

Abstract
Background
Earlier, we reported that the simultaneous exposure of pulmonary arterial smooth muscle cells to HIV proteins and cocaine results in the attenuation of antiproliferative bone morphogenetic protein receptor‐2 (BMPR2) protein expression without any decrease in its mRNA levels. Therefore, in this study, we aimed to investigate the micro RNA‐mediated posttranscriptional regulation of BMPR2 expression.
Methods and Results
We identified a network of BMPR2 targeting micro RNAs including miR‐216a to be upregulated in response to cocaine and Tat‐mediated augmentation of oxidative stress and transforming growth factor‐β signaling in human pulmonary arterial smooth muscle cells. By using a loss or gain of function studies, we observed that these upregulated micro RNAs are involved in the Tat‐ and cocaine‐mediated smooth muscle hyperplasia via regulation of BMPR2 protein expression. These in vitro findings were further corroborated using rat pulmonary arterial smooth muscle cells isolated from HIV transgenic rats exposed to cocaine. More importantly, luciferase reporter and in vitro translation assays demonstrated that direct binding of novel miR‐216a and miR‐301a to 3′UTR of BMPR2 results in the translational repression of BMPR2 without any degradation of its mRNA.
Conclusions
We identified for the first time miR‐216a as a negative modulator of BMPR2 translation and observed it to be involved in HIV protein(s) and cocaine‐mediated enhanced proliferation of pulmonary smooth muscle cells.
Keywords: cocaine, drug abuse, human immunodeficiency virus, human immunodeficiency virus tat, intravenous drug use, microRNA, pulmonary hypertension, smooth muscle cell, translational repression, vascular remodeling
Subject Categories: Pulmonary Hypertension, Smooth Muscle Proliferation and Differentiation, Vascular Biology, Gene Expression & Regulation, Remodeling
Clinical Perspective
What Is New?
Identified miR‐216a as a novel bone morphogenetic protein receptor‐2 targeting micro RNA that is upregulated in hyperproliferative pulmonary arterial smooth muscle cells upon combined exposure to cocaine and HIV‐Tat protein.
What Are the Clinical Implications?
MiR‐216a could be considered as a potential target for bone morphogenetic protein receptor‐2‐associated pulmonary arterial hypertension and future studies on the human patient samples may reveal its potential as an effective diagnostic biomarker.
Introduction
Pulmonary arterial hypertension (PAH) is known to be caused by a severe remodeling of pulmonary arteries followed by an increased vascular resistance, ultimately resulting in cardiac failure.1 Vascular remodeling involves endothelial dysfunction and aberrant smooth muscle cell proliferation/migration. However, the detailed mechanisms and pathogenesis implicated in PAH still remain elusive. Recent advances highlight multiple factors critical to the development of PAH.2 One of the crucial components associated with the condition of familial PAH is an impaired bone morphogenetic protein receptor (BMPR)‐2 signaling in pulmonary arteries. The BMPR‐2 gene was found to harbor mutations in 50% to 90% of patients diagnosed with familial PAH.3 Apart from the association of BMPR2 mutations with familial PAH, many studies also report the association of reduced BMPR2 expression with other forms of PAH, including idiopathic PAH.4, 5, 6, 7, 8
HIV‐associated PAH is a devastating and life‐threatening condition with recent cohort studies reporting prevalence ranging from 2.6% to 15.5%.9, 10, 11, 12, 13, 14, 15 Furthermore, a higher percentage of HIV‐infected individuals among intravenous drug users correlate with higher prevalence of HIV‐PAH in HIV‐infected patients.16, 17, 18, 19, 20, 21 Our previous published findings also extensively highlight the exacerbated pulmonary vascular remodeling in the presence of both HIV‐viral proteins and drugs of abuse such as cocaine and morphine, therefore supporting the multiple‐hit hypothesis of PAH.22, 23, 24, 25
We earlier reported reduction in the protein expression of BMPR2 in human pulmonary arterial smooth muscle cells (HPASMCs) on exposure to HIV proteins: Tat, Nef, or gp120.22, 23, 26, 27 Importantly, our findings support the additive role of cocaine in potentiating the HIV‐protein mediated proliferation of smooth muscle cells and pulmonary vascular remodeling22, 23, 26, 27 that correlated with the enhanced attenuation in the levels of BMPR‐2 on the combined exposure when compared with exposure to either HIV proteins or cocaine alone. In addition, we reported that treatment with Tat gives maximum downregulation of BMPR2 expression and increase in the SMC proliferation in the presence of cocaine when compared with other HIV proteins.22
Interestingly, we found a significant decrease in BMPR2 protein expression, with slight but significantly elevated levels of BMPR2 mRNA in the HPASMCs treated with cocaine and HIV‐Tat.22 Although studies found that ubiquitination and proteasomal degradation of BMPR2,28, 29 posttranscriptional regulation of the mRNA by either micro RNAs (miRNAs) and other noncoding RNAs could be another possible mechanism involved in the alterations in BMPR2 protein expression independent of its mRNA levels. We here hypothesize that the reduction in the levels of BMPR2 protein in response to HIV proteins and cocaine involves miRNA‐mediated translational repression. We identified miR‐216a as a novel BMPR2 targeting miRNA and investigated its role along with other known BMPR2 targeting miRNAs in regulating cocaine and/or HIV‐1 protein(s)‐mediated smooth muscle hyperplasia. Using miRNA mimics/antagomirs, 3′‐untranslated region (UTR) BMPR2 luciferase reporter assay and in vitro translation approaches, we identified miR‐216a having an ability to directly regulate the translation of BMPR2.
Materials and Methods
The data, analytic methods, and study materials except plasmid constructs will be made available to other researchers for purposes of reproducing the results or replicating the procedures on request. Various plasmid constructs used in this study are commercially available from other sources.
Cell Culture and Treatments
Primary HPASMCs (ScienCell Research Laboratories, Carlsbad, CA) were grown on smooth muscle cell media supplemented with 2% fetal bovine serum, smooth muscle cell growth supplements, and penicillin/streptomycin (ScienCell Research Laboratories). For premature or mature miRNA quantification or Western blot analysis of cocaine‐ and/or Tat‐treated HPASMCs, 5×104 cells were seeded per well in 6‐well plates with complete media. At 80% confluency, cells were made quiescent with serum‐free smooth muscle cell media for 48 hours and treated with cocaine at 1 μmol/L final concentration and/or HIV‐Tat protein at 25 ng/mL final concentration for 24, 48, and 72 hours before proceeding to RNA or protein extraction. For some experiments, HPASMCs were pretreated with antioxidant cocktail or transforming growth factor β (TGF‐β) ligand (at 10 ng/mL final concentration) or TGF‐β receptor 1 inhibitor (at 10 μmol/L final concentration) for 30 minutes followed by cocaine and Tat as indicated in the figures. The antioxidant cocktail included ascorbic acid, reduced glutathione, and α‐tocopherol at 200, 500, and 350 μmol/L final concentrations, respectively.
Primary rat pulmonary arterial smooth muscle cells (RPASMC) isolated from wild‐type (WT) or HIV‐transgenic rats treated with or without cocaine were grown using complete rat smooth muscle cell media (Cell Applications Inc, San Diego, CA) as described earlier.23 Animal studies were approved by Kansas University Medical Center's Institutional Animal Care and Use Committee.
Plasmids, miRNA Mimics/Antagomirs, and Transfections
Plasmid vectors of pEZX‐MT05 series containing BMPR2 3′UTR regions (Gene Accession NM_001204.5) under Gaussia luciferase reporter and independently expressing secreted alkaline phosphatase were obtained from Genecopoeia (Rockville, MD). The control plasmid with no BMPR2 3′UTR region was also obtained from Genecopoeia. The vectors pEZX‐MT05‐A and pEZX‐MT05‐B included different regions of 3′UTR of BMPR2 with miR‐301a and miR‐216a binding sites, respectively. Plasmid vector of pcDNA3.1+ series cloned with 6× repetitive binding sites for either miR‐216a‐5p or miR‐301a‐3p downstream to firefly luciferase reporter were custom designed and obtained from VectorBuilder (Cyagen Biosciences Inc, Santa Clara, CA) for in vitro transcription/translation assays. These vectors were named as pcDNA3.1+FL6X‐216a5p (FL216), pcDNA3.1+FL6X‐301a3p (FL301), or pcDNA3.1+FL0 (FL0). A plasmid vector pRL‐TK (renilla luciferase reporter mRNA) containing renilla luciferase was purchased from Promega (Cat#E2231). Both the firefly and renilla luciferases were under T7 promoter in their respective vectors to enable the preparation of their respective mRNA reporters by in vitro transcription.
Chemically modified single‐strand miRNA antagomirs against miR‐19a, miR‐21, and miR‐301a were purchased from Genecopoeia. Chemically modified single‐strand mirVana miRNA antagomirs for miR‐130a, ‐216a‐5p, and all the chemically modified double stranded mirVana mimics with an inactivated passenger strand (*strand) used in this study were received from ThermoFisher Scientific (Rockford, IL). HPASMC were reverse transfected with miRNA antagomirs or mimics using HiPerfect transfection reagent at the time of seeding and then after 24 hours were made quiescent by incubating cells for 48 hours in serum free medium followed by 48 hours treatment with cocaine and HIV Tat. The cells were then harvested for either WB or quantitative reverse transcription polymerase chain reaction (qRT‐PCR) analysis as described previously.22
Real‐Time RT‐PCR Analysis
Total RNA was isolated from HPASMC or cultured RPASMC (isolated from 4‐ and 9‐month‐old rats) by TRIzol (Invitrogen, Carlsbad, CA) as per manufacturer's instructions followed by the quantitative analysis of precursor (pre‐miR) or mature (miR) miRNA or mRNA by real‐time qRT‐PCR. In order to quantify the miRNAs, the cDNA was synthesized first from the isolated total RNA by using miScript‐II RT Kit (Qiagen, Cat#218161) followed by qRT‐PCR by using QuantiTect SYBR Green PCR Kit (Qiagen, Cat#204145). For qRT‐PCR analysis of pre‐miR‐216a, ‐301a, miR‐19a, miR‐20a, ‐miR‐21, miR‐301a, miR‐365b, miR‐454, miR‐656, and miR‐1291, SYBR Green primers were purchased from Qiagen (Table S1) and for the detection of miR‐216a and miR‐130a the TaqMan primers/probe combined assays were obtained from ThermoFisher Scientific (Table S1). For quantification of BMPR2 mRNA levels, qRT‐PCR was performed as mentioned previously.22 The primers for the qRT‐PCR of firefly and renilla luciferase reporter mRNAs were custom designed and purchased from Integrated DNA Technologies (Table S1). The custom primers were checked for their specificity by melt curve plot during the qRT‐PCR.
