

Abstract
Background
Hypercholesterolemia‐induced decreased availability of nitric oxide (NO) is a major factor in cardiovascular disease. We previously established that cholesterol suppresses endothelial inwardly rectifying K+ (Kir) channels and that Kir2.1 is an upstream mediator of flow‐induced NO production. Therefore, we tested the hypothesis that suppression of Kir2.1 is responsible for hypercholesterolemia‐induced inhibition of flow‐induced NO production and flow‐induced vasodilation (FIV). We also tested the role of Kir2.1 in the development of atherosclerotic lesions.
Methods and Results
Kir2.1 currents are significantly suppressed in microvascular endothelial cells exposed to acetylated–low‐density lipoprotein or isolated from apolipoprotein E–deficient (Apoe −/−) mice and rescued by cholesterol depletion. Genetic deficiency of Kir2.1 on the background of hypercholesterolemic Apoe −/−mice, Kir2.1 +/− /Apoe −/− exhibit the same blunted FIV and flow‐induced NO response as Apoe −/−or Kir2.1 +/− alone, but while FIV in Apoe −/− mice can be rescued by cholesterol depletion, in Kir2.1 +/− /Apoe −/− mice cholesterol depletion has no effect on FIV. Endothelial‐specific overexpression of Kir2.1 in arteries from Apoe −/− and Kir2.1 +/− /Apoe −/− mice results in full rescue of FIV and NO production in Apoe −/− mice with and without the addition of a high‐fat diet. Conversely, endothelial‐specific expression of dominant‐negative Kir2.1 results in the opposite effect. Kir2.1 +/− /Apoe −/−mice also show increased lesion formation, particularly in the atheroresistant area of descending aorta.
Conclusions
We conclude that hypercholesterolemia‐induced reduction in FIV is largely attributable to cholesterol suppression of Kir2.1 function via the loss of flow‐induced NO production, whereas the stages downstream of flow‐induced Kir2.1 activation appear to be mostly intact. Kir2.1 channels also have an atheroprotective role.
Keywords: atherosclerosis, endothelial dysfunction, endothelial shear stress, hypercholesterolemia, K channel, Kir channels, nitric oxide, vascular endothelium
Subject Categories: Animal Models of Human Disease, Vascular Disease, Endothelium/Vascular Type/Nitric Oxide, Ion Channels/Membrane Transport, Lipids and Cholesterol
Clinical Perspective
What Is New?
Vascular endothelial inwardly rectifying K+ (Kir) channels are functionally impaired in hypercholesterolemia, which results in reduced flow‐induced vasodilation.
Overexpression of endothelial Kir2.1 rescues the vasodilatory response to flow via recovery of nitric oxide production.
Kir2.1 deficiency exacerbates atherosclerotic lesion formation in mice fed a high‐fat diet, especially in otherwise atheroresistant regions (ie, descending aorta).
What Are the Clinical Implications?
Endothelial Kir channels may be clinically relevant targets in preventing/reversing hypercholesterolemia‐induced vascular dysfunction and atherosclerosis.
Under physiological conditions increases in shear stress imposed on the vascular wall by increased laminar blood flow promotes an active vasodilatory response mediated by the endothelium. Endothelial mechanosensors translate shear forces into chemical messengers represented primarily by nitric oxide (NO), prostacyclin, and endothelium‐derived hyperpolarizing factor.1, 2 We recently established a critical role for inwardly rectifying K+ (Kir) channels, specifically Kir2.1, in mediating flow‐induced vasodilation (FIV) in mouse mesenteric arteries.3 This effect is mediated by shear‐induced activation of Kir2.1 resulting in NO production via endothelial NO synthase (eNOS). In this study, we tested the role of endothelial Kir2.1 channels in dyslipidemia‐induced endothelial dysfunction.
Endothelial dysfunction is a hallmark predictor of advanced disease states with special reference to atherosclerosis. Hypercholesterolemia, a leading risk factor for atherosclerosis, induces endothelial dysfunction through an incompletely understood mechanism, although reduced production/bioavailability of NO has been established.4, 5, 6 Elevated low‐density lipoprotein levels correlate with reduced flow‐mediated dilation in humans in the brachial artery.7, 8 Mouse models of hypercholesterolemia, both genetic and diet‐induced, have also been shown to have impaired arterial dilations in response to the endothelium‐dependent vasodilators acetylcholine and bradykinin.9, 10, 11, 12, 13 In the present study we use an ex vivo mouse model of FIV to assess endothelial function in dyslipidemic mice.
Cholesterol‐lowering therapies can reverse endothelial dysfunction14 and our earlier studies demonstrated that Kir channels are sensitive to and inhibited by elevated cellular cholesterol.15, 16, 17 Our results indicate that impairment of FIV in resistance arteries of apolipoprotein E–deficient (Apoe −/−) mice, an established model of hypercholesterolemia,18 can be attributed to loss of Kir channel function independent of their expression. Remarkably, overexpression of Kir2.1 specifically in the endothelium results in full rescue of FIV in resistance arteries of Apoe −/− mice. We propose that hypercholesterolemia induces endothelial dysfunction through cholesterol‐induced suppression of Kir channels. In addition, we have further studied the effect of Kir2.1 channel deficiency on atherosclerosis in the Apoe −/− background.
Methods
The authors agree to make the data and materials available upon request. The corresponding author, Irena Levitan, PhD, at the University of Illinois at Chicago will maintain availability of such data and materials.
Animals
All mice were approved for use according to the University of Illinois at Chicago Animal Care Committee (ACC#16‐183). Mice were housed in the UIC Animal Care Vivarium, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and were provided food and water ad libitum. Unless otherwise stated, male mice 20 to 30 weeks old were euthanized by carbon dioxide asphyxiation followed by cervical dislocation before tissue dissection. FVB Kir2.1 +/− mice were originally purchased from Jackson Laboratory (005057) and crossed with FVB Apoe −/− mice provided by Dr Catherine Reardon at the University of Chicago. C57BL/6 (B6) Apoe −/− mice were originally purchased from Jackson Laboratory (002052). Appropriate breeding was performed to maintain the lines in house. A subset of B6 Apoe −/− mice were put on a high‐fat (42% kcal from fat), high‐cholesterol (0.2%) Western diet (Harlan, TD.88137) for 8 weeks to induce severe hypercholesterolemia.19
Endothelial Cell Isolation
Purification for cell culture
Whole mesenteric arcades were dissected from mice and cleaned of adipose tissue, and endothelial cells (ECs) were isolated and purified as previously described.3 Briefly, arteries were exposed to an enzyme cocktail consisting of neutral protease (0.5 mg/mL; Worthington) and elastase (0.5 mg/mL; Worthington) in digestion buffer for ≈30 minutes at 37°C. Added to this cocktail was collagenase type I (0.5 mg/mL final; Worthington) for another hour or until the arteries appeared dispersed. The digested tissue was then passed through a 70‐μm cell sieve, resuspended in magnetic‐activated cell sorting buffer (Miltenyi Biotec), and centrifuged at 300g for 10 minutes. Cells were then incubated with magnetic‐activated cell sorting buffer containing MicroBeads (Miltenyi Biotec) conjugated to anti‐mouse CD31 antibody for 20 minutes at 4°C. Separation of ECs (CD31+) from non‐ECs (CD31−) was performed by passing the cells through columns attached to a magnet, thereby trapping CD31+ cells in the column and allowing CD31− cells to pass through. The “flow through” of CD31− cells was passed through a new column to increase CD31+ yield. The CD31+ cells were passed through a new column to increase purity. Cells were then immediately cultured for Western blot and electrophysiology.
Fresh isolation for electrophysiology
ECs from WT and Apoe −/− mice were isolated as previously described3, 20 with minor modifications. Briefly, mesenteric arcades cleaned of adipose tissue are digested using an enzyme cocktail of neutral protease (0.5 mg/mL; Worthington) and elastase (0.5 mg/mL; Worthington) in digestion buffer for 1 hour at 37°C. Added to this cocktail was collagenase type I (0.5 mg/mL final; Worthington) for 2 to 2.5 minutes. Following incubation with enzymes, the arteries were broken down on ice in digestion buffer using syringe needles. Broken down arteries were pipetted several times using a glass pipette to mechanically disperse ECs into a single cell suspension and allowed to adhere on ice to appropriate dishes for patch clamp experiments.
General Electrophysiology
Soft glass pipettes (SG10 glass, Richland Glass) were pulled using a vertical pipette puller (Model PP‐830) and had resistances between 2 and 4 MΩ. The perforated patch technique was performed by adding amphotericin B (250 μg/mL) to the pipette solution. Perforated patches were obtained within 2 to 5 minutes after formation of a GΩ seal, and accepted recordings for offline analysis maintained a series resistance between 10 and 30 MΩ. Currents from ECs were recorded using an EPC9/10 amplifier and accompanying acquisition and analysis software (Pulse and PulseFit, HEKA Electronik). ECs were held at −30 mV and a voltage ramp of −120 to +40 mV applied over 400 ms. Currents were low‐pass filtered at 2 kHz and recordings were digitized at 10 kHz. When necessary, leak subtraction was performed offline to collect the most accurate data points at −100 mV for group analysis.
Cultured ECs
Mouse mesentery ECs were incubated for 24 hours with or without acetylated low‐density lipoprotein (acLDL; 50 μg/mL) on glass cover slips. ECs were rinsed in PBS and transferred to the minimally invasive flow device for recording currents before and after application of shear stress, as previously described.3 Shear stress was calculated using the equation τ=6 μQ/h2w, where τ is shear stress (0.7 dynes/cm2), μ is fluid viscosity (0.009 g/cm per second), Q is flow rate (0.3 mL/s applied by gravity perfusion), h is the height (0.1 cm), and w is the width (2.2 cm) of the minimally invasive flow device.
Fresh ECs
ECs liberated from WT or Apoe −/− mice were allowed to adhere to the surface of a petri dish. For experiments involving cholesterol depletion of Apoe −/− ECs using methyl‐β‐cyclodextrin (MβCD), ECs from each mouse were divided into 2 groups: control (no MβCD) and those exposed to MβCD (5×10−3 mol/L). Cells were incubated for 1 hour in serum‐free media (Lonza) in a standard cell culture incubator. Solutions were rapidly exchanged so that cells were bathed in a 60 mmol/L K+ solution for subsequent perforated patch clamp electrophysiology.
FIV in Mesenteric Arteries
Dilations to flow were measured as previously described.3, 21, 22, 23 First‐order mesenteric arteries were cannulated and equilibrated at 60 cm H2O for 1 hour in a chamber designed to accept external bath circulated by a peristaltic pump. The pump moves Krebs solution bubbled with O2 and passes it through a Langendorff to be warmed to 37°C before reaching the chamber. The chamber was also kept at 37°C by a separate circuit connected to a water bath incubator. Diameters were recorded by video microscopy and a video image measurement system calibrated for horizontal measurements (model VIA‐100, Boekeler). Baseline diameter was recorded before preconstricting the vessel to ≈50% baseline diameter with endothelin‐1 (1.2–2×10−10 mol/L). Arteries that did not constrict >40% to 2×10−10 mol/L endothelin‐1 were discarded from study. Flow was induced by simultaneously raising and lowering the reservoirs in equal and opposite directions to obtain a dose response to flow in the form of an increasing pressure gradient denoted as ΔX cm H2O (eg, elevating 1 reservoir up to 65 cm H2O and lowering the other reservoir to 55 cm H2O administers a flow of Δ10 cm H2O). The arteries were exposed to each step increase in flow in this manner for 5 minutes before a measurement was recorded. The effects of Ba2+ (3×10−5 mol/L), L‐NG‐Nitroarginine methyl ester (LNAME; 10−4 mol/L), and apamin (2×10−8 mol/L) were observed by adding the drug(s) to the circulating bath solution for 30 minutes before repeating the FIV protocol. Administering intraluminal flow in this way effectively isolates the effects of flow‐induced shear stress by keeping intraluminal pressure constant.24 Performing these experiments in this way fit into the physiological ranges of shear stress arteries of this size experience (≈10 and 50 dynes/cm2 at pressure gradient–induced flows between Δ40 and Δ100).3, 24 At the end of each protocol, papaverine (10−4 mol/L) was added to the bath to ensure intact arterial smooth muscle function before proceeding to the next protocol for the same vessel. For arteries that did not dilate to papaverine beyond 80% baseline diameter, especially in the case of experimental groups that exhibited reduced FIV, the experiment was stopped and no measurements were accepted for such an artery as to not include data that may not represent pure endothelial dysfunction in these mouse models. For representative traces, dilations to flow were continuously monitored through the experiment with a data point being collected every 30 seconds. For experiments involving cholesterol depletion of Apoe −/− arteries, first‐order mesenteric arteries from an individual mouse were separated into 2 groups: control (no MβCD) and those exposed to MβCD (5×10−3 mol/L) for 1 hour before testing FIV.
