [DOI] [PubMed] [Google Scholar]
- 40. Jouven X, Empana JP, Schwartz PJ, Desnos M, Courbon D, Ducimetiere P. Heart‐rate profile during exercise as a predictor of sudden death. N Engl J Med. 2005;352:1951–1958. [DOI] [PubMed] [Google Scholar]
- 41. Metra M, Torp‐Pedersen C, Swedberg K, Cleland JG, Di Lenarda A, Komajda M, Remme WJ, Lutiger B, Scherhag A, Lukas MA, Charlesworth A, Poole‐Wilson PA. Influence of heart rate, blood pressure, and beta‐blocker dose on outcome and the differences in outcome between carvedilol and metoprolol tartrate in patients with chronic heart failure: results from the COMET trial. Eur Heart J. 2005;26:2259–2268. [DOI] [PubMed] [Google Scholar]
- 42. Mulder P, Barbier S, Chagraoui A, Richard V, Henry JP, Lallemand F, Renet S, Lerebours G, Mahlberg‐Gaudin F, Thuillez C. Long‐term heart rate reduction induced by the selective I(f) current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation. 2004;109:1674–1679. [DOI] [PubMed] [Google Scholar]
- 43. Human genomics. The Genotype‐Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hague C, Uberti MA, Chen Z, Bush CF, Jones SV, Ressler KJ, Hall RA, Minneman KP. Olfactory receptor surface expression is driven by association with the beta2‐adrenergic receptor. Proc Natl Acad Sci USA. 2004;101:13672–13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dawson TM, Arriza JL, Jaworsky DE, Borisy FF, Attramadal H, Lefkowitz RJ, Ronnett GV. Beta‐adrenergic receptor kinase‐2 and beta‐arrestin‐2 as mediators of odorant‐induced desensitization. Science. 1993;259:825–829. [DOI] [PubMed] [Google Scholar]
- 46. Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. G protein‐coupled receptor kinase 3 (GRK3) gene disruption leads to loss of odorant receptor desensitization. J Biol Chem. 1997;272:25425–25428. [DOI] [PubMed] [Google Scholar]
- 47. Zaccolo M. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol. 2009;158:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jovancevic N, Dendorfer A, Matzkies M, Kovarova M, Heckmann JC, Osterloh M, Boehm M, Weber L, Nguemo F, Semmler J, Hescheler J, Milting H, Schleicher E, Gelis L, Hatt H. Medium‐chain fatty acids modulate myocardial function via a cardiac odorant receptor. Basic Res Cardiol. 2017;112:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Howard L, Nelson KK, Maciewicz RA, Blobel CP. Interaction of the metalloprotease disintegrins MDC9 and MDC15 with two SH3 domain‐containing proteins, endophilin I and SH3PX1. J Biol Chem. 1999;274:31693–31699. [DOI] [PubMed] [Google Scholar]
- 50. Thomsen AR, Plouffe B, Cahill TJ III, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ. GPCR‐G protein‐beta‐arrestin super‐complex mediates sustained G protein signaling. Cell. 2016;166:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin‐mediated endocytosis. Mol Biol Cell. 2005;16:2058–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shin N, Ahn N, Chang‐Ileto B, Park J, Takei K, Ahn SG, Kim SA, Di Paolo G, Chang S. SNX9 regulates tubular invagination of the plasma membrane through interaction with actin cytoskeleton and dynamin 2. J Cell Sci. 2008;121:1252–1263. [DOI] [PubMed] [Google Scholar]
- 53. Lundmark R, Carlsson SR. Sorting nexin 9 participates in clathrin‐mediated endocytosis through interactions with the core components. J Biol Chem. 2003;278:46772–46781. [DOI] [PubMed] [Google Scholar]
- 54. Wang Y, Lauffer B, Von Zastrow M, Kobilka BK, Xiang Y. N‐ethylmaleimide‐sensitive factor regulates beta2 adrenoceptor trafficking and signaling in cardiomyocytes. Mol Pharmacol. 2007;72:429–439. [DOI] [PubMed] [Google Scholar]
- 55. Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. [DOI] [PubMed] [Google Scholar]
- 56. Lundmark R, Carlsson SR. SNX9—a prelude to vesicle release. J Cell Sci. 2009;122:5–11. [DOI] [PubMed] [Google Scholar]
- 57. Lundmark R, Carlsson SR. The beta‐appendages of the four adaptor‐protein (AP) complexes: structure and binding properties, and identification of sorting nexin 9 as an accessory protein to AP‐2. Biochem J. 2002;362:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pearse BM, Smith CJ, Owen DJ. Clathrin coat construction in endocytosis. Curr Opin Struct Biol. 2000;10:220–228. [DOI] [PubMed] [Google Scholar]
- 59. Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The interaction of beta‐arrestin with the AP‐2 adaptor is required for the clustering of beta 2‐adrenergic receptor into clathrin‐coated pits. J Biol Chem. 2000;275:23120–23126. [DOI] [PubMed] [Google Scholar]
- 60. Shenoy SK, Lefkowitz RJ. Seven‐transmembrane receptor signaling through beta‐arrestin. Sci STKE. 2005;2005:cm10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Power Calculation for Whites and Blacks Involved in the Discovery Analysis
Table S2. Heart Rate Response to Atenolol and Hydrochlorothiazide in the PEAR Study Participants
Table S3. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Whites Treated With Atenolol Monotherapy in the PEAR Study
Table S4. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Blacks Treated With Atenolol Monotherapy in the PEAR Study
Table S5. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Whites Treated With Atenolol Add‐on Therapy in the PEAR Study
Table S6. Single Nucleotide Polymorphisms With P Value <0.00001 From the Genome‐Wide Association Analysis of β‐Blockers Heart Rate Response in Blacks Treated With Atenolol Add‐on Therapy in the PEAR Study
Figure S1. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR white participants treated with atenolol monotherapy using HR∆21 as the phenotype.
Figure S2. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR black participants treated with atenolol monotherapy using HR∆21 as the phenotype.
Figure S3. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR white participants treated with atenolol add‐on therapy using HR∆32 as the phenotype.
Figure S4. Manhattan plot and q‐q plot obtained from the genome‐wide association analysis conducted in PEAR black participants treated with atenolol add‐on therapy using HR∆32 as the phenotype.