Western Blot
Western blots using total protein extracts from HPASMCs exposed to different treatments were performed as described previously.22 BMPR2 antibody (cat#sc5682) was obtained from Santa Cruz Biotechnology (1:500 dilution) and PTEN antibody (cat#9559S) was obtained from Cell Signaling Technologies (1:1000 dilution).
Cell Proliferation Assay
HPASMCs (1×104 cells/well) were reverse transfected as indicated in the figures at the time of plating with mimics and/or antagomirs of selected miRNAs either individually or in combination in 96‐well plate by using HiPerfect transfection reagent. A BMPR2 rescue experiment was performed with transfection of BMPR2 small interfering RNA in combination with miR‐216a antagomir. At 24 hours posttransfection, cells were made quiescent for 24 hours using 0.1% serum media followed by cocaine and Tat treatment for 48 hours. RPASMC from 9‐month‐old WT and HIV‐transgenic rats with cocaine treatment were reverse transfected with miRNA antagomirs using HiPerfect transfection reagent. At 24 hours posttransfection, cells were replenished with 0.1% serum containing medium for 72 hours. At the end of treatments, proliferation was quantified using CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI) as described earlier.22
Luciferase Reporter Assay
HPASMCs were seeded in complete smooth muscle cell media at 0.5×105 cells per well of 24‐well plates. After 24 hours of seeding the cells, the cotransfection was performed with either the control pEZ‐MT05‐D or pEZX‐MT05‐B or pEZX‐MT05‐A plasmid at a concentration of 250 ng/well with various concentrations (5, 10, and 20 nmol/L) of either miRNA mimics and/or antagomirs for miR‐216a‐5p or miR‐301a‐3p. The transfection reaction mixtures for DNA and miRNAs were prepared separately using Genejuice and Ribojuice transfection reagents, respectively, and subsequently added together on the cells. The culture medium was collected at 24 and 48 hours post cotransfection and the amount of Gaussia luciferase and secreted alkaline phosphatase activities were measured by secrete pair dual luciferase assay kit (Genecopoeia, Rockville, MD) as per the manufacturer's instructions. Secreted alkaline phosphatase activity was used to normalize Gluc activity.
Preparation of mRNA Reporters by In Vitro Transcription
pcDNA3.1FL6X‐216a5p, pcDNA3.1FL6X‐301a3p, or pcDNA3.1FL0 plasmids were linearized with Nco‐I and pRL‐TK plasmid was linearized with Hind‐III to generate the internal control RL0 at 37°C for 2 hours. Linearized plasmids were purified using Qiaquick PCR cleanup kit (Qiagen, Cat#28104) and later resuspended in nuclease free water. In vitro transcription was performed with the linearized plasmid using the mMESSAGE mMACHINE kit (Ambion, #AM1344) for the capped mRNA synthesis according to manufacturer's protocol followed by purification by RNeasy mini kit (Qiagen, Cat#74104). Capped mRNAs were next polyadenylated in 100 μL reactions using Poly (A) tailing kit (ThermoFisher Scientific, Cat#AM1350) as per manufacturer's instructions with 4 times diluted Escherichia coli Poly A Polymerase enzyme, which adds <100 poly A nucleotide tail at this dilution. Reactions were terminated with 1 mL of 5 mmol/L EDTA.
In Vitro Translation Assay
The in vitro translation assay was designed by combining the protocols described earlier30, 31 with minor modifications. Briefly, 0.025 pmol of firefly reporter mRNA and 0.025 pmol of renilla control mRNA were mixed with mimics of either miR‐216a‐5p or miR‐301a‐3p (Thermo Fisher Scientific) in 1:6 (mRNA/miRNA) ratio in 11 μL. Reactions for vector and miRNA scrambled controls were set up separately to a total volume of 11 μL. The mixture was heated to 75°C for 3 minutes, cooled to room temperature for 5 minutes, and then kept on ice. To begin in vitro translation, 39 μL of nuclease‐treated rabbit reticulocyte lysate (Cat#L4960, Promega) master mix (35 μL rabbit reticulocyte lysate, 4–8 U [1 μL] RNasin [Promega], 20 mmol/L [1 μL] complete amino acid mixture [Promega]) was added to the 11 μL of reporter and mimic mix followed by incubation at 30°C for 1.5 hours. Reactions were stopped by transferring the reaction tubes to ice. The translation rates were indirectly measured by the activities of renilla and firefly luciferase by using the dual luciferase assay kit (Promega). The levels of renilla and firefly reporter mRNAs added for the in vitro translation assay reactions were monitored after the assay by qRT‐PCR. The final post qRT‐PCR reaction contents were also resolved on 1.5% Agarose gel to further assess the levels of mRNA reporters postassay.
Statistical Analysis
Statistical analysis was carried out on GraphPad prism software. We first analyzed the data for normality and equal distribution followed by 1‐way ANOVA for post hoc Bonferroni test with multiple comparisons. Data were averaged as mean±SEM from ≥4 independent experiments unless otherwise mentioned. A nonparametric Kruskal–Wallis test was performed on animal findings (n=3/group). The test results were considered significant when the corrected P values were ≤0.05.
Results
Increased Expression of BMPR2 Targeting miRNAs in HPASMCs Treated With Cocaine and HIV Tat
To validate our hypothesis of miRNA‐mediated translation repression of BMPR2, we first predicted miRNAs that could potentially target the BMPR‐2 gene using the TargetScan 7.1 tool.32 As shown in Table S2, miRNAs were ranked and selected according to their efficiency of targeting BMPR2 that was based primarily on the context++ score where higher negative context score indicates more efficient binding. Preferentially conserved targeting (PCT) score was obtained only for the highly conserved miRNA binding sites. The miRNAs miR‐130a, ‐301a, and ‐454 shared a single binding site of 7 mer in the 3′UTR, whereas other miRNAs had independent binding site of 7 to 8 mer seed match on different regions of the 3′UTR of BMPR‐2.
We quantified the expression levels of these selected miRNAs in HPASMCs exposed to either individual or combined treatment of HIV‐Tat and cocaine for a time period of 24, 48, and 72 hours. The qRT‐PCR analysis showed 3‐ to 5‐fold significant increase in the expression levels of 6 out of 12 miRNAs in the cells exposed to combined treatment or 1.5‐ to 2‐fold increases on monotreatment when compared with untreated controls (Figure 1A). Significant increase in the expression of miRNAs (miR‐19a, miR‐20a, ‐21, ‐130a, ‐216a, and ‐301a) was observed on combined treatment when compared with monotreatments or untreated control. However, miR‐454 showed a little but significant increase only at 24 hours of combined treatment when compared with untreated cells (Figure 1B). Comparison of normalized Ct values of mature miRNAs showed that miR‐19a and ‐130a are expressed at slightly higher levels compared with miR‐20a, ‐21, 301a, and 216a with an overall decrease in the normalized Ct values on C+T combined treatment (Figure S1). Quantification of miRNAs, namely, miR‐365b, ‐16‐2, ‐656, ‐26, and ‐1291, found no significant changes in their expression in response to any of the treatments (Figure 1B). Overall, we identified a network of miRNAs that are significantly upregulated under C+T combined and individual treatments that are predicted to target BMPR2.
Figure 1.
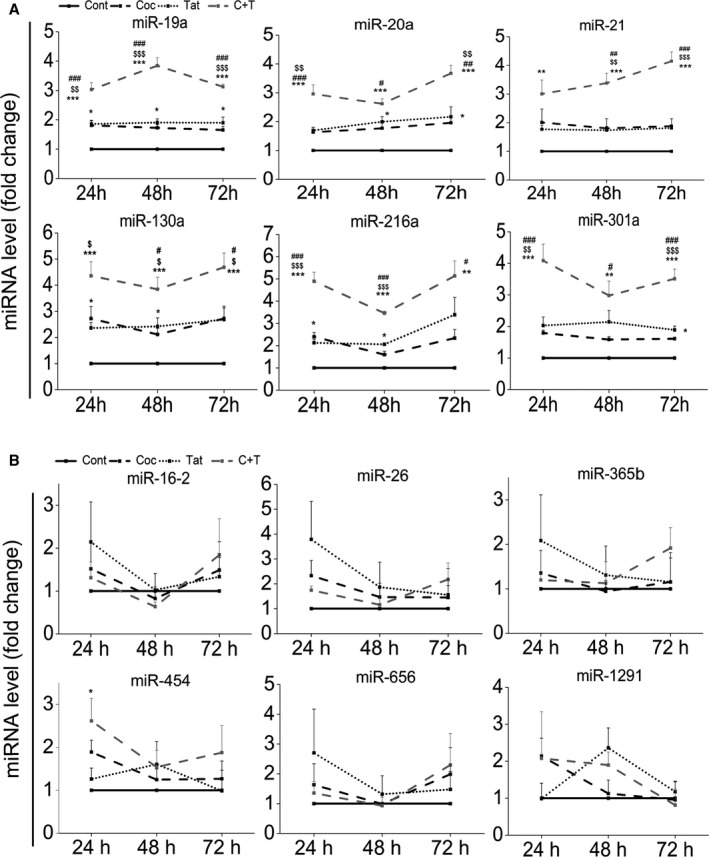
Quantitative real‐time (qRT) polymerase chain reaction (PCR) analyses showing significant upregulation (A) or no change (B) of microRNAs (miRNAs) that are predicted to target 3′untranslated region of bone morphogenetic protein receptor‐2 in human pulmonary arterial smooth muscle cells (HPASMCs) exposed to cocaine and/or Tat protein of HIV. HPASMCs were made quiescent with 0.1% fetal bovine serum containing medium for 48 hours followed by a treatment with cocaine at 1 μmol/L and/or HIV Tat at 25 ng/mL for 24, 48, and 72 hours. The total RNA was isolated and cDNA was prepared before proceeding to quantify miRNAs by SYBR green or Taqman‐based qRT‐PCR. *P<0.05, **P<0.01, ***P<0.001 vs Control (Cont), # P<0.05, ## P<0.01, ### P<0.001, vs Cocaine (Coc), $ P<0.05, $$ P<0.01, $$$ P<0.001 vs Tat. C+T: cocaine and Tat.