NO Detection in Mesenteric Arteries
To detect flow‐induced NO production in mouse mesenteric arteries, first order arteries were incubated in HEPES containing the NO‐specific fluorescent dye Diaminorhodamine‐4M (DAR4M, Enzo Scientific) at room temperature for 2 hours in the dark. Arteries were cannulated and pressurized as described above and then exposed to a pressure gradient of Δ60 cm H2O or maintained at 60 cm H2O (no flow control) for 30 minutes. Arteries were removed from the chambers and mounted on glass slides to be imaged by fluorescence microscopy (Nikon Eclipse 80i). No fewer than 5 images per artery were taken from each vessel at ×40 magnification. Fluorescence intensity from each image generated an average fluorescence intensity/artery. Arteries that received flow were compared with “no flow” controls treated at the same time. ImageJ software25 was used to measure FI.
Western Blot
Western blots on cultured ECs and whole mesenteric arcades were performed as previously described.3 ECs with or without 24‐hour exposure to acLDL (50 μg/mL) were rinsed in ice‐cold PBS and lysed with cold radioimmunoprecipitation assay buffer containing protease inhibitors (Calbiochem). Lysed samples were centrifuged to remove membrane lipids and supernatants stored at −20°C until use. Whole mesenteric arcades were sonicated on ice in cold radioimmunoprecipitation assay buffer containing protease inhibitors immediately upon dissection and cleaning of adipose tissue. Samples were centrifuged to form a pellet that was sonicated once more before a final centrifugation step to collect supernatant. Samples were stored at −80°C until use. Bradford assays were performed to ensure loading of equal protein. Samples were reduced and denatured by boiling at 90°C for 10 minutes with SDS sample buffer. Samples were run on 10% SDS gels and transferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% nonfat milk before overnight incubation with primary antibodies to Kir2.1 (rabbit monoclonal; Abcam), eNOS (rabbit polyclonal; Santa Cruz), or β‐actin (mouse monoclonal; Protein Tech) at 4°C. Following incubations with appropriate secondary antibodies, membranes were incubated with enhanced chemiluminescence and exposed to film. Developed films were scanned to produce TIFF digital images for analysis using ImageJ software.25
Adenoviral Transduction
hVE‐Cadherin Kir2.1 WT and dominant‐negative (DN) adenoviruses were generated by Vector Biolabs from constructs we provided. For transduction of adenovirus into ECs, WT Kir2.1 adenovirus or empty adenovirus was incubated under optimum culture conditions for ECs (full EGM2 media [LONZA], 5% CO2, 37°C) at 10 MOI for 48 hours before perforated patch clamp or imaging of hemagglutinin (HA)‐tag (WT Kir2.1) or green fluorescent protein (empty) fluorescence.3 For transduction of adenovirus into arteries, 5 to 10 first‐order mesenteric arteries were isolated and incubated in DMEM containing 100 MOI of either WT Kir2.1, DN Kir2.1, or empty adenovirus for 48 hours in a 5% CO2 cell culture incubator before FIV or detection of NO.
Fluorescent imaging of the HA‐tag
Mouse ECs were transduced with Kir2.1 adenovirus or empty adenovirus as described above and fixed with 4% paraformaldehyde. Fixed ECs were incubated overnight at 4°C with a primary HA‐tag antibody (goat polyclonal; Abcam) to detect an internal HA insert located in the Kir2.1 construct that presents as an extracellular epitope. A conjugated secondary antibody (Alexa Fluor 555; Life Technologies) was used to detect the presence of HA‐tag via fluorescence microscopy using a Zeiss microscope, AxioCam camera/AxioVision software, and XCite light source. As expected, HA‐tag expression was not detected in ECs transduced with empty adenovirus; however, to ensure successful transduction, we tested for the presence of green fluorescent protein, which is included in the empty adenovirus construct.
Lesion Analysis in En Face Aortas
Mice were fed a high‐fat diet beginning at 12 weeks of age for 6 or 9 months and provided food ad libitum. At 36 (6‐month diet) or 48 (9‐month diet) weeks of age, male Apoe −/− or Kir2.1 +/− /Apoe −/− mice were anesthetized by continuous delivery of 3.5% isoflurane in 96.5% oxygen throughout the procedure. An incision was made in the right ventricle to drain blood, and tissue fixation was performed by an initial perfusion via left ventricular puncture to administer saline to flush out the blood for ≈5 minutes. This was followed by perfusion with fixative (4% paraformaldehyde) for ≈15 to 20 minutes. The aorta was then isolated to the thoracic region before the diaphragm and cut specifically for en face staining. Staining for lesions was performed with an Oil Red O solution (0.3% Oil Red O in isopropanol diluted 3:2 in deionized H2O) for 20 minutes and washed 3 times with an isopropanol/water mixture. Images of stained aortas were then captured using a USB CAM30/50 camera and software (World Precision Instruments) attached to a stereoscope. All images were taken at the same magnification and pixel rates.
Solutions and reagents
Digestion solution contained (in mmol/L): NaCl (55), KCl (5.6), MgCl2 (2), Na‐glutamate (80), HEPES (10), and glucose (10),and pH 7.3. Perforated patch bath solution contained (in mm/L): NaCl (80), KCl (60), HEPES (10), MgCl2 (1), CaCl2 (2), glucose (10), and pH 7.4. Perforated patch pipette solution contained (in mmol/L): NaCl (5), KCl (135), EGTA (5), MgCl2 (1), glucose (5), HEPES (10), and pH 7.2. Krebs solution contained (in mmol/L): NaCl (123), KCl (4.7), MgSO4 (1.2), CaCl2 (2.5), NaHCO3 (16), EDTA (0.026), glucose (11), KH2PO4 (1.2), and pH 7.4. HEPES buffer contained (in mmol/L): NaCl (140), KCl (4), MgCl2 (1), glucose (5), HEPES (10), CaCl2 (2), and pH 7.4. Radioimmunoprecipitation assay buffer contained (in mmol/L): Tris (150, pH 7.6), 1% NP‐40, 0.5% Na‐DOC, NaCl (150), and EDTA (1). All reagents were from Sigma unless otherwise stated.
Statistical Analysis
A paired or unpaired Student t test, 1‐way ANOVA, or a 2‐way with or without repeated measures ANOVA were used where indicated. Significance, in all cases, was set to P<0.05. Bonferroni post hoc tests were used to determine where differences existed after significance was detected with ANOVA.
Results
Shear‐Induced Increase in Kir Current is Reduced in acLDL‐Treated Primary ECs
We previously reported the inhibitory effect of hypercholesterolemia and elevated cellular cholesterol on aortic EC Kir currents.15, 26 However, the effect of dyslipidemia on ECs isolated from arteries representing the resistance vasculature was not investigated. Using a modified low‐density lipoprotein (acLDL; 50 μg/mL), which is efficient in loading ECs with cholesterol,15, 21 we demonstrate here that acLDL‐mediated cholesterol loading results in strong suppression of basal activity and flow sensitivity of Kir channels in primary mesenteric ECs.
Both acLDL‐treated and control ECs exhibited Kir currents that, as expected, reversed around −20 mV (EK under experimental conditions ≈21 mV in 60 K+ bath and 135 K+ pipette; Figure 1A and 1B) but the current densities in cells exposed to acLDL were significantly lower than in control cells (Figure 1A, 1B, and 1E). Moreover, exposure to well‐defined shear stress (0.7 dynes/cm2) using a gravity perfused minimally invasive flow device resulted in a significant increase in Kir current density in control cells (Figure 1A and 1C), an effect that is reversible on stopping perfusion to the device (Figure S1). However, only a small increase in Kir current density was observed in acLDL‐treated cells (Figure 1A through 1D). To further isolate the shear stress–sensitive component of the current, we calculated IɅSS by subtracting the currents recorded under static conditions Istat from the currents recorded during the flow exposure Ishear in the same cell (Figure 1A and 1B, lower panels). This analysis shows that IɅSS of Kir current was significantly reduced by the exposure to acLDL (Figure 1F).
Figure 1.
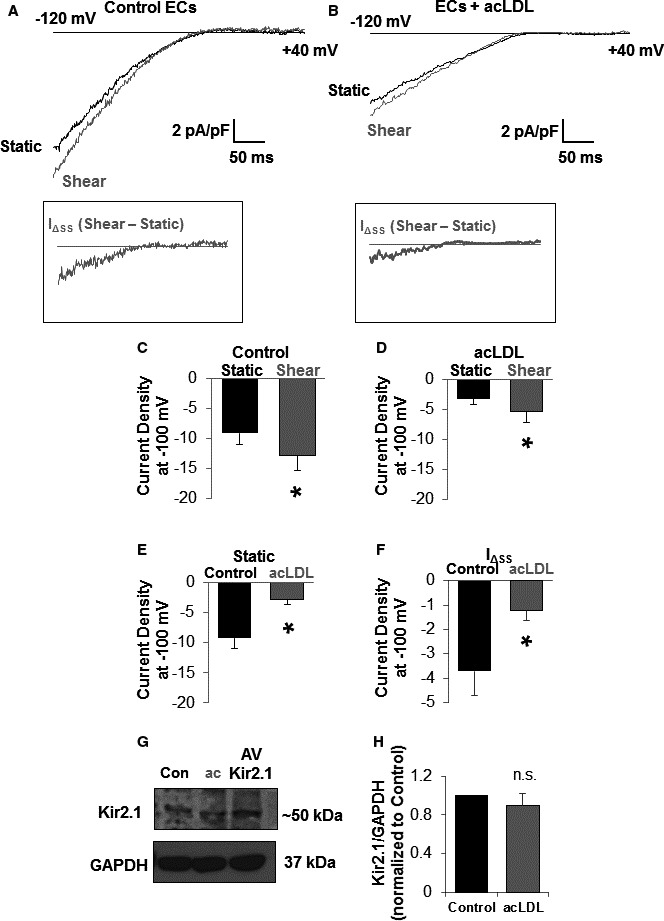
Dyslipidemia reduces shear stress–induced increases in endothelial cell (EC) inwardly rectifying K+ (Kir) current without influencing Kir2.1 channel expression in vitro. Representative traces of Kir currents from (A) control and (B) acetylated low‐density lipoprotein (acLDL; 50 μg/mL [24 hours])–treated mesenteric endothelial cells. Ramps from −120 to +40 mV elicited inward K+ currents that reversed around −21 mV. Insets represent the increase in current (IΔ SS) evoked by 0.7 dynes/cm2 in the respective traces. Shear significantly increased Kir current in (C) control (n=8) and (D) acLDL‐treated (n=7) ECs. Treating ECs with acLDL inhibited (E) baseline Kir currents recorded in a static bath (n=9 and 10 cells for control and acLDL, respectively) and (F) the shear‐induced increase in Kir current. For (E and F), a paired or unpaired Student t test was used where appropriate. Cell capacitance was similar between control and acLDL‐treated ECs (10.0±1.7 pF vs 11.8±1.5 pF, respectively). Note that Kir channel protein expression was not altered by treatment with acLDL in (G) a representative blot and (H) group data (n=5) normalized to the loading control, GAPDH. For accurate comparison of densitometry from separate blots, control samples were first normalized to 1. An unpaired Student t test was used to test for significant differences. For all group data, error bars represent SEM. *P<0.05.
We also tested the expression of Kir2.1, the most abundant and physiologically relevant Kir channel in this cell type,3 in ECs exposed to acLDL to determine whether a reduction in channel expression might contribute to the reduced baseline current. There was no effect of acLDL on Kir2.1 protein levels (Figure 1G) indicating that channel function, and not expression, is affected by cholesterol loading cells in vitro.
Decreased Kir Currents in ECs of Apoe −/− Mice are Recovered by Cholesterol Depletion
To determine the effects of hypercholesterolemia in vivo, we next tested the functional expression of Kir in ECs freshly isolated from mesenteric arteries of hypercholesterolemic Apoe −/− mice, as compared with WT controls. Mice were fed normal rodent chow and recordings were performed immediately (within a window of 2–3 hours) after EC isolation and with no extended exposure to culture conditions. As shown in Figure 2A and 2B, while freshly isolated ECs from Apoe −/− arteries exhibit typical Kir currents with pronounced inward rectification and predicted reversal potential of ≈−21 mV, these currents are much smaller than in cells isolated from WT arteries. Furthermore, to test whether reduced Kir currents in Apoe −/− ECs can be rescued by the removal of cholesterol, freshly isolated Apoe −/− ECs were exposed to MβCD (5×10−3 mol/L for 1 hour) known to efficiently sequester cholesterol from cells and tissues.27 Exposure to MβCD results in significant recovery of Kir currents (Figure 2C and 2D), thus supporting the notion that decreased activity of Kir channels in Apoe −/− mice is cholesterol dependent.
Figure 2.
