

Abstract
Advanced glycation end products (AGEs) play a role in pathogenesis of diabetic nephropathy (DN). Myo-inositol oxygenase (MIOX) has been implicated in tubulointerstitial injury in the context of DN. We investigated the effect of AGEs on MIOX expression and delineated mechanisms that lead to tubulointerstitial injury. The status of MIOX, RAGE, and relevant cellular signaling pathways activated following AGE:RAGE interaction was examined in tubular cells and kidneys of AGE-BSA-treated mice. A solid-phase assay revealed an enhanced binding of RAGE with AGE-BSA, AGE-laminin, and AGE-collagen IV. The cells treated with AGE-BSA had increased MIOX activity/expression and promoter activity. This was associated with activation of various signaling kinases of phosphatidylinositol 3-kinase (PI3K)-AKT pathway and increased expression of NF-κB, transforming growth factor (TGF)-β, and fibronectin, which was negated with the treatment of MIOX/RAGE- small interfering (si) RNA. Concomitant with MIOX upregulation, there was an increased generation of reactive oxygen species (ROS), which could be abrogated with MIOX/RAGE- siRNA treatment. The kidneys of mice treated with AGE-BSA had significantly high urinary A/C ratio, upregulation of MIOX, RAGE and NF-κB, along with influx of monocytes into the tubulointerstitium, increased the expression of MCP-1, IL-6, and fibronectin and increased the generation of ROS. Such perturbations were abrogated with the concomitant treatment of inhibitors MIOX or RAGE (d-glucarate and FPS-ZM1). These studies support a role of AGE:RAGE interaction in the activation of PI3K-AKT pathway and upregulation of MIOX, with excessive generation of ROS, increased expression of NF-κB, inflammatory cytokines, TGF-β, and fibronectin. Collectively, these observations highlight the relevance of the biology of MIOX in the contribution toward tubulointerstitial injury in DN.
Keywords: diabetes, glycation, myo-inositol oxygenase, reactive oxygen species
INTRODUCTION
The pathogenesis of diabetic nephropathy (DN) is multifactorial, and the advanced glycation end products (AGEs) have been postulated to play a vital role in the progression of this disease process (35). The AGEs bind to the receptor of advanced glycation end products (RAGE) and activate a multitude of intracellular signaling cascades (23, 26). The RAGE is a multiligand pattern recognition receptor, but binds to AGEs with high affinity. The activation of RAGE initiates certain inadvertent signaling pathways, such as PKC, JNK, and phosphatidylinositol 3-kinase (PI3K) (45). Apparently, this leads to increased cellular oxidative stress and activation of transcription factor NF-κB, and that, in turn, triggers an increased synthesis of extracellular matrix (ECM) proteins (23). As a consequence of these cellular perturbations, the tubular cells, besides the glomerulus, are likely to undergo apoptosis with worsening of tubular homeostasis, accentuation of renal injury and acceleration of the progression of diabetic nephropathy (DN). Conceivably, the AGEs adversely affect both the glomerular and tubular compartments of the kidney (59). However, much attention has been given to the glomerular compartment, whereas the tubular compartment has received relatively little attention, although it occupies >80% of renal parenchyma.
Myo-inositol oxygenase (MIOX) is a cytosolic enzyme expressed predominantly in the renal proximal kidney tubules, where it catabolizes myo-inositol (MI) and channels MI into glucuronate-xylulose pathway (2, 54). MIOX is a 33-kDa monomeric single-domain protein with a helical fold, which is distantly related to the diverse HD (histidine-aspartate) domain superfamily (6). MIOX is the only enzyme that catabolizes myo-inositol, and, thus, plausibly acts as an important regulator of plasma inositol concentration in mammals (10). Its upregulation is associated with an intrarenal myo-inositol deficiency, which has been implicated in the acceleration of diabetic nephropathy with increased expression of fibronectin (FN) (61). Previously, we observed upregulation of MIOX in proximal tubular epithelial cells (HK-2) via PI3K pathway under high glucose ambience (39). Its upregulation was also noted to be associated with proximal tubular injury in vivo and that was accompanied with mitochondrial fragmentation, cytochrome-c release and oxidative stress (63). Likewise, its fatty acid-induced upregulation is also associated with increased generation of reactive oxygen species (ROS), apoptosis, and tubular injury (55). Interestingly, overexpression of MIOX has been shown to accentuate the formation of ROS and exacerbation of injury under high glucose ambience in renal tubular cells (51). Moreover, mice overexpressing MIOX were noted to be susceptible to chemical injury that was confined to the proximal tubules and that seemed to be also mediated via excessive generation of ROS (15). Despite the wealth of knowledge available, the role of AGEs or AGE:RAGE interactions in the pathobiology of MIOX relevant to the progression of renal tubulointerstitial injury in the context of diabetic tubulopathy is unknown.
The aim of the present study was to investigate the effect of AGEs derived from modified albumin, laminin, and collagen IV on cellular MIOX expression and to delineate the underlying mechanisms that would identify MIOX’s potential role in the progression of diabetic nephropathy. To accomplish this objective, both in vitro and in vivo experiments were carried out, and the status of molecules involved in various signaling pathways specifically relevant to the pathogenesis of diabetic tubulopathy was examined.
MATERIALS AND METHODS
Antibodies and other reagents.
Antobodies and other reagents were purchased from the following vendors. Their catalog numbers are included in parentheses: Abcam: anti-RAGE (ab37647) and anti-TGF-β1 (ab66073) antibody; Cell Signaling Technology: anti-phospho-NF-κB p65 (Ser-536) (8242S), -PI3K p110α (4249S), -rabbit mAb Akt (9272S), -phospho-Akt (Ser-473) (9271S), -PDK1(3062S) and -phospho-PDK1 (3061S) antibody; Exocell: mouse albumin ELISA kit (1011); Bioassay System: creatinine assay kit (DICT-500); Life Technologies: TO-PRO-3-iodide (T3605); Sigma: purified fatty acid-free BSA (A4612), laminin (L2020), collagen IV (C5533), methylglyoxal (MO252), d-glucaric acid (21236), human kinase RAGE-small interfering (si) RNA (SIHK1924), siRNA universal negative control (SIC001), S-100B protein (S6677), wortmannin (W1628), calphostin (C6303), dihydroethidium (DHE, D7008), recombinant RAGE protein (SRP6051), 2′,7′-dichlorofluorescin diacetate (DCF-DA; D6883), anti-actin (A5441) and -fibronectin (F7387) antibody; OriGene Technologies: MIOX-siRNA (SR310776); Calbiochem: 4-chloro-N-cyclohexyl-N-(phenylmethyl)-benzamide (FPS-ZM1, RAGE antagonist; 553030); and Promega: dual-luciferase assay kit (E1910) and Fugene 6 (E269). The secondary antibodies were purchased from Sigma as follows: anti-rabbit IgG-horseradish peroxidase (HRP; A0545), -mouse IgG-HRP (A9917), -rabbit IgG-FITC (F9887), and -mouse IgG-FITC (F0257). The MIOX antibody is available in our laboratory, and its preparation has been described previously (40).
Preparation of glycated BSA, laminin, and collagen IV.
Glycation of BSA (AGE-BSA), laminin, and type IV collagen was performed, as described previously (17). Briefly, 2 mg of BSA or laminin or collagen was incubated with or without 10 μl of 40% (wt/vol) methylglyoxal in 5 ml 0.4 M phosphate buffer for 24 h. The treated sample was then dialyzed against 30 mM (NH4)HCO3 (pH 7.8) to remove free methylglyoxal. The content of advanced glycation end products (AGEs) in the samples was measured by fluorescence spectrometry with excitation at 325 nm and emission at 350 nm (Perkin Elmer, Waltham, MA).
Solid-phase binding assay.
To assess the binding of RAGE with glycated vs. nonglycated substrate proteins (albumin, collagen IV, and laminin), solid-phase binding assays were performed, as described previously (60). The binding affinity was compared with S-100B, which is known to bind RAGE (38). First, RAGE stock solution was prepared using a full-length RAGE recombinant protein (20 μg/ml) in a 50-mM carbonate/ bicarbonate buffer (1.59 g/liter Na2CO3 and 2.93 g/liter NaHCO3), pH 9.6. A 96-well MICROTEST assay plate (Falcon) was coated with 25 μl (2 mg/ml) of various substrate proteins (glycated or nonglycated) and allowed to adhere at 4°C overnight. The wells were then briefly rinsed with TBS-Tween (TBST) solution (20 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween 20). The protein-binding sites were blocked by adding 100 μl of blocking buffer (3% BSA in PBS) for 2 h at 22°C. 100 μl of RAGE with varying concentrations (0.5–5.0 μg/ml) in a binding buffer (20 mM Tris·HCl, pH 7.4, 0.15 M NaCl, 1 M CaCl2, 1 M MgCl2) was added and incubated at 4°C for 12 h. After washing with TBST, 50 μl of primary antibody directed against RAGE (1:200 dilution) was added to the plates and incubated at 4°C 12 h. This was followed by incubation with 50 μl of secondary antibody (anti-rabbit IgG- HRP (1:20,000 dilution) at 4°C for 12 h. The HRP enzymatic activity was detected by incubation of the antibody reaction mixture with 50 μl of 3,3,5,5-tetramethyl-benzidine for 5 min at 22°C. The reaction was terminated by adding 50 μl of 1 M HCl. The absorbance in various wells of the plate was measured at 450 nm using an iMark microplate absorbance reader (Bio-Rad, Hercules, CA), and readings were plotted against various concentrations of RAGE used in the assay procedure.
Cell culture.
Renal proximal tubular epithelial cell line HK-2 (human) was obtained from American Type Culture Collection (Manassas, VA). HK-2 cells were initially grown in a keratinocyte serum-free medium (Life Technologies) in the presence of 5 ng/ml recombinant EGF and 0.05 mg/ml bovine pituitary extract + 1× penicillin-streptomycin solution. The cells were then maintained in DMEM (Sigma), containing 5 mM glucose, 5% FBS, and 1× penicillin-streptomycin solution in an atmosphere of 5% CO2 at 37°C. The cells were used for substrate-, dose-, time-dependent expression of various proteins and MIOX activity. For inhibitor studies, HK-2 cells undergoing various treatments with BSA or AGE-BSA were subjected to concomitant treatment with various inhibitors, such as MIOX inhibitor d-glucaric acid (10 mM), PKC inhibitor calphostin (0.05 μM), and phosphoinositide-dependent protein kinase-1 (PDK1)/PI3K inhibitor wortmannin (0.5 μM). In a separate set of experiments, HK-2 cells were also transfected with MIOX- or RAGE-siRNA (50 μM).
Cell-matrix adhesion assay.