Inhibition or Overexpression of BMPR2 Targeting miRNAs Prevented or Potentiated the Cocaine‐ and Tat‐Mediated Decrease in BMPR2 Protein Levels, Respectively
Next, to determine whether the selected upregulated BMPR2 targeting miRNAs in Tat‐ and cocaine‐treated HPASMCs are involved in regulating the expression levels of BMPR2, we performed transfections using mimics or antagomirs for these specific miRNAs. Some of the upregulated miRNAs shown in Figure 1A have been reported earlier to target BMPR2 specifically.33, 34, 35 We investigated the expression of these reported miRNAs namely miR‐21, ‐130a or ‐19a as positive controls along with the analysis of novel miR‐216a and ‐301a in regulation of BMPR‐2 expression. As shown in Figure 2, we monitored the expression of mRNA and protein levels of BMPR2 under the inhibition or overexpression of these selected miRNAs. The transfection efficiency of mimic and antagomirs in HPASMCs was confirmed by qRT‐PCR for all miRNAs and the representative graph is shown for mimics and antagomirs against miR‐130a and ‐216a in Figure S2. The miRNA antagomir studies showed no change in the BMPR‐2 mRNA levels in cocaine‐ and Tat‐treated cells in the presence or absence of selected antagomirs when compared with scrambled control (Figure 2A). However, the cocaine‐ and Tat‐treated cells transfected with miRNA mimics showed reduction in the level of BMPR‐2 mRNA by ≈2‐fold compared with untransfected, cocaine‐, and Tat‐treated HPASMCs (Figure 2B). On the other hand, when we analyzed the BMPR2 protein expression, transfection of cells with antagomirs could prevent the cocaine and Tat mediated decrease in the protein levels of BMPR‐2 (Figure 2C). Conversely, the protein levels of BMPR2 further reduced in the cocaine‐ and Tat‐treated cells overexpressing selected miRNAs compared with untransfected treated cells (Figure 2D). Taken together, these results highlight a potential regulatory link between the putative BMPR2 targeting miR‐216a and ‐301a and the protein levels of BMPR2 in HPASMCs. In addition, all the selected upregulated miRNAs in C+T treated smooth muscle cells have also been reported to target phosphatase and tensin homolog (PTEN) earlier.36, 37, 38, 39, 40 In light of these findings, we also observed significant prevention of C+T‐mediated downregulation of PTEN in the presence of antagomirs against miR‐19, ‐216, and ‐130 as shown in Figure S3.
Figure 2.
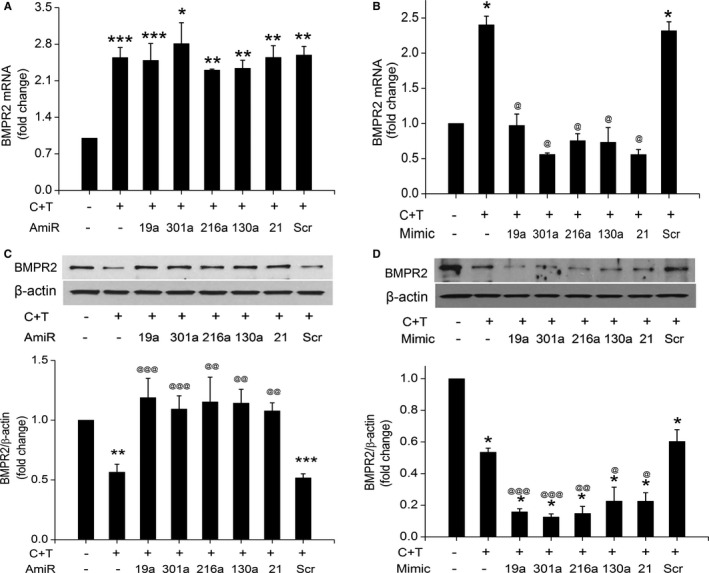
Alteration in bone morphogenetic protein receptor‐2 (BMPR2) expression in response to inhibition or overexpression of selective micro RNAs (miRNAs). A, Quantitative real‐time polymerase chain reaction analyses showing BMPR2 mRNA levels in human pulmonary arterial smooth muscle cells (HPASMCs) treated with cocaine and Tat (C+T) after transfections with respective antagomirs (AmiR) of miRNAs targeting BMPR2 3′untranslated region. *P<0.05, **P<0.01, ***P<0.001, vs control. B, BMPR2 mRNA levels in cocaine and Tat protein of HIV‐treated HPASMCs transfected with mimics for the selected miRNAs. *P<0.001 vs cont, @ P<0.001 vs C+T. C, BMPR2 protein levels in cocaine and HIV Tat protein treated, antagomir transfected HPASMCs. Upper panel shows the representative Western blot image and lower panel includes the densitometric analysis of the Western blots from ≥3 independent experiments by using ImageJ software. **P<0.01, ***P<0.0001 vs control, @@ P<0.001, @@@ P<0.001 vs C+T. D, BMPR2 protein levels in cocaine and HIV Tat‐treated cells in the presence or absence of miRNA mimics. Upper panel is the representative Western blot and lower panel shows average densitometric analysis. *P<0.001 vs cont, @ P<0.05, @ P<0.01, @@@ P<0.001 vs C+T. Scr indicates scrambled.
Inhibition or Overexpression of BMPR2 Targeting miRNAs Prevented or Potentiated the Cocaine‐ and Tat‐Mediated Increase in Smooth Muscle Proliferation, Respectively
Our previous studies indicate that either individual or combined treatments of cocaine and Tat result in enhanced proliferation of HPASMCs.22 So, we next determined whether alteration in the levels of selected BMPR2 targeting miRNAs is involved in cocaine‐ and Tat‐mediated smooth muscle hyperplasia by regulating BMPR2 protein levels. We performed proliferation assay in response to cocaine and Tat in cells overexpressed or knocked down with particular BMPR2 targeting miRNA. As shown in Figure 3A, while the cells exposed to C+T combined treatment in the presence of scrambled antagomir maintained the cocaine‐ and Tat‐mediated enhanced proliferation of HPASMCs, the cells transfected with specific miRNA antagomirs failed to show the cocaine‐ and Tat‐mediated induction in cell proliferation. On the contrary, cocaine and Tat treatment of HPASMC transfected with miRNA mimics for miR‐19a, ‐130a, ‐216a and ‐301a resulted in significantly enhanced proliferation compared with cocaine‐ and Tat‐treated untransfected or scrambled mimic transfected cells (Figure 3B). Also, the simultaneous transfection of HPASMC with all miRNA inhibitors showed further decrease in C+T‐mediated proliferation when compared with cells transfected with only miR‐216a or only miR‐301a (Figure 3C). In addition, C+T treatment of HPASMCs transfected with BMPR2 small interfering RNA not only significantly further enhanced the C+T‐mediated proliferation (Figure 3D) but also prevented the miR‐216a antagomir‐mediated abrogation of C+T‐induced proliferation (Figure 3D). Taken together, these results point toward the involvement of these selected miRNAs in regulating the cocaine and Tat mediated proliferation of HPASMCs, especially through BMPR2.
Figure 3.
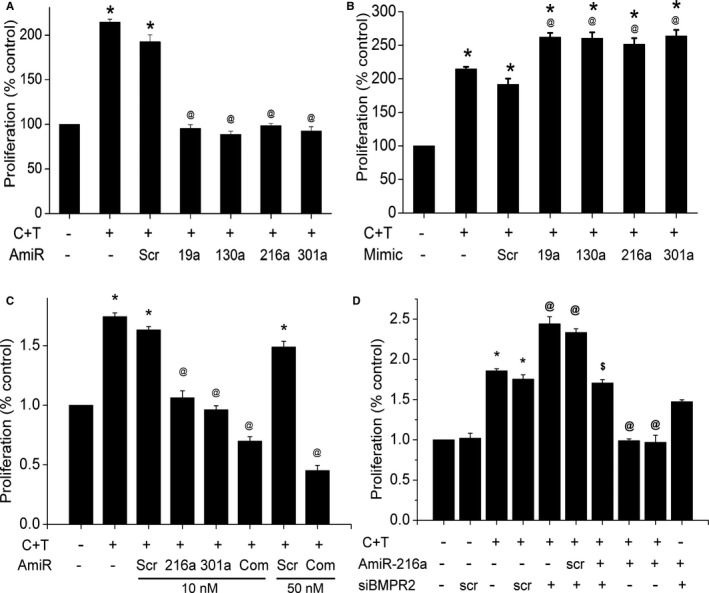
Effect of inhibition (A) or overexpression (B) of selected bone morphogenetic protein receptor‐2 (BMPR2) targeting micro RNAs (miRNAs) on the smooth muscle proliferation. Human pulmonary arterial smooth muscle cells (HPASMCs) (1×104 cells/well) were reverse transfected for 3 hours at the time of plating with antagomirs (A) or mimics (B) corresponding to the selected miRNAs in a 96‐well plate using HiPerfect transfection reagent. C, Effect of simultaneous inhibition of all selected miRNAs (miRNA ‐19a, ‐20a, ‐21, ‐130a, ‐216a and 301a) on HPASMC proliferation, Cells were transfected antagomir (AmiR) mix at final 10 or 50 nmol/L concentration. D, BMPR2 rescue experiment showing the cotransfection of small interfering RNA of BMPR2 (siBMPR2) and AmiR‐216 on C+T‐treated cells. At 24 hours posttransfection, the cells were made quiescent for 24 hours in 0.1% fetal bovine serum containing medium followed by cocaine and Tat (C+T) treatment for 48 hours before performing the cell proliferation assay. *P<0.0001 vs control, @ P<0.0001 vs C+T, P<0.001 vs Scr AmiR+(C+T)+ siBMPR2. Scr indicates scrambled.
