

Introduction
Sudden cardiac death (SCD) is a leading cause of mortality worldwide with an estimated 15% to 20% of all deaths. SCD mostly occurs in older adults with acquired structural heart disease.1 Among these structural heart diseases, coronary heart disease, presumed to be the most common pathology underlying SCD, accounts for 75% of SCD in Western countries and 50% to 60% in Japan, followed by cardiomyopathies of 10% to 15% in Western countries and 30% to 35% in Japan. Only 1% to 2% of SCD in Western countries compared with 10% in Japan occurs because of rare inherited cardiac arrhythmias without obvious structural change in the heart.1, 2 For these cases, a routine postmortem autopsy cannot explain their death, and these can be classified as sudden unexplained death (SUD).
Here, we focus on a specific SUD called sudden unexplained nocturnal death syndrome (SUNDS) and attempt to provide a comprehensive understanding of SUNDS. SUNDS occurs predominantly in Southeast Asia and has different academic terms but similar definitions in different countries, such as bangungut in Philippines, Lai Tai in Thailand, Pokkuri Death Syndrome (PDS) in Japan, and SUNDS in the United States and China.3, 4, 5, 6, 7 Despite these multiple terms, the common characteristics (modalities) of these victims are sudden death of young healthy individuals (the vast majority are males) during nocturnal sleep and postmortem routine autopsy that cannot explain their deaths.
It has been 100 years since the first description of SUNDS in 1917 in the Philippines.7 Since then, 2 major active periods of studies on SUNDS have accumulated vast information for further understanding the pathogenesis of SUNDS. As a disorder mainly prevalent in Southeast Asia, SUNDS did not attract worldwide attention until 1981, when its first report was submitted to the Centers for Disease Control and Prevention.5 Since then, numerous scholars have sought to understand the enigma of SUNDS. Over the next 20 years, studies mainly focused on collecting epidemiological characteristics to reveal probable pathogenesis associated with external environment factors. Since the first literature revealed the correlation between SUNDS and pathogenic mutation on SCN5A in 2002, the pathogenetic study on SUNDS has entered a new stage of molecular genetics aimed at uncovering the intrinsic gene susceptibility in different individuals (Figure 1). To better understand SUNDS, we conduct this comprehensive review and outline an academic history of SUNDS over the past 100 years, including epidemiology, pathology, and pathogenetic hypotheses, particularly the molecular genetics of SUNDS.
Figure 1.
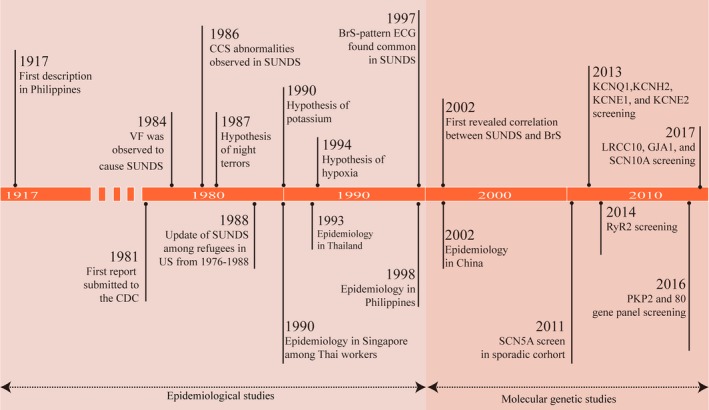
Milestones in SUNDS. It has been 100 years since the first description of SUNDS in 1917 in Philippines. There were 2 major active periods of studies on SUNDS: The early stage mainly focused on collecting the epidemiological characteristics and revealing the probable external environmental risk factors; the later stage was aimed at uncovering the intrinsic gene susceptibility. BrS indicates Brugada Syndrome; CCS, cardiac conduction system; CDC, Centers for Disease Control and Prevention; SUNDS, sudden unexplained nocturnal death syndrome; US, the United States; VF, ventricular fibrillation.
Epidemiology of SUNDS
SUNDS in the United States
Since the first report of 12 sudden unexplained nocturnal deaths among Laotian refugees in the United States was submitted, the Centers for Disease Control and Prevention had received 13 reports with a total number of 117 SUNDS cases in Southeast Asian refugees during 1981–1988. Interestingly, the incidence of SUNDS had dropped sharply from its peak of 59 to 92/100 000 in 1980 to only 1/100 000 in 1987.5, 8 The incidence of SUNDS in 1981 differed in different ethnicities (per 100 000): 92 for Hmong, 82 for Laotians, and 59 for Kampucheans. From the reports collected by the Centers for Disease Control and Prevention, several results might unmask SUNDS: (1) The mean age was 33 years old and 75% were 25 to 44 years old; (2) Gurgling, gasping, or labored respirations appeared in some victims before death; (3) None of the victims had a history of syncope episodes, epilepsy, or allergies; (4) Only 1 of 51 SUNDS cases was confirmed to have family history (Table 1).
Table 1.
Epidemiology of SUNDS in Different Countries
| Country | Ethnicity | Inclusion Criteria | SUNDS Cases | Inci.a | Mean Age | Peak Risk Age (y) | Sex Ratiob | Peak Risk Month | Occupation | Symptoms Before Death | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| USA (1981)c | South‐East Asian refugees in the USA |
25–63 y old SCD at night or during sleep postmortem autopsy Negatived |
25 | 59–92 | 33 | NA | NA | NA | NA | Choking, gurgling, gasping, or labored respirations, without wheezing or stridor | 5, 8 |
| USA (1987) | 5 | 1 | 33 | NA | 5/0 | NA | NA | NA | 8 | ||
| Singapore (1982–1990) | Thai workers in Singapore |
21–54 y old SCD during sleep Postmortem autopsy (57/161 done) negative |
161 | 78 | 34 | NA | 161/0 | No mo pattern | Construction worker | Noisy breathing, gasping, or groaning | 9 |
| Thailand (1990) | Thai‐Lao in the northeast |
20–49 y old SCD during night or daytime sleep Seemed to be healthy (Postmortem autopsies were not done in all cases) |
31 | 38 | 38 | 45–49 | 30/1 | Apr | Farmer or laborer | Groaning, muttering, choking or coughing, nightmare or night terror, restless sleep, spastic rigidity with or without contracting limbs, board‐like rigidity of abdominal wall, salivation, tongue biting | 10 |
| Philippines (1948–1982) | Filipinos in Manila |
25–44 y old SCD during sleep or nap Postmortem autopsy (95% done) negative |
772 | 10.8–26.3 | 33 | 30–34 | 24/1 | Dec–Jan | NA | NA | 11 |
| Philippines (2003) | Filipinos |
20–39 y old No postmortem autopsy |
8 | 43 | 29.75 | NA | 3/1 | NA | NA | No moaning | 12 |
| China (2001–2013) | Han population of China |
15–55 y old SCD during nocturnal sleep Postmortem autopsy negative |
1854 | 1 | 32 | 30 | 13.8/1 | Apr–Jun | Blue‐collar worker (97.7%) | Sudden tachypnea, strange groan or gasping, scream, abnormal snoring, sudden cough, abrupt tic of limbs, abrupt discomfort, white froth through mouth, feeling cold before retiring | 3, 13 |
NA indicates not available; Ref, references; SCD, sudden cardiac death; SUNDS, sudden unexplained nocturnal death syndrome.
Inci: incidence: 1/100 000.
Sex ratio: male/female.
The year SUNDS cases were collected from.
Postmortem autopsies were negative: no lethal acute coronary thrombosis, myocardial infarction, or myocarditis accounting for death.
Lai Tai in Thailand
The word “Lai” in Laotian means a loud groan occurring during sleep or a loud noise made while frightened and “Tai” is a Thai word meaning death. The term “Lai Tai” appeared at least 50 years earlier before it attracted public interests in Thailand because of the report of SUDs occurring among Thai workers in Singapore in 1990.14 Since then, several national surveys on the incidence of SUD during sleep (Lai Tai) were conducted. The results obtained from these retrospective studies were consistent with an incidence of 20.8/100 000 per year in Thailand and the highest incidence of 25.9 to 38/100 000 in the northeast area.4, 10, 15 However, compared with Lai Tai among Thai in Thailand, Thai migrant workers in Singapore had a higher incidence of 78/100 000.9 Several characteristics of Lai Tai can be drawn from the epidemiological survey: (1) The mean age of Lai Tai was 38 years old and the peak risk was 45 to 49 years; (2) Most occupations bore intense heavy labor such as farmers or laborers but earned low income; (3) There were more Lai Tai cases in the hot season than any other seasons; (4) Lai Tai appeared more frequently in SUDS victims' family members than in controls; (5) Some witnessed cases had respiratory signs, motor movements, or salivation a few minutes before death10 (Table 1).
Bangungut in the Philippines
Bangungut, a Tagalog word meaning “to rise and moan in sleep,” had long been well known to Filipinos before its first description in the literature in 1917.7 Despite the fact that the first report of SUNDS was in Filipinos, there was not a detailed epidemiological report on the incidence of SUNDS among Filipinos until 1998.11 In the report, Munger et al reviewed the autopsy records filed in Manila during 1948 to 1982 and obtained SUNDS rates varying from 10.8 to 26.3 per 100 000 person‐years. The rates showed little discrepancy with another national survey conducted in 2003 using a questionnaire.12 The similarities of these SUNDS cases were the following: (1) Ninety‐six percent of cases were males with an average age of 33 years; (2) The SUNDS cases were seasonal with a peak in December to January; (3) The mode time of death was 3:00 am (Table 1).
Pokkuri Death Syndrome in Japan
“Pokkuri” means suddenly and unexpectedly in Japanese, and Pokkuri Death Syndrome (PDS) refers to an unexplained type of SCD in Japan with unknown cause. Various epidemiological surveys showed that sudden death cases in Japan are about 100 000 annually, of which 60% to 70% are caused by cardiovascular causes. However, of the total number of sudden cardiovascular disease deaths, ≈10% were categorized as PDS.2, 6, 16 Although PDS has been studied for at least half a century in Japan, there still has not been a literature revealing the epidemiological characteristics of PDS in detail.
SUNDS in China
We conducted the first epidemiological survey of SUNDS in China in 2002 on peasant‐derived workers in Dongguan city.17 Since then, we have been collecting SUNDS cases and making a series of epidemiological reports on SUNDS.3, 13, 18, 19 Based on autopsy data collected in the city of Dongguan, and the Bao'an and Longgang Districts of the city of Shenzhen from 2001 to 2006, an incidence of 1/100 000 was obtained for SUNDS3 and confirmed by our following study based on data collected from 2007 to 2013.13 These surveys demonstrate similar characteristics of SUNDS: (1) The highest‐risk age was 30 years old, and >80% of victims were 21 to 40 years old; (2) More than 97% were blue‐collar workers (poorly educated workers engaged in manual labor, such as construction workers); (3) The monthly distribution of SUNDS was positively correlated with monthly emergency fever cases with peak rates in April to June; (4) Family history of SUNDS was rarely observed; (5) Seventy‐five percent of witnessed cases occurred with respiratory signs, abrupt tic, and other manifestations before death; (6) Seventy percent of witnessed cases died between 0:00 and 4:00 am 3, 13 (Table 1).
