

Abstract
Background
Previous experimental studies have shown that downstream microvascular thromboinflammation is involved in brain damage from acute ischemic stroke. Using intravital microscopy, we investigated and characterized the sequence of downstream microvascular thromboinflammation in an ischemia/reperfusion acute ischemic stroke model.
Methods and Results
Rats underwent transient monofilament middle cerebral artery (MCA) occlusion. Cerebral microcirculation in the MCA territory was exposed through a craniotomy and analyzed using real‐time intravital imaging coupled with laser Doppler interferometry. Leukocytes, platelets, fibrinogen, and blood–brain barrier permeability were analyzed by intravenous injection of fluorescent antibodies and bovine serum albumin. MCA occlusion induced a sudden and profound drop in downstream microvascular blood flow associated with leukocyte margination in the venous compartment. Leukocyte margination fostered fibrinogen deposition and thrombosis in postcapillary venules. Either in venules or arterioles, blood flow was not fully restored after MCA recanalization. Furthermore, venular thrombi persisted despite MCA recanalization, and leukocyte extravasation continued to develop in venules in association with blood–brain barrier disruption. Finally, microhemorrhages were occasionally observed, colocalizing with thrombosed venules characterized by marked leukocyte margination.
Conclusions
We showed that microvascular thrombosis in transient monofilament MCA occlusion and blood–brain barrier disruption are initiated immediately after occlusion and are propagated through the venous compartment in close association with marginating leukocytes. MCA occlusion–induced downstream microvascular thromboinflammation response was responsible for incomplete reperfusion after MCA recanalization and delayed microhemorrhages.
Keywords: blood–brain barrier, leukocyte, middle cerebral artery occlusion, no‐reflow, venous thrombosis
Subject Categories: Inflammation, Vascular Biology, Blood-Brain Barrier, Ischemic Stroke, Pathophysiology
Clinical Perspective
What Is New?
Downstream thromboinflammation is an early event triggered immediately after occlusion of a large proximal artery.
It is characterized by leukocyte margination in cortical microvessels where blood flow is markedly reduced despite collateral circulation from pial anastomoses.
Leukocyte margination fosters fibrinogen deposition and secondary venous thrombosis, as well as rupture of the blood–brain barrier in both cortical venules and arterioles.
Thrombi persisted and leukocyte extravasation continued progressing in venules after recanalization, which only partially corrected blood flow in cortical microvessels.
In arterioles, despite recanalization‐induced leukocyte washout, vascular leakage developed at sites where neutrophils had accumulated during the occlusion period.
What Are the Clinical Implications?
Our results underscore the possible difficulty of reversing the microvascular consequences of large vessel occlusion because they are only partially corrected by proximal recanalization.
The results further suggest that early actors of the thromboinflammatory cascade should be targeted as soon as possible and before proximal recanalization for maximal efficacy in the treatment of acute ischemic stroke caused by large vessel occlusion.
Introduction
Experimental studies using animal models of cerebral ischemia/reperfusion have provided broad evidence that proximal arterial occlusion causes deleterious secondary events that significantly contribute to acute ischemic stroke (AIS) pathophysiology.1 In particular, cerebral ischemia/reperfusion is known to trigger interactions among the vessel wall, platelets, leukocytes, and coagulation that can impair reperfusion and destabilize the blood–brain barrier (BBB).1, 2 How and when these interactions, grouped under the term thromboinflammation, take place remains unclear. At present, the dominant paradigm is that they would mostly develop during the reperfusion phase and contribute to reperfusion injury.3 We observed in a recent study that leukocyte accumulation, mainly neutrophils, in microvessels of the ischemic hemisphere occurred as early as 30 minutes after proximal artery occlusion.4 The latter phenomenon was also reported in earlier studies5 including models of permanent focal cerebral ischemia, indicating that the thromboinflammatory response to cerebral ischemia was not necessarily the consequence of reperfusion.6 Limiting thromboinflammation has been proposed as a possible strategy to improve penumbra salvage in AIS. A better understanding of AIS‐associated thromboinflammation could thus help in designing novel treatment for AIS. In this study, using intravital microscopy coupled with laser Doppler interferometry, we investigated and characterized the sequence of downstream microvascular responses to cerebral ischemia/reperfusion with particular respect to blood flow, microvascular thrombosis, and BBB damage.