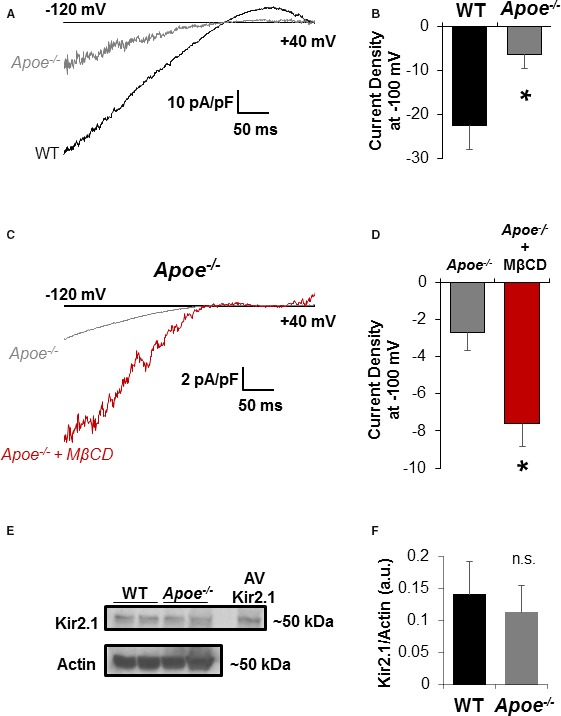
Cholesterol depletion recovers inwardly rectifying K+ (Kir) currents in apolipoprotein E–deficient (Apoe −/−) mice endothelial cells (ECs). A, Representative perforated patch recordings of ECs freshly isolated from wild‐type (WT) and Apoe −/− mice maintained on a standard rodent chow diet. As expected, Kir currents reverse at EK (≈−21 mV under the experimental conditions). Currents were evoked by a ramp from −120 to +40 mV over 400 ms. B, Group data reveal a significant decrease in currents detected in Apoe −/− ECs (9 cells from 5 mice; P<0.05) when compared with WT (6 cells from 4 mice). Cell capacitances of ECs from WT and Apoe −/− were similar (15.9±1.7 pF vs 17.2±3.4 pF, respectively). C, Representative traces of inwardly rectifying currents from ECs freshly isolated from Apoe −/− mice and incubated for 1 hour with or without methyl‐β‐cyclodextrin (MβCD; 5×10−3 mol/L). Cell capacitance was unaffected by MβCD (21.1±4.2 pF in control ECs vs 21.6±4.9 pF in cells exposed to MβCD). D, Group data reveal significant recovery of currents with MβCD (n=7 cells in each group from 3 mice; P<0.05). E, Representative blots comparing Kir2.1 channel expression between WT and Apoe −/− whole mesenteric arcades. ECs transduced with adenovirus (AV) Kir2.1 were used as a positive control for identifying bands at the appropriate molecular weight for Kir2.1 (≈50 kDa). F, Group data reveal no significant differences between WT and Apoe −/− Kir2.1 channel expression when normalized to actin expression. *P<0.05.
In contrast, no difference in Kir2.1 channel expression was observed in mesenteric arteries between WT and Apoe −/− mice via Western blot analysis (Figure 2E and 2F). The Western blot experiments were performed using whole mesenteric arteries rather than isolated ECs to get enough material for the analysis, but since we and others previously showed that Kir2.1 is not expressed in smooth muscle cells of murine mesenteric arteries,3, 20, 28 the source of Kir2.1 in these arteries is expected to be primarily ECs. A decrease in Kir currents with no effect on the channel expression in Apoe −/− mice shown here is consistent with our previous studies demonstrating that an increase in cellular cholesterol results in a decrease in the activity of Kir2 channels without having an effect on the channel expression.15, 17, 29
Contribution of Kir2.1 to FIV is Abolished in Mesenteric Arteries of Apoe −/− Mice
The findings in Figures 1 and 2 have critical implications for endothelial function and vasomotor response to increased flow/shear stress as we recently established a major role for endothelial Kir2.1 in mediating FIV. To test the role of Kir2.1 in dyslipidemia‐induced impairment of FIV, we generated a mouse with reduced Kir2.1 expression on the Apoe −/− background. This was done by crossing Kir2.1 +/− heterozygous mice, which we have recently shown to have significantly reduced functional expression of Kir2.1 in mesenteric endothelium3 with Apoe −/− mice, a well‐established model of dyslipidemia‐induced endothelial dysfunction.10 Since Kir2.1 +/− heterozygous mice were developed on the FVB background, Apoe −/− in the FVB background30 was used to generate the Kir2.1 +/− /Apoe −/− mice. We have already demonstrated that Kir2.1 +/− mice have significantly reduced FIV (shown again in Figure S2A through S2C), as compared with WT controls.3 Here, we address whether plasma dyslipidemia has differential effects on FIV in Apoe −/− versus Kir2.1 +/− /Apoe −/− mice. We expect that if cholesterol‐induced suppression of endothelial Kir channels leads to impaired FIV, arteries isolated from Apoe −/− mice should have reduced FIV response as compared with arteries isolated from WT mice, whereas Kir2.1 +/− /Apoe −/− mice are expected to exhibit the same FIV impairment as Apoe −/− alone. We demonstrate that this is indeed the case in murine mesenteric resistance arteries: FIV responses of arteries isolated from both Apoe −/− and Kir2.1 +/− /Apoe −/− mice are significantly reduced compared with WT arteries and the two completely overlap (Figure 3A and 3B).
Figure 3.
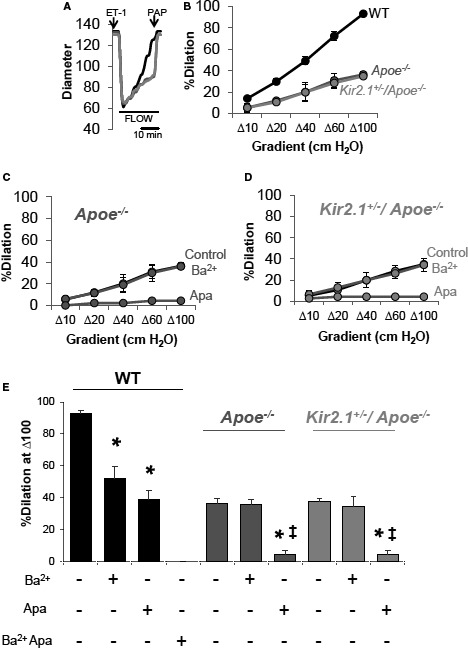
Hypercholesterolemia‐induced reduction of flow‐induced vasodilation (FIV) in inwardly rectifying K+ (Kir)–deficient mouse mesenteric arteries. A, Representative traces showing dilations to increasing intraluminal flow in arteries isolated from wild‐type (WT), apolipoprotein E–deficient (Apoe −/−), and Kir2.1 +/− /Apoe −/− mice. Arterial diameter was continuously monitored and a measurement was recorded every 30 seconds. Endothelin 1 (ET‐1) (1.2–2×10−10 mol/L) was used to preconstrict the vessels before an FIV protocol. At the end of an FIV protocol, papaverine (10−4 mol/L) was used to determine endothelium‐independent function and validity of the previous FIV protocol. In most cases, papaverine induced dilations >95%. B, Group data showing the full dose response to flow using the pressure gradient method in each model. For WT, 6 arteries from 4 mice; Apoe −/−, 4 arteries from 4 mice; and Kir2.1 +/− /Apoe −/−, 4 arteries from 4 mice. SEM was calculated using the number of mice. Inhibitors of Kir (Ba2+; 3×10−5 mol/L) and small conductance Ca2+‐activated K+ (apamin; 2×10−8 mol/L) channels reveal the contribution of Kir channels in (C) Apoe −/− and (D) Kir2.1 +/− /Apoe −/− mice is abolished while the function of small conductance Ca2+‐activated K+ remains intact in response to intraluminal flow. E, Group data highlighting differences in response to Δ100 cm H2O determined by Bonferroni correction after 2‐way repeated measures ANOVA. *P<0.05 vs control (no inhibitor) and ‡ P<0.05 vs Ba2+.
Briefly, mesenteric resistance arteries are cannulated on glass micropipettes and pressurized for 1 hour by gravity‐induced pressure (60 cm H2O) from solution‐filled reservoirs that perfuse the artery. Arteries are then preconstricted with endothelin‐1 and increasing levels of intraluminal flow administered by moving reservoirs in equal and opposite directions, thus maintaining intraluminal pressure while flow through the vessel increases.3, 22, 31 No differences are observed between baseline arterial diameters when comparing relevant experimental groups (Table S1). As expected, arteries from WT mice dilate in a dose‐dependent fashion to increases in flow delivered via the pressure gradient method (Figure 3A and 3B).3 A full dilatory response is observed at a pressure gradient of Δ100 cm H2O. However, arteries from Apoe −/− mice dilate only partially, reaching ≈40% of the full dilation at the same gradient, and no further decrease is observed in Kir2.1 +/− /Apoe −/− mice. After each protocol, papaverine (10−4 mol/L) was used to test the function of the artery to ensure that endothelium‐dependent function was being investigated independently of potential changes in smooth muscle function over time, especially in the Kir2.1 +/− and Apoe −/− mouse models where FIV was reduced (average % dilation to papaverine in arteries from Apoe −/− mice: 97.6±4.2; average % dilation to papaverine in arteries from Kir2.1 +/− /Apoe −/− mice: 95.1±3.6). Results from arteries that did not dilate to >80% in response to papaverine were discarded.
The loss of the Kir component of the FIV in Apoe −/− mesenteric arteries was confirmed by blocking the currents using Ba2+, a well‐known blocker of Kir channels. As previously shown,3 FIV in WT mesenteric arteries was reduced by blocking Kir channels with Ba2+ (3×10−5 mol/L) and by blocking small conductance Ca2+‐sensitive K+ channels with apamin (2×10−8 mol/L), whereas the combined application of Ba2+ and apamin abolished FIV (Figure S2D and S2E). In contrast, no effect of Ba2+ on FIV was observed in either Apoe −/− or Kir2.1 +/− /Apoe −/− arteries, indicating that the Kir‐dependent component of FIV was absent in both models (Figure 3C through 3E). The Kir‐independent FIV component was blocked in both dyslipidemic mouse models by apamin, suggesting that small conductance Ca2+‐activated K+ channel function remain intact in Apoe −/− mice (Figure 3C through 3E).
Recovery of FIV in Mesenteric Arteries of Apoe −/− Mice by Cholesterol Depletion: Role of Kir
Since we observed the recovery of inwardly rectifying currents in ECs from Apoe −/− mice depleted of cholesterol (Figure 2C and 2D), we tested whether removing cholesterol from mesenteric arteries of Apoe −/− mice could also recover FIV. Figure 4A and 4C shows that incubating arteries with MβCD (5×10−3 mol/L; 1 hour) rescues FIV and sensitivity to Ba2+, indicating that the rescue effect was mediated by recovering Kir channel function. In contrast, repeating these experiments on mesenteric arteries of Kir2.1 +/− /Apoe −/− mice does not rescue FIV, and Ba2+ also has no effect (Figure 4C and 4D), suggesting that recovery of FIV by cholesterol depletion in arteries is dependent on the full expression of Kir2.1.
Figure 4.
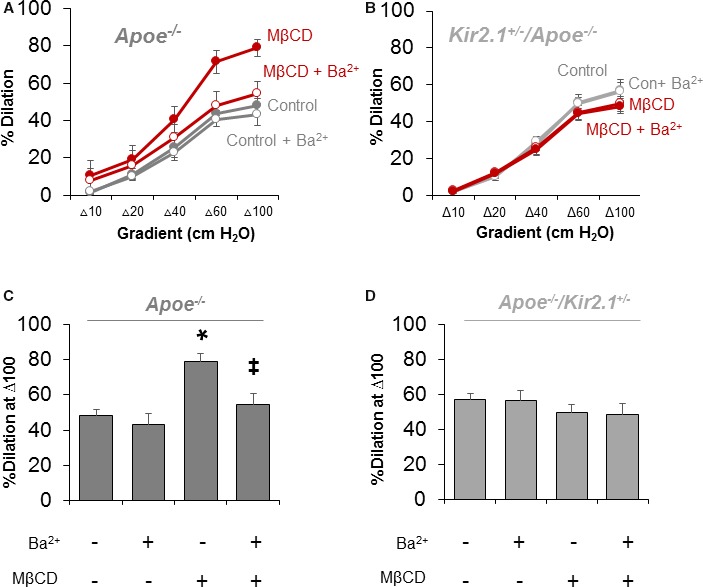
Recovery of flow‐induced vasodilation (FIV) in cholesterol‐depleted arteries is dependent on inwardly rectifying K+ (Kir) 2.1. Dilations to flow in mesenteric arteries isolated from (A) apolipoprotein E–deficient (Apoe −/−; 3 arteries per group from 3 mice) or (B) Kir2.1 +/− /Apoe −/− (5 arteries per group from 5 mice) mice incubated with or without methyl‐β‐cyclodextrin (MβCD; 5×10−3 mol/L) for 1 hour before testing FIV. Group data in (C) reveal significant recovery of FIV in arteries that is Ba2+ (3×10−5 mol/L)‐sensitive from Apoe −/− mice, while that in (D) shows no effect of MβCD in arteries from Kir2.1 +/− /Apoe −/− mice. Group data in (C) and (D) highlight differences in response to Δ100 cm H2O determined by Bonferroni correction after 2‐way repeated measures ANOVA. *P<0.05 vs control (no Ba2+ or MβCD) and ‡ P<0.05 vs+MβCD.