The incidence of SUNDS in Southeast Asia shows ethnic disparities, with the highest in Thai‐Lao. Some of these differences may be explained by their diverse inclusion criteria: a few deaths were not restricted to nocturnal sleep; not all postmortem autopsy results were available in some surveys with a higher incidence of SUNDS. Han Chinese have lower rates of SUNDS, likely because of our stricter inclusion criteria. Surprisingly, the occurrence of SUNDS is more frequent in refugees and immigrants when compared with the same ethnicities in local places.5, 8, 9, 10 Furthermore, most deaths occurred within the first 2 years after their immigration to the United States and declined rapidly after the peak mortality rates.8 Most victims were men in their 30s and served as the primary income earner in their families by engaging in heavy physical labors but earning low salaries. These are all indications that maladjustment to a new environment, separation from family, overwork, low income, and other social psychology and socioeconomic problems may be potential risk factors underlying SUNDS. From monthly distribution of SUNDS and symptoms before death, we can see that fever and respiratory, metabolic, and neuropathic or psychogenic abnormalities in nocturnal sleep may be involved in triggering SUNDS.
Definition and Pathological Studies of SUNDS
As the name suggests, SUNDS, as an exclusion diagnosis, is made when a postmortem examination cannot determine the lethal pathological changes to explain the cause of death. Over the past 100 years, the definition and pathological changes of SUNDS have varied in different countries and periods.
Initially, the diagnosis of SUNDS depended on clinical history and interview from relatives or witnesses of victims even without a gross autopsy,4, 10 which might overestimate the incidence. Indeed, a following review of autopsy reports of SCD in Manila showed that only 48% remain unexplained after a gross autopsy and more than half the deaths are caused by myocardial infarction and other ill‐defined heart diseases,11 further implying that the high incidence of SUNDS obtained from interview might be overrated.
In the 1980s, gross autopsy became a routine examination for SUD in the United States, and deaths with no lethal acute coronary thrombosis, myocardial infarction, or myocarditis were diagnosed as SUNDS. To accurately define the cardiac abnormalities underlying SUNDS, 18 hearts collected from these SUNDS cases were reevaluated by Kirschner et al.20 They found that 17/18 had cardiac conduction system (CCS) anomalies and 14 hearts had persistent fetal dispersion of the atrial ventricular node within the central fibrous body of the atrial ventricular septum under microscopic examination. As the CCS anomalies might provide the anatomic substrate for potentially lethal arrhythmia, this could explain some of the deaths. Cardiomegaly was also observed in most cases: 14/18 showed slight to significant ventricular hypertrophy with either enlargement or dilatation of the cardiac chambers.20 Although these findings implied that histological examination of CCS is essential for suspected SUNDS, the role of CCS anomalies in SUNDS victims still remained unclear because of the small sample of 18 hearts.
In the 1990s, Song et al from our department presented an accurate and simple method based on >1000 cases examining the CCS,21, 22 which aroused great interest in forensic medicine, both domestically and internationally. Moreover, a few SUDs could be explained by the lethal CCS anomalies observed with this method.23 Since then, the examination of CCS has been incorporated in our routine postmortem examinations. When fatal CCS abnormality was observed, we adopted a definite diagnosis, instead of SUNDS, as the cause of death. Despite the fact that sudden arrhythmia death may result from some cardiac diseases,24 our team considered SUNDS only when all unnatural deaths (including suicide, homicide, and accidental death) and natural deaths caused by other diseases (such as coronary artery disease, myocardial infarction, myocarditis, and fatal CCS abnormalities) were excluded. Based on this strict criterion, we collected 148 consecutive SUNDS cases for 17 years to investigate the pathological differences between SUNDS and controls. Compared with controls, SUNDS cases were prone to slightly increased heart weight and enlarged heart size (increased circumferences of all cardiac valves, especially mitral valves). Despite these variations within the normal range, slight but significant differences existed between the 2 groups.25 Coincidentally, Steinhaus et al found that mean cardiac mass was linearly associated with risk for sudden arrhythmia death in cases without significant coronary artery disease,24 further implying the correlation between SUD and a larger or heavier heart.
SUNDS diagnosis demands stringent review of the circumstances of death, the clinical history, gross autopsy, histological examination, and toxicological screening to exclude probable known diseases or unnatural deaths. Despite no obvious or lethal pathological changes, a slight but significantly larger and heavier heart may exist in SUNDS, hinting a new direction for pathogenetic research.
Pathogenetic Hypotheses About SUNDS
Over the past century, the pathogenesis of SUNDS has been extensively explored, mainly focusing on physiological abnormalities associated with electrical instability, respiratory problems, sleeping disorders, and metabolic and endocrine disturbance.
Primary Arrhythmia
In 1984, Otto et al observed recurrent ventricular fibrillation in 3 young, male, Southeast Asian immigrants who were resuscitated from ventricular fibrillation. Even though clinical evaluation revealed no significant coronary or structural cardiac disease, 1 patient who died suddenly after 4 months was suspected as SUNDS and they proposed that ventricular fibrillation appeared to be the mechanism of SUNDS.26 Many following surveys were conducted to explore arrhythmia‐associated environmental risk factors, such as potassium deficiency and excessive carbohydrate eating habit.27, 28, 29 In 1997, an abnormal ECG pattern with ST‐segment elevation in the right precordial leads (V1–V3) was observed in patients rescued from ventricular fibrillation or relatives of those who succumbed to SUNDS,30 resembling ECG in Brugada Syndrome (BrS) patients who were also predisposed to SCD caused by ventricular arrhythmia (VA) at rest or during sleep.31 Indeed, VA can explain the sudden demise and no obvious pathological abnormalities in the hearts of SUNDS cases. In the following decades, investigators have been engaged in revealing the relationship between SUNDS and BrS, but there still has not been a definitive conclusion.25, 32 Nevertheless, it is commonly recognized that VA is a major cause of SUNDS.32, 33 The following risk factors presumed to be involved in SUNDS may also trigger VA.
Potassium Deficiency
Hypokalemia causes not only VA, but also muscle weakness and paralysis. Coincidentally, Nimmannit et al found that SUND and hypokalemic paralysis were prevalent in the same population and the same geographical area. In addition, hypokalemic paralysis usually occurred in the middle of the night or in the early hours of the morning, which was consistent with SUNDS. Considering the risk factors of SUNDS, they postulated that eating excessive carbohydrate or other physical and mental stress stimulated the shift of potassium into cells at night, thus resulting in a disturbance in potassium homoeostasis, which contributed to sudden VA death.27, 28, 29
Hypoventilation, Hypoxemia, and Acidosis
From the above epidemiological studies, we can see that hypoxemia, caused by these respiratory signs (tachypnea, groan, gasping, and snoring), often appears before death. Significant nocturnal hypoxia was more common in cases with a previous history of near‐SUNDS or a familial history of SUNDS, implying that nocturnal hypoxia might be the primary abnormality in SUNDS.34 The high upper airways resistance and hypoventilation during rapid eye movement sleep can cause hypoxemia, which may explain why SUNDS occurs nocturnally.35 We reviewed the literatures associated with obstructive sleep apnea hypopnea syndrome and arrhythmia or SCD, and found that obstructive sleep apnea hypopnea syndrome increased risk of VA by a complex mechanism (involved in hypoxemia, oxidative stress, dysfunction of autonomic nervous system, secretion of vasoactive substances, and decrease of intrathoracic pressure).36
Acidosis often results from hypoventilation, hypoxemia, and chronic potassium deficiency,14, 29 suggesting that intracellular and extracellular acidosis may play an important role in SUNDS. Moreover, the PH of arterial blood in sudden infant death syndrome cases was reported below PH 7.0.37 Using the whole‐cell patch clamp method, we proved that intracellular and extracellular acidosis significantly decreases wild or mutant sodium currents in vitro co‐expression systems.38, 39
Night Terrors
Night terrors are a sleep disorder that is characterized by vocalization, clonic movements, an unarousable state, and severe autonomic discharge.40 These manifestations can also be observed in SUNDS cases before their deaths. The incidence of SUNDS among Southeast Asians refugees in the United States decreased from 59/100 000 in 1981 to 1/100 000 in 1987.8 Night terrors might be a reasonable explanation for the sharp decline of SUNDS rate: Southeast Asian refugees at 1.5 years after migration had high levels of depression, anxiety, and hostility, which was similar to sufferers of adult night terrors. At 3.5 years after migration, all of these symptoms improved, and so did the occurrence of SUNDS.40 Victims discovered in the night terrors are unarousable, and in the few successfully aroused patients, terrifying dreams were often experienced.40 In addition, frequent experiences of “dab tsog (frightening night spirit pressing on chest),” nightmares, sleep paralysis, and hypnogogic hallucinations still exist in Hmong after immigrating to the United States for decades, probably putting Hmong at high risk for SUNDS.41 The hypothesized mechanism is that night terrors cause sympathetic discharge, resulting in increased cardiac vulnerability to VA in patients with cardiac conduction defects.40
Remnant Lipoproteins and Coronary Artery Vasospasm
Nakajima et al observed elevated plasma remnant lipoprotein levels and narrowed circumferences of coronary arteries in PDS cases.2 They proposed the hypothesis that elevated plasma remnant lipoprotein levels might impair endothelium‐dependent vasorelaxation in narrowed coronary arteries as an early event, and activated LOX‐1 receptor and Rho‐kinase pathway in smooth muscle cells then cause coronary artery vasospasm and cardiac arrest. Coronary artery vasospasm could result in multiple necroses of cardiomyocytes, with potassium released from necrotic sites causing VA.42
Others risk factors, such as excessive vanadium intake, inhibition of Na, K‐ATPase and H, K‐ATPase activity,43 rapid eye movement bursts,14, 44 plasma testosterone,45 epilepsy,46, 47 diabetes mellitus,48 melioidosis bacteria,49 toxic shock,50 etc, may also be involved in the pathogenesis of SUNDS. In general, SUNDS specifically happens during nocturnal sleep, suggesting that the unique Hmong sleep disorder profile of a high prevalence of sleep apnea, sleep paralysis, nightmares, and other rapid eye movement–related sleep abnormalities41 plays an important role in SUNDS attacks.
Molecular Genetics of SUNDS
A most striking characteristic in the epidemiology of SUNDS is that it mainly attacks the Southeast Asian population, suggesting a hereditary susceptibility in Southeast Asians. Molecular genetics studies of SUNDS began with clues from BrS.32, 33, 51 Since the first literature discussing the correlation between SUNDS and BrS was published in 2002,32 SUNDS research entered the stage of molecular genetics.
In this review, we summarize the molecular genetic studies of SUNDS. As most of the variants have not been performed with functional experiments, we evaluate the pathogenicity of variations by the predictive programs. All the variants detected in SUNDS cases are categorized as 3 major types: the novel mutations absent in all public databases; the rare variants with a minor allele frequency (MAF) ≤0.01 in the homologous population; and common single nucleotide polymorphisms (SNPs) with a MAF >0.01. When the common SNPs show significant difference in allele or genotype frequency between SUNDS and controls (homologous population), we define the SNPs as risk factors for SUNDS.
Sodium Channel Mutations
Cardiac sodium channel comprises a pore‐forming ion‐conducting α‐subunit Nav1.5 encoded by SCN5A gene and ancillary β‐subunits encoded by SCN1B‐SCN4B.52 Nav1.5 consists of a cytoplasmic N terminus, 4 structurally homologous domains (DI–DIV), and a cytoplasmic C terminus (Figure 2A). Each domain consists of 6 transmembrane α‐helical segments (S1–S6). Mutations of sodium channel resulting in decreased sodium current (INa) and increased or prolonged late sodium current (INaL) have been proven to be associated with BrS and long QT syndrome type 3, respectively.53, 54 Similar to SUNDS, both BrS and long QT syndrome type 3 tend to experience cardiac events at rest or during sleep. Therefore, altered sodium current caused by mutation on SCN5A or SCN1B‐SCN4B might explain some SUNDS.