Adhesion assays were performed as described previously with minor modifications (12). Tissue culture 96-well plates were coated for 2 h at 37°C with 25 μl (2 mg/ml) glycated or nonglycated BSA, collagen IV, laminin, or PBS. For the cell adhesion assay, a 100-μl medium was added to wells, and plates were kept at 22°C for 1 h. Then, the HK-2 cells (1 × 104) were seeded onto the 96-well plates in quadruplicate for 36 h at 37°C in an atmosphere of 5% CO2 and 95% air. Cells were extensively washed with PBS, fixed with 4% paraformaldehyde, stained with crystal violet for 10 min, and then lysed with 2% SDS and spectrometric readings made using an ELISA plate reader at a wavelength of 570 nm.
Cell viability assays.
Cell viability was assessed using a colorimetric assay, which is based on the conversion of 3-(4, 5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to purple formazan by NAD(P)H-dependent cellular oxidoreductase. About 1 × 104 HK-2 cells in 100 μl of media were seeded in 96-well plates coated with various glycated and nonglycated ECM proteins (laminin or collagen) or BSA (as described above in solid-phase binding assay), and incubated for 36 h at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Following various treatments, 10 μl of MTT (5 mg/ml; Sigma) solution was added and incubated in the dark for 4 h. The reaction was terminated by the addition of a 100-μl stop buffer (15% SDS in 40% dimethylformamide). After stabilization of the reaction for 2 h, absorbance readings were recorded at a wavelength of 560 nm, using an ELISA reader. Absorbance readings were also made at 630 nm, which served as background controls. The relative viability of cells was calculated as A560 – A630 test/A560 – A630 control. The viability of control (PBS) untreated cells was designated as 100%.
MIOX assay.
MIOX activity was measured as reported previously (39). Mice kidney cortices or HK-2 cells undergoing various treatments were suspended in a buffer containing 50 mM sodium acetate, 1 mM ferrous ammonium sulfate, 2 mM l-cysteine, 1 mM glutathione, 1 mM PMSF, 0.2 mM sodium orthovanadate, and 50 mM sodium fluoride followed by homogenization and a brief sonication. After centrifugation at 10,000 g for 5 min at 4°C, the supernatant was collected, and the protein concentration was adjusted to 100 μg/ml. MIOX assay was carried out at 30°C for 30 min in a 500-μl reaction volume containing 50 mM sodium acetate, 1 mM ferrous ammonium sulfate, 2 mM l-cysteine and 60 mM myo-inositol. Fifty microliters (100 μg/ml) of supernatant was added into the reaction mixture for MIOX activity assay. The reaction was terminated by boiling followed by precipitation with 3% TCA. Following a centrifugation at 1,000 g for 5 min, d-glucuronate content was determined in the supernatant by the addition of double volume of freshly prepared Orcinol reagent (40 mg of Orcinol and 9 mg of FeCl3·6H2O dissolved in 10 ml of concentrated HCl). Colorimetric readings were made at A660 nm. MIOX activity was averaged from four different experiments.
Specificity of RAGE in cell-matrix adhesion assay.
Adhesion assays were also performed in tissue culture 96-well plates coated with glycated or nonglycated BSA, and cells were allowed to adhere for a varying time period ranging from 15 min to 24 h. A comparative adherence to AGE-BSA vs. BSA substrates was assessed. To assess the specificity of RAGE-dependent adherence, the cells were treated with RAGE-siRNA, and cell-matrix adhesion assays were performed as described above.
Transfection and promoter activity luciferase assay.
The reporter plasmid construct (pGL3-2512-1) was transfected into exponentially growing HK-2 cells. The cells were seeded onto 24-well culture plates at a density of 1 × 105 cells/well and incubated for ~18 h to achieve ~80% confluence for transfection. The transfection was carried out with 1.0 μl of Fugene 6 (Invitrogen) and 1 μg of reporter plasmid construct. Cotransfection of 100 ng of pcDNA-rLUC (Renilla luciferase) was used as an optimized equalization control. The cells were then subjected to various treatments. Assays for both Renilla and firefly luciferase activity were carried out 48 h posttransfection using the commercial dual-luciferase kit (Promega) and a TD 20/20 luminometer (Promega), according to the manufacturer’s guidelines. Basal promoter activity was determined in cells transfected with reporter construct pGL3-basic only.
Real-time RT-PCR.
For quantification of RAGE, MIOX, and NF-κB expression, total RNA was extracted from the cells by using TRIzol reagent (Invitrogen), and contaminated genomic DNA was removed with RNase-free DNase treatment (50 U/ml) in the presence of RNasin (1 unit/μl). Two μg of total RNA was then reverse-transcribed by using Superscript II reverse transcriptase (25 units/l) and 24-mer oligo(dT) as the primer. The synthesized cDNA was analyzed in a sequence detection system (model 7000; Applied Biosystems, by using specific MIOX, RAGE, NF-κB, MCP-1, IL-6, 18s, and β-actin primers and absolute quantitative PCR SYBR Green mixtures (ABgene, Rochester, NY). Table 1 includes the description of specific primers used for RT-PCR.
Table 1.
Primers used for RT-PCR analyses
| Gene | Species | Sense | Antisense |
|---|---|---|---|
| RAGE | Human | 5′-GCT GTC AGC ATC AGC ATC AT-3′ | 5′-TCC TCC TCT TCC TCC TGG-3′ |
| MIOX | Human | 5′-CGC AGA GAA ACC CGA CTA AG-3′ | 5′-CAA AGC TCAGAGAGC CCA TC-3′ |
| NF-κB | Human | 5′-ATG GCT TCT ATG AGG CTG AG-3′ | 5′-GTT GTT GTT GGT CTG GAT GC-3′ |
| Actin | Human | 5′-AGG CAC CAG GGC GTG AT-3′ | 5′-GCC CAC ATA GGA ATC CTT CTG AC-3′ |
| MIOX | Mice | 5′-CTG GTG GAC GAA TCT GAC CCA G-3′ | 5′-GCC ACG GAT AGA AGG AGT GGA ATC G-3′ |
| RAGE | Mice | 5′-GGA CCC TTA GCT GGC ACT TAG A-3′ | 5′-GAG TCC CGT CTC AGG GTG TCT-3′ |
| NF-κB | Mice | 5′-TGT GCG ACA AGG TGC AGA AA-3′ | 5′-ACA ATG GCC ACT TGC CGA T-3′ |
| 18s | Mice | 5′-AAA CGG CTA CCA CAT CCA AG-3′ | 5′-CCT CCA ATG GAT CCT CGT TA-3′ |
| MCP-1 | Mice | 5′-CCC AAT GAG TAG GCT GGAGAG-3′ | 5′-TGG TTG AAA AGG TAG TGG ATG-3′ |
| IL-6 | Mice | 5′-CGG AGA GGA GAC TTC ACA G-3′ | 5′-CAG AAT TGC CAT TGC ACA AC-3′ |
See the text for definitions.
Western blot analysis.
Cells were treated with BSA or AGE-BSA or RAGE-siRNA or various inhibitors, as described in Cell culture, for 24 h and were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Nonidet P-40) containing a protease inhibitor mixture (Sigma) on ice for 30 min. The lysate was centrifuged at 12,000 g at 4°C for 10 min, and the supernatant was collected for protein expression studies by immunoblot analyses following protein determination by the Bradford method. Equal amounts of protein (30 μg) from various samples were subjected to SDS-10% PAGE and then transferred onto nitrocellulose membranes by electroblotting procedures. The immunoblots were individually probed with various primary antibodies, as described in Antibodies and other reagents. The specificity of each of the antibodies used have been validated by respective vendors and have been cited in our and others’ previous publications (13, 15, 32, 39, 40, 48, 52, 60, 64). The blots were then incubated with secondary antibodies, which included an anti-rabbit or anti-mouse IgG antibody. The autoradiograms of Western blots were prepared using the ECL system.
Analysis of intracellular ROS generation.
Intracellular ROS were detected by DCF-DA in HK-2 cells, as described previously (56). The HK-2 cells were grown in multiwell plates. After treatment with different concentrations of AGEs or BSA, the culture medium was replaced by HBSS containing 50 μM DCF-DA (dissolved in DMSO), and cells were further incubated in the dark for 1 h at 37°C. The cells were then washed with HBSS and solubilized in 1 ml of SDS (0.01% in 0.2 M NaOH). The generation of the fluorescent product 2′,7′-dichlorofluorescin (DCF) was monitored in a spectrofluorometer (F-2500; Hitachi, Tokyo, Japan) using an excitation wavelength of 485 nm and emission at 530 nm. The final fluorescent intensity was normalized to the protein content, and the results were expressed in terms of fluorescence arbitrary units (AU)/mg protein.
Immunofluorescence microscopy.
HK-2 cells (1 × 106) were seeded on coverslips and treated with BSA, AGE-BSA (200 and 400 μg/ml), and MIOX-siRNA. They were maintained for 24 h in low glucose culture medium (5 mM). They were washed with PBS twice at 5-min intervals followed by fixation with 4% paraformaldehyde at 22°C for 30 min. The cells were rewashed 3 times with PBS. The cells were then permeabilized with 0.25% Triton-X100 in PBS for 10 min. After blocking with 1% BSA in PBS containing Triton-X100 for 30 min, the cells were incubated with primary antibodies (rabbit anti-MIOX, anti-mouse anti-fibronectin for 12–15 h at 4°C. After three washes with PBS, cells were incubated with FITC-conjugated secondary antibodies in 1% BSA for 1 h at 22°C in the dark. After washing with PBS, the cells were counterstained with TO-PRO-3 iodide to visualize nuclei. The coverslips were then mounted on the glass slides, and the cells examined by a Zeiss microscope equipped with UV epi-illumination. In addition, the cells undergoing various treatments as above were stained with DCF-DA to assess the generation of ROS.
Animal experimental design.
An established protocol for the AGE overload model in mice with minor modifications was employed (50). A total of 40 male CD1 mice at 8 wk of age [body weight (BW) = 25–30 g] were divided into two major groups: 1-wk (1W) and 2-wk (2W) treatment groups. These two groups were further subdivided into five subgroups: 1) control (without any treatment), 2) BSA, 3) AGE-BSA, 4) AGE-BSA+FPS-ZM1, and 5) AGE-BSA+d-glucarate. Thus a total of 10 groups with 4 animals each were used. BSA and AGE-BSA (20 mg/kg BW ip) were given daily for 1 to 2 wk. The FPS-ZM1 (1 mg/kg BW) and d-glucarate (20 mg/kg BW ip) were administered intraperitoneally daily as well, concomitantly with AGE-BSA. The FPS-ZM1 is a RAGE antagonist, while d-glucarate/glucaric acid is a MIOX inhibitor (28, 30, 63). Before euthanasia, the urine was collected in metabolic cages to determine the albumin/creatinine ratio using mouse Exocell kits. The kidneys were collected for various morphological, biochemical, and molecular biology studies. The CD1 mice were used in these studies, since they have been described to yield consistent results in terms of development of proteinuria and renal morphological changes in the experimental diabetic state (25). All animal studies were approved by the Animal Care and Use Committee of Northwestern University.