Loss of Flow‐Induced Production of NO in Mesenteric Arteries of Apoe −/− and Kir2.1 +/− /Apoe −/− Mice
We previously showed that shear‐induced activation of Kir2.1 in mesenteric arterial ECs promotes NO production.3 Therefore, we predicted that the reduced FIV in Apoe −/− mice, which we suggest is attributed to deficiency in functional EC Kir2.1, would have reduced detectable NO levels during intraluminal flow stimulation. WT arteries showed a robustly detectable fluorescent signal, while arteries from both Apoe −/− and Kir2.1 +/− /Apoe −/− mice had significantly reduced signal (fold change relative to no flow control: 2.8±0.8 versus 1.1±0.1 versus 1.1±0.2, respectively; Figure 5A and 5B).
Figure 5.
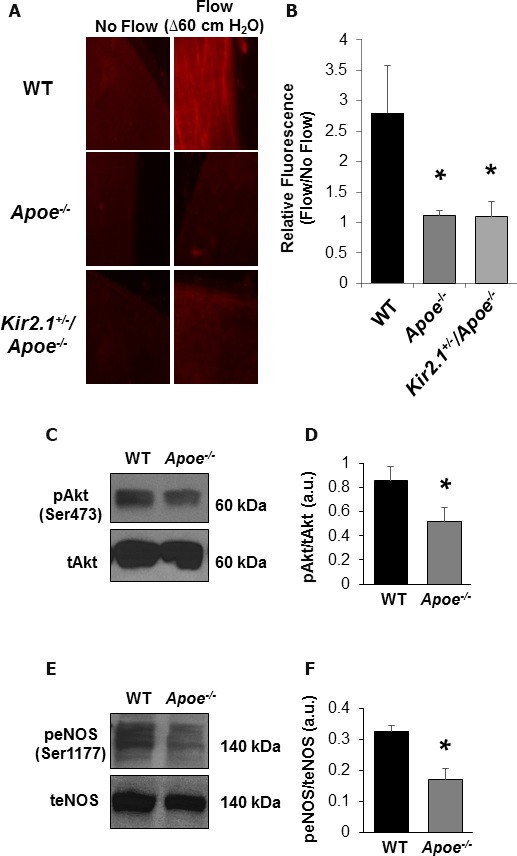
Hypercholesterolemia‐induced reduction in nitric oxide (NO) detection following intraluminal flow is inwardly rectifying K+ (Kir) dependent in mouse mesenteric arteries. A, Representative images (×40 magnification) of mesenteric arteries exposed to flow (Δ60 cm H2O for 30 minutes; right column) or arteries incubated under no flow conditions for the same duration (left column). An increase in NO detection by the NO‐specific dye, diaminorhodamine‐4M, is only present in the wild‐type (WT; top row) artery, while no noticeable increase is observed in apolipoprotein E–deficient (Apoe −/−; middle row) or Kir2.1 +/− /Apoe −/− (bottom row) arteries. B, Group data were generated by taking the fold increase from arteries that received flow to those arteries that did not. Arteries analyzed in this way were always from the same mouse (4 pairs of arteries from 4 mice for all groups). Significant differences exist in NO detection after flow when comparing either Apoe −/− model with WT. Bonferroni post hoc analysis followed 1‐way ANOVA. *P<0.05 vs WT. C, Representative blot showing phosphorylated Akt (Ser473) in WT and Apoe −/− whole mesenteric arcades. D, Group data revealing a significant decrease in Apoe −/− pAkt after normalizing to respective total Akt bands. E, Representative blots showing phosphorylated endothelium NO synthase (eNOS) (Ser1177) in WT and Apoe −/− whole mesenteric arcades. F, Group data revealing a significant decrease in Apoe −/− peNOS after normalizing to respective total eNOS bands.
We recently identified a mechanistic link between endothelial Kir2.1 activation by shear stress and FIV via subsequent Akt and eNOS activation.3 In accordance with these findings, both phosphorylated‐Akt (Ser437; Figure 5C and 5D) and phosphorylated eNOS (Ser1177; Figure 5E and 5F) were decreased in Apoe −/− compared with WT mesenteric arteries, indicating the previously identified pathway to be significantly impaired by dyslipidemia and thus resulted in reduced NO production, shown in Figure 5A and 5B. In addition, the expression of total eNOS was not reduced in Apoe −/− mice (Figure S3A and S3B), further supporting that inhibition of NO production should be attributed to the loss of the upstream regulatory event and not to the loss of eNOS itself. Moreover, we also show that dilation induced by the Ca2+ ionophore A23187, which is known to promote vasodilation via activation of eNOS,32, 33 is also fully intact in Apoe −/− arteries (Figure S3C). These data suggest that the reduced FIV observed in Apoe −/− mice is not attributable to changes in eNOS expression in Apoe −/− mice but to reduced cell signaling beginning with blunted Akt activation likely caused by cholesterol‐induced suppression of endothelial Kir channels.
Rescue of Hypercholesterolemia‐Induced Loss of FIV by EC‐Specific Expression of Kir2.1
We sought to determine whether Kir2.1 function in cholesterol‐loaded primary ECs could be recovered by overexpression of Kir2.1 using adenoviral techniques. To do this, ECs were first treated with acLDL for 24 hours and then transduced with adenovirus containing a WT Kir2.1 construct with expression driven by an hVE‐Cadherin promoter or as control, an empty adenovirus, for 48 hours.3, 34 Transduction of ECs was unaffected by cholesterol loading as determined by staining for HA‐tag that is on the extracellular portion of the Kir2.1 construct (Figure S4Ai). Perforated patch clamp analysis revealed significant recovery of baseline Kir current densities in ECs treated with Kir2.1 adenovirus versus ECs treated with empty adenovirus (−11.3±1.3 pA/pF versus −2.3±0.6 pA/pF, respectively; Figure 6A and 6B). Notably, Kir2.1 currents in acLDL‐treated cells transduced with Kir2.1 adenovirus are similar to the level of the endogenous Kir2.1 currents in cells not exposed to acLDL. Shear‐induced increases in Kir current densities were also rescued in the Kir2.1 adenovirus‐treated cholesterol‐loaded cells (average IΔSS of −4.2±1.5 pA/pF; Figure 6A and 6B) indicating that overexpression of Kir2.1 in cholesterol‐loaded ECs in vitro restores channel function to the level of endogenous currents in cells with normal cholesterol.
Figure 6.
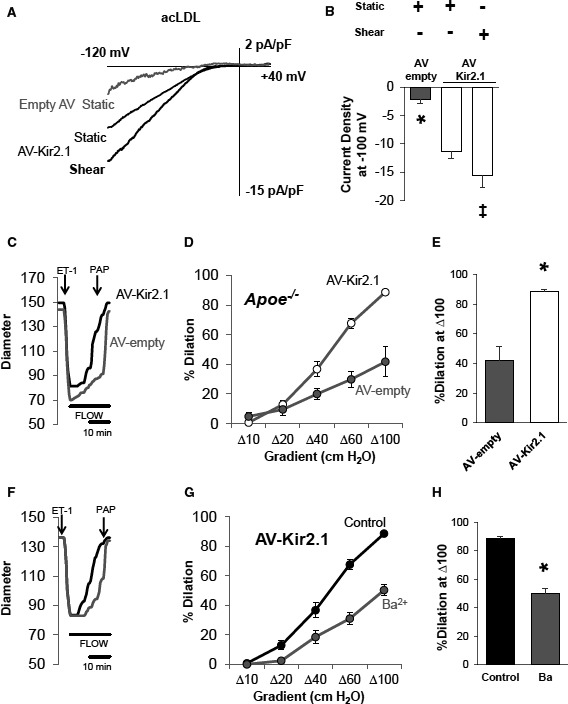
Overexpression of endothelial inwardly rectifying K+ (Kir) 2.1 recovers shear‐sensitive Kir currents and flow‐induced vasodilation (FIV) in models of dyslipidemia. A, Representative perforated patch recordings of Kir currents from endothelial cells (ECs) transduced with an adenovirus (AV)‐empty or endothelial‐specific (hVE‐Cadherin) AV‐Kir2.1 in a static bath show an increase in Kir current in AV‐Kir21–transduced ECs despite the presence of acetylated low‐density lipoprotein (acLDL; 50 μg/mL). Also shown is the rescued shear‐sensitivity of Kir current in ECs transduced with AV‐Kir2.1 in the presence of acLDL from the same cell. B, Group data indicate significant differences in currents between acLDL‐treated ECs transduced with either AV‐empty or AV‐Kir2.1 under static conditions (8 cells for each group; unpaired Student t test, *P<0.05). Group data also reveal that shear‐induced increases in EC Kir current were rescued by AV‐Kir2.1 transduction in the presence of acLDL (5 cells; paired Student t test, ‡ P<0.05). No statistical difference was detected in cell capacitances between cells treated with AV‐Kir2.1 or empty AV (25.58±6.1 pF vs 19.53±3.02 pF, respectively). Overexpression of endothelial Kir2.1 with AV‐Kir2.1 also rescued FIV as shown in (C) representative traces of the pressure gradient method of inducing intraluminal flow, (D) FIV curves showing group data responses to increases in flow (4 arteries per AV group from 4 mice), and (E) dilations to Δ100 cm H2O showing a significant rescue of FIV in the AV‐Kir2.1–treated arteries. Sensitivity to Ba2+ (3×10−5 mol/L) is also rescued in arteries transduced with AV‐Kir2.1 as shown in (F) representative traces, (G) FIV curves (4 arteries from 4 mice), and (H) dilations to Δ100 cm H2O showing a significant inhibition by Ba2+. For (E and H), Bonferroni correction following 2‐way repeated measures ANOVA was performed; *P<0.05, SEM was calculated using number of mice. ET‐1 indicates endothelin 1; PAP, pulmonary artery pressure.
Most importantly, EC‐specific overexpression of Kir2.1 in mesenteric arteries harvested from Apoe −/− mice resulted in the full rescue of the FIV response (Figure 6C through 6E). The rescue is performed ex vivo by infecting the arteries isolated from Apoe −/− mice with the hVE‐Cadherin–driven Kir2.1 adenovirus immediately after the harvest and the functional activity is tested after 48 hours while arteries are maintained in normal growth medium. We have established the validity of this ex vivo transduction protocol with EC‐specific Kir2.1 adenovirus and verified that endothelial‐induced vasodilation of freshly isolated mesenteric arteries is maintained for the 48‐hour window.3 No recovery was observed in arteries that were transduced with the empty adenovirus in parallel. In addition, to further verify that the rescue effect is the result of restoring the Kir‐dependent FIV component, the arteries transduced with EC‐Kir2.1 adenovirus or with empty adenovirus were exposed to Ba2+. The sensitivity to Ba2+ was recovered in those arteries transduced with Kir2.1 adenovirus (Figure 6F through 6H), while no Ba2+ effect was observed in arteries transduced with empty adenovirus (Figure S4B through S4D).
Rescue of Hypercholesterolemia‐Induced Loss of NO Production by EC‐Specific Expression of Kir2.1
To test the capacity of overexpressing endothelial Kir2.1 in rescuing shear‐induced production of NO, we transduced arteries from Kir2.1 +/− /Apoe −/− mice with Kir2.1 adenovirus. A clear recovery of flow‐induced NO production was observed when comparing Kir2.1 adenovirus transduced arteries with those transduced with empty adenovirus (fold change relative to no flow control: 2.5±0.3 versus 1.1±0.1, respectively; Figure 7A and 7B). The level in the Kir2.1 adenovirus–transduced Kir2.1 +/− /Apoe −/− arteries was comparable to the level observed in WT arteries (Figure 5A and 5B).
Figure 7.