Increased Expression of BMPR2 Targeting miRNAs in PASMC From HIV‐Transgenic Rats Exposed to Cocaine
We recently reported decreased protein expression of BMPR2 in hyperproliferative RPASMCs isolated from HIV transgenic (HIV‐Tg) rats exposed to cocaine that corresponded with increased mean pulmonary arterial pressure, right ventricular systolic pressure, and pulmonary vascular remodeling in these rats.23 In order to further validate our cell‐culture findings, we next investigated the expression of the above mentioned selected miRNAs in RPASMCs isolated from 4‐ and 9‐month‐old WT or HIV‐Tg rats treated with or without cocaine as described earlier.23 As shown in Figure 4, RPASMCs from both 4‐ and 9‐month‐old cocaine‐treated HIV‐Tg rats (HIV+Coc) had significant additive increase in the levels of miR‐19a, ‐21, ‐216a, and ‐301a when compared with RPASMCs isolated from saline‐treated WT or HIV‐Tg and from cocaine‐treated WT rats. A significant increase in the expression of miR‐130a was observed in cells isolated from 9 months old HIV+Coc group rats when compared with the WT+Sal group (Figure 4B) although we didn't find any significant change in the levels of miR‐130a in RPASMCs obtained from 4 months old HIV+Coc rats (Figure 4A). We next assessed the role of novel miR‐216a and ‐301a miRNAs in the downmodulation of BMPR2 protein expression in RPASMCs of HIV+Coc group rats. Selective inhibition of miR‐216a and miR‐301a in RPASMC from both WT and HIV+Coc group rats by transfection of specific antagomirs resulted in no change in the BMPR2 mRNA levels when compared with corresponding scrambled controls (Figure 5A) as was observed in HPASMCs (Figure 2A). However, the BMPR2 protein levels were found to be higher in HIV+Coc as well as WT group cells transfected with miR‐216a and ‐301a antagomirs compared with their respective scrambled antagomir controls (Figure 5B). Furthermore, analysis of proliferation of RPASMCs upon inhibiting miR‐216a and ‐301a showed reduced proliferation in both HIV+Coc and WT groups compared with their respective controls transfected with scrambled miRNA (Figure 5C). Thus, these results on HIV‐Tg rats corroborate our in vitro findings and suggest an important role of BMPR2 targeting miRNAs in cocaine‐ and HIV proteins‐mediated hyperproliferation of pulmonary smooth muscle cells.
Figure 4.
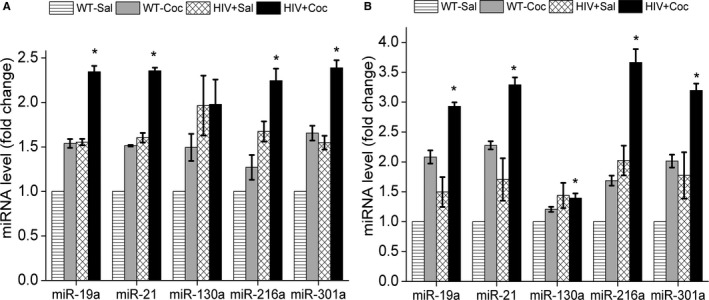
Increased levels of bone morphogenetic protein receptor‐2 (BMPR2) targeting micro RNAs (miRNAs) in rat pulmonary arterial smooth muscle cells (RPASMCs) isolated from human immunodeficiency virus–transgenic rats treated with cocaine. Quantitative real‐time polymerase chain reaction analyses of BMPR2 targeting miRNAs in RPASMC isolated from 4‐month‐old (A) and 9‐month‐old (B) wild‐type (WT) or HIV‐transgenic rats (HIV) treated with cocaine (Coc) or saline (Sal). Data are averaged as mean±SEM of n=3 rats/group; *P<0.05 vs WT+Sal.
Figure 5.
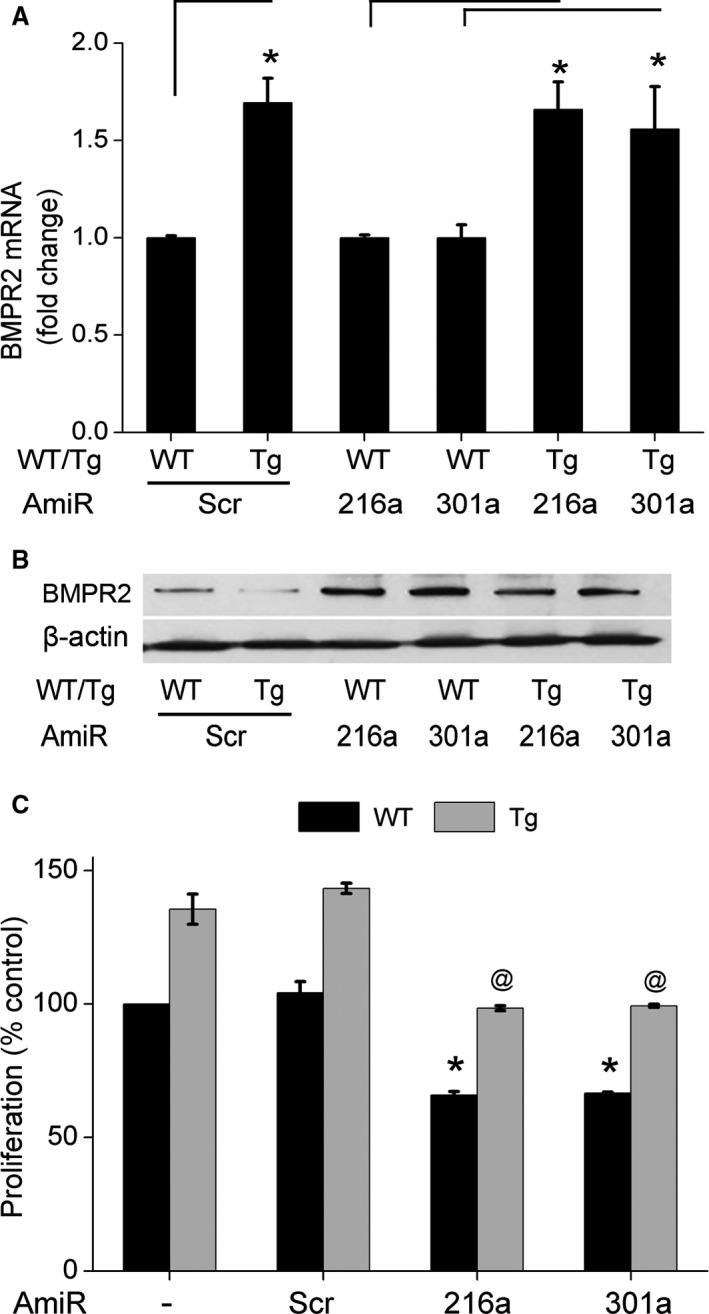
Effect of alterations in the levels of miR‐216a and miR‐301a on bone morphogenetic protein receptor‐2 (BMPR2) mRNA levels (A), BMPR2 protein levels (B), and proliferation (C) of rat pulmonary arterial smooth muscle cells (RPASMCs) isolated from 9‐month‐old wild‐type (WT) or HIV‐transgenic (Tg) rats treated with cocaine (n=3/group). RPASMCs (5×104 cells/well) were reverse transfected for 3 hours with antagomirs (AmiR) for miR‐216a and miR‐301a or with scrambled control (Scr) followed by quantitative real time polymerase chain reaction, Western blot, or proliferation analyses as described earlier.23 A, *P<0.05, vs WT. B, For Western blot analysis, sample for each group was prepared by pooling RPASMC from 3 different animals transfected with AmiRs against miR‐216a or ‐301a. Data are averaged as mean±SEM (n=3 rats/group) treated with the AmiRs. C, *P<0.05 vs WT Scr and @ P<0.05 vs HIV+Coc Scr.
MiR‐216a and miR‐301a Mediate Inhibition of BMPR2 Expression by Directly Binding to Its 3′ UTR
Next we checked whether putative BMPR2 targeting miRNAs—miR‐216a‐5p and miR‐301a‐3p—directly bind to 3′UTR of BMPR2 by performing a luciferase reporter assay. A plasmid containing the 3′UTR of BMPR2 downstream to firefly luciferase gene was used for this assay (Figure 6A) as mentioned in detail in the Materials and Methods section. Cotransfection of BMPR2 3′UTR‐luciferase plasmid with different concentrations of mimics for either miR‐216a or ‐301a resulted in decreased luciferase reporter expression at both 24 hours (Figure 6B and 6C) and 48 hours (Figure S4A and S4B) posttransfection when compared with the cells transfected with either the BMPR2 3′‐UTR construct alone or in combination with scrambled control. This miR‐216a or ‐301a mimics‐mediated decrease in luciferase expression was prevented by the co‐transfection of mimics with their corresponding antagomirs (Figure 6B and 6C). On the other hand, the co‐transfection of control luciferase reporter plasmid (lacking BMPR2 3′UTR) with mimics and/or antagomirs showed no change in the luciferase expression levels (Figure S4C). These observations support that the miRNAs miR‐216a and ‐301a are able to bind to 3′UTR of BMPR2 specifically and negatively affect its expression in HPASMCs.
Figure 6.
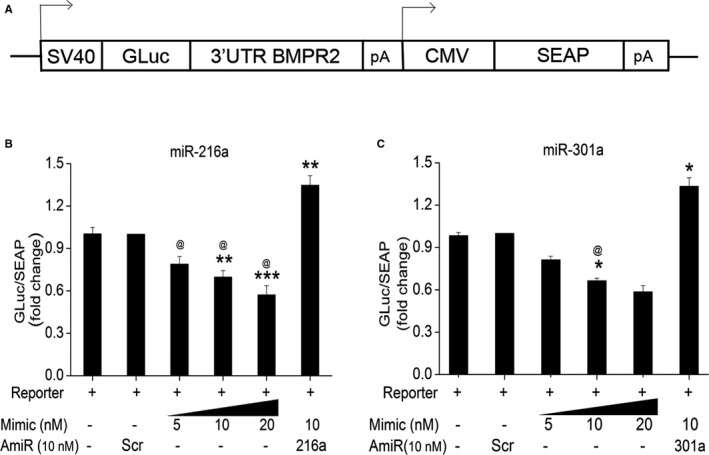
Luciferase reporter assay showing the outcome of miR‐216a and miR‐301a binding to 3′untranslated region (UTR) of bone morphogenetic protein receptor‐2. A, Scheme of luciferase construct. Human pulmonary arterial smooth muscle cells were seeded at 5×105 per well on 24‐well plates 24 hours before the cotransfection of either pEZX‐MT05‐A/B plasmid containing the predicted binding sequence for miR‐216a‐5p (B) or miR‐301a‐3p (C) with or without various concentrations of corresponding mimics (5, 10, and 20 nmol/L) and/or 10 nmol/L antagomirs (AmiR). The culture medium was collected at 24 hours post cotransfection and the amount of Gaussia luciferase (GLuc) and secreted alkaline phosphatase (SEAP) activities were measured. GLuc to SEAP ratio was calculated as a direct effect of miRNA binding mediated inhibition. *P<0.05, **P<0.01, ***P<0.001 vs scrambled control (Scr cont). @ P<0.0001 vs AmiR+Mimic combined treatment. CMV indicates cytomegalovirus; pA, poly‐adenylation; and SV40, simian virus 40.