Assessment of expression of MIOX and fibronectin and ROS generation in renal tissues.
MIOX and fibronectin expression was evaluated by immunofluorescence microscopy. Briefly, 4-μm-thick cryostat kidney sections from mice undergoing various treatments were prepared and mounted on glass slides. They were then air-dried, which was followed by fixation with 10% phosphate-buffered formalin for 20 min. After washing the formalin with PBS, the sections were stained with MIOX and fibronectin antibodies, as described above. Likewise, the status of ROS generation was monitored by staining the cryostat sections with dihydroethidium (DHE). In addition, kidney sections were prepared from paraffin-embedded tissues for light microscopy to assess the influx of inflammatory cells in mice undergoing various treatments.
Statistical analysis.
Statistical analyses were carried out using GraphPad Prism (version 5.01). The significance of the results was determined using one-way ANOVA (the Tukey multiple-comparison test) for all experiments. Results are expressed as means ± SD of quadruplicate samples.
RESULTS
Protein glycation increases interaction with RAGE and enhances cellular adherence and MIOX activity.
Solid-phase binding assays were performed to assess the binding of glycated vs. nonglycated ECM proteins and albumin with RAGE. S-100B was used as a known positive control. A dose-dependent increase in the binding with RAGE of all the glycated proteins was observed, and the binding was saturated at a concentration of 4.0 μg/ml (Fig. 1A). S-100B exhibited strong binding among various proteins, but it was lower than glycated laminin and albumin. The glycated albumin demonstrated a noteworthy binding with RAGE at all the concentrations from 0.5 to 4.0 μg/ml, and it was comparable to glycated laminin. Glycated collagen IV exhibited comparatively weak binding. Interestingly, the nonglycated albumin exhibited higher binding than collagen despite the latter being glycated. Because glycated albumin had a higher binding with RAGE than S-100B and it was comparable to glycated laminin, glycated BSA was considered as a potential candidate for later experiments.
Fig. 1.

Glycation and advanced glycation end products (AGE):receptor of advanced glycation end products (RAGE) interaction enhances cellular adhesiveness and myo-inositol oxygenase (MIOX) activity. A: solid-phase binding assays revealed a dose-dependent increase in the binding of glycated proteins with RAGE (0.5–4.0 μg/ml). The control S-100B exhibited a strong binding among various proteins. However, the glycated albumin had higher binding with RAGE, and it was comparable to glycated laminin. B: cell adhesion assays revealed that cellular adherence of HK-2 renal cells was glycation-dependent, and results with AGE-BSA and AGE-laminin were comparable. C: no significant differences were observed in viability of cells plated onto glycated vs. nonglycated substrata. D: interestingly, the cells maintained on glycated proteins had higher levels of MIOX activity and the highest being with AGE-BSA. E: adherence of cells plated on AGE-BSA substratum was time-dependent, and it increased over a period of 24 h. RAGE-small interfering (si) RNA treatment significantly reduced the adherence of cells maintained on AGE-BSA, suggesting AGE:RAGE interaction facilitates cellular adhesiveness. Lam, laminin; Col. IV, collagen IV. #P < 0.05 compared with cells treated with BSA or Col. IV or laminin (B–D). *P < 0.01 compared with cells treated with BSA or Col. IV or laminin (B–D). #P < 0.05 compared with cells treated with BSA for 15–60 min (E). *P < 0.01 compared with cells treated with AGE-BSA or RAGE-siRNA for 15–60 min (E).
To translate the observed increased binding of glycated proteins with RAGE into cellular functionality, adherence assays were performed using renal HK-2 cells. In this regard, the RAGE has been reported to promote adherence and can modulate cellular interactions in terms of adhesion and viability; however, the reports describing the differential effect of various glycated proteins on the behavior of renal cells are somewhat limited (47). Cell adhesion assays were performed with all the glycated and nonglycated proteins, and incubation was carried out for 24 and 48 h. The adhesive capacity of HK-2 renal cells was glycation dependent on a given protein (Fig. 1B). Marginal differences with respect to adhesive capacity of cells were observed between 24 vs. 48 h of incubation, and the results of 48 h are included in Fig. 1B. Overall, all of the glycated proteins exhibited increased adhesive capacity compared with the nonglycated proteins. Interestingly, the cellular adherence observed with glycated BSA was comparable with glycated laminin (Fig. 1B). With respect to viability of cells, no significant differences were observed in cells plated onto glycated vs. nonglycated substrata. The viability was marginally decreased for cells plated on glycated substrata compared with nonglycated proteins; however, it was not statistically significant (Fig. 1C). Besides adherence and viability, another aspect of cellular functionality of HK-2 cells, relating to MIOX activity, was assessed. The cells maintained on glycated proteins yielded higher levels of MIOX activity. The highest increase in MIOX activity was observed in cells plated on AGE-BSA substratum, although an increase in enzyme activity was also noted in cells maintained on AGE-collagen IV and -laminin substrata (Fig. 1D). Because glycated BSA exerts a maximal beneficial effect on the functionality of HK-2 cells, therefore, AGE-BSA was selected for the subsequent experiments. Next, we evaluated the time dependency (15 min–24 h) of cellular adherence using glycated BSA and RAGE-siRNA to disrupt AGE:RAGE interaction. In general, adherence of cells increased over a period of 24 h commencing at ~12 h following plating on AGE-BSA substratum (Fig. 1E). Since treatment of RAGE-siRNA resulted in the inhibition of adherence in cells maintained on AGE-BSA substratum, this suggested that increased adhesiveness of renal cells, to a large extent, is facilitated by glycation of albumin and is dependent on AGE:RAGE interaction (Fig. 1E).
Treatment of AGEs induces MIOX activity and its expression in tubular cells in a time- and dose-dependent manner.
The HK-2 cells were exposed to BSA or glycated BSA (AGE-BSA, 200 μg/ml) included in the culture media for 6–48 h, and MIOX activity was assessed. The activity increased in a time-dependent manner, and a significant increase was discernible at 24 h (Fig. 2A). A minor increase in the activity was also observed with the treatment of BSA alone. For dose dependency, the HK-2 cells were exposed to various concentrations of AGE-BSA ranging from 50 to 400 μg/ml. A remarkable increase in MIOX activity was observed at a concentration of 200 μg/ml, with a mild further increase at 400 μg/ml of AGE-BSA (Fig. 2C). No significant concentration-dependent increase in MIOX activity was observed with the BSA treatment alone. With respect to expression, a dose-dependent increase in MIOX mRNA expression, as assessed by quantitative PCR, was observed (Fig. 2D). Time-dependency experiments revealed a notable increase in MIOX protein expression at 24 h with a mild further increase at the 36- or 48-h time point, as assessed by Western blot analyses (Fig. 2B). Interestingly, a congruent time-dependent increase in the expression of RAGE was also observed with a significant increase at 24 h, suggesting AGE:RAGE interaction, possibly modulates MIOX expression and its enzymatic activity.
Fig. 2.
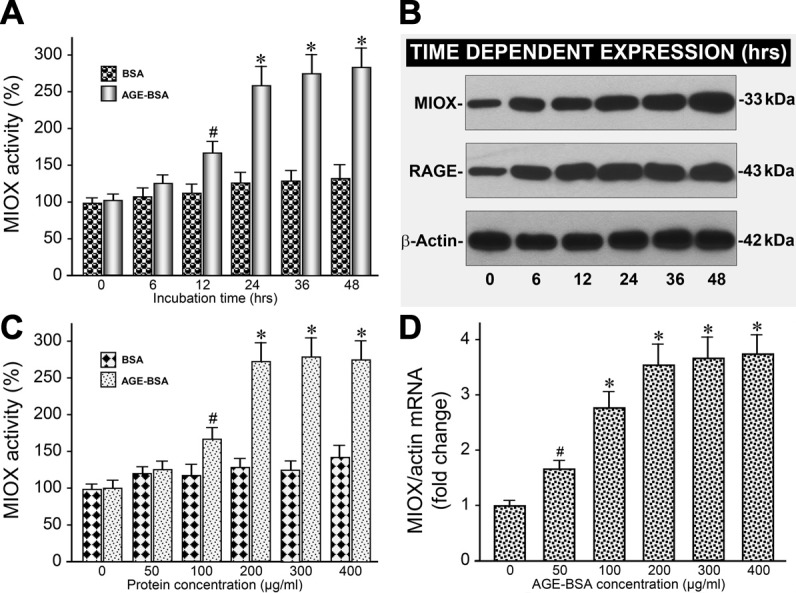
AGE-BSA increases MIOX activity and its expression in a time- and dose-dependent manner. A: AGE-BSA increased MIOX activity in a time-dependent (6-48 h) manner, and it was readily discernible at 24 h. B: likewise, there was a time-dependent increase in the expression of MIOX and RAGE. C and D: also, AGE-BSA (50–400 μg/ml) induced a remarkable increase in MIOX activity and its mRNA expression in a concentration-dependent manner commencing at 200 μg/ml. No significant concentration-dependent increase in MIOX activity was observed at 300 or 400 μg/ml. #P < 0.05 compared with cells treated with BSA (A and C). *P < 0.01 compared with cells treated with BSA (A and C). #P < 0.05 compared with untreated cells (D). *P < 0.01 compared with untreated cells (D).
AGE-BSA induces expression of RAGE and MIOX, phosphorylation, and expression of downstream molecules in a dose-dependent manner.
Because AGE:RAGE interaction modulates MIOX expression, this led us to evaluate whether such an interaction regulates phosphorylation of downstream molecules. The phosphorylating enzymes that are activated following AGE:RAGE interactions were examined. Besides a dose-dependent upregulated expression of RAGE and MIOX, an increased phosphorylation of PI3K, PDK, and AKT was observed (Fig. 3A). Incidentally, the increased phosphorylation of MIOX by these enzymes has been shown to accentuate MIOX activity as well (39). In addition, AGE:RAGE interaction has been shown to activate the PI3K/AKT pathway, possibly by the generation of ROS, which downstream would likely affect NF-κB activation (14, 22). Along these lines, we observed dose-dependent increased expression of NF-κB and the downstream molecules, including TGF-β and fibronectin (Fig. 3A). These sequences of events seem to be specific to AGE:RAGE interaction since its disruption with RAGE-siRNA reduces the expression of RAGE, as well as the expression of the molecules relevant to the downstream events (Fig. 3A). Interestingly, the increased generation of ROS is also associated with increased expression and activity of MIOX (51). This would mean that there is likely to be further increased activation of PI3K/AKT, NF-κB, and downstream molecules. The excessive generation of ROS was assessed by measuring the intensity of DCF-DA fluorescence in cells treated with AGE-BSA. A dose-dependent increase in the fluorescence was observed following treatment with AGE-BSA, which could be dampened by the treatment of RAGE- or MIOX-siRNA (Fig. 3B). While no significant change was observed following BSA treatment alone, suggesting that the AGE:RAGE interaction leads to the generation of ROS. The ROS can also increase the expression of MIOX, since it includes oxidant/antioxidant response elements in its promoter, and overexpression of MIOX leads to generation of ROS; this would set up a cyclic generation of ROS and increased activation the PI3K/AKT/NF-κB pathway with enhanced upregulation of downstream events (39).