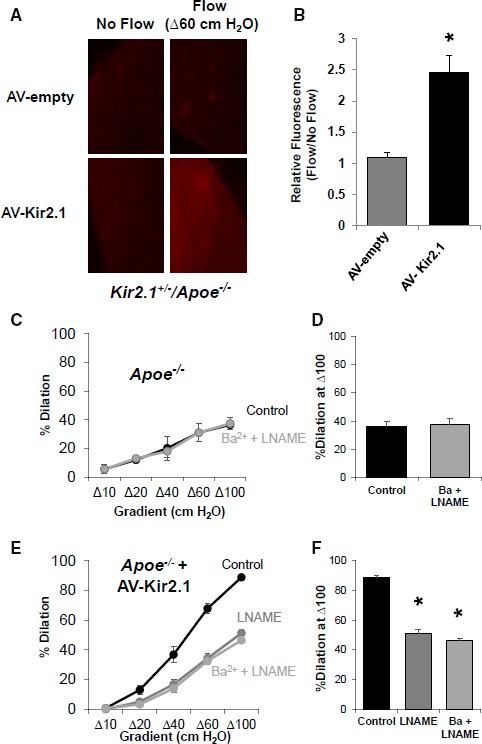
Overexpression of endothelial inwardly rectifying K+ (Kir) 2.1 recovers flow‐induced nitric oxide (NO) production in models of dyslipidemia. A, Representative images (×40 magnification) of mesenteric arteries from Kir2.1 +/− /apolipoprotein E–deficient (Apoe −/−) mice transduced with empty adenovirus (AV) or AV‐Kir2.1 and exposed to Δ60 cm H2O intraluminal flow or maintained under no flow conditions. An increase in detectable flow‐induced NO by the NO‐specific dye, diaminorhodamine‐4M, is not observed in arteries that received empty AV (top row); however, strong fluorescent detection of flow‐induced NO is observed in arteries transduced with AV‐Kir2.1 (bottom row). B, Group data were generated by taking the fold increase in flow‐induced NO fluorescence from the no flow condition of an artery treated with the same AV (4 pairs of arteries from 3 mice); *P<0.05. C and D, flow‐induced vasodilation (FIV) in arteries from Apoe −/− mice are not sensitive to combined application of Ba2+ (30 μmol/L) or L‐NG‐Nitroarginine methyl ester (LNAME; 100 μmol/L) (4 arteries from 4 mice); however, (E) overexpression of endothelial Kir2.1 by transduction with AV‐Kir2.1 recovers FIV sensitivity to LNAME alone and no further inhibition is observed in combination with Ba2+ (4 arteries per AV group from 4 mice), as previously shown in wild‐type (WT) mice.3 F, Group data showing dilations to Δ100 cm H2O indicate significant recovery of FIV sensitivity to LNAME in arteries transduced with AV‐Kir2.1. *P<0.05. For all bar graphs, SEM was calculated with number of mice.
To further investigate whether infecting the arteries with EC‐Kir2.1 restores the NO‐dependent component of FIV, we tested the effect of LNAME (10−4 mol/L), on NO production. Consistent with the loss of the Ba2+ sensitivity of FIV and of the flow‐induced NO production in Apoe −/− mesenteric arteries shown above, FIV of these arteries was not sensitive to LNAME and Ba2+ (Figure 7C and 7D). A similar lack of an effect was observed in Kir2.1 +/− /Apoe −/− mice (Figure S5A and S5B). Transduction with Kir2.1 adenovirus, however, fully restored the sensitivity of FIV to LNAME, while application of Ba2++LNAME had no further effect than LNAME alone (Figure 7E and 7F), as was established in arteries of WT mice (Figure S5C and S5D).3 Together, these data indicate that overexpression of exogenous Kir2.1 in the endothelium can recover the full complement of the Kir2.1/eNOS/NO pathway we recently identified.3 We suggest the loss of functional Kir2.1 channels leading to decreased bioavailability of NO to be a critical component of the development of hypercholesterolemia‐induced endothelial dysfunction.
EC‐Kir2.1 Rescues FIV in Both Atheroresistant and Atheroprone Apoe −/− Mice
Previous studies have demonstrated that Apoe −/− mice on the FVB background are relatively atheroresistant, whereas Apoe −/− mice on the C57BL/6 (B6) background are significantly more atheroprone.30, 35 We tested, therefore, whether EC‐specific overexpression of Kir2.1 is able to rescue the FIV response not only in Apoe −/−/FVB mice but also in Apoe −/−/B6 mice. Mesenteric arteries from WT/B6 mice responded similarly to intraluminal flow as the arteries from WT/FVB mice, including inhibition of FIV by application of Ba2+ (Figure S6A and S6B). In addition, the contribution of endothelial Kir2.1 to FIV in WT/B6 mice was confirmed using an EC‐specific DN Kir2.1 adenovirus construct (Figure S6C and S6D). Infecting arterial ECs with the DN Kir2.1 mutant results in the inclusion of DN mutant subunits into Kir2.1 tetramers, rendering the channels inactive. Subsequently, nonfunctional channels are incorporated into the membrane. In both Apoe −/− models, the FIV responses were Ba2+ insensitive (Figures 3C, 3E and S6E, S6F for Apoe −/−/FVB and Apoe −/−/B6, respectively). Most importantly, a full rescue of FIV by the overexpression of EC‐Kir2.1 was observed in mesenteric arteries of atheroprone Apoe −/−/B6 mice (Figure 8A and 8E). In contrast, overexpression of EC DN Kir2.1 in Apoe −/−/B6 arteries had no effect (Figure 8B and 8E).
Figure 8.
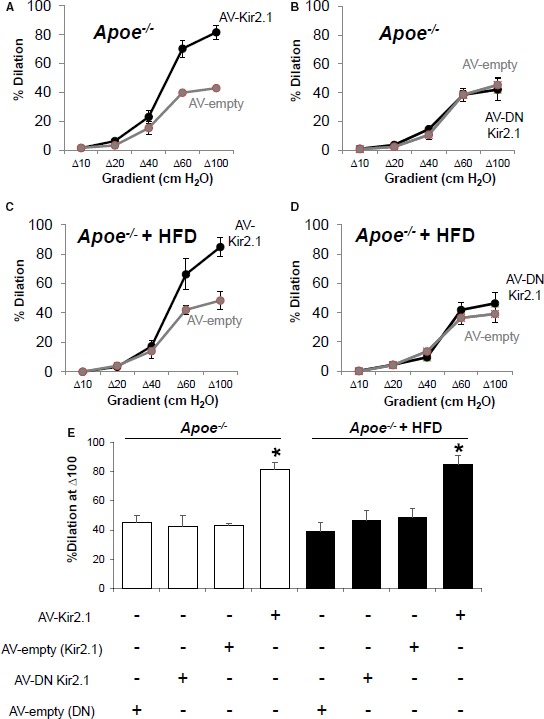
Overexpression of endothelial inwardly rectifying K+ (Kir) 2.1 recovers flow‐induced vasodilation (FIV) in apolipoprotein E–deficient (Apoe −/−)/B6 mice fed a high‐fat diet (HFD). A, FIV curves generated via the pressure gradient method show that transducing mesenteric arteries of Apoe −/−/B6 mice with adenovirus (AV) Kir2.1 recovers FIV compared with those that were transduced with empty AV (3 arteries per AV group from 3 mice). B, In contrast, transducing arteries with AV‐dominant negative (DN) Kir2.1 has no effect compared with empty AV (arteries per AV group from 4 mice). C, Arteries isolated from Apoe −/− /B6 mice fed a high‐fat, high‐cholesterol diet for 8 weeks also exhibit a recovery of FIV when transduced with AV‐Kir2.1 (4 arteries per AV group from 4 mice), whereas (D) arteries transduced with AV DN Kir2.1 respond similarly to intraluminal flow as compared with those arteries transduced with empty AV (4 arteries per AV group from 4 mice). E, Group data show significant differences to Δ100 cm H2O intraluminal flow observed in both normal and Apoe −/− /B6 mice fed an HFD in those arteries transduced with AV‐Kir2.1 when compared with the appropriate AV‐empty group. Bonferroni correction following 2‐way repeated measures ANOVA was performed; *P<0.05, SEM was calculated with number of mice.
Exacerbating Apoe −/− Phenotype With High‐Fat Diet Does Not Prevent EC‐Kir2.1 Rescue of FIV
To determine the extent to which overexpression of endothelial Kir2.1 could reverse the hypercholesterolemia‐induced endothelial dysfunction, we fed Apoe −/−/B6 mice a Western‐type high‐fat, high‐cholesterol diet for 8 weeks. We were surprised to find that, regardless of diet‐induced contributions to endothelial dysfunction, overexpression of endothelial Kir2.1 rescued FIV. Arteries from Apoe −/− mice fed a high‐fat diet and receiving the Kir2.1 adenovirus had a significant recovery of FIV compared with those arteries that received the empty adenovirus (Figure 8C and 8D). Furthermore, and similar to arteries isolated from Apoe −/− mice maintained on a low‐fat diet, transduction of endothelial‐specific DN Kir2.1 adenovirus had no effect on FIV in arteries from Apoe −/− mice fed a high‐fat diet (Figure 8D and 8E). These data strongly: (1) support the role for endothelial Kir2.1 in hypercholesterolemia‐induced endothelial dysfunction, and (2) implicate the potential for endothelial Kir2.1 in recovering vascular function even in the presence of severe hypercholesterolemia.
Kir2.1 Deficiency Promotes the Formation and Alters the Distribution of Atherosclerotic Lesions in Mouse Aortas
Endothelial dysfunction is a hallmark of disease progression and promotes more severe and advanced disease states such as atherosclerosis. Our present findings link cholesterol‐induced suppression of endothelial Kir2.1 to endothelial dysfunction; therefore, we tested the role of Kir2.1 in atherosclerosis and lesion formation in FVB mice. Although this strain is known to be relatively resistant to atherosclerosis compared with B6 mice with lesions forming only after an extended duration on a high‐fat diet,30, 35, 36 this is the genetic strain in which the Kir2.1 +/− model exists. Apoe −/− and Kir2.1 +/− /Apoe −/− mice on the FVB background were fed the high‐fat Western diet for extended periods (6 or 9 months), and lesion formation was subsequently detected by en face Oil Red O staining of the aortic arch and descending aorta (DA) taken immediately before the diaphragm. Lesions were measured as a percentage of aortic area containing lesion. After 6 months of feeding (Figure S7), minimal lesion formation was present and no differences between mouse models were detected. In contrast, after 9 months of high‐fat feeding (Figure 9), robust lesion formation was detected in both groups but was significantly elevated in Kir2.1 +/− /Apoe −/− (Figure 9B) mice. Furthermore, the distribution of the plaques in Kir2.1 +/− /Apoe −/− mice was significantly altered compared with Apoe −/− mice with a notable increase in the DA (29.3±2.9 versus 44.4±5.9 in aortic arch and 7.6±0.9 versus 24.0±5.8 in DA for Apoe −/− and Kir2.1 +/− /Apoe −/− mice, respectively), an otherwise atheroresistant region. This finding implicates an important role for Kir2.1 in the progression of vascular disease, especially when considering potential contributions to distinct regions that experience disturbed flow and are atheroprone versus laminar flow, atheroresistant regions of the aorta.
Figure 9.
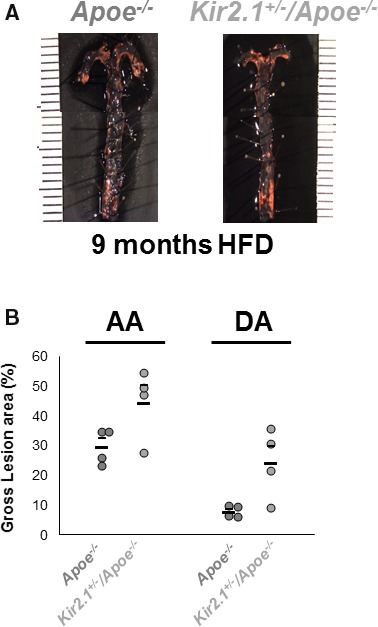
Inwardly rectifying K+ (Kir) 2.1 deficiency promotes the formation and alters the distribution of atherosclerotic lesions in mouse aortas. A, Representative images of en face aortas from apolipoprotein E–deficient (Apoe −/−; left) and Kir2.1 +/− /Apoe −/− (right) stained with Oil Red O for lesion detection. Mice were fed a Western, high‐fat, high‐cholesterol diet (HFD) for 9 months before analysis. Aortas were isolated into the thoracic region immediately before the diaphragm. B, Group analyses of the lesions detected show lesion formation in the aortic arch (AA) and descending aorta (DA) as measured as percentage of the total area containing lesions. Larger, lower bars represent the group average and smaller, upper bars represent the standard error. For all groups, n=4. Significance was tested using 2‐way ANOVA. P<0.05 when comparing Apoe −/− and Kir2.1 +/− /Apoe −/−.
Discussion
Hypercholesterolemia is a leading risk factor in the progression of cardiovascular disease and hypercholesterolemia‐induced endothelial dysfunction is a precursor to cardiovascular disease development. A hallmark of endothelial dysfunction is a reduction in NO production in response to increased flow4; however, the mechanisms governing the development of endothelial dysfunction under hypercholesterolemic conditions are unclear. We previously showed that hypercholesterolemia suppresses endothelial Kir channels,15, 26 putative mechanosensors.37, 38 We recently identified endothelial Kir2.1 channels as a critical component of FIV whereby activation of Kir2.1 promotes Akt phosphorylation and ultimately results in NO production and vasodilation in resistance arteries.3 In the current study, we show for the first time that suppression of Kir channels is a key contributor to endothelial dysfunction in Apoe −/− mice, a major mouse model of atherosclerosis. More specifically, we tested the hypothesis that hypercholesterolemia suppresses shear‐induced activation of endothelial Kir2.1 via cholesterol‐induced inhibition of channel activity thus preventing NO production and inhibiting FIV.