Figure 2.
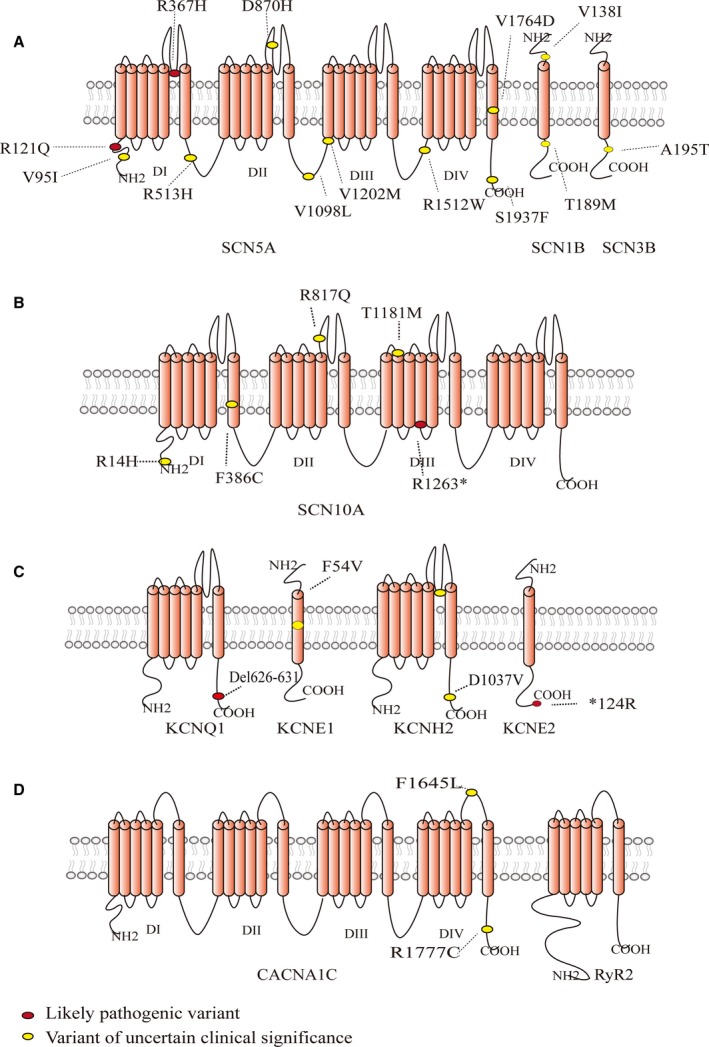
Predicted protein topology of ion channels and the location of variations responsible for SUNDS. A, Cardiac sodium channel comprises a pore‐forming ion‐conducting α‐subunit Nav1.5 and ancillary β‐subunits. B through D, Predicted protein topology of Nav1.8 (SCN10A), Kv11.1 (KCNH2), Kv7.1 (KCNQ1), MinK (KCNE1), MiRP1 (KCNE2), CaV1.2 (CACNA1C), and RyR2. Pathogenicity is determined by ACMG guideline. ACMG indicates American College of Medical Genetics; SUNDS, sudden unexplained nocturnal death syndrome.
Vatta et al first revealed the genetic basis of SUNDS by screening patients with family history of sudden death for mutations in SCN5A in 2002. Three of 10 families had identified SCN5A mutations, namely, R367H, A735V, and R1192Q (now termed R1193Q). Biophysical analysis in Xenopus oocytes showed that R367H mutation, located in the P loop between DIS5 and DIS6, did not express any inward current and the peak INa was thought to be decreased because of the loss of 1 functional allele. The second mutation, A735V, located in DIIS1, shifted steady‐state activation towards more positive potentials. The third mutation, R1193Q, located in DIII, accelerated the decay of INa. Both A735V and R1193Q, also observed in BrS, could cause reduced INa. On the basis of these characteristics, they proposed that SUNDS and BrS were phenotypically, genetically, and functionally the same disorder.32 Nevertheless, patients enrolled in this study were actually more likely BrS rather than SUNDS, because they were relatives of sudden death victims, and were chosen with an ECG pattern of ST‐segment elevation in leads V1–V3. Soon after, van den Berg et al reported another SCN5A mutation (1795insD) in a large family characterized by nocturnal SCD. ECG showed combined features of long QT syndrome type 3, BrS, and familial CCS disease, often in the same individuals.33 These 2 studies were based on familial SUNDS cases. However, some patients involved in the study might not actually be SUNDS cases because only rare people who succumb to SUNDS have a family history. Their conclusions, then, inevitably present limitations in explaining the mechanism of SUNDS and warrant further investigation into mutations in sporadic cases. We conducted a study with 123 sporadic SUNDS cases to screen SCN5A in the Han population of China. A total of 7 unique putative pathogenic mutations in 8/123 and 3 rare variants of uncertain clinical significance in 3/123 cases were identified on SCN5A. Of the 7 unique putative pathogenic mutations, 2 (V95I and R121Q) localized to the intracellular N‐terminus, 3 to the Nav1.5 channel pore region (R367H, DIS5‐DIS6; D870H, DIIS5‐DIIS6; and V1764D, DIVS6), 1 within the DI‐DII linker (R513H), and 1 (S1937F) in the C‐terminus of SCN5A. In the 3 rare nonsynonymous variants (V1098L, V1202M, and R1512W), R1512W was previously reported in BrS.55, 56 We also tested SCN1B‐SCN4B and another 2 Nav1.5 regulatory proteins encoding gene, namely, MOG1 and GPD1‐L. However, no putative pathogenic mutations were found in SCN1B‐SCN4B, MOG1, or GPD1‐L, except for 2 rare nonsynonymous variants (V138I and T189M) on SCN1B and 1 (A195T) on SCN3B. Interestingly, V95I‐SCN5A and T189M‐SCN1B were detected in the same individual. Two SNPs (1141‐3C>A and C3269>T) of SCN5A showed significant differences in genotype or allele frequencies between SUNDS and controls.57 However, H558R and R1193Q, which were previously reported to be associated with BrS and SUNDS, did not have significant difference in this cohort.32, 58 We further conducted a target‐captured next‐generation sequencing‐based (NGS) study on 44 consecutive individuals who had SUNDS to study 80 channelopathy‐ and cardiomyopathy‐related genes, and a variant (V1340I) on SCN5A was detected in 1 case.25
Besides Nav1.5, another tetrodotoxin‐sensitive Na+ channel, Nav1.8, encoded by SCN10A, was also reported to be associated with BrS.59 SCN10A, located adjacent to SCN5A, also has a structure and amino acid sequences similar to Nav1.5 (Figure 2B).60 We detected 6 rare variations and 16 common SNPs on SCN10A in 105 SUNDS cases. Of the 6 rare variations, only 1 nonsense variant (R1263*) was novel. As no pathogenic rare variants in 80 arrhythmia‐associated genes were found in the SCN10A variation carriers, R1263* was thought to be the putative pathogenic mutation. Two rare missense variants (R14H and F386C) were predicted as likely pathogenic, while the remaining 3 (R817Q, T1181M, and P1683S) were benign. As for the 16 common SNPs, only c.2884A>G (I962V) had a statistically significant difference in allele frequency, with an odds ratio of 1.551for G allele, between SUNDS and the controls.61
Potassium Channel Mutations
The voltage‐gated potassium channel (Kv) consists of 6 transmembrane regions, and the α subunit generally interacts with accessory β subunits to recapitulate the biophysical properties of K+ currents (Figure 2C). Mutations in 2 different potassium channels, namely, Kv7.1 encoded by KCNQ1 and Kv11.1 encoded by KCNH2, caused >60% Long QT Syndrome (LQTS). To form functional Kv11.1 and Kv7.1 channels, 1 accessory subunit called MinK encoded by KCNE1 and another called MiRP1 encoded by KCNE2 are necessary.53, 62 Since LQTS can cause SCD, mutations in KCNQ1, KCNH2, and their accessory KCNE1, KCNE2 may explain some SUNDS. A genotyping result of the 4 LQTS‐susceptibility genes was obtained by us, in which 14 rare genetic variants were identified but only 2 mutations were nonsynonymous: F54V in the KCNE1 gene, and a terminator codon mutation *124R of KCNE2 gene. Four nonsynonymous, not statistically significant SNPs were also identified.63 In another cohort of 44 SUNDS, we detected 4 variants in potassium channels, namely, G626_P631del and a splice site 1128+5G>A in KCNQ1, D1037V in KCNH2, and N179S in KCNJ5.25 So, to summarize, potassium channel disorders might be one possible, but not closely related, pathogenic cause of SUNDS.
Calcium Channel Mutations
CACNA1C‐encoded L‐type voltage‐gated calcium channels (LTCC) CaV1.2 share structural homology with voltage‐gated sodium channel Nav1.5 (Figure 2D), and also interacts with auxiliary subunits including CACNB2 encoded β2b subunits, which promotes trafficking of CaV1.2 to the sarcolemmal membrane. Gain and loss of function mutations in CaV1.2 are associated with cardiac arrhythmia and SCD.64, 65 In the 44 SUNDS cohort, we identified 3 people who carried CaV1.2 or its auxiliary subunits' rare nonsynonymous variants. One individual merely carried a mutation of F1465L on CACNA1C, which was deleterious by Condel prediction and absent in all public databases, implying that the mutation was responsible for his death. Another person had R1777C on CACNA1C as well as V1098L on SCN5A, both of which were predicted to be neutral by Condel. The last had 3 rare nonsynonymous variants on 3 gene loci, namely, R452C on CACNB2, R1502W on MYH6, and R293C on TNNT2.25 Thus, LTCC may play a role in SUNDS, and compound variations are potential mechanisms underlying SUNDS.
Mutations in the RyR2 gene were reported to be associated with catecholaminergic polymorphic ventricular tachycardia and arrhythmogenic right ventricular cardiomyopathy,66, 67, 68 implying that the RyR2 gene may be a susceptible gene for SUNDS. We screened 29 of the 105 coding exons on the RyR2 gene previously reported to be associated with catecholaminergic polymorphic ventricular tachycardia and arrhythmogenic right ventricular cardiomyopathy in 127 sporadic Chinese SUNDS cases. Only 2 novel synonymous but no nonsynonymous variants were identified.69 These results suggested that RyR2 may not be a key susceptible gene for Chinese SUNDS.
Mutations in the Connexome
The rapid depolarizing electrical impulse propagation is dependent on a highly specialized structure called intercalated disc (ID). At the ID, fascia adherens junctions and desmosomes participate in mechanical connection, while gap junctions and sodium‐channel complexes are involved in electrical coupling.70 Emerging evidence shows that each component might act together as a single structural and functional entity, known as the connexome. This concept has become essential to explain the overlapping of 2 inherited diseases based on disturbance of the ID: arrhythmogenic cardiomyopathy (ACM) and BrS.71 ACM is considered to be a cardiac structural disease with monomorphic arrhythmias, whereas BrS is considered to be a typical electrical disorder without (or with only mild) structural abnormalities. Recently numerous studies have shown that the 2 diseases not only have some common manifestations (ACM has typical BrS‐ECG, BrS has structural defects), but also share common mutations in proteins present at the ID.72 The desmosomal genes account for 88% of the pathogenic mutations described in ACM patients and some desmosomal components, such as desmoglein‐2 and plakophilin‐2, have been proven to interact with Nav1.5.73, 74 Mutations in the desmosomal genes have been found in BrS patients and affect Nav1.5 function directly. In addition, mutations in SCN5A have been related to ACM.75 Since SUNDS was thought to be the same allelic disorder as BrS, all these imply that mutations in proteins involved in the connexome may be the genetic causes of some SUNDS cases.