MiR‐216 and ‐301a miRNAs Specifically Inhibit Translation of BMPR2 Without Any Effect on RNA Levels
To further assess the ability of miR‐216a‐5p and miR‐301a‐3p to inhibit translation independent of mRNA degradation, we investigated translation in a cell‐free environment with a controlled mRNA amount. We designed an in vitro translation experiment where we utilized different luciferase reporter mRNAs with and without the specific miRNA binding sites positioned downstream to the firefly luciferase (Figure 7A). Renilla luciferase reporter mRNA (RL0) was used as an internal control and was added to all the in vitro translation reactions. Four different combinations of in vitro translation reactions were used for comparison: Renilla luciferase reporter mRNA RL0 was combined either with control firefly luciferase reporter with no miRNA binding sites (FL0) or with a firefly luciferase reporter with 6 times repetitive sequence of miRNA binding sites specific for 216a (FL216) or 301a (FL301). The same experimental conditions were maintained for both miR‐216a and miR‐301a, with mRNA reporter to the respective mimic's molar ratio of 1:6. The results showed 50% to 60% significant inhibition of translation of firefly luciferase in the test reaction containing the specific mRNA reporter (FL216 or FL301) with binding site for either miR‐216a or ‐301a along with its corresponding miRNA mimic (Figure 7B). Whereas all the 3 controls, FL0 alone, or FL216/FL301 incubated with scrambled mimic or FL0 incubated with either miR‐216a/301a mimic, showed no significant change in the firefly luciferase levels and were comparable to each other (Figure 7B) as demonstrated by dual luciferase assay. Analysis of RNA recovered after in vitro translation reactions by qRT‐PCR showed no change in the levels of mRNA (Figure 7C) in all sets of experimental reactions. The final PCR reaction when loaded on an agarose gel also showed no apparent changes in the firefly luciferase or renilla luciferase amplicons, indicating that the mRNAs reporters added in the reaction remained unchanged (Figure S5A and S5B). Therefore these findings suggest that the observed decrease in the BMPR2 expression in response to miR‐216a or miR‐301a is specific to the translational inhibition mediated by the binding of these specific miRNAs to the BMPR2 3′UTR rather than the possible degradation of a BMPR2 mRNA.
Figure 7.
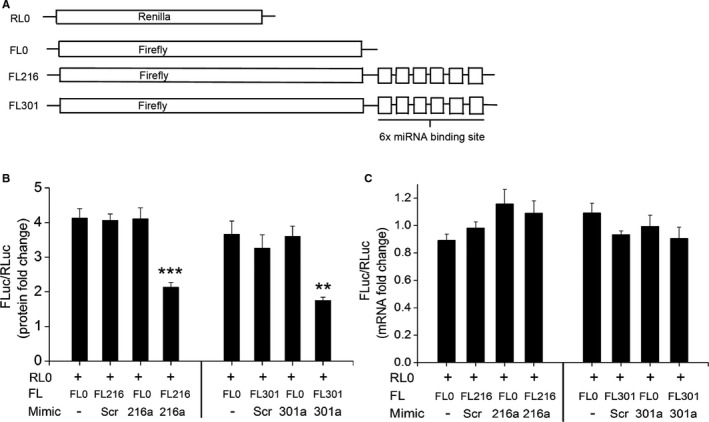
In vitro translation studies for miRNA‐mediated inhibition of translation. A, Scheme of mRNA reporters used for an experiment. B, Firefly mRNA reporter with the binding site for either miR‐216a (FL216) or miR‐301a (FL301) was incubated with the corresponding micro RNA (miRNA) mimics during in vitro translation reaction. Analyses show the firefly luciferase activity normalized to the renilla luciferase activity as measured by dual luciferase assay. C, Quantitative real‐time polymerase chain reaction for firefly luciferase mRNA normalized to renilla luciferase mRNA isolated from the in vitro translation reactions. **P<0.05, ***P<0.001 vs other reaction controls. FL0 indicates firefly and RL0 indicates renilla luciferase reporters with no miRNA binding sites, respectively; Scr, scrambled control.
Transcriptional Upregulation of miR‐216a in Cocaine‐ and Tat‐Treated HPASMC
Next we measured the pre‐miR levels of miR‐216a in order to determine whether the observed C+T‐mediated upregulation of miR‐216a is at the transcriptional level or at the miRNA processing level. We observed moderate but significant upregulation in the level of pre‐miR‐216a in HPASMC after combined C+T treatment for 24 hours (Figure 8A). However, the expression levels of pre‐miR‐216a in the individual treatment of either cocaine or Tat protein did not change. Since miRNA expression is known to alter in response to oxidative stress or viral proteins41, 42, 43 and both Tat44, 45, 46, 47 and cocaine48, 49, 50 are known to induce oxidative stress, we next investigated whether alteration in the levels of miR‐216a in response to Tat and/or cocaine is through reactive oxygen species. As shown in Figure 8B, treatment of cells with cocaine and Tat in the presence of antioxidants prevented the C+T‐mediated increase in the precursor or mature miR‐216a, indicating the involvement of reactive oxygen species signaling in the transcriptional regulation of miR‐216a. Given that miR‐216a has been reported earlier to be regulated in response to TGF‐β signaling,51 we decided to check the levels of miR‐216a in cells treated with C+T in the presence of TGF‐β receptor‐1 inhibitor. Indeed, we observed inhibition in the C+T‐mediated augmentation of miR‐216a levels in the presence of TGFβ receptor‐1 inhibitor (Figure 8C). Contrary to this, addition of TGF‐β ligand upregulated the levels of miR‐216a like that of cocaine and Tat treatment (Figure 8C). So, we conclude here that the observed cocaine‐ and Tat‐induced increase in the levels of miR‐216a involves TGF‐β and reactive oxygen species–mediated transcriptional regulation. In parallel, we also investigated expression of miR‐301a in response to TGF‐β receptor‐1 inhibition of cocaine‐ and Tat‐treated cells but observed no significant alterations in the levels of miR‐301a (data not shown). Nevertheless, we did find a significant drop in the C+T‐mediated increase in pre‐ and mature‐miR301a (Figure S6).
Figure 8.
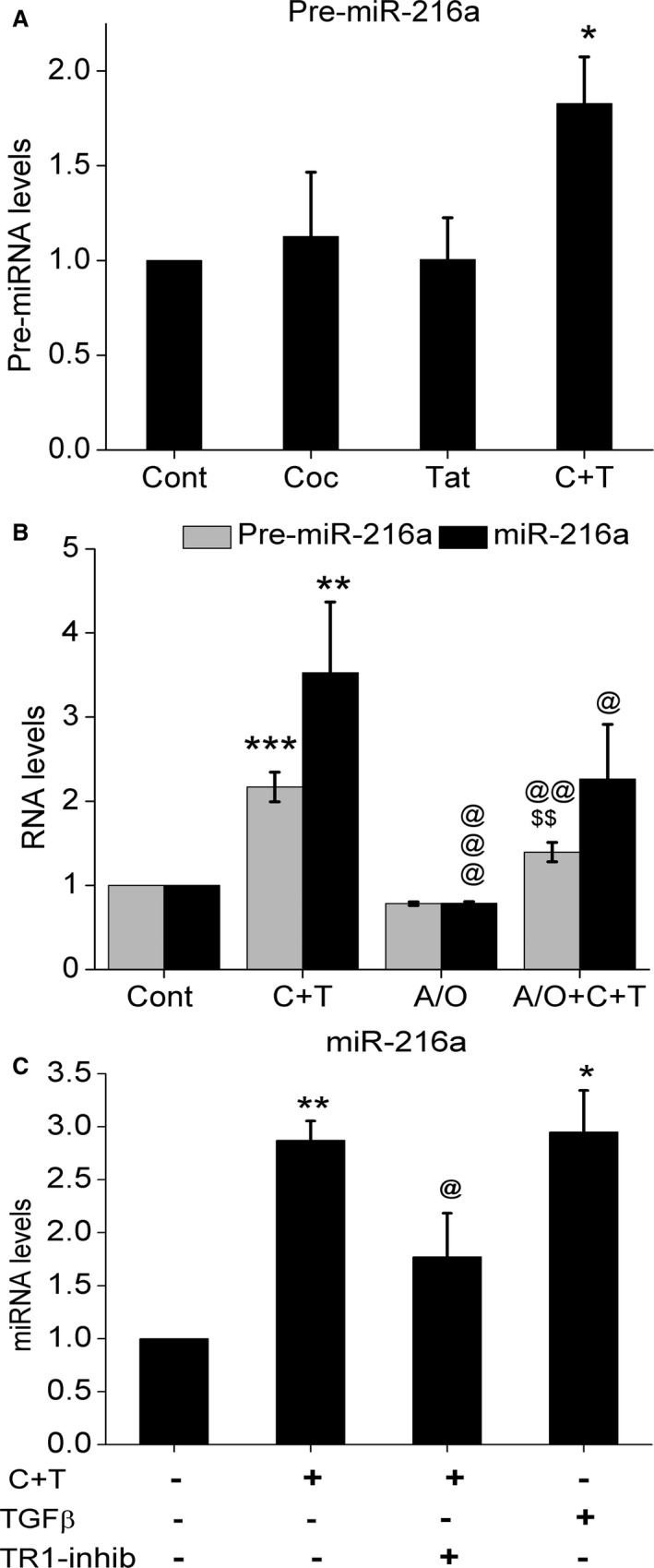
Oxidative stress and transforming growth factor (TGF)‐β signaling mediated regulation of micro RNA‐216a expression. A, Levels of pre‐miR216a in cocaine and/or Tat‐treated human pulmonary arterial smooth muscle cells (HPASMCs). B, Levels of pre‐ and mature‐miR216a in cocaine‐ and Tat‐treated HPASMCs with or without antioxidant pretreatment. C, Levels of mature miR‐216a in response to TGF‐β ligand treatment or TGF‐β receptor‐1 inhibition of C+T‐treated cells. *P<0.05, **P<0.01, ***P<0.001 vs Cont (control), @ P<0.05, @@ P<0.01, @@@ P<0.0001 vs C+T (cocaine and Tat treatment), $$P<0.05 vs A/O (Antioxidant cocktail).