Our new findings demonstrate that hypercholesterolemia results in the loss of Kir‐dependent NO production and FIV in mesenteric arteries of Apoe −/− dyslipidemic mice, an effect reversed by cholesterol depletion. Furthermore, this impairment of vascular function can be fully rescued by the overexpression of Kir2.1 in the endothelium. These findings point to Kir2.1 channels as important potential targets in combating vascular dysfunction in hypercholesterolemic conditions and provide a novel mechanistic basis for decreased availability of NO, which is well known to be associated with the progression of atherosclerosis.
First, we establish that loading the cells with cholesterol using modified low‐density lipoproteins (acLDL) suppresses the shear‐induced increase in Kir current in ECs purified from the resistance vasculature. These observations are consistent with our previous studies showing that cholesterol loading inhibits Kir channels in aortic endothelium.15, 26 The function of the Kir2.1 channel, the major Kir channel in the endothelium of mesenteric resistance arteries,3, 20 is suppressed by cholesterol via specific sterol‐protein interactions between the cholesterol molecule and a hydrophobic pocket located between the transmembrane domains of the channels.39, 40 This is the first study, however, to examine the functional implications of cholesterol‐suppression of endothelial Kir channels. To directly assess whether cholesterol suppresses Kir channels in a model of hypercholesteremia in vivo, we isolated ECs from WT and Apoe −/− mice and performed perforated patch clamp analyses. ECs freshly isolated from Apoe −/− mice had reduced currents compared with WT ECs, whereas cholesterol depletion of Apoe −/− ECs using MβCD results in significant recovery. This supports our previous findings in freshly isolated aortic ECs of diet‐induced, hypercholesterolemic pigs15 and provides direct evidence that cholesterol inhibits functional Kir channels in a setting of hypercholesterolemia in vivo.
To investigate the impact of these findings on vascular function, we tested endothelial function in response to increases in intraluminal flow, a stimulus in which we have established a crucial role for endothelial Kir2.1 in mesenteric arteries. We show that impairment of FIV and of flow‐induced NO production in resistance arteries of Apoe −/− mice, a well‐established model for hypercholesterolemia, critically depends on endothelial Kir2.1 channels. Importantly, small conductance Ca2+‐activated K+ channel function appears largely intact, thus providing an additional line of evidence for the exclusive impairment of an NO‐mediated pathway as opposed to the NO‐independent pathway we previously showed to involve small conductance Ca2+‐activated K+ channel activation.3
Previous studies found that agonist‐induced (eg, acetylcholine) dilations of resistance arteries in Apoe −/− mice on normal low‐fat diets appear mostly intact10, 41, 42, 43 but the sensitivity to LNAME is reduced.44 This would suggest that compensatory mechanisms, most likely endothelium‐derived hyperpolarizing factor, exist in absence of NO during receptor‐mediated endothelium‐dependent vasodilation in the Apoe −/− mouse.9, 43, 44 In contrast, Apoe −/− mice fed a high‐fat, high‐cholesterol diet have reduced dilations to such agonists, suggesting a threshold whereby an overall reduction in endothelial function exists.45, 46, 47 Our present results indicate, however, that FIV, a mechanical stimulus, is impaired in the resistance arteries of Apoe −/− mice independent of diet and with no observable vasodilator compensation for a lack of NO. This indicates that the level of hypercholesterolemia in Apoe −/− mice fed a normal chow diet, although less than that of Apoe −/− mice fed a high‐fat diet, is enough to sufficiently suppress Kir activity. The following lines of evidence indicate that FIV impairment in this model is attributed to the loss of endothelial Kir activity: (1) creating a Kir2.1 downregulated mouse model on the background of Apoe −/− shows that FIV in the arteries from Apoe −/− mice lacks the Kir2.1‐dependent component; (2) this conclusion is verified by blocking Kir activity with Ba2+ or downregulating its functional expression with DN Kir2.1, both of which significantly suppress FIV in WT arteries but have no effect on arteries isolated from Apoe −/− mice; (3) cholesterol depletion of mesenteric arteries of Apoe −/− mice results in recovery of FIV, an effect dependent on Kir and not observed in arteries of Kir2.1 +/− /Apoe −/− mice; and finally (4) FIV in Apoe −/− arteries is fully restored by EC‐specific overexpression of Kir2.1. In particular, 2 lines of evidence highlight a central role for Kir channels in hyper‐cholesterolemia‐induced endothelial dysfunction: (1) recovery of FIV with cholesterol depletion of mesenteric arteries of Apoe −/− mice, and (2) recovery of FIV/NO production with overexpression of endothelial Kir2.1 in arteries of Apoe −/− mice and Apoe −/− mice crossed with Kir2.1‐deficient mice. Together, these findings suggest that cholesterol‐induced suppression of the endothelial Kir2.1 is the underlying mechanisms of reduced FIV and NO production, and that the threshold of such inhibition of the channel by cellular cholesterol can be overcome by additional, functional channels specifically expressed in vascular endothelium.
It is important to note, however, that the loss of the Kir2.1‐dependent component of FIV is attributed to the loss of functional activity of Kir channels and not to a decrease in Kir2.1 protein expression. This is supported in the present article by both the in vitro and in vivo models. Treating mesenteric ECs in culture with acLDL does not affect Kir2.1 protein expression despite significantly reducing both baseline currents and the response to shear stress. Similarly, Apoe −/− mice do not have altered channel expression when compared with WT arteries, again, despite having comparatively reduced currents. This observation is fully consistent with our previous findings showing that cholesterol loading suppresses Kir2.1 activity but has no effect on the level of channel expression.15, 17, 29 The question arises as to how it is possible to rescue FIV by Kir2.1 overexpression in arteries in which the endogenous Kir activity is suppressed. We showed in multiple previous studies,15, 17, 26, 48, 49 as well as here, that cholesterol loading does not fully prevent Kir2.1 activation, rather it only decreases the activity relative to that observed under low cholesterol conditions. Therefore, overexpression of Kir2.1 in cells loaded with cholesterol is expected to overcome the cholesterol‐induced inhibition of Kir activity and result in a significant increase in Kir currents relative to the endogenous currents observed in cholesterol‐loaded cells. Our present observations show this is indeed the case.
In addition, we show that hypercholesterolemia in 2 distinct mouse strains, the atheroresistant FVB and the atheroprone B6, exhibit similarly blunted FIV. The mesenteric arteries in Apoe deficiency in both genetic backgrounds lost sensitivity to Ba2+ and LNAME. The differences in atherogenicity between FVB and B6 mice were shown to correlate to differences in macrophage mobilization and recruitment, whereas no difference was observed in vascular cholesterol efflux.35, 50 This evidence supports the notion that endothelial cellular cholesterol is probably similarly elevated in both FVB and B6 Apoe −/− mice and perhaps explains why endothelial dysfunction is similarly observed in both despite differences in atherogenesis.30 To further address the ability of endothelial Kir2.1 overexpression to override inhibition by membrane cholesterol, we put atheroprone Apoe −/− B6 mice on a high‐fat, high‐cholesterol diet to induce severe hypercholesterolemia19 and endothelial dysfunction.12 Overexpression of endothelial Kir2.1 overcame the suppressive effects of severe hypercholesterolemia in Apoe −/− B6 mice fed a high‐fat diet, despite potential elevations in plasma cholesterol reaching almost 4 times that of Apoe −/− mice on a normal diet.19 We interpret these findings as indicating that functional Kir2.1 channels expressed in the endothelium can rescue impaired FIV and NO production regardless of the severity of hypercholesterolemia or the extent of atherosusceptibility.
Another critical point to address is whether the loss of the Kir‐dependent FIV component in Apoe −/− arteries may be a result of decreased eNOS expression, the downstream target of Kir2.1 activation. We show, however, that this is not the case because eNOS expression is unaffected in Apoe −/− arteries as compared with WT controls. These findings are in line with that previously reported for eNOS expression in carotid arteries and coronary resistance arteries of Apoe −/− mice.9, 51 We also assessed dilations to the Ca2+ ionophore, A23187, in arteries of Apoe −/− mice and observed comparable responses to that of WT arteries, indicating intact eNOS activity. We suggest that eNOS is regulated by an upstream mediator that is likely Kir channels because NO production is fully restored by endothelial‐specific Kir2.1 overexpression. In terms of the link between Kir activity and eNOS activation, we have recently shown that it is mediated by phosphorylation of Akt1.3 Therefore, we tested the phosphorylation status of Akt1 and eNOS in Apoe −/− mice and detected a significant decrease in both relative to WT supporting the notion that the Kir2.1/Akt/eNOS/NO pathway is functionally impaired, likely beginning with suppression of the upstream mediator, Kir2.1. These findings provide in vivo evidence for hypercholesterolemia‐induced suppression of eNOS, mostly mediated by the suppression of Kir channel activity. This is an effect that was previously reported in cultured aortic ECs loaded with cholesterol and exposed to shear stress.52 In addition, a decrease in Akt phosphorylation was previously reported in various hypercholesterolemic models.53, 54, 55
Another possible connection between inhibition of Kir activity and a decrease in the availability of NO is eNOS uncoupling via elevated production of reactive oxygen species. Indeed, several studies proposed that endothelial dysfunction in hypercholesterolemia results from an increase in reactive oxygen species production, which reduced the level of tetrahydrobiopterin (BH4), an essential cofactor for eNOS and the production of NO.56, 57 In support of this, administering BH4 recovers endothelial function in an NO‐dependent fashion in humans and rodents with hypercholesterolemia comparable to responses observed in normocholesterolemic controls.58, 59, 60 Moreover, studies in lung ischemia suggest that membrane depolarization, which occurs as a result of a decrease in Kir channel activity under ischemic conditions, induces activation of NADPH oxidase 2 and an increase in reactive oxygen species production.61 However, if a similar mechanism occurs during hypercholesterolemia in the mesenteric microvasculature to contribute to a decrease in NO bioavailability, it is likely secondary to the more direct decrease in Kir‐induced NO production we report in the present study. Further studies are needed to elucidate the mechanistic links between shear‐induced Kir channel activity, Akt phosphorylation, elevated reactive oxygen species, and BH4 levels in vascular endothelium as a mechanism for endothelial dysfunction.
Conclusions
The endothelial dysfunction present in hypercholesterolemia has larger clinical implications to advanced cardiovascular disease. Our data show that aortas from Kir2.1 +/− /Apoe −/− mice have elevated lesions compared with aortas from Apoe −/− mice. This effect was observed after 9 months of feeding. Furthermore, aortas from Kir2.1 +/− /Apoe −/− mice exhibited an altered distribution of plaques with a more diffuse distribution between the aortic arch and the DA. It is interesting to note that hypercholesterolemia affects FIV similarly in the atheroprone B6 and atheroresistant FVB strains. This is plausible because hypercholesterolemia is similarly elevated in both strains. In fact, Apoe −/−/FVB mice are well established to have greater plasma cholesterol levels.30, 35, 36 Taken together, these data suggest that neither the cholesterol responsiveness of the Kir channel nor suppression of FIV are contributors to the differences in the atherosusceptibility of these 2 strains. Further studies are needed to explore these underlying differences. Interestingly, Kir deficiency manifests its differential atherosclerotic effects mostly in the DA, an area of the aorta that does not readily develop atherosclerosis. The redistribution of lesions to an otherwise atheroresistant region of the aorta implicates a critical role for Kir2.1 in contributing to an atheroresistant profile in the DA region. Moreover, since it is well known that the difference in atherosusceptibility between aortic arch and DA regions is related to hemodynamic forces,62, 63, 64 our observations are consistent with the proposed role of Kir channels in endothelial mechanotransduction.
Sources of Funding
This work was supported by NIH R01 HL073965 (to Levitan), NIH R01 HL130513 (to Phillips), NIH R01 HL68661 (to Getz and Reardon), and an American Heart Association postdoctoral fellowship 16POST27000011 (to Fancher).
Disclosures
None.
Supporting information
Table S1. Baseline Internal Diameter at 60 cm H2O (Number of Vessels)
Figure S1. Stopping flow returns currents to static baseline and acetylated low‐density lipoprotein (acLDL). Representative perforated patch recording showing static inwardly rectifying K+ (Kir) currents (black), shear stress (0.7 dynes/cm2)–induced activation of Kir currents using the gravity perfused minimally invasive flow (MIF) device (gray), and the return to static Kir currents upon stopping flow (SF) to the MIF device (red). A voltage ramp from −120 to +40 mV was used to elicit Kir current in a 60‐mmol/L K+ bath (EK ≈−20 mV).
Figure S2. Mesenteric arteries from inwardly rectifying K+ (Kir) 2.1 +/− exhibit reduced, Ba2+‐insensitive dilations to flow. Representative flow‐induced vasodilation (FIV) traces from (A) Kir2.1 +/− and (D) wild‐type (WT) mice generated using the pressure gradient method to induce intraluminal flow through mesenteric arteries. Endothelin‐1 (ET‐1) (1.2–2×10−10 mol/L) was used to preconstrict arteries, and papaverine (10−4 mol/L) was used to test arterial function at the end of each protocol. FIV curves from (B) Kir2.1 +/− and (E) WT mice show the differences in Ba2+ (3×10−5 mol/L) sensitivity. Furthermore, FIV in WT mice is sensitive to apamin (2×10−8 mol/L) and abolished by the combination of Ba2+ and apamin. C, Kir2.1 +/− group data highlighting FIV before (control) and after application of Ba2+ measured at Δ100 cm H2O. Six arteries from 4 mice for both WT and Kir2.1 +/− groups. All error bars represent the SEM, which was calculated using number of mice.