Fig. 3.
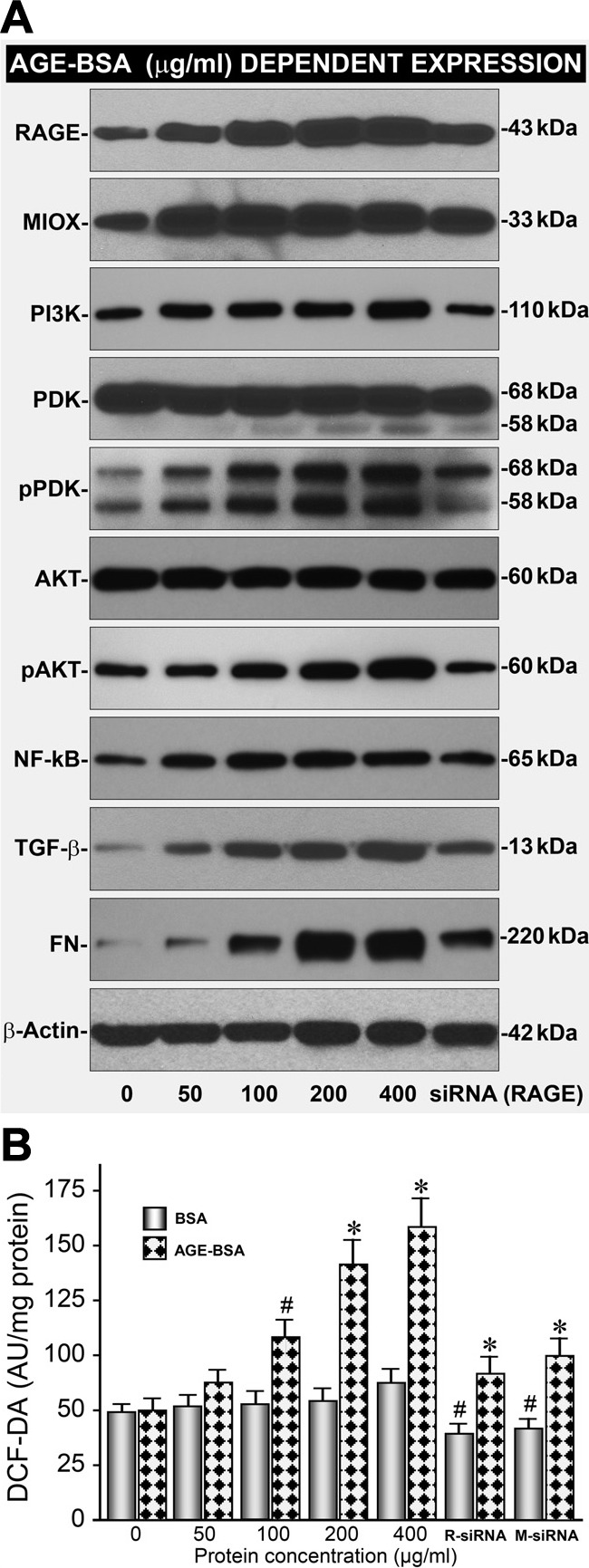
AGE-BSA increases expression of RAGE & MIOX concomitant with phosphorylation and expression of downstream signaling molecules. A: AGE-BSA upregulated the expression of RAGE and MIOX along with increased phosphorylation of PI3K, PDK, and AKT in a dose-dependent manner (50–400 μg/ml). The increased phosphorylation of MIOX by these enzymes has also been shown to accentuate MIOX activity (39). A: AGE-BSA also increased the expression of NF-κB, TGF-β, and fibronectin, which was possibly related to increased generation of reactive oxygen species (ROS). RAGE-siRNA reduced the expression of RAGE, as well as the expression of molecules relevant to the downstream events, suggesting a specific modulation following AGE:RAGE interaction. B: also, a dose-dependent increase in the generation of ROS, as assessed by DCF-DA staining, was observed following treatment with AGE-BSA, and it was dampened by the treatment of RAGE- or MIOX-siRNA. The ROS mediated increased MIOX expression and MIOX phosphorylation enhances its activity with generation of additional ROS; henceforth, this would set up a cyclic generation of ROS and increased activation of PI3K/AKT/NF-κB pathway with marked acceleration of downstream events. PI3K, phosphatidylinositide 3-kinase; PDK, pyruvate dehydrogenase kinase; AKT, protein kinase B; TGF-β, transforming growth factor-β; FN, fibronectin; DCF-DA, 2′,7′-dichlorofluorescin diacetate. R-siRNA, RAGE-siRNA; M-siRNA, MIOX-siRNA. #P < 0.05 compared with cells treated with BSA or RAGE- or MIOX-siRNA (B). *P <0.01 compared with cells treated with BSA or RAGE- or MIOX-siRNA (B).
AGEs treatment induces cellular expression of MIOX and concomitantly increases oxidant stress and de novo synthesis of fibronectin in a dose-dependent manner.
Immunofluorescence studies confirmed the AGE-BSA-induced cellular expression of MIOX and modulation of certain downstream events. A notable increase in the expression of MIOX was observed at a concentration range of 200–400 μg/ml of AGE-BSA (Fig. 4, B and C). Concomitant treatment of MIOX-siRNA remarkably reduced MIOX expression (Fig. 4D). Likewise, a remarkable increased generation of ROS was observed, as reflected by an increase in the DCF-DA staining of the cells exposed to AGEs (Fig. 4, F and G). Similarly, a parallel increased de novo synthesis of fibronectin (green fluorescence) was observed following AGE-BSA treatment (Fig. 4, J and K). It was readily noticeable at a concentration of 400 μg/ml of AGE-BSA. The concomitant treatment of MIOX-siRNA reduced fibronectin expression. Because MIOX-siRNA downregulated the cellular expression of MIOX and fibronectin while reducing ROS generation (Fig. 4, D, H, and L), this would suggest that these signaling events are interrelated and they commence with the upstream AGE:RAGE interaction, and MIOX modulation is downstream of such an interactive event.
Fig. 4.

AGE-BSA induces cellular expression of MIOX with increased generation of ROS and de novo synthesis of fibronectin. An increase in the expression of MIOX was notated at a concentration range of 200–400 μg/ml of AGE-BSA (B and C). Associated with it was a marked increased generation of ROS, as assessed by DCF-DA staining (F and G). Along with it a parallel increased de novo synthesis of fibronectin (green immuno-fluorescence) was observed (J and K). The increased MIOX expression, generation of ROS, and synthesis of fibronectin was readily discernible at a concentration of 400 μg/ml of AGE-BSA. Treatment with MIOX-siRNA reduced the expression of MIOX and fibronectin and ROS generation (D, H, and L). This suggests that these signaling events are interrelated, and they commence following AGE:RAGE interaction.
Inhibition of AGEs-induced expression of RAGE, MIOX, and NF-κB.
Various inhibitors were used to inhibit the differential expression of RAGE, MIOX, and transcription factor NF-κB to assess whether there is a correlation among these molecules as far as their activity and expression are concerned. First of all, RAGE-siRNA inhibited the protein expression of RAGE, MIOX, and NF-κB (Fig. 5E). RAGE-siRNA also inhibited the gene expression of RAGE, MIOX, and NF-κB (Fig. 5, A and D). In addition, inhibition of enzymatic and promoter activity of MIOX was observed (Fig. 5, B and C). MIOX activity and its protein expression, but not gene expression, were affected by the various inhibitors of phosphorylation, i.e., calphostin and wortmannin (Fig. 5, A, B, and E). Interestingly, inhibitors of phosphorylation also affected the protein expression of MIOX and NF-κB, but not of RAGE (Fig. 5E). Of note, MIOX-siRNA also inhibited gene expression of MIOX and NF-κB but not of RAGE, suggesting RAGE is upstream, while NF-κB is downstream of MIOX with respect to various signaling events (Fig. 5D). MIOX and NF-κB expression and activity of MIOX, but not of RAGE, were also affected by the MIOX inhibitor, i.e., d-glucaric acid (Figs. 5, B and E). Finally, the increase in MIOX promoter activity by AGE-BSA was comparable to that induced by methylglyoxal (MGO) and was inhibited by N-acetyl cysteine (NAC) (Fig. 5C); this suggested that most likely MIOX-mediated ROS generation in addition to AGE:RAGE-induced oxidant stress; probably both in tandem contribute to the activation of NF-κB transcription factor and inflammatory cytokines and TGF-β ultimately, resulting in excessive synthesis of ECM proteins, such as fibronectin and consequential fibrosis. This issue was addressed in in vivo studies.
Fig. 5.