Materials and Methods
The data and analytic methods and the study materials will be made available to other researchers on request for purposes of reproducing the results or replicating the procedure.
Middle Cerebral Artery Occlusion and Reperfusion
Male Sprague‐Dawley rats (Janvier, France) underwent 120 minutes of transient monofilament middle cerebral artery occlusion (MCAO), as described previously.7 All experimental procedures were declared to the French ministry of research and authorized after ethics review (no. 20160125164052; APAFIS 3792).
Real‐Time Intravital Imaging Coupled With Laser Doppler Interferometry
Distal branches of the middle cerebral artery (MCA) of rats were exposed through a 4×4‐mm craniotomy performed in the right temporoparietal cortex with a hand‐held drill, as described previously.7 Cerebral microcirculation was directly visualized using a fluorescence macroscope (MacroFluo; Leica Microsystems) equipped with a heating plate with a thermostat and a ×5 objective and connected to a scientific CMOS camera (ORCA‐Flash4.0; Hamamatsu Photonics). Data acquisition and analysis were done using Metamorph software (Molecular Devices).
All fluorescent markers were administered intravenously into the tail vein. Rhodamine 6G was used to label leukocytes and platelets, FITC (fluorescein isothiocyanate)–conjugated polyclonal rabbit anti–human fibrinogen was used to stain fibrin(ogen), Alexa 555–conjugated hamster anti–rat CD42d was used to stain platelets, and Alexa Fluor 647–conjugated BSA was used to assess vascular permeability. Fluorescent BSA was injected intravenously 5 minutes before recanalization.
Cortical vessel diameter before MCA occlusion and 1 hour after recanalization was measured in intravital video microscopy images using ZEN software (Zeiss).
Blood cell velocity in microvessels was measured using a single‐point laser Doppler vibrometer with an integrated CCD video camera (CLV‐2534; Polytec) and mounted on the macroscope to monitor laser spot positioning. To allow specific measurement of blood cell velocity, breathing‐related and flow‐induced vessel wall vibrations were identified by their bidirectional associated signal and eliminated by applying a low‐pass filter set at 2 kHz. Frequencies due to the unidirectional out‐of‐plane vibrations caused by circulating blood cells were converted to speed according to the formula v=(Δf×λ)/(2×cosα), where v is the blood cell velocity, Δf is the Doppler‐frequency shift, λ is the wavelength of the emitted wave (633 nm), and α is the angle between the blood cell direction and the incident laser beam, which was estimated at 80° (Figure 1). Data were recorded and treated using the Polytec Vibrometer Software.
Figure 1.
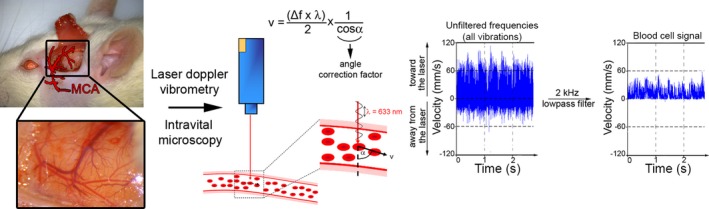
Schematic representation of the experimental design used for intravital imaging and laser Doppler vibrometry of pial microvessels downstream of the middle cerebral artery (MCA). Blood flow and cell interactions in cortical pial microvessels downstream of the MCA were analyzed by intravital microscopy and laser Doppler interferometry through a cranial window. To enable measurement of red blood cell velocity, a low‐pass filter set at 2 kHz was applied to eliminate environmental noise, including breathing‐related movement. Using these settings, cell interactions and blood flow were monitored before MCA occlusion, during occlusion, and after MCA recanalization.
Statistical Analysis
Red blood cell velocities were compared using the nonparametric Wilcoxon signed rank test for paired samples. Values of P<0.05 were considered statistically significant.
Results
MCAO Induces Sudden and Profound Blood Flow Anomalies Associated With Marked Leukocyte Margination in Downstream Microvessels
We first analyzed the impact of middle cerebral artery occlusion (MCAO) on blood flow in downstream pial microvessels. MCAO caused a sudden and profound drop in venous blood flow that was concomitant with a marked leukocyte margination (Figure 2A and 2B) that continued to develop throughout the occlusion period (Figure 2A). In downstream arterioles, MCAO led to a drop in blood flow (Figure 2A and 2C) that was also accompanied by leukocyte adhesion, albeit to a much lesser extent than in venules (Figure 2A). Remarkably, during MCAO, momentary and recurrent inversions of blood flow direction were systematically observed in downstream arterioles because of competitive flow coming from pial anastomoses (Figure 2A, Video S1).