Plakophilin‐2
Plakophilin‐2 (PKP2), as an armadillo‐repeat protein, contributes to the protein anchor of desmosomal cadherins.76, 77 Disease‐causing mutations in PKP2 have been detected in nearly 40% of ACM patients.72 In 2016, we first reported the genetic phenotype of PKP2 gene in Chinese SUNDS.78 Three novel mutations and 9 reported SNPs were identified. Of the 3 mutations, c.600G>A (p.V200V) was a synonymous mutation, while the other 2—c.475G>A (p.A159T) and c.794G>A (p.G265E) —together with the variation c.2169A>G (p.T723T), were identified in the same case. Although silico prediction showed that A159T or G265E alone did not damage the function of PKP2, the effect of the compound mutation is still unknown.
Desmoplakin
Desmoplakin (DSP), also one of the desmosomal components, connects the desmosomal complex to intermediate filaments.76 Disease‐causing mutations in DSP have been detected in 10% to 15% of ACM patients.72 Seven DSP gene rare missense variants were identified in 8 of 40 SUNDS cases: 2 (A373>T, A1562>C) were absent in dbSNP and the database of Beijing Genomics Institute; 1 (G7735>C) was absent in dbSNP but present in the database of Beijing Genomics Institute with an MAF of 0.0004; 2 variants (Q90R and R2639Q) were previously proved to be pathogenic for cardiomyopathy patients; the remaining 2 variants (E1357D and R1308Q) both located on exon 23, and E1357D were predicted as pathogenic in silico, while R1308Q was benign.79, 80, 81 All of the 7 variants seemed to be responsible for SUNDS. Thus, DSP might have a strong association with SUNDS.
Connexin 43
Connexin 43 (Cx43), encoded by the GJA1 gene, is the most abundant connexin present in gap junctions of cardiomyocytes.82 GJA1 mutations have been proved to be pathogenic for ACM.83 We performed a gene screening on GJA1 in 124 sporadic SUNDS cases to obtain 1 novel homozygous variant, c.169C>T (Q57*), and 1 heterozygous synonymous variant, c.624C>T (I208I), respectively, in 2 cases. Q57* led to termination of Cx43 protein production on the first conserved extracellular loop, which was thought to be the cause of this SUNDS case. The variant I208I had a significantly lower MAF in the East Asian population than in SUNDS, suggesting that allele T of this SNP might be a SUNDS risk allele.84
Vinculin
Vinculin (VCL) is a membrane‐cytoskeletal protein that connects integrin adhesion molecules to the actin cytoskeleton to form adhesion junctions at ID.85 Several mutations in VCL have been identified to be pathogenic for dilated cardiomyopathy or hypertrophic cardiomyopathy.86 We identified a rare variant M94I in 1/44 SUNDS cases39 and an East Asian common variant D841H in 8/120 SUNDS cases.87 Co‐expression of Nav1.5 with M94I‐VCL showed a reduction in peak INa and a further reduction in acid conditions. VCL was proved to directly interact and co‐localize with Nav1.5 at the IDs. Moreover, the variation M94I did not change the interaction or co‐localization. The rare variation M94I might cause this death through reducing INa in acidotic conditions by altering Nav1.5 electrical properties but not expression levels.39 The H841 allele was more frequent in SUNDS and gave an odds ratio of 5.226 compared with controls. Similar to M94I, D841H also reduced peak INa without changing the interaction or co‐localization.87
Other Genes Associated With SUNDS
Nitric oxide synthase 1 adaptor protein (NOS1AP), a regulator of neuronal nitric oxide synthase, interacts with neuronal nitric oxide synthase or ion channels (especially potassium channels). Several common SNPs on the NOS1AP gene had been reported to be associated with LQTS.88 Among these SNPs, 5 (rs10918594, rs12143842, rs16847548, rs12567209, and rs10494366) were chosen to detect the correlation between SUNDS and NOS1AP in 123 sporadic Chinese SUNDS cases.89 Of the 5 SNPs, only rs12567209 had significant difference between SUNDS and controls in allele but not genotype frequency. These indicated that the 5 common NOS1AP SNPs had only limited effect in SUNDS, further implying that defects of potassium channels or LQTS might not have strong associations with SUNDS.
Leucine‐rich repeat containing 10 (LRRC10) is a cardiac‐specific and highly conserved protein that is crucial for proper cardiac development, and knockout of LRRC10 results in dilated cardiomyopathy in mice.90 As for SUNDS, 3 LRRC10 genetic variants were detected in 4/113 sporadic SUNDS cases including 1 noncoding region mutation (c.‐2G>T), 1 missense mutation (c.385G>A, p.E129K), and 1 missense rare variant (c.206C>T, p.P69L). Of the 3 variants, c.‐2G>T was absent in any Asian population, suggesting it might be a risk factor for SUNDS. Since P69 and E129 are evolutionarily conserved among different species and no pathogenic rare variants of 80 candidate genes were detected in the 2 carriers, P69L and E129K might be the genetic causes of the 2 SUNDS.91 In addition, P69L was also detected in 2/10 BrS, further indicating likely pathogenic potential.91
From the results of the NGS of 80 genes conducted in 44 consecutive SUNDS cases, 22 of 44 people hosted at least 1 rare nonsynonymous variant and 7 of 44 hosted ultrarare variants. Compound rare nonsynonymous variants were obtained in 12 of 22 people in whom at least 2 rare nonsynonymous variants were simultaneously detected.25 It is commonly accepted that compound heterozygotes may confer a worse prognosis in family members of LQTS, BrS, and those affected by sick sinus syndrome.92, 93, 94 Common polymorphism H558R was found to have higher frequency in BrS, but no differences in sporadic SUNDS cohort and controls.57 However, electrical study showed that SCN5A‐H558R not only reversed the decrease of peak INa amplitude caused by VCL‐D841H, but also significantly increased the late INa for VCL‐D841H in HEK293 cells. In addition, SCN5A‐H558R and VCL‐D841H were detected in 1 SUNDS case.87 These suggest that compound variants might play a critical role in SUNDS.
Through these genetic studies conducted on SUNDS, we have identified a spectrum of nearly 100 rare variants on 33 genes, including genes associated with channelopathies and cardiomyopathies, to be potentially associated with SUNDS (Table 2). Although we mainly focused on rare variants of potential clinical diagnostic utility, a total of 5 common SNPs have also been identified as risk factors for SUNDS (Table S1), implying that common SNPs also participate in pathogenesis of SUNDS. Currently, the main challenge in molecular genetics is the clinical interpretation of the genetic variants detected in SUNDS. Therefore, molecular autopsy is recommended as a routine examination for SUNDS to collect sufficient evidence.
Table 2.
Spectrum of Genes Identified in SUNDS
| Gene | Locus | Protein | Current | Associated Disease |
|---|---|---|---|---|
| Na+ channels | ||||
| SCN5A | 3p22.2 | Sodium voltage‐gated channel α‐subunit 5 (Nav1.5) | INa | BrS |
| SCN1B | 19q13.11 | Sodium voltage‐gated channel β‐subunit 1 | INa | BrS |
| SCN3B | 11q24.1 | Sodium voltage‐gated channel β‐subunit 3 | INa | BrS |
| SCN10A | 3p22.2 | Sodium voltage‐gated channel α‐subunit 10 (Nav1.8) | INa | BrS |
| K+ channels | ||||
| KCNQ1 | 11p15.4‐15.5 | Potassium voltage‐gated channel subfamily Q member 1 (Kv7.1) | IKs | LQTS |
| KCNH2 | 7q36.1 | Potassium voltage‐gated channel subfamily H member 2 (Kv11.1) | IKr | LQTS |
| KCNE1 | 21q22.12 | Potassium voltage‐gated channel subfamily E regulatory subunit 1 (mink) | IKs | LQTS |
| KCNE2 | 21q22.11 | Potassium voltage‐gated channel subfamily E regulatory subunit 1 (MiRP1) | IKr | LQTS |
| KCNJ5 | 11q24.3 | Potassium voltage‐gated channel subfamily J member 5 (Kir3.4) | IKACh | LQTS |
| Ca2+ channels | ||||
| CACNA1C | 12p13.33 | Calcium voltage‐gated channel subunit α1 C (CaV1.2) | ICa,L | LQTS |
| CACNB2 | 10p12.31‐12.33 | Calcium voltage‐gated channel auxiliary subunit β‐2 | ICa,L | LQTS |
| RyR2 | 1q43 | Ryanodine receptor 2 | Ca2+ release | CPVT, ARVC |
| Connexome | ||||
| PKP2 | 12p11.21 | Plakophilin 2 | INa | ARVC |
| DSP | 6p24.3 | Desmoplakin | NA | ARVC |
| GJA1 | 6q22.31 | Gap junction protein α 1/Connexin 43 | NA | HLHS |
| VCL | 10q22.2 | Vinculin | INa | DCM, HCM |
| Other genes | ||||
| LRRC10 | 12q15 | Leucine‐rich repeat containing 10 | NA | DCM |
| ABCC9 | 12p12.1 | ATP binding cassette subfamily C member 9 | NA | AF |
| ACTN2 | 1q43 | α2‐Actinin | INa | DCM |
| AKAP9 | 7q21.2 | A‐kinase anchoring protein 9 | NA | LQTS |
| ANKRD | 10q23.31 | Ankyrin repeat domain | NA | DCM |
| DMPK | 19q13.32 | DM1 protein kinase | NA | LVNC |
| EYA4 | 6q23.2 | EYA transcriptional coactivator and phosphatase 4 | NA | DCM |
| GATA4 | 8p23.1 | GATA binding protein 4 | NA | AF |
| JUP | 17q21.2 | Junction plakoglobin | NA | ARVC |
| LDB3 | 10q23.2 | LIM domain‐binding protein 3 | NA | DCM, LVNC |
| LMNA | 1q22 | Lamin A/C | NA | DCM |
| MYBPC3 | 11p11.2 | Myosin‐binding protein C, cardiac‐type | NA | HCM, DCM, LVNC |
| MYH6 | 14q11.2 | Myosin 6 | NA | DCM, HCM |
| MYH7 | 14q11.2 | Myosin heavy chain 7 | NA | DCM, HCM, RCM |
| NUP155 | 5p13.2 | Nucleoporin 155 | NA | AF |
| SGCD | 5q33.2‐33.3 | Sarcoglycan delta | NA | DCM |
| TNNT2 | 1q32.1 | Troponin T2, cardiac type | NA | DCM |
AF indicates atrial fibrillation; ARVC, arrhythmogenic right ventricular cardiomyopathy; BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; HLHS, hypoplastic left heart syndrome; LQTS, long QT syndrome; LVNC, left ventricular noncompaction; NA, not available; SUNDS, sudden unexplained nocturnal death syndrome.