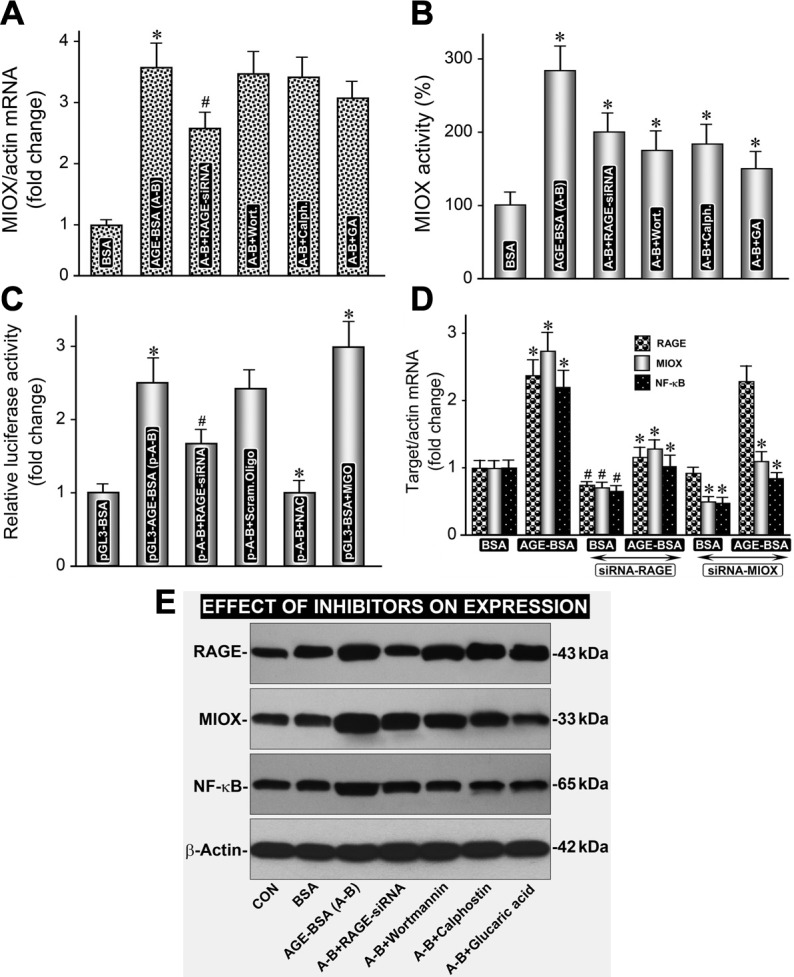
AGE-BSA-induced expression of MIOX and NF-κB, and MIOX activity is inhibited by inhibitors of phosphorylation, and RAGE- and MIOX-siRNA. A, D, and E: RAGE-siRNA inhibited the protein expression of RAGE, MIOX and NF-κB. B and C: in addition, RAGE-siRNA inhibited enzymatic, as well as promoter activity of MIOX. D: MIOX-siRNA affected the expression of MIOX and NF-κB only. A, B, and E: MIOX activity and its protein expression, but not the gene expression, were affected by the various inhibitors of phosphorylation, i.e., calphostin and wortmannin. A, B, and D: E: inhibitors of phosphorylation also affected the protein expression of MIOX and NF-κB, but not of RAGE. B and E: MIOX and NF-κB expression and activity of MIOX, but not of RAGE, were also affected by the MIOX inhibitor, i.e., d-glucaric acid. C: finally, the increase in MIOX promoter activity by AGE-BSA was comparable to that induced by methylglyoxal (MGO; a strong oxidant), and it was inhibited by N-acetyl cysteine (NAC), suggesting that MIOX-mediated ROS generation in addition to AGE:RAGE-induced oxidant stress together contribute to the activation of downstream signaling. A-B, AGE-BSA; Wort., Wortmannin; Calph, calphostin; GA, glucaric acid. #P < 0.05 compared with cells treated with BSA or RAGE-siRNA (A–C). *P < 0.01 compared with cells treated with BSA or RAGE-siRNA (A–C). *P < 0.01 compared with cells treated with BSA (D). #P < 0.05 compared with cells treated with BSA or RAGE- or MIOX-siRNA (D).
Effect of AGE-BSA in vivo administration on renal functional parameters and expression of RAGE, MIOX, NF-κB, and TGF-β.
The albumin-creatinine ratio (ACR) rose steeply following AGE-BSA administration for 1 wk, and a mild further increase was observed after 2 wk of treatment, and it was highly significant compared with the basal values (Fig. 6, A and B). The rise in ACR was associated with an increase in MIOX activity at 1 and 2 wk (Fig. 6, C and D). A parallel increase in the expression of RAGE, MIOX, NF-κB, and TGF-β was also observed following 1 and 2 wk of treatment of AGE-BSA (Fig. 6, E–G). The rise in ACR or increase in the expression of RAGE, MIOX, NF-κB, and TGF-β was notably reduced with the administration of RAGE inhibitor FPS-ZM1 (Fig. 6, A–G). In addition, a decrease in mRNA expression of MIOX, NF-κB, and TGF-β was observed with the treatment of MIOX inhibitor, i.e., d-glucaric acid, while expression of RAGE was unaffected (Fig. 6, E and G). This strongly suggested that signaling molecules downstream of MIOX are affected by the d-glucaric acid treatment, whereas RAGE inhibitor FPS-ZM1 modulates expression of all of the molecules that are affected following AGE-BSA administration.
Fig. 6.

In vivo administration of AGE-BSA affects renal functions, MIOX activity, and expression of RAGE, MIOX, NF-κB, and TGF-β. A and B: a rise in albumin/creatinine ratio was (ACR) observed following administration of AGE-BSA for 1 and 2 wk. C–G: it was associated with an increase in MIOX activity and expression of RAGE, MIOX, NF-κB, and TGF-β. C: A–G: rise in ACR or increase in the expression of RAGE, MIOX, NF-κB, and TGF-β was remarkably reduced with the administration of RAGE inhibitor, FPS-ZM1. E–G: a decrease in mRNA expression of MIOX, NF-κB, and TGF-β was also observed with the administration of MIOX inhibitor, i.e., glucaric acid, while expression of RAGE was unaffected. These results indicate that RAGE inhibitor, FPS-ZM1, modulates expression of all of the downstream signaling molecules, while d-glucaric acid, an inhibitor of MIOX, affects molecules downstream of MIOX following AGE-BSA treatment. GA, glucaric acid; FPS-ZM1, 4-chloro-N-cyclohexyl-N-(phenylmethyl)-benzamide. #P < 0.05 compared with mice treated with BSA (A–F) *P < 0.01 compared with mice treated with BSA (A–F).
Contribution of MIOX in AGE-induced expression of inflammatory cytokines and influx of inflammatory cells in the tubulointerstitial compartment.
In view of the increased ROS generation and expression of the master regulator of cytokines, NF-κB, following AGE:RAGE interaction, we proceeded to assess the contribution of MIOX in the modulation of various signaling molecular targets. A significant increase in the expression of MCP-1 and IL-6 mRNA was observed in mice administered AGE-BSA for 1 wk (Fig. 7, A and B).The mRNA was reduced with the concomitant administration of RAGE inhibitor FPS-ZM1. A decreased expression was also seen following treatment with MIOX inhibitor d-glucaric acid, although the degree of inhibition of mRNA was comparatively less than that seen with FPS-ZM1 administration. This AGE-induced increase in the expression of the inflammatory cytokines paralleled the increased influx of inflammatory cells in the tubulointerstitial compartment (Fig. 7, D vs. C, arrowheads). Like with inflammatory cytokines, the influx was reduced with the administration of FPS-ZM1 or d-glucaric acid (Fig. 7, E and F, arrowheads). Qualitatively, reduction in the influx of cells was somewhat less with the administration of d-glucaric acid compared with FPS-ZM1. Nevertheless, the fact that there was a reduction in the influx of inflammatory cells with the treatment of d-glucaric acid suggests a definitive contribution of MIOX in this process initiated by AGE:RAGE interaction.
Fig. 7.

In vivo administration of AGE-BSA leads to increased expression of inflammatory cytokines and influx of inflammatory cells in the tubulointerstitium. A and B: an increase in the expression of MCP-1 and IL-6 mRNA was observed in mice following administration of AGE-BSA for 1 wk, and it was reduced with the concomitant treatment of the RAGE inhibitor FPS-ZM1 or MIOX inhibitor d-glucaric acid. The AGEs-induced increase expression paralleled with the increased influx of inflammatory cells in the tubulointerstitial compartment (C vs. D, arrowheads). The influx was reduced with the administration of FPS-ZM1 or d-glucaric acid (E and F, arrowheads). The reduction in the expression of cytokines and influx of inflammatory cells with the treatment of glucaric acid suggest a contribution of MIOX in this process initiated by AGE:RAGE interaction. MCP-1, monocyte chemotactic protein-1; IL-6, interleukin 6. #P < 0.05 compared with mice treated with BSA (A and B). *P < 0.01 compared with mice treated with BSA (A and B).
AGE-induced cellular expression of MIOX, fibronectin, and generation of ROS and relevance of MIOX in the expression of the target molecules.
In these sets of experiments, the effect of AGE-BSA on MIOX expression and its contribution in the expression of fibronectin and generation of intracellular ROS was assessed. Like in vitro studies, MIOX expression significantly increased in the renal cortical tubules following 2 wk of treatment with AGE-BSA (Fig. 8, B vs. A). No MIOX expression was seen in the glomerular compartment (arrowheads). The AGEs-induced MIOX expression was reduced with concomitant administration of RAGE inhibitor FPS-ZM1 or MIOX inhibitor d-glucaric acid (Fig. 8, C and D). AGE-BSA also induced a remarkable increase in the generation of ROS, as highlighted with increased staining of nuclei with DHE, and ROS generation was reduced with treatment of FPS-ZM1 or d-glucaric acid (Fig. 8, E–H). Notably, in parallel with the increased expression of MIOX expression and generation of ROS, AGE-BSA induced a remarkable increase in the expression of fibronectin (Fig. 8, J vs. I). The expression of fibronectin was confined to the tubulointerstitial compartment. Fibronectin expression in the glomerular compartment could be seen as well; however, an increase was not readily discernible (arrowheads). Interestingly, the administration of FPS-ZM1 or d-glucaric acid significantly reduced the expression of fibronectin in the tubulointerstitial compartment (Fig. 8, K and L), which was in parallel to the decreased MIOX expression and generation of ROS. This suggests a bona fide contribution of MIOX in the signaling events that are initiated by AGE:RAGE interactions.
Fig. 8.

In vivo administration of AGE-BSA leads to increased expression of MIOX, fibronectin, and generation of ROS. An increased MIOX expression was observed in the renal cortical tubules following 2 wk of treatment with AGE-BSA (B vs. A). No MIOX expression was seen in the glomerular compartment (arrowheads). C and D: MIOX expression was reduced with concomitant administration of RAGE inhibitor, FPS-ZM1, or MIOX inhibitor, d-glucaric acid. E–H: an altered MIOX expression was associated with an increase in the generation of ROS, as assessed by dihydroethidium (DHE) staining, and it was reduced with the treatment of FPS-ZM1 or d-glucaric acid. AGE-BSA treatment also induced an increase in fibronectin expression in the tubulointerstitium, and it was reduced with the administration of FPS-ZM1 or d-glucaric acid (J vs. I, and K and L). Fibronectin expression in the glomerulus was not affected (arrowheads). The changes in ROS and fibronectin paralleled the altered MIOX expression, suggesting contribution of MIOX in the signaling events that are initiated following AGE:RAGE interaction. GA, glucaric acid; FPS-ZM1, 4-chloro-N-cyclohexyl-N-(phenylmethyl)-benzamide.