Discussion
HIV infection and cocaine abuse have been identified as independent factors that cause pulmonary hypertension52 and other cardiovascular complications such as atherosclerosis and hypertension,21, 53 with simultaneous exposure to both resulting in further increase in the prevalence of these complications with worse outcomes.17, 18, 19 We earlier reported enhanced pulmonary vascular remodeling with accelerated smooth muscle proliferation in lungs from HIV+intravenous drug user patients who were mainly cocaine abusers and in lungs from HIV‐Tg rats treated with cocaine that had elevated mean pulmonary arterial pressure and right ventricular systolic pressure.22, 23, 26, 54 Defect in the expression or function of BMPR2 protein contributes to the enhanced proliferation of vascular smooth muscle cells and development of hereditary and idiopathic pulmonary hypertension.54, 55 Likewise, we also reported attenuation in the expression of BMPR2 protein levels in lungs from HIV+intravenous drug users and in the pulmonary smooth muscle cells isolated from HIV‐Tg rats exposed to cocaine. Interestingly, all our in vivo, ex vivo, and in vitro findings revealed that although the hyperproliferative pulmonary smooth muscle cells on exposure to cocaine and HIV proteins had reduced expression of BMPR2 protein, BMPR2 mRNA levels remained significantly high.22 BMPR2 downregulation has been reported to occur through multiple ways such as posttranscriptional and posttranslational levels in different model systems. Ubiquitinylated degradation of BMPR2 by SMURF and viral K5 ubiquitin ligases have been reported previously.28, 29, 56 Considering the overall reduction of ubiquitination process under HIV infection,57 the miRNA pathway may play an alternative role in the observed BMPR2 downregulation in response to HIV‐1 and cocaine exposure. In extension to our previous findings, in the present study we demonstrate the role of miRNAs in regulating the translation expression of BMPR2 independent of its mRNA degradation in HPASMCs when exposed to cocaine and HIV‐1 protein Tat.
While we found here that both cocaine and HIV‐Tat monotreatments are able to induce a differential expression of 7 out of 12 miRNAs in HPASMCs, we observed a significant additive effect of the combined treatment on the expression of these miRNAs. By using a loss or gain of function studies with miRNA antagomirs or mimics for selected miRNAs, we demonstrated that the BMPR2 targeting miRNAs are capable of either limiting or enhancing the proliferation of HPASMCs, respectively. We observed a significant inverse correlation in the expression levels of miRNAs (miR‐19a, ‐20a, ‐21, ‐130a, ‐216a, and ‐301a) and BMPR2 protein levels in cells treated with cocaine and Tat. However, the mRNA levels of BMPR2 in cells transfected with antagomirs of these specific miRNAs did not change in response to treatments with cocaine and Tat when compared with untransfected controls. Nevertheless, the transfection of cells with miRNA mimics resulted in significantly decreased BMPR2 mRNA levels. This could be attributed to the supraphysiological levels of miRNAs after transfection of mimics that can have an adverse effect on the mRNA levels, unlike the endogenously expressed miRNAs.58 These results were confirmed in RPASMCs isolated from WT and HIV‐Tg rats treated with or without cocaine. An additive increase in the expression of BMPR2 targeting miRNAs was observed in HIV‐Tg rats treated with cocaine compared with HIV‐Tg rats treated with saline or WT rats treated with cocaine. Although miR‐20a has conserved sequence in both rats and humans, no amplification was observed in qRT‐PCR. It may be possible that miR‐20a is expressed at very low levels in RPASMCs so as to make it undetectable by qRT‐PCR.
Of the miRNAs that were selected for analysis, miR‐19a and ‐20a belong to the miRNA 17 to 92 clusters, and they have been previously shown to regulate cell proliferation by targeting BMPR2 and PTEN genes.59 Here we now suggest miR‐130a and miR‐216a also as regulators of PTEN expression. MiR‐21 has also been demonstrated previously to be able to target 3′UTR of the BMPR2 gene in 293T cells. However, contrary to these findings, there are reports that ignore or do not support the ability of miRNAs miR‐19a, ‐21, and ‐130a to target BMPR2 in some cell types.35, 60 Although the miR‐130/301 family of miRNAs has been shown to be induced by multiple triggers of pulmonary hypertension, and antagomirs against these miRNAs reversed the condition of pulmonary hypertension in animal models,33, 34, 61, 62 the ability of miR‐130/301 to directly target BMPR2 mRNA was not evaluated before.
More importantly, we here identify a new BMPR2 targeting miRNA, miR‐216a, which has previously been reported to increase in response to TGF‐β in renal glomerular mesangial cells.51 In addition, miR‐216 has been reported to target SMAD7 and PTEN, which in turn stimulates TGF‐β and PI3K/Akt signaling, respectively, and facilitates hepatocarcinogenesis.40 It is interesting to note that we also observed earlier an increase in the levels of TGF‐β and activation of downstream TGF‐β receptor‐SMAD2/3 signaling in HPASMCs in response to combined treatment of cocaine and Tat.63 Our current experiments with TGF‐β ligand or TGF‐β receptor‐1 inhibitor in cocaine‐ and Tat‐treated hyperproliferative smooth muscle cells also support the regulation of miRNA‐216a by TGF‐β signaling. It is intriguing to note that genomic analysis of miR‐216a gene (Genecards ID: GC02M055988) also showed the presence of an enhancer element for STAT3 transcription factor. JAK/STAT3 is known to be activated by TGF‐β signaling,64 therefore further suggesting transcriptional regulation of miR216a expression. Hence, it is possible that miR‐216a acts as a critical component of the crosstalk between the proliferative TGF‐β receptor arm and antiproliferative BMPR arm of TGF‐β superfamily signaling to regulate proliferation. Alternatively, expression of miRNAs is also known to be regulated in response to changes in cellular redox balance, which plays an important role in cellular growth and differentiation.41, 42, 43 Corroborating to these findings, we also found antioxidant‐mediated partial abrogation of pre‐ and mature miRNA levels of 216a and 301a in HPASMC, therefore suggesting a cumulative effect of reactive oxygen species and TGF‐β signaling in the transcriptional regulation of these miRNAs.
In this study we demonstrate that the binding of miR‐216a and miR‐301 to the 3′UTR of BMPR2 leads to the translational inhibition of BMPR2. The luciferase reporter assay performed in HPASMCs confirmed that the BMPR2 3′UTR is the direct target of miR‐216a and miR‐301a and these miRNAs are capable of efficiently regulating the expression of the target sequence. The suppression of firefly luciferase expression by miR‐216a and miR‐301a mimics was observed to be concentration dependent. Combination of specific mimics along with the corresponding antagomirs reversed the miR‐216a and miR‐301a mimics‐mediated reduced expression of luciferase reporter. The outcome of the binding of miRNAs to its targets has been reported to result in translational inhibition with or without its target mRNA degradation. The ability of the miRNAs to degrade its mRNAs is in part predicted by the sequence complementarity between miRNA and mRNA. However, not necessarily all miRNAs function that way.65 Previous studies used in vitro translation assays to delineate the mechanisms of miRNA‐mediated posttranscriptional regulation where they observed that the complete complementarity of miRNAs negatively impact the levels of the target mRNA, whereas the partially complement miRNA was shown to elicit only the translational repression.30, 31 The sequences of miR‐216a and miR‐301a were predicted to be partially complementary to 3′UTR of BMPR2 by TargetScan 7.0, and we also used a cell‐free in vitro translation system with firefly luciferase mRNA reporters with or without the binding sites for either miR‐216a or ‐301a to deduce the early steps involved in the miRNA‐mediated suppression of BMPR2 protein expression in an RNA controlled environment. Our findings suggest that the observed inhibition in the protein levels of BMPR2 in cocaine‐ and Tat‐treated HPASMCs is not mediated by the degradation of mRNA on binding of miRNAs miR‐216a and ‐301a to its 3′UTR but is a result of translational inhibition.
In summary, exposure of pulmonary arterial smooth muscle cells to cocaine and HIV proteins results in induction of BMPR2 targeting miRNA levels including miR‐19a, ‐20a, ‐21, ‐130a, 216a, and ‐301a. Inhibition of these miRNAs prevented the cocaine and HIV‐Tat mediated reduction in the BMPR2 protein expression and hyperproliferation of HPASMCs, whereas overexpression of miRNAs further potentiated the cocaine‐ and Tat‐mediated effects. Finally, we for the first time demonstrate the ability of miR‐216a and ‐301a to efficiently bind to 3′UTR of BMPR2 and inhibit translation without the degradation of BMPR2 mRNA, and suggest involvement of oxidative stress and TGF‐β‐mediated signaling in cocaine‐ and Tat‐mediated upregulation of miRNA‐216a expression. Further studies, using in vivo models of PAH, with antagomirs targeting miR‐216a alone or in combination with other proliferative miRNAs, are vouched to validate effectiveness of miRNA targeted therapy in reversing the condition of pulmonary arterial hypertension.
Sources of Funding
This work was supported by NIH grants: R01DA034542, R01DA042715, R01HL129875, and American Heart Association's Scientist Development grant: 11SDG7500016 awarded to Dhillon.
Disclosures
None.
Supporting information
Table S1. Primers Used for the qRT‐PCR
Table S2. List of BMPR2 Targeting miRNAs Selected Using TargetScan 7.1 Software
Figure S1. Normalized threshold cycle (Ct) values of the miRNAs in untreated control and cocaine‐ and Tat‐treated human pulmonary arterial smooth muscle cells (HPASMCs) at 48 hours. *P<0.05, **P<0.01 vs Cont.
Figure S2. Levels of miRNAs in cells transfected with antagomirs (A) and mimics (B) of miR‐130a, and ‐216a. Quantitative real‐time (qRT) polymerase chain reaction was performed using total RNA isolated after 48‐hour treatment of transfected human pulmonary arterial smooth muscle cells (HPASMCs) with cocaine and Tat. The graph represents the average of 2 independent experiments.