Interpreting the Molecular Genetic Evidence
The studies have adopted various approaches and MAFs in different control groups to filtrate for rare variants, thus potentially resulting in different diagnostic yields and limiting comparability. First, early studies used Sanger sequencing of single genes and MAF in a local control group, while later studies adopted NGS with large panels of 80 genes and MAFs in many public databases. Second, some gene screenings are only conducted in a small cohort with NGS, whereas SCN5A, SCN1B, SCN3B, KCNQ1, KCNH2, KCNE1, KCNE2, and PKP2 are in 2 cohorts with Sanger sequencing or NGS. Finally, although nonsense mutations are more likely to be pathogenic, the majority in our studies are missense variants of uncertain significance without functional experiments or linkage studies. Therefore, we reanalyzed variants from all the studies of sporadic cases and filtered for nonsynonymous and splice site rare variants with a MAF <0.01 in the East Asian (4327 and 9345 individuals, respectively) in Exome Aggregation Consortium and Genome Aggregation Database (Table S2).
If we consider SUNDS as a Mendelian disorder, manual curation against the American College of Medical Genetics guidelines95 yields 6 likely pathogenic variants with an overall yield of 4% (Figure 2, Table S2). The low diagnostic yield indicates that Mendelian disorder might merely have limited significance in explaining sporadic SUNDS. Furthermore, most of these variants detected in SUNDS are not pathogenic according to the American College of Medical Genetics, while compound variants are detected in nearly 1/3 cases. Hence, we tend to think of SUNDS as a multigene disorder. In order to evaluate the clinical diagnostic utility of each gene, these variants were then aggregated per gene and compared with the same variants in the same genes in East Asian by χ2 tests (Table 3).
Table 3.
Yields of Gene Variants in SUNDS Versus EAS in ExAC or gnomAD
| Gene | Alleles/Total SUNDS Cases (%) | ExAC | gnomAD | ||
|---|---|---|---|---|---|
| Alleles/Total EAS Exomes (%) | P Valuea | Alleles/Total EAS Exomes (%) | P Value | ||
| Na+ channels | |||||
| SCN5A | 13/167 (7.78) | 19/4327 (0.43) | <0.001b | 45/9345 (0.48) | <0.001b |
| SCN1B | 5/167 (2.99) | 63/4327 (1.46) | 0.214 | 144/9345 (1.53) | 0.249 |
| SCN3B | 1/167 (0.60) | 3/4327 (0.07) | 0.141 | 5/9345 (0.05) | 0.101 |
| SCN10A | 5/105 (4.76) | 152/4327 (3.51) | 0.696 | 338/9345 (3.58) | 0.736 |
| K+ channels | |||||
| KCNQ1 | 2/152 (1.32) | 5/4327 (0.12) | 0.022b | 17/9345 (0.18) | 0.037b |
| KCNH2 | 3/152 (1.97) | 6/4327 (0.14) | 0.003b | 15/9345 (0.16) | 0.003b |
| KCNE1 | 1/149 (0.67) | 0/4327 (0) | 0.034b | 0/9345 (0.00) | 0.016b |
| KCNE2 | 1/148 (0.68) | 0/4327 (0) | 0.034b | 0/9345 (0.00) | 0.016b |
| KCNJ5 | 1/44 (2.27) | 2/4327 (0.05) | 0.031b | 4/9345 (0.04) | 0.024b |
| Ca2+ channels | |||||
| CACNA1C | 2/44 (4.55) | 5/4327 (0.12) | 0.002b | 17/9345 (0.18) | 0.004b |
| CACNB2 | 1/44 (2.27) | 0/4327 (0) | 0.010b | 2/9345 (0.02) | 0.014b |
| Connexome | |||||
| PKP2 | 2/144 (1.39) | 0/4327 (0) | 0.001b | 0/9345 (0.00) | <0.001b |
| DSP | 5/40 (12.50) | 84/4327 (1.94) | 0.002b | 197/9345 (2.09) | 0.002b |
| GJA1 | 2/124 (1.61) | 0/4327 (0) | 0.001b | 0/9345 (0.00) | <0.001b |
| VCL | 1/44 (2.72) | 2/4327 (0.05) | 0.031b | 5/9345 (0.05) | 0.028b |
| Other genes | |||||
| LRRC10 | 3/113 (2.65) | 18/4327 (0.42) | 0.016b | 31/9345 (0.33) | 0.008b |
| ABCC9 | 1/44 (2.27) | 10/4327 (0.23) | 0.107 | 27/9345 (0.29) | 0.126 |
| ACTN2 | 2/44 (4.55) | 4/4327 (0.09) | 0.002b | 12/9345 (0.13) | 0.002b |
| AKAP9 | 2/44 (4.55) | 89/4327 (2.06) | 0.241 | 195/9345 (2.07) | 0.244 |
| ANKRD | 1/44 (2.27) | 11/4327 (0.25) | 0.117 | 22/9345 (0.23) | 0.104 |
| DMPK | 1/44 (2.27) | 0/4327 (0) | 0.01b | 9/9345 (0.10) | 0.047b |
| EYA4 | 1/44 (2.27) | 0/4327 (0) | 0.01b | 0/9345 (0.00) | 0.005b |
| GATA4 | 2/44 (4.55) | 0/4327 (0) | <0.001b | 0/9345 (0.00) | <0.001b |
| JUP | 1/44 (2.27) | 19/4327 (0.44) | 0.187 | 34/9345 (0.36) | 0.155 |
| LDB3 | 2/44 (4.55) | 10/4327 (0.23) | 0.007b | 26/9345 (0.28) | 0.008b |
| LMNA | 1/44 (2.27) | 0/4327 (0) | 0.01b | 3/9345 (0.03) | 0.019b |
| MYBPC3 | 3/44 (6.82) | 8/4327 (0.18) | <0.001b | 19/9345 (0.20) | <0.001b |
| MYH6 | 3/44 (6.82) | 20/4327 (0.46) | 0.002b | 50/9345 (0.53) | 0.002b |
| MYH7 | 2/44 (4.55) | 6/4327 (0.14) | 0.003b | 10/9345 (0.11) | 0.001b |
| NUP155 | 1/44 (2.27) | 1/4327 (0.02) | 0.02b | 3/9345 (0.03) | 0.019b |
| SGCD | 1/44 (2.27) | 55/4327 (1.27) | 0.438 | 143/9345 (1.52) | 0.497 |
| TNNT2 | 1/44 (2.27) | 0/4327 (0) | 0.01b | 0/9345 (0.00) | 0.005b |
| Total | 107.11 | 13.68 | 14.37 | ||
EAS indicates East Asian; MAF, minor allele frequency; SUNDS, sudden unexplained nocturnal death syndrome.
Nonsynonymous and splice site variants with MAF <0.01 in all SUNDS cases were compared with EAS in ExAC or gnomAD by χ2 tests using the SPSS 20.0. Continuity‐adjusted χ2 test or Fisher exact text are also chosen according to expected frequencies of the cells. A 2‐sided P<0.05 was considered significant.
P<0.05.
A total of 24 genes show significant differences between SUNDS and East Asian yields, which can be candidate genes for future molecular diagnosis or risk stratification. Among these genes, DSP and SCN5A seem to be closely related with SUNDS with higher yields than others. Although DSP (with a yield of 12.50%) seems to be the foremost gene here, DSP gene screening is only conducted on 40 SUNDS cases, which may have significant sampling error. Nevertheless, 2 different cohorts of SUNDS have confirmed the importance of SCN5A in SUNDS, which accounts for nearly 8%. Moreover, mutations regulatory proteins of SCN5A are also responsible for SUNDS, indicating that the defects of NaV1.5 play the vital role of pathogenesis of SUNDS. Compared with sodium channels, defects in potassium and calcium channels seem to be less common in SUNDS because potentially pathogenic variations of potassium and calcium channels–associated genes are rarely detected in SUNDS. The total yields of variants in SUNDS cases was substantially higher than in East Asian (107.11% versus 13.68% or 14.37%), and the yield in SUNDS exceeds the value of 100% because of the compound variants in 1 case.
In summary, genetic studies support SUNDS as a multifactorial entity with involvement of channelopathies and cardiomyopathy‐associated genetic variants, but current evidences are far from sufficient to provide a definite molecular diagnosis of SUNDS.
SUNDS: Identical to BrS?
SUNDS and BrS, 2 diseases predisposed to SCD secondary to ventricular tachycardia in the absence of cardiac structural abnormality, share several common characteristics. First, both prefer to attack young healthy male individuals in nocturnal sleep, and are more prevalent in Southeast Asian than Western countries.96 Second, ECG of survived SUNDS is similar to that of BrS.30 Third, in a follow‐up study on BrS families, polymorphic ventricular tachycardia would occur after large meals, especially with foods rich in carbohydrates,97 which is consistent with SUNDS.27, 28, 29 Finally, fever may cause the appearance of BrS ECG pattern and trigger episodes of ventricular tachycardia in affected patients,98 which is not in conflict with SUNDS.3 SUNDS then appears to be the same disorder as BrS in epidemiology and phenotype. The initial molecular genetic study proposed that SUNDS was the same as BrS in genotype and function defects. However, patients enrolled in this study were more likely to be diagnosed as BrS. Thus, whether SUNDS is allelic to BrS in genetics and functional defects requires further evaluation.
In epidemiology, only rare SUNDS cases have family history, while nearly 40% of BrS is familial,99 implying that there are different degrees of genetic and environmental contribution to the 2 disorders. In pathology, SUNDS is prone to a larger heart size, especially increased circumference of mitral valves.25 However, BrS is thought to be caused by depolarization–repolarization defects in the right ventricular outflow tract, often with an enlarged circumference. In phenotype, the ECG patterns of SUNDS are usually collected from first relatives or VA survivors of people who died of SUNDS.30 This is incompatible with the fact that SUNDS rarely displays familial aggregation and SUNDS attacks healthy people during nocturnal sleep, which is difficult to discover and rescue in time. Considering that BrS can cause SCD in nocturnal sleep, we postulate that the families included in SUNDS studies are most likely BrS patients. In genetics, we identified a primary genetic spectrum of 33 genes associated with SUNDS. SUNDS has an obviously lower yield of SCN5A mutations with nearly 8%, while BrS is 30%.25 Moreover, the DSP gene has not been reported to be associated with BrS but has the highest yield of variation in SUNDS, further suggesting genetic discrepancy. SUNDS, then, is not identical to BrS in epidemiology, pathology, phenotype, and molecular genetics, but closely related with cardiomyopathy in genetics.
Our Hypothesis
Normal heart rhythm requires the finely orchestrated activity of several ion channels and the orderly propagation of electrical impulses throughout the cardiomyocytes. Disruption of either action potential or propagation can result in potentially lethal arrhythmia.100 Hence, mutations in ion channels or structure proteins can cause SCD. Additionally, emerging evidence reveals the overlap between ACM and BrS in genetic, molecular, and pathophysiological mechanisms.72 For example, a DSG2‐N271S transgenic mouse model showed reduced conduction velocity and increased arrhythmia susceptibility preceding the onset of necrosis and replacement fibrosis.74 Moreover, in cardiac‐myocyte‐specific VCL knock‐out mice, 2 stages of phenotype were observed: SCD caused by ventricular tachycardia attacked 49% mice younger than 3 months despite well‐preserved systolic cardiac function, while the surviving mice developed dilated cardiomyopathy and died of heart failure around 6 months of age.101 Similar effects were also observed in heterozygous JUP and truncated PKP2 mice, in which cardiac arrhythmia appeared preceding the structural abnormalities.102, 103 Interestingly, reduced expression of Nav1.5 in cell membrane also appeared in these models.101, 102, 103 These findings indicate that mutations in connexome can affect ion‐channel functions or ID structures, thus disturbing propagation of electrical impulses and causing arrhythmia in the absence of structural abnormalities. From Table 3, we can see that primary arrhythmia‐associated or cardiomyopathy‐associated genes seem to share equal proportions in SUNDS. Furthermore, most of these variants detected in SUNDS are not pathogenic in ClinVar interpretation while compound variants are detected in nearly 1/3 cases, suggesting that compound multigenic variations other than monogenic mutation might be the underlying genetic basis of SUNDS. We conducted a protein‐to‐protein interaction using STRING (https://string-db.org) and found that most of these proteins have a direct or indirect interaction with SCN5A (Figure 3). Combining all these studies, we propose an assumption that SUNDS is a multigenic disorder and a subtype or early stage of cardiomyopathies. Mutations in the predisposing genes (encoding ion channels or structure proteins) may affect SCN5A function and then disturb the action potential production or propagation, finally resulting in ventricular arrhythmia and SCD with no or only slight cardiac structural changes.