DISCUSSION
The results of this investigation yield certain insights into the intricate participation of MIOX, a tubular enzyme, in the cellular events initiated following AGE:RAGE interaction that are likely to lead to tubulointerstitial injury. Such an injury into the tubulointerstitial compartment can occur in a variety of ways, and one of the recently described mechanisms relates to oxidant stress (23, 24, 42, 49). Especially, this may be the case in settings in which there is an upregulation of MIOX or accentuation of its activity (39, 40, 48, 51, 55, 61, 63). Because AGE-RAGE interaction is known to induce generation of ROS, the premise of this investigation was to delineate the events where MIOX would contribute toward the oxidant stress injury to the tubular compartment, using in vitro and in vivo model systems.
For in vitro studies, we have to select the AGE that would mimic the in vivo system having comparable binding to RAGE with its major high-affinity binding ligand S-100B; the latter belongs to the family of calcium-binding proteins (38). Another consideration needs to be kept in perspective for designing the AGE ligand as it relates to the ECM, since the latter undergoes notable transformation in the form of thickening of tubular basement membranes (TBMs) in tubulointerstitial injury in diabetic nephropathy (20). The major TBM or GBM proteins include type IV collagen and laminin, and the AGEs are known to modulate the turnover and expression of these ECM proteins and of proteases in glomerular cells, resulting in nodular sclerosis—a hallmark characteristic of diabetic nephropathy (5). In any instance, the glycated ligand of interest should have comparable binding affinity with RAGE as that of ECM proteins and S-100B. AGE-BSA had binding with RAGE higher than S-100B and glycated type IV collagen, and it was comparable to that of AGE-laminin; thus, we considered AGE-BSA as a suitable candidate for our studies (Fig. 1). In fact, numerous past studies have used AGE-BSA to investigate the pathobiology of proximal tubules or tubulointerstitial compartment (17, 31, 36, 44, 57). The AGE-BSA also has been used to investigate mesangial cell biology, and in addition, its utility to study AGE-induced dysfunctions of endothelial and retinal pigment epithelial cells in vivo has been well described (5, 11, 50). The effect of AGE:RAGE interaction on cellular functionality was gauged by adherence assays, where it was noted that AGE-BSA increases adhesiveness of cells in a time-dependent manner with no effect on cell viability (Fig. 1). Likewise, increased adherence of alveolar epithelial cells has been described with the activation or overexpression of RAGE (12). The fact that AGE-BSA increased the expression of RAGE would be supportive of the previous observations made in pulmonary alveolar cells (Fig. 2). Also, cellular adherence could be reduced with the treatment of RAGE siRNA, and this would attest to the specificity of the effect observed following AGE:RAGE interaction (Fig. 1).
The most interesting finding of this investigation with respect to MIOX pathobiology is that, concomitant with the activation of RAGE following exposure of AGE-BSA, there was an increase in dose- and time-dependent activity and expression of MIOX (Figs. 1 and 2). Although this increase could be seen with AGE-collagen IV or -laminin, the degree of stimulation of the activity was highest with AGE-BSA (Fig. 1). The exact mechanism by which AGE-BSA increases the expression of MIOX is unknown, but potentially, it could be related to the ROS that are known to be generated following AGE:RAGE interaction, with the release of arachidonic acid and activation of cytosolic phospholipase A2 (cPLA2) (17). It is worth mentioning here that the MIOX promoter includes osmotic-, carbohydrate-, and both oxidant and antioxidant response elements, and its transcription is heavily influenced by organic osmolytes, high glucose ambience, and oxidant stress (39, 40, 55). Indeed, previously, we observed that various molecules, such as high glucose, sterol response elements, oxidant, and antioxidant response elements can influence MIOX’s transcription. It is likely that ROS produced following AGE:RAGE interaction led to an upregulation of MIOX, since under oxidant stress states, MIOX upregulation has been seen associated with nuclear translocation of oxidant-responsive transcription factor Nrf2 (40). Another potential mechanism responsible for the increased activity of MIOX could be related to the upregulation of various kinases involved in the PI3K/AKT signaling pathway that are activated upon AGE:RAGE interaction (14, 36, 41, 62). In this regard, we observed increased expression of PI3K and the phosphorylated form of pPDK and pAKT (Fig. 3). Their upregulated expression is likely to enhance the phosphorylation of MIOX and, thereby, its activity, as alluded to in our previous publications (39, 55). The upregulation seems to be specific to AGE:RAGE interaction since RAGE-siRNA could reduce the expression of MIOX, concomitant with the downregulation of kinases involved in the PI3K/AKT signaling pathway (Fig. 3).
The activation of the PI3K/AKT pathway can lead to upregulation of NF-κB and its nuclear translocation. In this regard, there are a multitude of mechanisms whereby AGE:RAGE interaction could lead to increased activity of NF-κB and consequential downstream events (19, 45). First, a strong biological link or cross talk between NF-κB and PI3K/AKT survival pathways has been reported in the the literature (22). Activated PI3K is known to phosphorylate serine/threonine kinase AKT (PKB), which, in turn, would phosphorylate the p65 subunit of NF-κB at its Ser-534 residue with a consequential increase in its activity (3). Likewise, another posttranslational modification that follows AGE:RAGE interaction, i.e., sirtuin-1 (SIRT1)-dependent acetylation of p65, has been reported to increase NF-κB activity since administration of pyridoxamine, an inhibitor of AGE formation, restores SIRT-1 deacteylase activity and reduces p65 acetylation in db/db mice (34). Third, besides PI3K, other kinases that can modulate AKT phosphorylation include PKA, and consequently, it can increase the NF-κB activation (4). Along these lines, the activation of NF-κB may also occur by the activation of PKC since AGE:RAGE interaction activates PKC (46). However, in this scenario, the NF-κB activation, most likely, would occur via the increased generation of ROS. The ROS-induced activation of NF-κB activation, along with that of various inflammatory cytokines, via the classic IKK-dependent pathway, has been well documented in the literature (18). The fact that we observed notable reduction of NF-κB expression along with the decreased generation of AGE-induced ROS with the treatment of MIOX- or RAGE-siRNA would support the notion that ROS played a major role in the activation of ROS-induced activation of NF-κB with a substantial contribution of ROS generated following the upregulation of MIOX (Figs. 3 and 5). Also, the participation of kinases in the upregulation of MIOX and consequential increased generation of ROS and activation of NF-κB is further supported by the fact that their inhibitors of PI3K (wortmannin) and PKC (calphostin) could reduce the expression and activity of MIOX (Fig. 5). The role of MIOX in its upregulation, as well as generation of ROS, is strengthened by our observations that MGO, a potent oxidant, could induce the MIOX promoter activity, which could be significantly dampened by NAC, a strong antioxidant (Fig. 5). These changes in promoter activity would likely affect the expression/activity of MIOX and generation of ROS.
Besides NF-κB, the ROS are known to activate a number of cytokines that are believed to be intimately involved in the progression of diabetic nephropathy (14, 23, 26). They include proinflammatory, e.g., IL-6 and TNF-α, and growth-promoting cytokines, e.g., IGF-1 and TGF-β1. Among various renal cell types, the activation of TGF-β1 by AGE-BSA, via the generation of ROS, has been described in the literature for mesangial and tubular cells (19). Another important antioxidant response element (ARE) pathway, which supports the notion that ROS are relevant to the activation of TGF-β1, entails the involvement of transcription factor Nrf2 and sirtuin1 (21). Nrf2 nuclear accumulation is positively regulated by Sirtuin1, and the latter is inhibited following AGE-BSA treatment of mesangial cells with a concomitant increase in ROS and TGF-β1 activity. This suggests the involvement of Nrf2/ARE pathway signaling events that ultimately modulate the expression of TGF-β1. Another intriguing concept that has emerged in recent years indicates that there is reciprocal modulation of TGF-β and ROS (33). In such a scenario, there would be a perverse cycle of generation of ROS with sustained activation of TGF-β with consequential modulation of various downstream signaling events, ultimately resulting in the increased synthesis of ECM proteins. These cyclic events would be akin to the reciprocal activation between ROS and MIOX as well, meaning that ROS, once generated, would have sustained activation of downstream signaling, for instance, TGF-β1 activation. The fact that ROS generation could be reduced with the treatment of RAGE- or MIOX-siRNA along with downregulation of TGF-β1 would be supportive of our contention that upregulation of MIOX not only generates ROS but also maintains their relentless production with a consequential increase in ECM fibronectin (Fig. 3). Also, our cell culture studies indicating decreased cellular expression of MIOX, DCF-DA, and fibronectin following MIOX-siRNA treatment would strengthen the notion that ROS, at least partially derived as the by products of MIOX activation, play an essential role in the upregulation of profibrogenic cytokines and consequential excessive synthesis of ECM fibronectin (Fig. 4).
To authenticate the validity of the results of in vitro and in vivo studies, experiments related to renal functionality were performed. Administration of AGE-BSA resulted in a notable rise in ACR, which increased over a period of 2 wk (Fig. 6). Likewise, there was an increase in the tubular MIOX activity. The fact that there was a reduction of ACR and MIOX activity following either the inhibitor of RAGE, i.e., FPS-ZM1, or of MIOX, i.e., glucaric acid (GA), suggests that AGE:RAGE interaction while targeting the tubular compartment, possibly via the generation of ROS, can compromise renal functions. Interestingly, these findings suggest that AGE-BSA per se can perturb the renal homeostasis under euglycemic conditions, which would support the previous observations, although they pertain to the pathobiology of intrinsic glomerular cells modulated by ROS, proinflammatory cytokines, and growth factors (19, 43). In regard to the increase in ACR or albuminuria, some of the investigators believe that these functional derangements may be related to the excessive overload of AGE-BSA or fatty acid contamination of the BSA, which led to podocytes or possibly tubular injury (1). This probably is not the case in our studies, since we measured the excretion of endogenous mouse albumin, not BSA, to calculate the ACR, and BSA preparations essentially free of fatty acids were used. The most significant parallel between the results of in vitro studies and in vivo experiments is the activation of downstream signaling events, which most likely would be mediated via ROS (Fig. 6). Among the multitude of molecules that are amenable to activation by ROS include NF-κB, proinflammatory cytokines, and growth factors (7, 19). The fact that we observed upregulation NF-κB and TGF-β along with RAGE and MIOX would support the contention that ROS generated following AGE-RAGE interaction or due to increased activity of MIOX led to such downstream effects. Moreover, administration of glucaric acid, inhibitor of MIOX, led to downregulation of NF-κB and TGF-β1, indeed, strengthening our notion.