Figure 2.
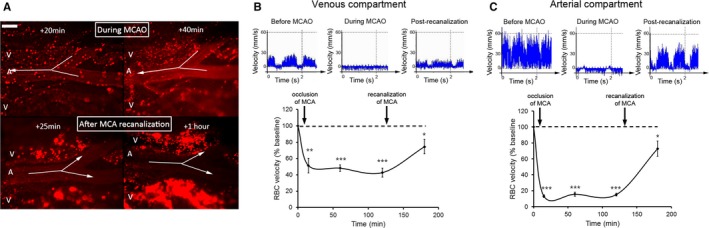
Early microvascular consequences of transient middle cerebral artery occlusion (MCAO). A, Fluorescent intravital microscopy images illustrating the marked adhesion of rhodamine 6G–labeled leukocytes in pial postcapillary venules (V) occurring early after occlusion of the middle cerebral artery (MCA; 20 and 40 minutes after occlusion; top panels), and their later extravasation still developing despite MCA recanalization (25 minutes and 1 hour after recanalization; lower panels). Leukocyte margination was also observed in pial arterioles (A) during occlusion but did not persist after MCA recanalization, leading to the almost complete washout of adhering leukocytes in those vessels. Arrows indicate blood flow direction in a pial arteriole and show reverse blood flow during occlusion and correction of blood flow direction after MCA recanalization. Bar=100 μm. B and C. Evolution of red blood cell velocity in pial venules (B) and arterioles (C) at baseline, during MCAO (15, 60, and 120 minutes after occlusion), and 60 minutes after recanalization. Results are expressed as percentages relative to baseline values. n=5 rats, 3 different vessels (100‐ to 250‐μm diameter) analyzed per rat for a total of 15 measurements for each time point. Error bars represent SEM. *P<0.05, **P<0.01, and ***P<0.001 compared with baseline.
MCA recanalization instantaneously restored downstream blood flow in both venules and arterioles, which corrected blood flow direction anomalies in arterioles (Figure 2A, Video S1). Nevertheless, blood flow was not fully restored by recanalization in either venules or arterioles, as red blood cell velocities after MCAO only reached ≈70% of their baseline values measured before MCAO (Figure 2B and 2C). The mean red blood cell velocity in venules was 7.0±1.9 mm·s−1 before MCAO and of 5.1±2.4 SD mm·s−1 at 1 hour after MCA recanalization. In arterioles, it was 33.4±13.8 SD mm·s−1 before MCAO and 24.2±15.2 SD mm·s−1 at 1 hour after MCA recanalization. Theoretically, to maintain blood flow, when assimilating the vessel lumen to a cylinder, vasodilatation of 20% could compensate for the observed reduction in red blood cell velocity to 70% of its initial value. A slight increase in vessel diameter was observed in both venules and arterioles at 1 hour after MCA recanalization (diameter [mean±SD] of 106.9±10.7% of initial diameter in arterioles and 106.7±13.3% in venules, n=15). These results indicate that, although insufficient for restoration of normal blood flow, a compensatory microvacular vasodilatory response occurs in transient monofilament MCAO.
Notably, MCA recanalization invariably led to the immediate detachment and almost complete washout of adhering leukocytes in arterioles (Figure 2A). In contrast, in all rats analyzed (n>10), leukocyte recruitment and extravasation continued to develop in venules despite recanalization (Figure 2A).
Leukocyte Margination Fosters Downstream Thrombosis and BBB Disruption
The fact that blood flow in downstream microvessels was only partially restored by MCA recanalization indicated that secondary responses to MCAO likely impaired full reperfusion. We thus investigated signs of secondary thrombosis in downstream microvessels. Fibrin(ogen) deposits were consistently observed on marginating leukocytes in postcapillary venules (Figure 3A). Fibrinogen deposition onto leukocytes was initiated during the MCAO period and eventually led to the formation of occlusive thrombi in postcapillary venules (Figure 3B). Of note, these MCAO‐induced secondary thrombi persisted despite MCA recanalization (Figure 3B).
Figure 3.