Figure 3.
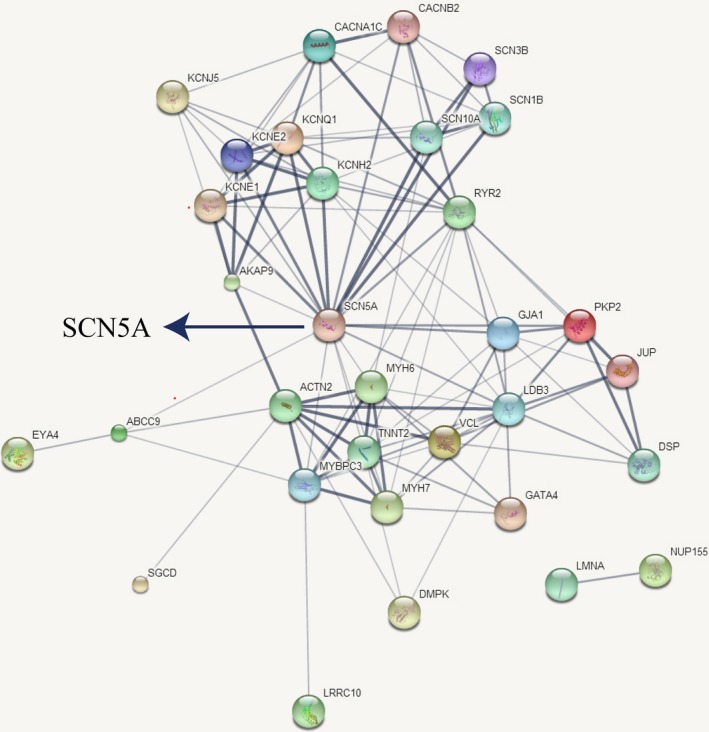
Protein‐to‐protein interaction of the 33 genes responsible for SUNDS. We conducted a protein‐to‐protein interaction using STRING (https://string-db.org) and found that most of these proteins have a direct or indirect interaction with SCN5A. Line thickness indicates the strength of data support. SUNDS indicates sudden unexplained nocturnal death syndrome.
Referring to the triple risk hypothesis of sudden infant death syndrome,104 we set a similar multifactorial model to organize current knowledge, endeavoring to elucidate the rough mechanism of SUNDS. We propose that SUNDS occurs when 3 factors coincide: a vulnerable young person carrying susceptible variants; stressors caused by environmental or social factors; and finally, a disturbance in action potential production and propagation in nocturnal sleep. The triple risk hypothesis postulates that SUNDS will attack the young individual only if all the 3 factors present simultaneously in nocturnal sleep. The final pathway of SUNDS is commonly accepted as lethal VA. The triple risk model indicates that channelopathies and cardiomyopathies‐associated variants may confer vulnerability to SUNDS; this vulnerability then is exacerbated when there is an environmental or social challenge, such as heavy labor, potassium deficiency, hypoventilation, night terrors, rapid eye movement burst, infection, and plasma testosterone exposure. Nonetheless, the interaction between genetic and environmental or social factors remains unclear. Subtle cardiac structural changes (increased heart weight and enlarged heart size) may also be potential but unnecessary structure substrates for SUNDS. However, we speculate that if ultrastructural cardiac changes are also taken into consideration, cardiac structural abnormalities may be a fourth risk factor. Whether the structural changes (mild increase in heart weight and size) are a primary cause or a secondary change is still unknown. If they are secondary, can genetic risk factors or impaired action potential propagation result in these changes (Figure 4)?
Figure 4.
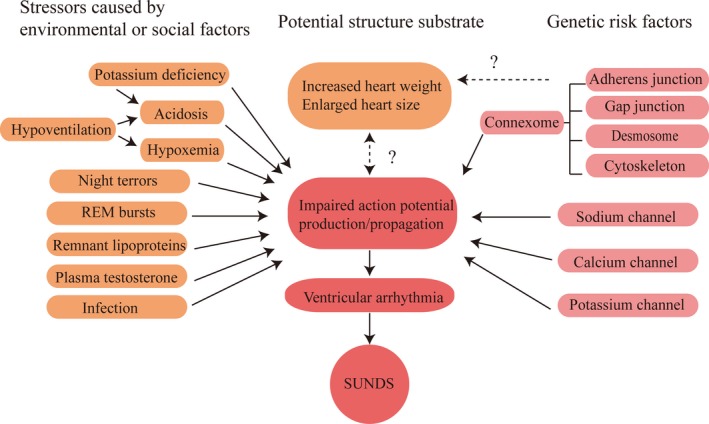
Triple‐risk hypothesis of SUNDS. We propose that SUNDS occurs when 3 factors coincide: a vulnerable young person carrying susceptible variants; stressors caused by environmental or social factors; and finally a disturbance in action potential production and propagation in nocturnal sleep. The final pathway of SUNDS is commonly accepted as lethal ventricular arrhythmia. Cardiac structural changes may also be potential but unnecessary structure substrates for SUNDS. Whether the structural changes are a primary cause or a secondary change is still unknown. Certain associations between potential structure substrate, genetic risk factors, and impaired action potential production and propagation have yet to be demonstrated (dashed arrow). AP indicates action potential; REM, rapid eye movement; SUNDS, sudden unexplained nocturnal death syndrome.
In this model, we set a supposed threshold for the multifactorial disease. Different genetic backgrounds show diverse intrinsic genetic susceptibility to SUNDS (Figure 5). Familial SUNDS cases might carry ultra‐rare variants that confer them genetic susceptibility. The variants severely damage the action potential production and propagation, and slight stressors can trigger lethal arrhythmia. This can be regarded as a monogenic disorder, such as the SCN5A‐1795insD causing SUNDS, studied in a large family.33 A few people carrying the rare variations and common polymorphisms in susceptible genes, such as these individuals who succumbed to sporadic SUNDS, show limited production or propagation defects but can be compensated in normal conditions. When the compensation is destroyed by increasing stressors, such as heavy labor, the electrical activity disturbance might spread throughout cardiac cells in an amplifying cascading manner, followed immediately by cardiac arrest. This is why SUNDS does not occur in all pathogenic variation carriers and most carriers are sporadic who survive to adulthood. Most people carry common polymorphisms in susceptible genes and show minute genetic susceptibility to SUNDS. Despite similar lifestyles, susceptible variations carriers are much more vulnerable to SUNDS than noncarriers. Based on this assumption, we can also explain the high incidence and sharp reduction of SUNDS among Southeast Asian refugees in the United States that might result from their adaption and decreased stressors, which is consistent with a survey on 747 Hmong immigrants in Wisconsin.41
Figure 5.
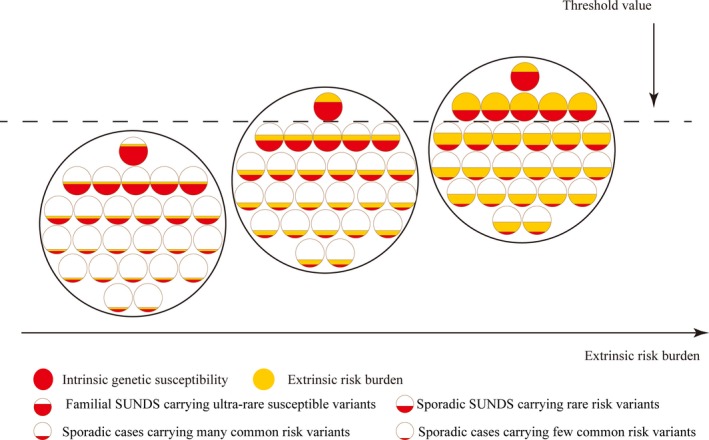
The multifactorial model of SUNDS. In this model, we set a supposed threshold for the multifactorial disease. The 2 major parts, namely, intrinsic genetic susceptibility and extrinsic risk burden, determine whether SUNDS occurs or not. People with different genetic background show different intrinsic genetic susceptibility to SUNDS. SUNDS indicates sudden unexplained nocturnal death syndrome.
If we consider SUNDS as a subtype or early stage of cardiomyopathies, many phenomena can be explained. First, SUNDS is mostly identified in people younger than 60 years old, because structural changes caused by arrhythmogenic cardiomyopathy are more apparent in people over 60 years old and can be diagnosed after routine postmortem autopsy. Second, SUNDS cases were prone to have a slight but significantly increased heart weight and enlarged heart size (increased circumferences of all cardiac valves, especially mitral valves), suggesting a developing process in cardiac remolding. Therefore, SUNDS in young people may be deemed as a transitory stage for cardiomyopathy. Finally, stressors of individuals younger than 15 years are usually not severe enough to cause SCD. Extensive studies are needed to prove or disprove our hypothesis. Individual systematic risk stratification may become possible with appropriate knowledge of SUNDS.
Conclusions
SUNDS is a multifactorial disorder with racial and ethnic disparities in incidence, affected by susceptible variants and stressors caused by environmental and social factors. Currently there are 33 genes and nearly 100 variations and polymorphisms potentially associated with SUNDS. Among these 33 genes, channelopathies and cardiomyopathies‐associated variants seem to share equal proportions in SUNDS. We assume that SUNDS is merely a subtype or early stage of cardiomyopathies. However, current evidence cannot support or disprove our hypothesis. Our ongoing work on both establishing the molecular pathological spectrum of SUNDS and elucidating the mechanism underlying SUNDS may benefit the risk stratification of individuals with high risk for sudden unexplained death.
Sources of Funding
This work was supported by the Key Program 81430046 from the National Natural Science Foundation of China, National Key R&D Program (2017YFC0803502) of China, and the grant (17ykzd03) from Sun Yat‐sen University.
Disclosures
None.