The ROS-induced activation of transcription factor NF-κB is likely to induce the expression of a multitude of inflammatory cytokines, including IL-6 and TNF-α (14, 16). In the context of diabetic nephropathy, a low-grade inflammation (microinflammation) is well known as a downstream signaling event of hyperglycemia (53, 58). Besides hyperglycemia per se, the AGEs, both exogenous and endogenous, also can generate the microinflammatory response and upregulation of inflammatory cytokines and, thereby, the modulation of downstream events (41). The fact that the upregulation of inflammatory cytokines is suppressed by the administration of neutralizing RAGE antibody or pyridoxamine, an inhibitor of protein glycation, indicates that AGEs play an important role in the pathobiology of inflammation (14, 16). Likewise, we observed increased expression of IL-6 following exogenous AGE administration and its suppression with administration of RAGE inhibitor FPS-ZM1 and MIOX inhibitor glucaric acid (Fig. 7). The fact that glucaric acid could inhibit the IL-6 expression suggests that MIOX also plays a significant role in the AGE-induced inflammatory response. Besides IL-6, another inducible chemokine, i.e., MCP-1, was also concomitantly induced following the administration of exogenous AGEs (Fig. 7). The MCP-1/CCL2 has been regarded as a diagnostic biomarker in the progression of diabetic nephropathy, and since its expression can be suppressed with in vivo administration of RAGE-Aptamer, this suggests that most likely its upregulation is related to AGE:RAGE interaction (37, 53). Interestingly, the upregulation of MCP-1, besides ROS/NF-κB, may be induced by the activation of TGF-β-PI3K signaling pathway since it could be suppressed by the treatment of SB431542, a TGF-β I receptor inhibitor, or LY294002, a kinase inhibitor (29, 41). Because we observed an upregulation of TGF-β following in vivo administration of AGEs (Fig. 7), this would suggest that a multitude of signaling events is involved in the upregulation of inflammatory cytokines. More importantly, our data suggest that upregulated expression of MIOX is also an important contributory factor in the increased expression of this chemokine. The net result of the increased expression of MCP-1 would lead to an increased influx of inflammatory cells into the interstitium, as documented in Fig. 7, with ensuing anticipated tubulointerstitial injury.
Conceivably, as indicated above, the AGE-induced ROS modulate a multitude of signaling pathways, e.g., ERK-1/ERK-2 and PI3-kinase with the generation of proinflammatory cytokines, while evoking a chronic subclinical microinflammatory response (5, 8, 20, 62). In addition, there is an upregulation of profibrogenic cytokines such as TGF-β1, with activation of their putative receptors and consequential increased synthesis of ECM, leading eventually to renal fibrosis, i.e., glomerulosclerosis and tubulointerstitial fibrosis (9, 21, 44). A majority of the studies that focus on renal fibrosis relate to the biology of intrinsic glomerular cells, including mesangial cells and podocytes, that are affected by AGEs, although the information concerning tubular cells is somewhat limited (44). Exposure of mesangial cells to AGEs has been shown to increase in the synthesis of ECM proteins, such as fibronectin, laminin, and collagen (5, 8, 21, 43, 62). A similar increase in the synthesis of ECM proteins has been observed in glomerular podocytes, which are believed to undergo epithelial-mesenchymal transformation triggered by AGEs (26, 27). Likewise, exposure of proximal tubular cells (NRK-52E) with AGEs has been reported to induce generation of ROS and increased synthesis of fibronectin (44). Along the lines of these in vitro observations, we noted an increased in vivo synthesis of fibronectin in the tubulointerstitial compartment in mice treated with AGE-BSA with concomitant increased expression of MIOX and generation of ROS, as assessed by DHE staining (Fig. 8). The fact that the increased synthesis of fibronectin and the generation of ROS could be abrogated with the simultaneous treatment with FPS-ZM1, a RAGE antagonist, or glucaric acid, a MIOX inhibitor, suggest that the ROS-mediated tubulointerstitial injury is induced by the increased expression of MIOX following AGE:RAGE interaction (Fig. 8).
In summary, this investigation highlights the role of a proximal tubule-specific enzyme, i.e., MIOX, in the causation and amplification of oxygen radical-mediated tubulointerstitial fibrosis. It is worth mentioning here that following AGE:RAGE interaction, there is generation of ROS with upregulation MIOX, via transcriptional and posttranslational mechanisms, and additional generation of ROS, and, thereby, accentuation of tubulointerstitial injury in the context of diabetic nephropathy (Fig. 9).
Fig. 9.

Schematic drawing depicting the amplification of ROS-mediated responses by MIOX following AGE:RAGE interaction. Following AGE:RAGE interaction there is a generation of ROS, which leads to the transcriptional upregulation of MIOX, as well as activation of PKC. In addition, AGE:RAGE interaction leads to sequential activation and phosphorylation of various kinases, including PI3K, PDK, and AKT. They then phosphorylate various transcription factors with their translocation into the nucleus and target various genes that are responsible for profibrogenic and proinflammatory processes consequentially leading to fibrosing injury. As a sidearm of these signaling pathways, the activated kinases can phosphorylate MIOX, thereby increasing its enzymatic activity and cyclic generation of ROS. This pervasive generation of ROS leads to additional phosphorylation of transcription factors with amplification of transcription of target genes and worsening of fibrosing injury in the renal tubulointerstitial compartment, conceivably in settings of hyperglycemic or diabetic state. ECM, extracellular matrix. The drawing has taken into account the data of previous work of various authors (Refs. 38, 39, 47, 50, and 60).
GRANTS
This work was supported by grants from the National Institutes of Health (DK-60635) and Indian Council of Medical Research-International Fellowship (Young Biomedical Scientist) to R. S. Tupe.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.S. and R.S.T. conceived and designed research; I.S., R.S.T., and A.K.W. performed experiments; I.S. and R.S.T. interpreted results of experiments; A.K.W. and Y.S.K. edited and revised manuscript; A.K.W. and Y.S.K. approved final version of manuscript; Y.S.K. prepared figures.
REFERENCES
- 1.Agrawal S, Guess AJ, Chanley MA, Smoyer WE. Albumin-induced podocyte injury and protection are associated with regulation of COX-2. Kidney Int 86: 1150–1160, 2014. doi: 10.1038/ki.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arner RJ, Prabhu KS, Krishnan V, Johnson MC, Reddy CC. Expression of myo-inositol oxygenase in tissues susceptible to diabetic complications. Biochem Biophys Res Commun 339: 816–820, 2006. doi: 10.1016/j.bbrc.2005.11.090. [DOI] [PubMed] [Google Scholar]
- 3.Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer 125: 2863–2870, 2009. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balwani S, Chaudhuri R, Nandi D, Jaisankar P, Agrawal A, Ghosh B. Regulation of NF-κB activation through a novel PI-3K-independent and PKA/Akt-dependent pathway in human umbilical vein endothelial cells. PLoS One 7: e46528, 2012. doi: 10.1371/journal.pone.0046528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berrou J, Tostivint I, Verrecchia F, Berthier C, Boulanger E, Mauviel A, Marti HP, Wautier MP, Wautier JL, Rondeau E, Hertig A. Advanced glycation end products regulate extracellular matrix protein and protease expression by human glomerular mesangial cells. Int J Mol Med 23: 513–520, 2009. doi: 10.3892/ijmm_00000159. [DOI] [PubMed] [Google Scholar]
- 6.Brown PM, Caradoc-Davies TT, Dickson JM, Cooper GJ, Loomes KM, Baker EN. Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc Natl Acad Sci USA 103: 15,032–15,037, 2006. doi: 10.1073/pnas.0605143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J Immunol 165: 1013–1021, 2000. doi: 10.4049/jimmunol.165.2.1013. [DOI] [PubMed] [Google Scholar]
- 8.Chen JL, Francis J. Pyridoxamine, advanced glycation inhibition, and diabetic nephropathy. J Am Soc Nephrol 23: 6–8, 2012. doi: 10.1681/ASN.2011111097. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Kasama Y, Lee JS, Jim B, Marin M, Ziyadeh FN. Podocyte-derived vascular endothelial growth factor mediates the stimulation of α3(IV) collagen production by transforming growth factor-β1 in mouse podocytes. Diabetes 53: 2939–2949, 2004. doi: 10.2337/diabetes.53.11.2939. [DOI] [PubMed] [Google Scholar]
- 10.Croze ML, Soulage CO. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 95: 1811–1827, 2013. doi: 10.1016/j.biochi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Dahrouj M, Desjardins DM, Liu Y, Crosson CE, Ablonczy Z. Receptor mediated disruption of retinal pigment epithelium function in acute glycated-albumin exposure. Exp Eye Res 137: 50–56, 2015. doi: 10.1016/j.exer.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res 323: 475–488, 2006. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 13.Du L, Chen X, Cao Y, Lu L, Zhang F, Bornstein S, Li Y, Owens P, Malkoski S, Said S, Jin F, Kulesz-Martin M, Gross N, Wang XJ, Lu SL. Overexpression of PIK3CA in murine head and neck epithelium drives tumor invasion and metastasis through PDK1 and enhanced TGFβ signaling. Oncogene 35: 4641–4652, 2016. doi: 10.1038/onc.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Münch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J Neurochem 87: 44–55, 2003. doi: 10.1046/j.1471-4159.2003.01988.x. [DOI] [PubMed] [Google Scholar]
- 15.Dutta RK, Kondeti VK, Sharma I, Chandel NS, Quaggin SE, Kanwar YS. Beneficial effects of myo-inositol oxygenase deficiency in cisplatin-induced AKI. J Am Soc Nephrol 28: 1421–1436, 2017. doi: 10.1681/ASN.2016070744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elseweidy MMES, Elswefy SE, Younis NN, Zaghloul MS. Pyridoxamine, an inhibitor of protein glycation, in relation to microalbuminuria and proinflammatory cytokines in experimental diabetic nephropathy. Exp Biol Med (Maywood) 238: 881–888, 2013. doi: 10.1177/1535370213494644. [DOI] [PubMed] [Google Scholar]
- 17.Gallicchio MA, Bach LA. Advanced glycation end products inhibit Na+ K+ ATPase in proximal tubule epithelial cells: role of cytosolic phospholipase A2α and phosphatidylinositol 4-phosphate 5-kinase gamma. Biochim Biophys Acta 1803: 919–930, 2010. doi: 10.1016/j.bbamcr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72: 1493–1505, 2006. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Heidland A, Sebekova K, Schinzel R. Advanced glycation end products and the progressive course of renal disease. Am J Kidney Dis 38, Suppl 1: S100–S106, 2001. doi: 10.1053/ajkd.2001.27414. [DOI] [PubMed] [Google Scholar]