Leukocyte margination in the ischemic brain is associated with fibrinogen accumulation, thrombosis, increased permeability, and microhemorrhage. A and B, Fluorescent intravital microscopy images showing fibrinogen deposits (Fg, green) in areas of leukocyte adhesion (red) during middle cerebral artery (MCA) occlusion (A) and after recanalization (B). A, Note the striking contrast between the venule and the arteriole, with respect to leukocyte adhesion and fibrinogen deposits. B, Early leukocyte and fibrinogen accumulation eventually led to the occlusion of small venules (left image, bar=100 μm) and larger venules (right panel, bar=200 μm), which persisted despite MCA recanalization. Insets show higher magnification of the areas highlighted by arrows. C, Five minutes before recanalization, fluorescent BSA was injected intravenously to assess vascular permeability. Merged images showing the leakage of fluorescent BSA (magenta) outside a pial venule (upper panel) and arteriole (lower panel) at sites of leukocyte adhesion and extravasation (red). Whereas adherent leukocytes were still present in venules despite recanalization, they were washed out in arterioles. The images were taken 20 minutes after MCA recanalization. Bar=100 μm. D, Merged image demonstrating bleeding (asterisks) originating from a pial venule occluded by leukocytes (red) and platelets (green). This image was acquired 2.5 hours after MCA recanalization. Bar=200 μm. A indicates arteriole; V, venule.
In addition to foster intravascular fibrin(ogen) accumulation, sites of leukocyte adhesion and/or extravasation in venules and arterioles were also associated with BBB disruption, as indicated by the leakage of intravenously injected fluorescent BSA (Figure 3C). Whereas increased permeability to BSA from downstream venules and arterioles occurred systematically, microhemorrhages were much more occasional. Microhemorrhage was observed in 2 of 10 rats and developed from occluded postcapillary venules (Figure 3D).
Discussion
In this study, we showed that leukocyte margination is an early event that is triggered by arterial occlusion. Leukocyte margination starts concomitantly with the abrupt and profound drop in downstream arterial and venous blood flow that occurs despite collateral circulation provided by pial anastomoses. MCAO‐induced leukocyte adhesion and extravasation mostly affects postcapillary venules and drives secondary venous thrombosis by promoting the formation of intravascular fibrin(ogen) deposits onto the surface of adherent leukocytes. The latter observation supports previous findings showing that neutrophil depletion improved microvascular perfusion following MCAO.8
In agreement with an earlier study by del Zoppo and Mabuchi,1 we showed recently that neutrophils represented the vast majority of leukocytes being recruited to the ischemic hemisphere microvasculature during AIS.4, 7 A previous study using intravital microscopy to investigate leukocyte–endothelium interactions also reported that neutrophil recruitment to the ischemic brain was predominant in the venous compartment.6 The thrombosis pattern we described in this study, with a phenomenon of venous thrombosis developing in a context of stasis and neutrophil margination, strongly recalls that observed in animal models of deep vein thrombosis.9 Strengthening the resemblance with deep vein thrombosis, in which VWF (von Willebrand factor) and neutrophil extracellular traps have been shown to play an important role, recent studies have shown that both VWF and extracellular DNA contribute to MCAO‐associated thromboinflammation in mice.10, 11 Although previous studies have shown a clear benefit of targeting neutrophil–endothelial interactions in experimental models of AIS, this strategy has not successfully translated to the clinic, with several failed or interrupted trials.12 In those trials, drugs targeting neutrophils were administered ≈4 hours after stroke onset. Our results showing that neutrophil recruitment is initiated immediately after occlusion suggest that, to be effective, drugs targeting neutrophils should likely be administered as soon as possible and before recanalization.
Importantly, MCA recanalization only partially corrected MCAO‐induced microvascular anomalies. In fact, at 1 hour after recanalization, red blood cell velocities in cortical arterioles and venules remained below baseline values. Although compensatory vasodilation occurred in both types of vessel, its limited magnitude was not sufficient to restore normal blood flow following transient monofilament MCAO. Moreover, venous thrombi persisted and leukocyte extravasation continued progressing in venules after recanalization. In arterioles, despite the almost complete washout of adhering leukocytes following recanalization, vascular leakage developed at sites where neutrophils had accumulated during the occlusion period. These results underscore a possible difficulty in reversing MCAO‐triggered downstream microvascular responses.