Supporting information
Table S1. SNPs With Significant Difference in Genotype and Allele Frequencies
Table S2. Mutation/Rare Variants Detected in SUNDS*
(J Am Heart Assoc. 2018;7:e007837 DOI: 10.1161/JAHA.117.007837.)29502107
References
- 1. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116:1887–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nakajima K, Takeichi S, Nakajima Y, Fujita MQ. Pokkuri Death Syndrome; sudden cardiac death cases without coronary atherosclerosis in South Asian young males. Forensic Sci Int. 2011;207:6–13. [DOI] [PubMed] [Google Scholar]
- 3. Cheng J, Makielski JC, Yuan P, Shi N, Zhou F, Ye B, Tan BH, Kroboth S. Sudden unexplained nocturnal death syndrome in Southern China: an epidemiological survey and SCN5A gene screening. Am J Forensic Med Pathol. 2011;32:359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chokevivat V, Warintrawat S, Choprapawan C. Epidemiology of Lai Tai in Thailand. Desire Press: 1993;38–50. [Google Scholar]
- 5. Baron RC, Thacker SB, Gorelkin L, Vernon AA, Taylor WR, Choi K. Sudden death among Southeast Asian refugees. An unexplained nocturnal phenomenon. JAMA. 1983;250:2947–2951. [PubMed] [Google Scholar]
- 6. Gotoh K. A histopathological study on the conduction system of the so‐called “Pokkuri disease” (sudden unexpected cardiac death of unknown origin in Japan). Jpn Circ J. 1976;40:753–768. [DOI] [PubMed] [Google Scholar]
- 7. Guazon MP. Algunas notas sobre bangungut [in Spanish]. Revista Filipina de Medicina Y Farmacia. 1917;8:437–442. [Google Scholar]
- 8. The Center for Disease Control . Leads from the MMWR. Update: sudden unexplained death syndrome among Southeast Asian refugees—United States. JAMA. 1988;260:2033. [PubMed] [Google Scholar]
- 9. Goh KT, Chao TC. Sudden nocturnal deaths among Thai construction workers in Singapore [Letter]. Lancet. 1990;335:1154. [DOI] [PubMed] [Google Scholar]
- 10. Tungsanga K, Sriboonlue P. Sudden unexplained death syndrome in north‐east Thailand. Int J Epidemiol. 1993;22:81–87. [DOI] [PubMed] [Google Scholar]
- 11. Munger RG, Booton EA. Bangungut in Manila: sudden and unexplained death in sleep of adult Filipinos. Int J Epidemiol. 1998;27:677–684. [DOI] [PubMed] [Google Scholar]
- 12. Gervacio‐Domingo G, Punzalan FE, Amarillo ML, Dans A. Sudden unexplained death during sleep occurred commonly in the general population in the Philippines: a sub study of the National Nutrition and Health Survey. J Clin Epidemiol. 2007;60:567–571. [DOI] [PubMed] [Google Scholar]
- 13. Zheng J, Huang E, Tang S, Wu Q, Huang L, Zhang D, Quan L, Liu C, Cheng J. A case‐control study of sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Am J Forensic Med Pathol. 2015;36:39–43. [DOI] [PubMed] [Google Scholar]
- 14. Tanchaiswad W. Is sudden unexplained nocturnal death a breathing disorder? Psychiatry Clin Neurosci. 1995;49:111–114. [DOI] [PubMed] [Google Scholar]
- 15. Tatsanavivat P. Sudden Unexplained Deaths in Sleep (Lai Tai) of Young Men in Rural Northeastern Thailand. Desire Press: 1993;51–63. [DOI] [PubMed] [Google Scholar]
- 16. Bito S, Matsumura S, Singer MK, Meredith LS, Fukuhara S, Wenger NS. Acculturation and end‐of‐life decision making: comparison of Japanese and Japanese‐American focus groups. Bioethics. 2007;21:251–262. [DOI] [PubMed] [Google Scholar]
- 17. Cheng JD, Chen YC, Zeng JL. A preliminary study of epidemiology of sudden manhood death syndrome in the peasant derived workers in Dongguan city. Fa Yi Xue Za Zhi. 2002;3:135–136. [PubMed] [Google Scholar]
- 18. Cheng J, Li HX, Li J, Lu CY, Tang SB, Zhou F, Xing HW. The current epidemic circumstance of sudden unexplained nocturnal death syndrome in Chinese. Int J Intern Med. 2008;3:125–128. [Google Scholar]
- 19. Cheng J, Shi N, Makielski J. Epidemiology and genetic cause of sudden unexplained nocturnal death syndrome in Southern China. Circulation. 2007;16:791. [Google Scholar]
- 20. Kirschner RH, Eckner FA, Baron RC. The cardiac pathology of sudden, unexplained nocturnal death in Southeast Asian refugees. JAMA. 1986;256:2700–2705. [PubMed] [Google Scholar]
- 21. Song Y, Yao Q, Zhu J. Age‐related variation in the interstitial tissues of the cardiac conduction system: an autopsy study of 230 Han Chinese. Forensic Sci Int. 1999;104:133–142. [DOI] [PubMed] [Google Scholar]
- 22. Luo B, Song Y, Zhu J. Computerized microimage analysis of age‐related changes of human sinoatrial node. Forensic Sci Int. 1995;75:149–155. [DOI] [PubMed] [Google Scholar]
- 23. Luo B, Song Y, Zhu J. Research progress of sinoatrial node in cardiac conduction system. New Chinese Medicine. 1994;S1:52–53. [Google Scholar]
- 24. Steinhaus DA, Vittinghoff E, Moffatt E, Hart AP, Ursell P, Tseng ZH. Characteristics of sudden arrhythmic death in a diverse, urban community. Am Heart J. 2012;163:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang L, Tester DJ, Lang D, Chen Y, Zheng J, Gao R, Corliss RF, Tang S, Kyle JW, Liu C, Ackerman MJ, Makielski JC, Cheng J. Does sudden unexplained nocturnal death syndrome remain the autopsy‐negative disorder: a gross, microscopic, and molecular autopsy investigation in Southern China. Mayo Clin Proc. 2016;91:1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otto CM, Tauxe RV, Cobb LA, Greene HL, Gross BW, Werner JA, Burroughs RW, Samson WE, Weaver WD, Trobaugh GB. Ventricular fibrillation causes sudden death in Southeast Asian immigrants. Ann Intern Med. 1984;101:45–47. [DOI] [PubMed] [Google Scholar]
- 27. Nimmannit S, Malasit P, Chaovakul V, Susaengrat W, Nilwarangkur S. Potassium and sudden unexplained nocturnal death. Lancet. 1990;336:116–117. [DOI] [PubMed] [Google Scholar]
- 28. Feest TG, Wrong O. Potassium deficiency and sudden unexplained nocturnal death. Lancet. 1991;338:1406. [DOI] [PubMed] [Google Scholar]
- 29. Nimmannit S, Malasit P, Chaovakul V, Susaengrat W, Vasuvattakul S, Nilwarangkur S. Pathogenesis of sudden unexplained nocturnal death (Lai Tai) and endemic distal renal tubular acidosis. Lancet. 1991;338:930–932. [DOI] [PubMed] [Google Scholar]
- 30. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595–2600. [DOI] [PubMed] [Google Scholar]
- 31. Corrado D, Buja G, Basso C, Nava A, Thiene G. What is the Brugada syndrome? Cardiol Rev. 1999;7:191–195. [DOI] [PubMed] [Google Scholar]
- 32. Vatta M, Dumaine R, Varghese G, Richard TA, Shimizu W, Aihara N, Nademanee K, Brugada R, Brugada J, Veerakul G, Li H, Bowles NE, Brugada P, Antzelevitch C, Towbin J. A genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–345. [DOI] [PubMed] [Google Scholar]
- 33. van den Berg MP, Viersma JW, Beaufort‐Krol GC, Bink‐Boelkens MT, Bezzina CR, Veldkamp MW, Brouwer J, Haaksma J, van Tintelen JP, van Langen IM, Wouda AA, Wilde AA. A large family characterised by nocturnal sudden death. Neth Heart J. 2002;10:304–312. [PMC free article] [PubMed] [Google Scholar]
- 34. Charoenpan P, Muntarbhorn K, Boongird P, Puavilai G, Ratanaprakarn R, Indraprasit S, Tanphaichitr V, Likittanasombat K, Varavithya W, Tatsanavivat P. Nocturnal physiological and biochemical changes in sudden unexplained death syndrome: a preliminary report of a case control study. Southeast Asian J Trop Med Public Health. 1994;25:335–340. [PubMed] [Google Scholar]
- 35. Flenley DC. Breathing during sleep. Ann Acad Med Singapore. 1985;14:479–484. [PubMed] [Google Scholar]
- 36. Wu YD, Zhang LY, Cheng JD. Research progress of the relationship between SUNDS and OSAHS. J Forensic Med. 2017;1:52–57. [DOI] [PubMed] [Google Scholar]
- 37. Peters CH, Abdelsayed M, Ruben PC. Triggers for arrhythmogenesis in the Brugada and long QT 3 syndromes. Prog Biophys Mol Biol. 2016;120:77–88. [DOI] [PubMed] [Google Scholar]
- 38. Zheng J, Zhou F, Su T, Huang L, Wu Y, Yin K, Wu Q, Tang S, Makielski JC, Cheng J. The biophysical characterization of the first SCN5A mutation R1512W identified in Chinese sudden unexplained nocturnal death syndrome. Medicine (Baltimore). 2016;95:3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng J, Kyle J, Wiedmeyer B, Lang D, Vaidyanathan R, Makielski J. Vinculin variant M94I identified in sudden unexplained nocturnal death syndrome decreases cardiac sodium current. Sci Rep. 2017;7:42953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Melles RB, Katz B. Sudden, unexplained nocturnal death syndrome and night terrors. JAMA. 1987;257:2918–2919. [PubMed] [Google Scholar]
- 41. Young E, Xiong S, Finn L, Young T. Unique sleep disorders profile of a population‐based sample of 747 Hmong immigrants in Wisconsin. Soc Sci Med. 2013;79:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishiyama I, Kamiya M, Rose M, Komuro E, Takatsu A. Fulminant deletion of myoglobin from myocardial fibres in a state of acute cardiac failure inducing sudden cardiac arrest. Lancet. 1982;2:1468–1469. [DOI] [PubMed] [Google Scholar]
- 43. Sitprija V, Tungsanga K, Tosukhowong P, Leelhaphunt N, Kruerklai D, Sriboonlue P, Saew O. Metabolic problems in northeastern Thailand: possible role of vanadium. Miner Electrolyte Metab. 1993;19:51–56. [PubMed] [Google Scholar]
- 44. Guilleminault C, Pool P, Motta J, Gillis AM. Sinus arrest during REM sleep in young adults. N Engl J Med. 1984;311:1006–1010. [DOI] [PubMed] [Google Scholar]
- 45. Tsunoda K, Watanabe T, Tokutome S. Concentrations of plasma testosterone in sudden manhood death syndrome. Nihon Hoigaku Zasshi. 1997;51:26–31. [PubMed] [Google Scholar]
- 46. Saussu F, van Rijckevorsel K, de Barsy T. Bradycardia: an unrecognized complication of some epileptic crises. Rev Neurol. 1998;154:250–252. [PubMed] [Google Scholar]
- 47. Lee J, Devinsky O. The role of autonomic dysfunction in sudden unexplained death in epilepsy patients. Rev Neurol Dis. 2005;2:61–69. [PubMed] [Google Scholar]
- 48. Parekh B. The mechanism of dead‐in‐bed syndrome and other sudden unexplained nocturnal deaths. Curr Diabetes Rev. 2009;5:210–215. [DOI] [PubMed] [Google Scholar]
- 49. Yap EH, Chan YC, Goh KT, Chao TC, Heng BH, Thong TW, Tan HC, Thong KT, Jacob E, Singh M. Sudden unexplained death syndrome—a new manifestation in melioidosis? Epidemiol Infect. 1991;107:577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Al Madani O, Gordon AE, Weir DM, Raza MW, Busuttil A, Blackwell C. Pyrogenic toxins of Staphylococcus aureus in sudden unexpected nocturnal deaths in adults and older children: factors influencing the control of inflammatory responses to toxic shock syndrome toxins. FEMS Immunol Med Microbiol. 1999;25:207–219. [DOI] [PubMed] [Google Scholar]
- 51. Sangwatanaroj S, Yanatasneejit P, Sunsaneewitayakul B, Sitthisook S. Linkage analyses and SCN5A mutations screening in five sudden unexplained death syndrome (Lai‐Tai) families. J Med Assoc Thai. 2002;85(suppl 1):S54–S61. [PubMed] [Google Scholar]