Figure S3. Endothelial nitric oxide synthase (eNOS) expression and function are intact in apolipoprotein E–deficient (Apoe −/−) mice. A, Representative Western blot showing total eNOS expression in whole mesenteric arcades from wild‐type (WT) and Apoe −/− mice and (B) group data normalized to actin expression (n=4). C, No differences were observed in eNOS functional capacity as measured in arteries isolated from WT (6 arteries from 3 mice) and Apoe −/− (9 arteries from 5 mice) mice using a dose‐response to the Ca2+ ionophore, A23187. In all cases, the SEM was calculated as number of mice.
Figure S4. Incubating ECs with acetylated low‐density lipoprotein (acLDL) does not prevent adenoviral overexpression of inwardly rectifying K+ (Kir) 2.1, and empty adenovirus (AV) transduction has no effect on FIV in apolipoprotein E–deficient (Apoe −/−) mice. Ai, Endothelial cells (ECs) transduced with AV‐Kir2.1 show detectable hematgglutinin (HA)‐tag fluorescence in the presence (middle panel) or absence (top panel) of acLDL (50 μg/mL; 24‐hour incubation). HA‐tag fluorescence was not detected in those ECs transduced with empty AV (bottom panel); however, green fluorescent protein fluorescence is observed to indicate successful transduction of cells. Considerable changes in morphology were observed with adenoviral transduction; therefore, we show (Aii) photomicrographs confirming the appropriate morphology and expected staining of cultured ECs by platelet‐endothelial cell adhesion molecule (left) and Von Willebrand factor (middle) in nontransduced ECs in culture. B, Representative flow‐induced vasodilation (FIV) trace from a mesenteric artery transduced with empty AV. Endothelin‐1 (ET‐1) and papaverine were used as described above. C, FIV curves show that AV‐empty transduction has no effect on Apoe −/− response to flow. D, Group data highlighting the dilations to flow at Δ100 cm H2O. Only apamin (2×10−8 mol/L) inhibits FIV in Apoe −/− arteries transduced with empty AV (n=4). *P<0.05 vs control, ‡ P<0.05 vs Ba2+ (3×10−5 mol/L). Error bars represent the SEM.
Figure S5. The effect of L‐NG‐Nitroarginine methyl ester (LNAME) on flow‐induced vasodilation (FIV) in arteries from inwardly rectifying K+ (Kir) 2.1 +/− / apolipoprotein E–deficient (Apoe −/−) and wild‐type (WT) mice. FIV curves generated from (A) Kir2.1 +/− /Apoe −/− and (C) WT mesenteric arteries and exposed to either LNAME (10−4 mol/L) or a combination of Ba2+ (3×10−5 mol/L) and LNAME. Group data highlighting dilations to flow at Δ100 cm H2O in (B) Kir2.1 +/− /Apoe −/− (4 arteries from 4 mice) and (D) WT mice (6 arteries from 4 mice). *P<0.05 vs control. Error bars represent the SEM, which was calculated as number of mice. E, Representative images providing good positive controls for the detection of nitric oxide (NO) via the dye diaminorhodamine‐4M. Arteries from WT mice exposed to intraluminal flow (Δ60 for 30 minutes) have robust NO production (left) that is prevented by incubating arteries with LNAME (10−4 mol/L, right) for 20 minutes before and during intraluminal flow (Δ60 for 30 minutes).
Figure S6. Endothelial inwardly rectifying K+ (Kir) 2.1 is a major contributor to flow‐induced vasodilation (FIV) in mesenteric arteries from C57BL/6 (B6) mice. FIV curves generated from C57/BL6 (B6) wild‐type (WT) mice reveal sensitivity to (A) Ba2+ (3×10−5 mol/L; 5 arteries from 4 mice), and (C) transduction with endothelial‐specific adenovirus (AV) dominant‐negative (DN) Kir2.1 (4 pairs of arteries from 4 mice). E, B6 apolipoprotein E–deficient (Apoe −/−) mice (9 arteries from 5 mice), however, lack sensitivity to Ba2+. B, D, and F, Group data highlighting the differences in FIV measured at Δ100 cm H2O for each of (A, C, and E), respectively. *P<0.05 vs (B) control and (D) empty AV. Error bars represent the SEM using number of mice.
Figure S7. Lesion formation in apolipoprotein E–deficient (Apoe −/−) and inwardly rectifying K+ (Kir) 2.1 +/− /Apoe −/− mice aortas after 6 months of high‐fat feeding. A, Representative images of en face aortas from Apoe −/− (left) and Kir2.1 +/− /Apoe −/− (right) stained with Oil Red O for lesion detection. Mice were fed a Western, high‐fat, high‐cholesterol diet for 6 months before analysis. B, Group analyses of the lesions detected in aortic arch (AA) and descending aorta (DA) and measured as percentage of the total area containing lesions in Apoe −/− (n=4) and Kir2.1 +/− /Apoe −/− mice (n=4). Larger, lower bars represent the group average and smaller, upper bars represent the standard error. No significant differences were found between Apoe −/− and Kir2.1 +/− /Apoe −/− groups using 2‐way ANOVA.
(J Am Heart Assoc. 2018;7:e007430 DOI: 10.1161/JAHA.117.007430.)29502106
Contributor Information
Ibra S. Fancher, Email: ifancher@uic.edu.
Irena Levitan, Email: levitan@uic.edu.
References
- 1. Rubanyi GM, Freay AD, Kauser K, Johns A, Harder DR. Mechanoreception by the endothelium: mediators and mechanisms of pressure‐ and flow‐induced vascular responses. Blood Vessels. 1990;27:246–257. [DOI] [PubMed] [Google Scholar]
- 2. Vanhoutte PM, Boulanger CM, Mombouli JV. Endothelium‐derived relaxing factors and converting enzyme inhibition. Am J Cardiol. 1995;76:3E–12E. [PubMed] [Google Scholar]
- 3. Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA, Levitan I. Inwardly rectifying K+ channels are major contributors to flow‐induced vasodilatation in resistance arteries. J Physiol. 2017;595:2339–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. 2012;23:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J Clin Invest. 1999;103:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wever R, Stroes E, Rabelink TJ. Nitric oxide and hypercholesterolemia: a matter of oxidation and reduction? Atherosclerosis. 1998;137(suppl):S51–S60. [DOI] [PubMed] [Google Scholar]
- 7. Lewandowski P, Romanowska‐Kocejko M, Wegrzyn A, Chmara M, Zuk M, Limon J, Wasag B, Rynkiewicz A, Gruchala M. Noninvasive assessment of endothelial function and vascular parameters in patients with familial and nonfamilial hypercholesterolemia. Pol Arch Med Wewn. 2014;124:516–524. [DOI] [PubMed] [Google Scholar]
- 8. Vlahos AP, Naka KK, Bechlioulis A, Theoharis P, Vakalis K, Moutzouri E, Miltiadous G, Michalis LK, Siamopoulou‐Mavridou A, Elisaf M, Milionis HJ. Endothelial dysfunction, but not structural atherosclerosis, is evident early in children with heterozygous familial hypercholesterolemia. Pediatr Cardiol. 2014;35:63–70. [DOI] [PubMed] [Google Scholar]
- 9. Godecke A, Ziegler M, Ding Z, Schrader J. Endothelial dysfunction of coronary resistance vessels in apoE−/− mice involves NO but not prostacyclin‐dependent mechanisms. Cardiovasc Res. 2002;53:253–262. [DOI] [PubMed] [Google Scholar]
- 10. Meyrelles SS, Peotta VA, Pereira TM, Vasquez EC. Endothelial dysfunction in the apolipoprotein E‐deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. 2011;10:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. d'Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1017–1022. [DOI] [PubMed] [Google Scholar]
- 12. Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky HM. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high‐fat diet fed C57BL/6J mice. Circ Res. 2005;96:1178–1184. [DOI] [PubMed] [Google Scholar]
- 13. Ketonen J, Pilvi T, Mervaala E. Caloric restriction reverses high‐fat diet‐induced endothelial dysfunction and vascular superoxide production in C57Bl/6 mice. Heart Vessels. 2010;25:254–262. [DOI] [PubMed] [Google Scholar]
- 14. Landmesser U, Hornig B, Drexler H. Endothelial dysfunction in hypercholesterolemia: mechanisms, pathophysiological importance, and therapeutic interventions. Semin Thromb Hemost. 2000;26:529–537. [DOI] [PubMed] [Google Scholar]
- 15. Fang Y, Mohler ER III, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–1071. [DOI] [PubMed] [Google Scholar]
- 16. Mohler ER III, Fang Y, Shaffer RG, Moore J, Wilensky RL, Parmacek M, Levitan I. Hypercholesterolemia suppresses Kir channels in porcine bone marrow progenitor cells in vivo. Biochem Biophys Res Commun. 2007;358:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reardon CA, Getz GS. Mouse models of atherosclerosis. Curr Opin Lipidol. 2001;12:167–173. [DOI] [PubMed] [Google Scholar]
- 19. Plump AS, Smith JD, Hayek T, Aalto‐Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E‐deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. [DOI] [PubMed] [Google Scholar]
- 20. Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT. Inward rectifier potassium (Kir2.1) channels as end‐stage boosters of endothelium‐dependent vasodilators. J Physiol. 2016;594:3271–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voyta JC, Via DP, Butterfield CE, Zetter BR. Identification and isolation of endothelial cells based on their increased uptake of acetylated‐low density lipoprotein. J Cell Biol. 1984;99:2034–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zinkevich NS, Fancher IS, Gutterman DD, Phillips SA. Roles of NADPH oxidase and mitochondria in flow‐induced vasodilation of human adipose arterioles: ROS induced ROS release in coronary artery disease. Microcirculation. 2017;24 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuo L, Davis MJ, Chilian WM. Endothelium‐dependent, flow‐induced dilation of isolated coronary arterioles. Am J Physiol. 1990;259:H1063–H1070. [DOI] [PubMed] [Google Scholar]
- 24. Robinson AT, Fancher IS, Sudhahar V, Bian JT, Cook MD, Mahmoud AM, Ali MM, Ushio‐Fukai M, Brown MD, Fukai T, Phillips SA. Short‐term regular aerobic exercise reduces oxidative stress produced by acute in the adipose microvasculature. Am J Physiol Heart Circ Physiol. 2017;312:H896–H906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward‐rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crane GJ, Walker SD, Dora KA, Garland CJ. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J Vasc Res. 2003;40:159–168. [DOI] [PubMed] [Google Scholar]
- 29. Rosenhouse‐Dantsker A, Noskov S, Han H, Adney SK, Tang QY, Rodriguez‐Menchaca AA, Kowalsky GB, Petrou VI, Osborn CV, Logothetis DE, Levitan I. Distant cytosolic residues mediate a two‐way molecular switch that controls the modulation of inwardly rectifying potassium (Kir) channels by cholesterol and phosphatidylinositol 4,5‐bisphosphate (PI(4,5)P(2)). J Biol Chem. 2012;287:40266–40278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dansky HM, Charlton SA, Sikes JL, Heath SC, Simantov R, Levin LF, Shu P, Moore KJ, Breslow JL, Smith JD. Genetic background determines the extent of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1960–1968. [DOI] [PubMed] [Google Scholar]
- 31. Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow‐mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol. 2014;307:H1587–H1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vilahur G, Casani L, Mendieta G, Lamuela‐Raventos RM, Estruch R, Badimon L. Beer elicits vasculoprotective effects through Akt/eNOS activation. Eur J Clin Invest. 2014;44:1177–1188. [DOI] [PubMed] [Google Scholar]
- 33. Chen AF, O'Brien T, Tsutsui M, Kinoshita H, Pompili VJ, Crotty TB, Spector DJ, Katusic ZS. Expression and function of recombinant endothelial nitric oxide synthase gene in canine basilar artery. Circ Res. 1997;80:327–335. [DOI] [PubMed] [Google Scholar]
- 34. Zanetti M, Katusic ZS, O'Brien T. Adenoviral‐mediated overexpression of catalase inhibits endothelial cell proliferation. Am J Physiol Heart Circ Physiol. 2002;283:H2620–H2626. [DOI] [PubMed] [Google Scholar]
- 35. Sontag TJ, Krishack PA, Lukens JR, Bhanvadia CV, Getz GS, Reardon CA. Apolipoprotein A‐I protection against atherosclerosis is dependent on genetic background. Arterioscler Thromb Vasc Biol. 2014;34:262–269. [DOI] [PubMed] [Google Scholar]
- 36. Shim J, Handberg A, Ostergren C, Falk E, Bentzon JF. Genetic susceptibility of the arterial wall is an important determinant of atherosclerosis in C57BL/6 and FVB/N mouse strains. Arterioscler Thromb Vasc Biol. 2011;31:1814–1820. [DOI] [PubMed] [Google Scholar]
- 37. Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–170. [DOI] [PubMed] [Google Scholar]