Our results also suggest that the deleterious role played by neutrophils at the acute phase of ischemic stroke might not be limited to thrombosis propagation and may expand to BBB disruption and hemorrhagic transformation. In fact, we showed in this study that BBB disruption originates from sites of neutrophil recruitment. As reported previously, hemorrhagic transformation in the form of petechiae is promoted by hyperglycemia, which primes neutrophils for activation and interactions with endothelial cells.4 In the same study, hemorrhagic transformation originated from areas showing persistent hypoperfusion in magnetic resonance imaging despite recanalization. Taken together, these observations suggest that hemorrhagic transformation might be a consequence of persisting occlusion rather than reperfusion. Supporting this hypothesis, using intravital microscopy in the present study, we observed microhemorrhages developing from occluded postcapillary venules.
In conclusion, we showed that microvascular thrombosis and BBB disruption in transient monofilament MCAO are initiated immediately after occlusion and preferentially develop in the venous compartment in close association with leukocyte margination.
Sources of Funding
Dr Desilles is the recipient of a grant poste d'accueil INSERM. This work was supported by INSERM, La Fondation pour la Recherche sur les AVC (grant no. FR‐AVC‐003), and by La Fondation pour la Recherche Médicale (grant no. DPC20171138959). Dr Di Meglio is the recipient of a PhD grant from La Fondation de L'Avenir.
Disclosures
None.
Supporting information
Video S1. Microvascular consequences of middle cerebral artery (MCA) occlusion and recanalization. Intravital microscopy observation of pial microvessels downstream of the MCA during MCA occlusion and after MCA recanalization. Circulating leukocytes and platelets were labeled by intravenous injection of rhodamine 6G. Note that MCA occlusion causes a drop in venous blood flow and inversions of blood flow direction in downstream arterioles. Both phenomena are associated with leukocyte margination. Although recanalization partially corrected blood flow anomalies, leukocyte margination and extravasation continued to develop in venules. A indicates arterioles; V, venules.
(J Am Heart Assoc. 2018;7:e007804 DOI: 10.1161/JAHA.117.007804.)29496683
References
- 1. del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–894. [DOI] [PubMed] [Google Scholar]
- 2. De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. 2016;47:1165–1172. [DOI] [PubMed] [Google Scholar]
- 3. Nour M, Scalzo F, Liebeskind DS. Ischemia‐reperfusion injury in stroke. Interv Neurol. 2013;1:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desilles JP, Syvannarath V, Ollivier V, Journe C, Delbosc S, Ducroux C, Boisseau W, Louedec L, Di Meglio L, Loyau S, Jandrot‐Perrus M, Potier L, Michel JB, Mazighi M, Ho‐Tin‐Noé B. Exacerbation of thromboinflammation by hyperglycemia precipitates cerebral infarct growth and hemorrhagic transformation. Stroke. 2017;48:1932–1940. [DOI] [PubMed] [Google Scholar]
- 5. Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 6. Kataoka H, Kim SW, Plesnila N. Leukocyte‐endothelium interactions during permanent focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2004;24:668–676. [DOI] [PubMed] [Google Scholar]
- 7. Desilles JP, Loyau S, Syvannarath V, Gonzalez‐Valcarcel J, Cantier M, Louedec L, Lapergue B, Amarenco P, Ajzenberg N, Jandrot‐Perrus M, Michel JB, Ho‐Tin‐Noé B, Mazighi M. Alteplase reduces downstream microvascular thrombosis and improves the benefit of large artery recanalization in stroke. Stroke. 2015;46:3241–3248. [DOI] [PubMed] [Google Scholar]
- 8. Dawson DA, Ruetzler CA, Carlos TM, Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and microcirculatory perfusion in acute stroke in the SHR. Keio J Med. 1996;45:248–252; discussion 252‐243 [DOI] [PubMed] [Google Scholar]
- 9. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32:1884–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor‐cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114:3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Enlimomab Acute Stroke Trial Investigators . Use of anti‐ICAM‐1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57:1428–1434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Microvascular consequences of middle cerebral artery (MCA) occlusion and recanalization. Intravital microscopy observation of pial microvessels downstream of the MCA during MCA occlusion and after MCA recanalization. Circulating leukocytes and platelets were labeled by intravenous injection of rhodamine 6G. Note that MCA occlusion causes a drop in venous blood flow and inversions of blood flow direction in downstream arterioles. Both phenomena are associated with leukocyte margination. Although recanalization partially corrected blood flow anomalies, leukocyte margination and extravasation continued to develop in venules. A indicates arterioles; V, venules.