- 52. Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–828. [DOI] [PubMed] [Google Scholar]
- 53. Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. 2014;10:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Priori SG, Ruan Y, Liu N. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. [DOI] [PubMed] [Google Scholar]
- 55. Rook MB, Bezzina AC, Groenewegen WA, van Gelder IC, van Ginneken AC, Jongsma HJ, Mannens MM, Wilde AA. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507–517. [DOI] [PubMed] [Google Scholar]
- 56. Deschenes I, Baroudi G, Berthet M, Barde I, Chalvidan T, Denjoy I, Guicheney P, Chahine M. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. [DOI] [PubMed] [Google Scholar]
- 57. Liu C, Tester DJ, Hou Y, Wang W, Lv G, Ackerman MJ, Makielski JC, Cheng J. Is sudden unexplained nocturnal death syndrome in Southern China a cardiac sodium channel dysfunction disorder? Forensic Sci Int. 2014;236:38–45. DOI: 10.1016/j.forsciint.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 58. Chen JZ, Xie XD, Wang XX, Tao M, Shang YP, Guo XG. Single nucleotide polymorphisms of the SCN5A gene in Han Chinese and their relation with Brugada syndrome. Chin Med J (Engl). 2004;117:652–656. [PubMed] [Google Scholar]
- 59. Behr ER, Savio‐Galimberti E, Barc J, Holst AG, Petropoulou E, Prins BP, Jabbari J, Torchio M, Berthet M, Mizusawa Y, Yang T, Nannenberg EA, Dagradi F, Weeke P, Bastiaenan R, Ackerman MJ, Haunso S, Leenhardt A, Kaab S, Probst V, Redon R, Sharma S, Wilde A, Tfelt‐Hansen J, Schwartz P, Roden DM, Bezzina CR, Olesen M, Darbar D, Guicheney P, Crotti L, Jamshidi Y. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res. 2015;106:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zimmer T, Haufe V, Blechschmidt S. Voltage‐gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract. 2014;2014:449–463. DOI: 10.5339/gcsp.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang L, Zhou F, Huang L, Wu Q, Zheng J, Wu Y, Yin K, Cheng J. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int J Legal Med. 2017;131:53–60. [DOI] [PubMed] [Google Scholar]
- 62. El‐Sherif N, Turitto G, Boutjdir M. Congenital long QT syndrome and torsade de pointes. Ann Noninvasive Electrocardiol. 2017;22:e12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu C, Zhao Q, Su T, Tang S, Lv G, Liu H, Quan L, Cheng J. Postmortem molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2 genes in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic Sci Int. 2013;231:82–87. [DOI] [PubMed] [Google Scholar]
- 64. Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage‐dependent L‐type calcium channel. Circ Res. 2011;108:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Betzenhauser MJ, Pitt GS, Antzelevitch C. Calcium channel mutations in cardiac arrhythmia syndromes. Curr Mol Pharmacol. 2015;8:133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Betzenhauser MJ, Marks AR. Ryanodine receptor channelopathies. Pflügers Arch Eur J Physiol. 2010;460:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 68. Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10:189–194. [DOI] [PubMed] [Google Scholar]
- 69. Huang L, Liu C, Tang S, Su T, Cheng J. Postmortem genetic screening of SNPs in RyR2 gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Forensic Sci Int. 2014;235:14–18. [DOI] [PubMed] [Google Scholar]
- 70. Kleber AG, Saffitz JE. Role of the intercalated disc in cardiac propagation and arrhythmogenesis. Front Physiol. 2014;5:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Agullo‐Pascual E, Cerrone M, Delmar M. Arrhythmogenic cardiomyopathy and Brugada syndrome: diseases of the connexome. FEBS Lett. 2014;588:1322–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moncayo‐Arlandi J, Brugada R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat Rev Cardiol. 2017;14:744–756. [DOI] [PubMed] [Google Scholar]
- 73. Cerrone M, Lin X, Zhang M, Agullo‐Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin‐2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein‐2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. [DOI] [PubMed] [Google Scholar]
- 75. Te RA, Agullo‐Pascual E, James CA, Leo‐Macias A, Cerrone M, Zhang M, Lin X, Lin B, Sobreira NL, Amat‐Alarcon N, Marsman RF, Murray B, Tichnell C, van der Heijden JF, Dooijes D, van Veen TA, Tandri H, Fowler SJ, Hauer RN, Tomaselli G, van den Berg MP, Taylor MR, Brun F, Sinagra G, Wilde AA, Mestroni L, Bezzina CR, Calkins H, Peter V, Bu L, Delmar M, Judge DP. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non‐canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. [DOI] [PubMed] [Google Scholar]
- 77. Schmidt A, Jager S. Plakophilins—hard work in the desmosome, recreation in the nucleus? Eur J Cell Biol. 2005;84:189–204. [DOI] [PubMed] [Google Scholar]
- 78. Huang L, Tang S, Peng L, Chen Y, Cheng J. Molecular autopsy of desmosomal protein plakophilin‐2 in sudden unexplained nocturnal death syndrome. J Forensic Sci. 2016;61:687–691. [DOI] [PubMed] [Google Scholar]
- 79. Zhao Q, Chen Y, Peng L, Gao R, Liu N, Jiang P, Liu C, Tang S, Quan L, Makielski JC, Cheng J. Identification of rare variants of DSP gene in sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Int J Legal Med. 2016;130:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, van den Wijngaard A, Houweling AC, Jongbloed JD, Jordaens L, Cramer MJ, Doevendans PA, de Bakker JM, Wilde AA, van Tintelen JP, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index‐patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype‐phenotype follow‐up study. Circulation. 2011;123:2690–2700. [DOI] [PubMed] [Google Scholar]
- 81. Sato T, Nishio H, Suzuki K. Identification of arrhythmogenic right ventricular cardiomyopathy‐causing gene mutations in young sudden unexpected death autopsy cases. J Forensic Sci. 2015;60:457–461. [DOI] [PubMed] [Google Scholar]
- 82. Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309–322. [DOI] [PubMed] [Google Scholar]
- 83. Noorman M, Hakim S, Kessler E, Groeneweg JA, Cox MG, Asimaki A, van Rijen HV, van Stuijvenberg L, Chkourko H, van der Heyden MA, Vos MA, de Jonge N, van der Smagt JJ, Dooijes D, Vink A, de Weger RA, Varro A, de Bakker JM, Saffitz JE, Hund TJ, Mohler PJ, Delmar M, Hauer RN, van Veen TA. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu Q, Wu Y, Zhang L, Zheng J, Tang S, Cheng J. GJA1 gene variations in sudden unexplained nocturnal death syndrome in the Chinese Han population. Forensic Sci Int. 2017;270:178–182. [DOI] [PubMed] [Google Scholar]
- 85. Bays JL, DeMali KA. Vinculin in cell‐cell and cell‐matrix adhesions. Cell Mol Life Sci. 2017;74:2999–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vasile VC, Will ML, Ommen SR, Edwards WD, Olson TM, Ackerman MJ. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab. 2006;87:169–174. [DOI] [PubMed] [Google Scholar]
- 87. Cheng J, Kyle JW, Lang D, Wiedmeyer B, Guo J, Yin K, Huang L, Vaidyanathan R, Su T, Makielski JC. An East Asian common variant vinculin P.Asp841His was associated with sudden unexplained nocturnal death syndrome in the Chinese Han population. J Am Heart Assoc. 2017;6:e005330 DOI: 10.1161/JAHA.116.005330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Earle N, Yeo HD, Pilbrow A, Crawford J, Smith W, Shelling AN, Cameron V, Love DR, Skinner JR. Single nucleotide polymorphisms in arrhythmia genes modify the risk of cardiac events and sudden death in long QT syndrome. Heart Rhythm. 2014;11:76–82. [DOI] [PubMed] [Google Scholar]
- 89. Huang L, Yu Y, Chen Y, Tester DJ, Tang S, Ackerman MJ, Yuan Z, Cheng J. Association of common variants in NOS1AP gene with sudden unexplained nocturnal death syndrome in the southern Chinese Han population. Int J Legal Med. 2014;128:933–938. [DOI] [PubMed] [Google Scholar]
- 90. Brody MJ, Hacker TA, Patel JR, Feng L, Sadoshima J, Tevosian SG, Balijepalli RC, Moss RL, Lee Y. Ablation of the cardiac‐specific gene leucine‐rich repeat containing 10 (Lrrc10) results in dilated cardiomyopathy. PLoS One. 2012;7:51621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huang L, Tang S, Chen Y, Zhang L, Yin K, Wu Y, Zheng J, Wu Q, Makielski JC, Cheng J. Molecular pathological study on LRRC10 in sudden unexplained nocturnal death syndrome in the Chinese Han population. Int J Legal Med. 2017;131:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cordeiro JM, Barajas‐Martinez H, Hong K, Burashnikov E, Pfeiffer R, Orsino AM, Wu YS, Hu D, Brugada J, Brugada P, Antzelevitch C, Dumaine R, Brugada R. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006;114:2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long‐QT syndrome. Circulation. 2004;109:1834–1841. [DOI] [PubMed] [Google Scholar]
- 94. Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AJ. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003;112:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C, Ackerman M, Belhassen B, Estes NR, Fatkin D, Kalman J, Kaufman E, Kirchhof P, Schulze‐Bahr E, Wolpert C, Vohra J, Refaat M, Etheridge SP, Campbell RM, Martin ET, Quek SC. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace. 2013;15:1389–1406. [DOI] [PubMed] [Google Scholar]
- 97. Ikeda T, Abe A, Yusu S, Nakamura K, Ishiguro H, Mera H, Yotsukura M, Yoshino H. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17:602–607. [DOI] [PubMed] [Google Scholar]
- 98. Mizusawa Y, Morita H, Adler A, Havakuk O, Thollet A, Maury P, Wang DW, Hong K, Gandjbakhch E, Sacher F, Hu D, Amin AS, Lahrouchi N, Tan HL, Antzelevitch C, Probst V, Viskin S, Wilde AA. Prognostic significance of fever‐induced Brugada syndrome. Heart Rhythm. 2016;13:1515–1520. [DOI] [PubMed] [Google Scholar]
- 99. Schulze‐Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. [DOI] [PubMed] [Google Scholar]
- 100. Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. [DOI] [PubMed] [Google Scholar]
- 101. Zemljic‐Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac‐myocyte‐specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kirchhof P, Fabritz L, Zwiener M, Witt H, Schafers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, Breithardt G, Ruiz P, Wichter T, Levkau B. Age‐ and training‐dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin‐deficient mice. Circulation. 2006;114:1799–1806. [DOI] [PubMed] [Google Scholar]
- 103. Moncayo‐Arlandi J, Guasch E, Sanz‐de L, Casado M, Garcia NA, Mont L, Sitges M, Knoll R, Buyandelger B, Campuzano O, Diez‐Juan A, Brugada R. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Hum Mol Genet. 2016;25:3676–3688. [DOI] [PubMed] [Google Scholar]
- 104. Moon RY, Horne RS, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578–1587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. SNPs With Significant Difference in Genotype and Allele Frequencies
Table S2. Mutation/Rare Variants Detected in SUNDS*