- 20.Hu C, Sun L, Xiao L, Han Y, Fu X, Xiong X, Xu X, Liu Y, Yang S, Liu F, Kanwar YS. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr Med Chem 22: 2858–2870, 2015. doi: 10.2174/0929867322666150625095407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang K, Huang J, Xie X, Wang S, Chen C, Shen X, Liu P, Huang H. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med 65: 528–540, 2013. doi: 10.1016/j.freeradbiomed.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 22.Hussain AR, Ahmed SO, Ahmed M, Khan OS, Al Abdulmohsen S, Platanias LC, Al-Kuraya KS, Uddin S. Cross-talk between NFκB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One 7: e39945, 2012. [Erratum in PLoS One 9: e92484, 2014] doi: 10.1371/journal.pone.0039945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol 6: 395–423, 2011. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato M, Castro NE, Natarajan R. MicroRNAs: potential mediators and biomarkers of diabetic complications. Free Radic Biol Med 64: 85–94, 2013. doi: 10.1016/j.freeradbiomed.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitada M, Ogura Y, Koya D. Rodent models of diabetic nephropathy: their utility and limitations. Int J Nephrol Renovasc Dis 9: 279–290, 2016. doi: 10.2147/IJNRD.S103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar PA, Chitra PS, Reddy GB. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol Concepts 7: 293–309, 2016. doi: 10.1515/bmc-2016-0021. [DOI] [PubMed] [Google Scholar]
- 27.Kumar PA, Welsh GI, Raghu G, Menon RK, Saleem MA, Reddy GB. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch Biochem Biophys 590: 10–19, 2016. doi: 10.1016/j.abb.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Kwak T, Drews-Elger K, Ergonul A, Miller PC, Braley A, Hwang GH, Zhao D, Besser A, Yamamoto Y, Yamamoto H, El-Ashry D, Slingerland JM, Lippman ME, Hudson BI. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 36: 1559–1572, 2017. doi: 10.1038/onc.2016.324. [DOI] [PubMed] [Google Scholar]
- 29.Lee EY, Chung CH, Khoury CC, Yeo TK, Pyagay PE, Wang A, Chen S. The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-β, increases podocyte motility and albumin permeability. Am J Physiol Renal Physiol 297: F85–F94, 2009. doi: 10.1152/ajprenal.90642.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Park J-R, Kim WJ, Sundar IK, Rahman I, Park S-M, Yang SR. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J 31: 2076–2089, 2017. doi: 10.1096/fj.201601155R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao YC, Lee YH, Chuang LY, Guh JY, Shi MD, Huang JS. Advanced glycation end products-mediated hypertrophy is negatively regulated by tetrahydrobiopterin in renal tubular cells. Mol Cell Endocrinol 355: 71–77, 2012. doi: 10.1016/j.mce.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 32.Lin HJ, Hsu FY, Chen WW, Lee CH, Lin YJ, Chen YY, Chen CJ, Huang MZ, Kao MC, Chen YA, Lai HC, Lai CH. Helicobacter pylori activates HMGB1 expression and recruits RAGE into lipid rafts to promote inflammation in gastric epithelial cells. Front Immunol 7: 341, 2016. doi: 10.3389/fimmu.2016.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu R-M, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol 6: 565–577, 2015. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu R, Zhong Y, Li X, Chen H, Jim B, Zhou MM, Chuang PY, He JC. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 63: 2440–2453, 2014. doi: 10.2337/db13-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallipattu SK, Uribarri J. Advanced glycation end product accumulation: a new enemy to target in chronic kidney disease? Curr Opin Nephrol Hypertens 23: 547–554, 2014. doi: 10.1097/MNH.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masola V, Gambaro G, Tibaldi E, Onisto M, Abaterusso C, Lupo A. Regulation of heparanase by albumin and advanced glycation end products in proximal tubular cells. Biochim Biophys Acta 1813: 1475–1482, 2011. doi: 10.1016/j.bbamcr.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Matsui T, Higashimoto Y, Nishino Y, Nakamura N, Fukami K, Yamagishi SI. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 66: 1683–1695, 2017. doi: 10.2337/db16-1281. [DOI] [PubMed] [Google Scholar]
- 38.Meijer B, Gearry RB, Day AS. The role of S100A12 as a systemic marker of inflammation. Int J Inflamm 2012: 907078, 2012. doi: 10.1155/2012/907078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nayak B, Kondeti VK, Xie P, Lin S, Viswakarma N, Raparia K, Kanwar YS. Transcriptional and post-translational modulation of myo-inositol oxygenase by high glucose and related pathobiological stresses. J Biol Chem 286: 27594–27611, 2011. doi: 10.1074/jbc.M110.217141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nayak B, Xie P, Akagi S, Yang Q, Sun L, Wada J, Thakur A, Danesh FR, Chugh SS, Kanwar YS. Modulation of renal-specific oxidoreductase/myo-inositol oxygenase by high-glucose ambience. Proc Natl Acad Sci USA 102: 17952–17957, 2005. doi: 10.1073/pnas.0509089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol 2: 411–429, 2014. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng J, Li X, Zhang D, Chen JK, Su Y, Smith SB, Dong Z. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int 87: 137–150, 2015. doi: 10.1038/ki.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pugliese G, Pricci F, Romeo G, Pugliese F, Mene P, Giannini S, Cresci B, Galli G, Rotella CM, Vlassara H, Di Mario U. Upregulation of mesangial growth factor and ECM synthesis by advanced glycation end products via a receptor-mediated mechanism. Diabetes 46: 1881–1887, 1997. doi: 10.2337/diab.46.11.1881. [DOI] [PubMed] [Google Scholar]
- 44.Qi W, Niu J, Qin Q, Qiao Z, Gu Y. Glycated albumin triggers fibrosis and apoptosis via an NADPH oxidase/Nox4-MAPK pathway-dependent mechanism in renal proximal tubular cells. Mol Cell Endocrinol 405: 74–83, 2015. doi: 10.1016/j.mce.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 45.Rojas A, Delgado-López F, González I, Pérez-Castro R, Romero J, Rojas I. The receptor for advanced glycation end-products: a complex signaling scenario for a promiscuous receptor. Cell Signal 25: 609–614, 2013. doi: 10.1016/j.cellsig.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 46.Scivittaro V, Ganz MB, Weiss MF. AGEs induce oxidative stress and activate protein kinase C-β(II) in neonatal mesangial cells. Am J Physiol Renal Physiol 278: F676–F683, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Sessa L, Gatti E, Zeni F, Antonelli A, Catucci A, Koch M, Pompilio G, Fritz G, Raucci A, Bianchi ME. The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS One 9: e86903, 2014. doi: 10.1371/journal.pone.0086903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma I, Dutta RK, Singh NK, Kanwar YS. High glucose-induced hypomethylation promotes binding of Sp-1 to myo-inositol oxygenase: implications in the pathobiology of diabetic tubulopathy. Am J Pathol 187: 724–739, 2017. doi: 10.1016/j.ajpath.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma K. Obesity and diabetic kidney disease: role of oxidant stress and redox balance. Antioxid Redox Signal 25: 208–216, 2016. doi: 10.1089/ars.2016.6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soro-Paavonen A, Zhang WZ, Venardos K, Coughlan MT, Harris E, Tong DC, Brasacchio D, Paavonen K, Chin-Dusting J, Cooper ME, Kaye D, Thomas MC, Forbes JM. Advanced glycation end-products induce vascular dysfunction via resistance to nitric oxide and suppression of endothelial nitric oxide synthase. J Hypertens 28: 780–788, 2010. doi: 10.1097/HJH.0b013e328335043e. [DOI] [PubMed] [Google Scholar]
- 51.Sun L, Dutta RK, Xie P, Kanwar YS. Myo-inositol oxygenase overexpression accentuates generation of reactive oxygen species and exacerbates cellular injury following high glucose ambience. J Biol Chem 291: 5688–5707, 2016. doi: 10.1074/jbc.M115.669952. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Sutton NR, Hayasaki T, Hyman MC, Anyanwu AC, Liao H, Petrovic-Djergovic D, Badri L, Baek AE, Walker N, Fukase K, Kanthi Y, Visovatti SH, Horste EL, Ray JJ, Goonewardena SN, Pinsky DJ. Ectonucleotidase CD39-driven control of postinfarction myocardial repair and rupture. JCI Insight 2: e89504, 2017. doi: 10.1172/jci.insight.89504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 294: F697–F701, 2008. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 54.Thorsell AG, Persson C, Voevodskaya N, Busam RD, Hammarström M, Gräslund S, Gräslund A, Hallberg BM. Structural and biophysical characterization of human myo-inositol oxygenase. J Biol Chem 283: 15209–15216, 2008. doi: 10.1074/jbc.M800348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tominaga T, Dutta RK, Joladarashi D, Doi T, Reddy JK, Kanwar YS. Transcriptional and translational modulation of myo-inositol oxygenase (MIOX) by fatty acids: implications in renal tubular injury induced in obesity and diabetes. J Biol Chem 291: 1348–1367, 2016. doi: 10.1074/jbc.M115.698191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tupe RS, Agte VV. Effect of water soluble vitamins on Zn transport of Caco-2 cells and their implications under oxidative stress conditions. Eur J Nutr 49: 53–61, 2010. doi: 10.1007/s00394-009-0048-4. [DOI] [PubMed] [Google Scholar]
- 57.Verbeke P, Perichon M, Friguet B, Bakala H. Inhibition of nitric oxide synthase activity by early and advanced glycation end products in cultured rabbit proximal tubular epithelial cells. Biochim Biophys Acta 1502: 481–494, 2000. doi: 10.1016/S0925-4439(00)00071-5. [DOI] [PubMed] [Google Scholar]
- 58.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 124: 139–152, 2013. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 59.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D’Agati VD, Schmidt AM. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162: 1123–1137, 2003. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie P, Kondeti VK, Lin S, Haruna Y, Raparia K, Kanwar YS. Role of extracellular matrix renal tubulo-interstitial nephritis antigen (TINag) in cell survival utilizing integrin (α)vβ/focal adhesion kinase (FAK)/phosphatidylinositol 3-kinase (PI3K)/protein kinase B-serine/threonine kinase (AKT) signaling pathway. J Biol Chem 286: 34131–34146, 2011. doi: 10.1074/jbc.M111.241778. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Xie P, Sun L, Oates PJ, Srivastava SK, Kanwar YS. Pathobiology of renal-specific oxidoreductase/myo-inositol oxygenase in diabetic nephropathy: its implications in tubulointerstitial fibrosis. Am J Physiol Renal Physiol 298: F1393–F1404, 2010. doi: 10.1152/ajprenal.00137.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu D, Kyriakis JM. Phosphatidylinositol 3′-kinase-dependent activation of renal mesangial cell Ki-Ras and ERK by advanced glycation end products. J Biol Chem 278: 39349–39355, 2003. doi: 10.1074/jbc.M302771200. [DOI] [PubMed] [Google Scholar]
- 63.Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol 26: 1304–1321, 2015. doi: 10.1681/ASN.2014050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Spiegelman NA, Nelson OD, Jing H, Lin H. SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation. Elife 6: e25158, 2017. doi: 10.7554/eLife.25158. [DOI] [PMC free article] [PubMed] [Google Scholar]