- 38. Gerhold KA, Schwartz MA. Ion channels in endothelial responses to fluid shear stress. Physiology (Bethesda). 2016;31:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenhouse‐Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol‐binding regions in Kir2 channels. J Biol Chem. 2013;288:31154–31164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosenhouse‐Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100:381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arruda RM, Peotta VA, Meyrelles SS, Vasquez EC. Evaluation of vascular function in apolipoprotein E knockout mice with angiotensin‐dependent renovascular hypertension. Hypertension. 2005;46:932–936. [DOI] [PubMed] [Google Scholar]
- 42. Pereira RB, Vasquez EC, Stefanon I, Meyrelles SS. Oral P. gingivalis infection alters the vascular reactivity in healthy and spontaneously atherosclerotic mice. Lipids Health Dis. 2011;10:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wolfle SE, de Wit C. Intact endothelium‐dependent dilation and conducted responses in resistance vessels of hypercholesterolemic mice in vivo. J Vasc Res. 2005;42:475–482. [DOI] [PubMed] [Google Scholar]
- 44. Stapleton PA, Goodwill AG, James ME, Frisbee JC. Altered mechanisms of endothelium‐dependent dilation in skeletal muscle arterioles with genetic hypercholesterolemia. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1110–R1119. [DOI] [PubMed] [Google Scholar]
- 45. d'Uscio LV, Barton M, Shaw S, Luscher TF. Chronic ET(A) receptor blockade prevents endothelial dysfunction of small arteries in apolipoprotein E‐deficient mice. Cardiovasc Res. 2002;53:487–495. [DOI] [PubMed] [Google Scholar]
- 46. Lamping KG, Nuno DW, Chappell DA, Faraci FM. Agonist‐specific impairment of coronary vascular function in genetically altered, hyperlipidemic mice. Am J Physiol. 1999;276:R1023–R1029. [DOI] [PubMed] [Google Scholar]
- 47. Xu X, Gao X, Potter BJ, Cao JM, Zhang C. Anti‐LOX‐1 rescues endothelial function in coronary arterioles in atherosclerotic ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2007;27:871–877. [DOI] [PubMed] [Google Scholar]
- 48. Rosenhouse‐Dantsker A, Epshtein Y, Levitan I. Interplay between lipid modulators of Kir2 channels: cholesterol and PIP2. Comput Struct Biotechnol J. 2014;11:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han H, Rosenhouse‐Dantsker A, Gnanasambandam R, Epshtein Y, Chen Z, Sachs F, Minshall RD, Levitan I. Silencing of Kir2 channels by caveolin‐1: cross‐talk with cholesterol. J Physiol. 2014;592:4025–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein O, Dabach Y, Ben‐Naim M, Halperin G, Stein Y. Lower macrophage recruitment and atherosclerosis resistance in FVB mice. Atherosclerosis. 2006;189:336–341. [DOI] [PubMed] [Google Scholar]
- 51. d'Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium‐dependent relaxations in common carotid arteries of apolipoprotein E‐deficient mice. Stroke. 2001;32:2658–2664. [DOI] [PubMed] [Google Scholar]
- 52. Andrews AM, Muzorewa TT, Zaccheo KA, Buerk DG, Jaron D, Barbee KA. Cholesterol enrichment impairs capacitative calcium entry, eNOS phosphorylation & shear stress‐induced NO production. Cell Mol Bioeng. 2017;10:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharma S, Prasanthi RPJ, Schommer E, Feist G, Ghribi O. Hypercholesterolemia‐induced Abeta accumulation in rabbit brain is associated with alteration in IGF‐1 signaling. Neurobiol Dis. 2008;32:426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, Pant R, Menon VP, Otani H, Maulik N. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol. 2007;42:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma LL, Zhang FJ, Qian LB, Kong FJ, Sun JF, Zhou C, Peng YN, Xu HJ, Wang WN, Wen CY, Zhu MH, Chen G, Yu LN, Liu XB, Wang JA, Yan M. Hypercholesterolemia blocked sevoflurane‐induced cardioprotection against ischemia‐reperfusion injury by alteration of the MG53/RISK/GSK3beta signaling. Int J Cardiol. 2013;168:3671–3678. [DOI] [PubMed] [Google Scholar]
- 56. Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. [DOI] [PubMed] [Google Scholar]
- 57. Wang Q, Yang M, Xu H, Yu J. Tetrahydrobiopterin improves endothelial function in cardiovascular disease: a systematic review. Evid Based Complement Alternat Med. 2014;2014:850312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tamura Y, Naemura A, Inoue A, Ijiri Y, Seki J, Yada T, Goto M, Shinohara M, Kawashima S, Giddings JC, Yamamoto J. Impaired endothelial function may be due to decreased aortic tetrahydrobiopterin, assessed by a new flow‐mediated vasodilation in vivo in hypercholesterolemic/atherogenic mice. Blood Coagul Fibrinolysis. 2009;20:699–705. [DOI] [PubMed] [Google Scholar]
- 59. Fukuda Y, Teragawa H, Matsuda K, Yamagata T, Matsuura H, Chayama K. Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart. 2002;87:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wyss CA, Koepfli P, Namdar M, Siegrist PT, Luscher TF, Camici PG, Kaufmann PA. Tetrahydrobiopterin restores impaired coronary microvascular dysfunction in hypercholesterolaemia. Eur J Nucl Med Mol Imaging. 2005;32:84–91. [DOI] [PubMed] [Google Scholar]
- 61. Browning E, Wang H, Hong N, Yu K, Buerk DG, DeBolt K, Gonder D, Sorokina EM, Patel P, De Leon DD, Feinstein SI, Fisher AB, Chatterjee S. Mechanotransduction drives post ischemic revascularization through K(ATP) channel closure and production of reactive oxygen species. Antioxid Redox Signal. 2014;20:872–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tarbell JM, Shi ZD, Dunn J, Jo H. Fluid mechanics, arterial disease, and gene expression. Annu Rev Fluid Mech. 2014;46:591–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang YZ, Manduchi E, Jimenez JM, Davies PF. Endothelial epigenetics in biomechanical stress: disturbed flow‐mediated epigenomic plasticity in vivo and in vitro. Arterioscler Thromb Vasc Biol. 2015;35:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline Internal Diameter at 60 cm H2O (Number of Vessels)
Figure S1. Stopping flow returns currents to static baseline and acetylated low‐density lipoprotein (acLDL). Representative perforated patch recording showing static inwardly rectifying K+ (Kir) currents (black), shear stress (0.7 dynes/cm2)–induced activation of Kir currents using the gravity perfused minimally invasive flow (MIF) device (gray), and the return to static Kir currents upon stopping flow (SF) to the MIF device (red). A voltage ramp from −120 to +40 mV was used to elicit Kir current in a 60‐mmol/L K+ bath (EK ≈−20 mV).
Figure S2. Mesenteric arteries from inwardly rectifying K+ (Kir) 2.1 +/− exhibit reduced, Ba2+‐insensitive dilations to flow. Representative flow‐induced vasodilation (FIV) traces from (A) Kir2.1 +/− and (D) wild‐type (WT) mice generated using the pressure gradient method to induce intraluminal flow through mesenteric arteries. Endothelin‐1 (ET‐1) (1.2–2×10−10 mol/L) was used to preconstrict arteries, and papaverine (10−4 mol/L) was used to test arterial function at the end of each protocol. FIV curves from (B) Kir2.1 +/− and (E) WT mice show the differences in Ba2+ (3×10−5 mol/L) sensitivity. Furthermore, FIV in WT mice is sensitive to apamin (2×10−8 mol/L) and abolished by the combination of Ba2+ and apamin. C, Kir2.1 +/− group data highlighting FIV before (control) and after application of Ba2+ measured at Δ100 cm H2O. Six arteries from 4 mice for both WT and Kir2.1 +/− groups. All error bars represent the SEM, which was calculated using number of mice.
Figure S3. Endothelial nitric oxide synthase (eNOS) expression and function are intact in apolipoprotein E–deficient (Apoe −/−) mice. A, Representative Western blot showing total eNOS expression in whole mesenteric arcades from wild‐type (WT) and Apoe −/− mice and (B) group data normalized to actin expression (n=4). C, No differences were observed in eNOS functional capacity as measured in arteries isolated from WT (6 arteries from 3 mice) and Apoe −/− (9 arteries from 5 mice) mice using a dose‐response to the Ca2+ ionophore, A23187. In all cases, the SEM was calculated as number of mice.
Figure S4. Incubating ECs with acetylated low‐density lipoprotein (acLDL) does not prevent adenoviral overexpression of inwardly rectifying K+ (Kir) 2.1, and empty adenovirus (AV) transduction has no effect on FIV in apolipoprotein E–deficient (Apoe −/−) mice. Ai, Endothelial cells (ECs) transduced with AV‐Kir2.1 show detectable hematgglutinin (HA)‐tag fluorescence in the presence (middle panel) or absence (top panel) of acLDL (50 μg/mL; 24‐hour incubation). HA‐tag fluorescence was not detected in those ECs transduced with empty AV (bottom panel); however, green fluorescent protein fluorescence is observed to indicate successful transduction of cells. Considerable changes in morphology were observed with adenoviral transduction; therefore, we show (Aii) photomicrographs confirming the appropriate morphology and expected staining of cultured ECs by platelet‐endothelial cell adhesion molecule (left) and Von Willebrand factor (middle) in nontransduced ECs in culture. B, Representative flow‐induced vasodilation (FIV) trace from a mesenteric artery transduced with empty AV. Endothelin‐1 (ET‐1) and papaverine were used as described above. C, FIV curves show that AV‐empty transduction has no effect on Apoe −/− response to flow. D, Group data highlighting the dilations to flow at Δ100 cm H2O. Only apamin (2×10−8 mol/L) inhibits FIV in Apoe −/− arteries transduced with empty AV (n=4). *P<0.05 vs control, ‡ P<0.05 vs Ba2+ (3×10−5 mol/L). Error bars represent the SEM.
Figure S5. The effect of L‐NG‐Nitroarginine methyl ester (LNAME) on flow‐induced vasodilation (FIV) in arteries from inwardly rectifying K+ (Kir) 2.1 +/− / apolipoprotein E–deficient (Apoe −/−) and wild‐type (WT) mice. FIV curves generated from (A) Kir2.1 +/− /Apoe −/− and (C) WT mesenteric arteries and exposed to either LNAME (10−4 mol/L) or a combination of Ba2+ (3×10−5 mol/L) and LNAME. Group data highlighting dilations to flow at Δ100 cm H2O in (B) Kir2.1 +/− /Apoe −/− (4 arteries from 4 mice) and (D) WT mice (6 arteries from 4 mice). *P<0.05 vs control. Error bars represent the SEM, which was calculated as number of mice. E, Representative images providing good positive controls for the detection of nitric oxide (NO) via the dye diaminorhodamine‐4M. Arteries from WT mice exposed to intraluminal flow (Δ60 for 30 minutes) have robust NO production (left) that is prevented by incubating arteries with LNAME (10−4 mol/L, right) for 20 minutes before and during intraluminal flow (Δ60 for 30 minutes).
Figure S6. Endothelial inwardly rectifying K+ (Kir) 2.1 is a major contributor to flow‐induced vasodilation (FIV) in mesenteric arteries from C57BL/6 (B6) mice. FIV curves generated from C57/BL6 (B6) wild‐type (WT) mice reveal sensitivity to (A) Ba2+ (3×10−5 mol/L; 5 arteries from 4 mice), and (C) transduction with endothelial‐specific adenovirus (AV) dominant‐negative (DN) Kir2.1 (4 pairs of arteries from 4 mice). E, B6 apolipoprotein E–deficient (Apoe −/−) mice (9 arteries from 5 mice), however, lack sensitivity to Ba2+. B, D, and F, Group data highlighting the differences in FIV measured at Δ100 cm H2O for each of (A, C, and E), respectively. *P<0.05 vs (B) control and (D) empty AV. Error bars represent the SEM using number of mice.
Figure S7. Lesion formation in apolipoprotein E–deficient (Apoe −/−) and inwardly rectifying K+ (Kir) 2.1 +/− /Apoe −/− mice aortas after 6 months of high‐fat feeding. A, Representative images of en face aortas from Apoe −/− (left) and Kir2.1 +/− /Apoe −/− (right) stained with Oil Red O for lesion detection. Mice were fed a Western, high‐fat, high‐cholesterol diet for 6 months before analysis. B, Group analyses of the lesions detected in aortic arch (AA) and descending aorta (DA) and measured as percentage of the total area containing lesions in Apoe −/− (n=4) and Kir2.1 +/− /Apoe −/− mice (n=4). Larger, lower bars represent the group average and smaller, upper bars represent the standard error. No significant differences were found between Apoe −/− and Kir2.1 +/− /Apoe −/− groups using 2‐way ANOVA.