Figure S3. Expression levels of phosphatase and tensin homolog (PTEN) protein in human pulmonary arterial smooth muscle cells (HPASMCs) in the presence or absence of selected antagomirs. Bone morphogenetic protein receptor‐2 (BMPR2) blots were re‐probed to analyze the effect of antagomirs on PTEN expression. A, Representative Western blot image and (B) densitometry analysis of PTEN using ImageJ software. @ P≤0.05 vs Scr+C+T. *P<0.05 vs Cont.
Figure S4. Luciferase reporter assay showing the outcome of miR‐216a (A) and miR‐301a (B) binding to 3′ untranslated region (UTR) of BMPR2. Human pulmonary arterial smooth muscle cells (HPASMCs) were seeded at 0.5×105 per well on 24‐well plates 24 hours before the cotransfection of pEZX‐MT05‐B or pEZX‐MT05‐A plasmid containing the binding sequence for miR‐216a‐5p (A) or miR‐301a‐3p (B) or with a plasmid without bone morphogenetic protein receptor‐2 (BMPR2) 3′UTR (C) with various concentrations of corresponding mimics (5, 10, and 20 nmol/L) and/or 10 nmol/L antagomirs. The culture medium was collected at 48 hours post co‐transfection and the amount of Gaussia luciferase (GLuc) and secreted alkaline phosphatase (SEAP) activities were measured. Gluc‐to‐SEAP ratio was calculated as a direct effect of miRNA binding mediated inhibition. *P<0.05, **P<0.01vs Scr cont. @ P<0.05, @@ P<0.001 vs AmiR and MmiR combined treatment.
Figure S5. Representative agarose gel image showing the quantitative real‐time (qRT) polymerase chain reaction (PCR) products of the mRNA reporters that were isolated after in vitro translation reactions specific to miR‐216a (A) and miR‐301a (B). qRT‐PCR was performed using renilla or firefly primers for each PCR reaction, and samples obtained post‐PCR were resolved on 1.5% Agarose gel. Scr indicates scrambled.
Figure S6. Oxidative stress and transforming growth factor (TGF)‐β signaling mediated regulation of miRNA‐301a expression. A, Levels of pre‐miR‐301a in cocaine‐ and/or Tat‐treated human pulmonary arterial smooth muscle cells (HPASMCs) (A); and of pre‐ and mature ‐miR301a in cocaine‐ and Tat‐treated HPASMCs with or without antioxidant pretreatment (B). *P<0.05, ***P<0.01, ****P<0.001 vs Cont, @@ P<0.01 vs C+T, $ P<0.05, $$ P<0.01 vs A/O.
(J Am Heart Assoc. 2018;7:e008472 DOI: 10.1161/JAHA.117.008472.)29478969
Preliminary findings from the related work were published as an abstract for the American Thoracic Society International Conference, May 19 to 24, 2017, in Washington, DC.
References
- 1. Montani D, Günther S, Dorfmüller P, Perros F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud‐Macari E, Price LC. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013;8:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. [DOI] [PubMed] [Google Scholar]
- 4. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. [DOI] [PubMed] [Google Scholar]
- 5. Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–1063. [DOI] [PubMed] [Google Scholar]
- 6. King KE, Iyemere VP, Weissberg PL, Shanahan CM. Kruppel‐like factor 4 (KLF4/GKLF) is a target of bone morphogenetic proteins and transforming growth factor beta 1 in the regulation of vascular smooth muscle cell phenotype. J Biol Chem. 2003;278:11661–11669. [DOI] [PubMed] [Google Scholar]
- 7. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor‐beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790–795. [DOI] [PubMed] [Google Scholar]
- 8. Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, West J. Interaction of interleukin‐6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–L1479. [DOI] [PubMed] [Google Scholar]
- 9. Niakara A, Drabo YJ, Kambire Y, Nebie LV, Kabore NJ, Simon F. [Cardiovascular diseases and HIV infection: study of 79 cases at the National Hospital of Ouagadougou (Burkina Faso)]. Bull Soc Pathol Exot. 2002;95:23–26. [PubMed] [Google Scholar]
- 10. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud‐Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–1030. [DOI] [PubMed] [Google Scholar]
- 11. Morris A, Gingo MR, George MP, Lucht L, Kessinger C, Singh V, Hillenbrand M, Busch M, McMahon D, Norris KA, Champion HC, Gladwin MT, Zhang Y, Steele C, Sciurba FC. Cardiopulmonary function in individuals with HIV infection in the antiretroviral therapy era. AIDS. 2012;26:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isasti G, Moreno T, Perez I, Cabrera F, Palacios R, Santos J. High prevalence of pulmonary arterial hypertension in a cohort of asymptomatic HIV‐infected patients. AIDS Res Hum Retroviruses. 2013;29:231–234. [DOI] [PubMed] [Google Scholar]
- 13. Isasti G, Perez I, Moreno T, Cabrera F, Palacios R, Santos J. Echocardiographic abnormalities and associated factors in a cohort of asymptomatic HIV‐infected patients. AIDS Res Hum Retroviruses. 2013;29:20–24. [DOI] [PubMed] [Google Scholar]
- 14. ten Freyhaus H, Vogel D, Lehmann C, Kummerle T, Wyen C, Fatkenheuer G, Rosenkranz S. Echocardiographic screening for pulmonary arterial hypertension in HIV‐positive patients. Infection. 2014;42:737–741. [DOI] [PubMed] [Google Scholar]
- 15. Rasoulinejad M, Moradmand Badie S, Salehi MR, Seyed Alinaghi SA, Dehghan Manshadi SA, Zakerzadeh N, Foroughi M, Jahanjo Amin Abad F, Moradmand Badie B. Echocardiographic assessment of systolic pulmonary arterial pressure in HIV‐positive patients. Acta Med Iran. 2014;52:827–830. [PubMed] [Google Scholar]
- 16. Hind C. Pulmonary complications of intravenous drug misuse. 2. Infective and HIV related complications. Thorax. 1990;45:957–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwarze‐Zander C, Pabst S, Hammerstingl C, Ohlig J, Wasmuth JC, Boesecke C, Stoffel‐Wagner B, Carstensen A, Nickenig G, Strassburg CP, Rockstroh JK, Skowasch D, Schueler R. Pulmonary hypertension in HIV infection: a prospective echocardiographic study. HIV Med. 2015;16:578–582. [DOI] [PubMed] [Google Scholar]
- 18. Quezada M, Martin‐Carbonero L, Soriano V, Vispo E, Valencia E, Moreno V, de Isla LP, Lennie V, Almeria C, Zamorano JL. Prevalence and risk factors associated with pulmonary hypertension in HIV‐infected patients on regular follow‐up. AIDS. 2012;26:1387–1392. [DOI] [PubMed] [Google Scholar]
- 19. Opravil M, Sereni D. Natural history of HIV‐associated pulmonary arterial hypertension: trends in the HAART era. AIDS. 2008;22(suppl 3):S35–S40. [DOI] [PubMed] [Google Scholar]
- 20. Zuber J‐P, Calmy A, Evison JM, Hasse B, Schiffer V, Wagels T, Nuesch R, Magenta L, Ledergerber B, Jenni R. Pulmonary arterial hypertension related to HIV infection: improved hemodynamics and survival associated with antiretroviral therapy. Clin Infect Dis. 2004;38:1178–1185. [DOI] [PubMed] [Google Scholar]
- 21. Degano B, Guillaume M, Savale L, Montani D, Jais X, Yaici A, Le Pavec J, Humbert M, Simonneau G, Sitbon O. HIV‐associated pulmonary arterial hypertension: survival and prognostic factors in the modern therapeutic era. AIDS. 2010;24:67–75. [DOI] [PubMed] [Google Scholar]
- 22. Dalvi P, O'Brien‐Ladner A, Dhillon N. Downregulation of bone morphogenetic protein receptor axis during HIV‐1 and cocaine‐mediated pulmonary smooth muscle hyperplasia: implications for HIV‐related pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2013;33:2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dalvi P, Spikes L, Allen J, Gupta VG, Sharma H, Gillcrist M, Montes de Oca J, O'Brien‐Ladner A, Dhillon NK. Effect of cocaine on pulmonary vascular remodeling and hemodynamics in human immunodeficiency virus‐transgenic rats. Am J Respir Cell Mol Biol. 2016;55:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, O'Brien‐Ladner A, Dhillon NK. Enhanced pulmonary arteriopathy in simian immunodeficiency virus‐infected macaques exposed to morphine. Am J Respir Crit Care Med. 2012;185:1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dalvi P, Wang K, Mermis J, Zeng R, Sanderson M, Johnson S, Dai Y, Sharma G, Ladner AO, Dhillon NK. HIV‐1/cocaine induced oxidative stress disrupts tight junction protein‐1 in human pulmonary microvascular endothelial cells: role of Ras/ERK1/2 pathway. PLoS One. 2014;9:e85246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, Ladner AO. Effect of cocaine on human immunodeficiency virus‐mediated pulmonary endothelial and smooth muscle dysfunction. Am J Respir Cell Mol Biol. 2011;45:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dhillon N, Li F, Tawfik O, Cheney P, O'Brien‐Ladner A. Enhanced pulmonary arteriopathy in SIV‐infected macaques exposed to morphine [abstract]. Am J Respir Crit Care Med. 2010;181:A6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112:1159–1170. [DOI] [PubMed] [Google Scholar]
- 29. Murakami K, Mathew R, Huang J, Farahani R, Peng H, Olson SC, Etlinger JD. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp Biol Med. 2010;235:805–813. [DOI] [PubMed] [Google Scholar]
- 30. Wang B, Love TM, Call ME, Doench JG, Novina CD. Recapitulation of short RNA‐directed translational gene silencing in vitro. Mol Cell. 2006;22:553–560. [DOI] [PubMed] [Google Scholar]
- 31. Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS. MicroRNA inhibition of translation initiation in vitro by targeting the cap‐binding complex eIF4F. Science. 2007;317:1764–1767. [DOI] [PubMed] [Google Scholar]
- 32. Agarwal V, Bell GW, Nam J‐W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM. The microRNA‐130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem. 2015;290:2069–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qin W, Zhao B, Shi Y, Yao C, Jin L, Jin Y. BMPRII is a direct target of miR‐21. Acta Biochim Biophys Sin (Shanghai). 2009;41:618–623. [DOI] [PubMed] [Google Scholar]
- 35. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin‐6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3–microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. [DOI] [PubMed] [Google Scholar]
- 36. Pezzolesi MG, Platzer P, Waite KA, Eng C. Differential expression of PTEN‐targeting microRNAs miR‐19a and miR‐21 in Cowden syndrome. Am J Hum Genet. 2008;82:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dhar S, Kumar A, Rimando AM, Zhang X, Levenson AS. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomirs of the miR‐17 family in prostate cancer. Oncotarget. 2015;6:27214–27226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feng Y, Zhou S, Li G, Hu C, Zou W, Zhang H, Sun L. Nuclear factor‐kappaB‐dependent microRNA‐130a upregulation promotes cervical cancer cell growth by targeting phosphatase and tensin homolog. Arch Biochem Biophys. 2016;598:57–65. [DOI] [PubMed] [Google Scholar]
- 39. Pogribny IP, Muskhelishvili L, Tryndyak VP, Beland FA. The tumor‐promoting activity of 2‐acetylaminofluorene is associated with disruption of the p53 signaling pathway and the balance between apoptosis and cell proliferation. Toxicol Appl Pharmacol. 2009;235:305–311. [DOI] [PubMed] [Google Scholar]
- 40. Xia H, Ooi LL, Hui KM. MicroRNA‐216a/217‐induced epithelial‐mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. [DOI] [PubMed] [Google Scholar]
- 41. Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, Degraff W, Cook J, Harris CC, Gius D, Mitchell JB. Ionizing radiation‐induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30:716–723. [DOI] [PubMed] [Google Scholar]
- 43. Mukerjee R, Chang JR, Del Valle L, Bagashev A, Gayed MM, Lyde RB, Hawkins BJ, Brailoiu E, Cohen E, Power C, Azizi SA, Gelman BB, Sawaya BE. Deregulation of microRNAs by HIV‐1 Vpr protein leads to the development of neurocognitive disorders. J Biol Chem. 2011;286:34976–34985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andras IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, Toborek M. Signaling mechanisms of HIV‐1 Tat‐induced alterations of claudin‐5 expression in brain endothelial cells. J Cereb Blood Flow Metab. 2005;25:1159–1170. [DOI] [PubMed] [Google Scholar]
- 45. Price TO, Ercal N, Nakaoke R, Banks WA. HIV‐1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005;1045:57–63. [DOI] [PubMed] [Google Scholar]
- 46. Price TO, Uras F, Banks WA, Ercal N. A novel antioxidant N‐acetylcysteine amide prevents gp120‐ and Tat‐induced oxidative stress in brain endothelial cells. Exp Neurol. 2006;201:193–202. [DOI] [PubMed] [Google Scholar]
- 47. Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A. HIV‐Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem. 2003;84:169–179. [DOI] [PubMed] [Google Scholar]
- 48. Aksenov MY, Aksenova MV, Nath A, Ray PD, Mactutus CF, Booze RM. Cocaine‐mediated enhancement of Tat toxicity in rat hippocampal cell cultures: the role of oxidative stress and D1 dopamine receptor. Neurotoxicology. 2006;27:217–228. [DOI] [PubMed] [Google Scholar]
- 49. Mo W, Singh AK, Arruda JA, Dunea G. Role of nitric oxide in cocaine‐induced acute hypertension. Am J Hypertens. 1998;11:708–714. [DOI] [PubMed] [Google Scholar]
- 50. Lee YW, Hennig B, Fiala M, Kim KS, Toborek M. Cocaine activates redox‐regulated transcription factors and induces TNF‐alpha expression in human brain endothelial cells. Brain Res. 2001;920:125–133. [DOI] [PubMed] [Google Scholar]
- 51. Kato M, Wang L, Putta S, Wang M, Yuan H, Sun G, Lanting L, Todorov I, Rossi JJ, Natarajan R. Post‐transcriptional up‐regulation of Tsc‐22 by Ybx1, a target of miR‐216a, mediates TGF‐β‐induced collagen expression in kidney cells. J Biol Chem. 2010;285:34004–34015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Restrepo CS, Carrillo JA, Martínez S, Ojeda P, Rivera AL, Hatta A. Pulmonary complications from cocaine and cocaine‐based substances: imaging manifestations 1. Radiographics. 2007;27:941–956. [DOI] [PubMed] [Google Scholar]
- 53. Su J, Li J, Li W, Altura BT, Altura BM. Cocaine induces apoptosis in primary cultured rat aortic vascular smooth muscle cells: possible relationship to aortic dissection, atherosclerosis, and hypertension. Int J Toxicol. 2004;23:223–227. [DOI] [PubMed] [Google Scholar]
- 54. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF‐β receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. [DOI] [PubMed] [Google Scholar]
- 55. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor‐II gene. Am J Hum Genet. 2000;67:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Durrington HJ, Upton PD, Hoer S, Boname J, Dunmore BJ, Yang J, Crilley TK, Butler LM, Blackbourn DJ, Nash GB, Lehner PJ, Morrell NW. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J Biol Chem. 2010;285:37641–37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arora S, Verma S, Banerjea AC. HIV‐1 Vpr redirects host ubiquitination pathway. J Virol. 2014;88:9141–9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jin HY, Gonzalez‐Martin A, Miletic AV, Lai M, Knight S, Sabouri‐Ghomi M, Head SR, Macauley MS, Rickert RC, Xiao C. Transfection of microRNA mimics should be used with caution. Front Genet. 2015;6:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andreas E, Hoelker M, Neuhoff C, Tholen E, Schellander K, Tesfaye D, Salilew‐Wondim D. MicroRNA 17‐92 cluster regulates proliferation and differentiation of bovine granulosa cells by targeting PTEN and BMPR2 genes. Cell Tissue Res. 2016;366:219–230. [DOI] [PubMed] [Google Scholar]
- 60. Luo T, Cui S, Bian C, Yu X. Crosstalk between TGF‐β/Smad3 and BMP/BMPR2 signaling pathways via miR‐17‐92 cluster in carotid artery restenosis. Mol Cell Biochem. 2014;389:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO. Systems‐level regulation of microRNA networks by miR‐130/301 promotes pulmonary hypertension. J Clin Invest. 2014;124:3514–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang Y‐Y. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ‐miR‐130/301 circuit. Cell Rep. 2015;13:1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dalvi P, Sharma H, Konstantinova T, Sanderson M, O'Brien‐Ladner A, Dhillon N. Hyperactive TGF‐β signaling in smooth muscle cells exposed to HIV protein(s) and cocaine: Role in pulmonary vasculopathy. Sci Rep. 2017;7:10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tang LY, Heller M, Meng Z, Yu LR, Tang Y, Zhou M, Zhang YE. Transforming growth factor‐beta (TGF‐beta) directly activates the JAK1‐STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J Biol Chem. 2017;292:4302–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wilczynska A, Bushell M. The complexity of miRNA‐mediated repression. Cell Death Differ. 2015;22:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Used for the qRT‐PCR
Table S2. List of BMPR2 Targeting miRNAs Selected Using TargetScan 7.1 Software
Figure S1. Normalized threshold cycle (Ct) values of the miRNAs in untreated control and cocaine‐ and Tat‐treated human pulmonary arterial smooth muscle cells (HPASMCs) at 48 hours. *P<0.05, **P<0.01 vs Cont.
Figure S2. Levels of miRNAs in cells transfected with antagomirs (A) and mimics (B) of miR‐130a, and ‐216a. Quantitative real‐time (qRT) polymerase chain reaction was performed using total RNA isolated after 48‐hour treatment of transfected human pulmonary arterial smooth muscle cells (HPASMCs) with cocaine and Tat. The graph represents the average of 2 independent experiments.
Figure S3. Expression levels of phosphatase and tensin homolog (PTEN) protein in human pulmonary arterial smooth muscle cells (HPASMCs) in the presence or absence of selected antagomirs. Bone morphogenetic protein receptor‐2 (BMPR2) blots were re‐probed to analyze the effect of antagomirs on PTEN expression. A, Representative Western blot image and (B) densitometry analysis of PTEN using ImageJ software. @ P≤0.05 vs Scr+C+T. *P<0.05 vs Cont.
Figure S4. Luciferase reporter assay showing the outcome of miR‐216a (A) and miR‐301a (B) binding to 3′ untranslated region (UTR) of BMPR2. Human pulmonary arterial smooth muscle cells (HPASMCs) were seeded at 0.5×105 per well on 24‐well plates 24 hours before the cotransfection of pEZX‐MT05‐B or pEZX‐MT05‐A plasmid containing the binding sequence for miR‐216a‐5p (A) or miR‐301a‐3p (B) or with a plasmid without bone morphogenetic protein receptor‐2 (BMPR2) 3′UTR (C) with various concentrations of corresponding mimics (5, 10, and 20 nmol/L) and/or 10 nmol/L antagomirs. The culture medium was collected at 48 hours post co‐transfection and the amount of Gaussia luciferase (GLuc) and secreted alkaline phosphatase (SEAP) activities were measured. Gluc‐to‐SEAP ratio was calculated as a direct effect of miRNA binding mediated inhibition. *P<0.05, **P<0.01vs Scr cont. @ P<0.05, @@ P<0.001 vs AmiR and MmiR combined treatment.
Figure S5. Representative agarose gel image showing the quantitative real‐time (qRT) polymerase chain reaction (PCR) products of the mRNA reporters that were isolated after in vitro translation reactions specific to miR‐216a (A) and miR‐301a (B). qRT‐PCR was performed using renilla or firefly primers for each PCR reaction, and samples obtained post‐PCR were resolved on 1.5% Agarose gel. Scr indicates scrambled.
Figure S6. Oxidative stress and transforming growth factor (TGF)‐β signaling mediated regulation of miRNA‐301a expression. A, Levels of pre‐miR‐301a in cocaine‐ and/or Tat‐treated human pulmonary arterial smooth muscle cells (HPASMCs) (A); and of pre‐ and mature ‐miR301a in cocaine‐ and Tat‐treated HPASMCs with or without antioxidant pretreatment (B). *P<0.05, ***P<0.01, ****P<0.001 vs Cont, @@ P<0.01 vs C+T, $ P<0.05, $$ P<0.01 vs A/O.