

Abstract
The pancreatic β-cell plays a key role in glucose homeostasis by secreting insulin, the only hormone capable of lowering the blood glucose concentration. Impaired insulin secretion results in the chronic hyperglycemia that characterizes type 2 diabetes (T2DM), which currently afflicts >450 million people worldwide. The healthy β-cell acts as a glucose sensor matching its output to the circulating glucose concentration. It does so via metabolically induced changes in electrical activity, which culminate in an increase in the cytoplasmic Ca2+ concentration and initiation of Ca2+-dependent exocytosis of insulin-containing secretory granules. Here, we review recent advances in our understanding of the β-cell transcriptome, electrical activity, and insulin exocytosis. We highlight salient differences between mouse and human β-cells, provide models of how the different ion channels contribute to their electrical activity and insulin secretion, and conclude by discussing how these processes become perturbed in T2DM.
I. INTRODUCTION
We first met in a swimming pool, on a hot sunny day in Alicante in 1985, at the inaugural meeting of what was to become the Islet Study Group of the European Association for the Study of Diabetes. Since then, we have engaged in a scientific dance, collaborating at times, competing (in a friendly fashion) at others. It is now almost 30 yr since we wrote our first review on the electrophysiology of the pancreatic β-cell (31). Happily, it has not been, as T. S. Eliot once bemoaned, ‟twenty years largely wasted.ˮ Although our writing style may not have improved, the field has made enormous progress. We now know substantially more about the electrical activity of the β-cell and about the mechanisms of exocytosis. Transcriptome analysis of purified human (68, 475) and mouse (3, 56) β-cells is finally available.1 Genetic studies have identified the genes responsible for the most common forms of monogenic diabetes and numerous single nucleotide polymorphisms (SNPs) associated with an increased risk of type 2 diabetes (T2DM) (79, 519). We also have a better understanding of how insulin secretion is regulated in health and disease. Thus the aim of this review is to summarize these findings and attempt to synthesize them into a coherent picture of insulin secretion.2 Arguably, the adult human β-cell and its physiological regulation represent the ‟gold standardˮ for new therapies based on surrogate β-cells (546).
An updated review of the physiology of insulin secretion is particularly timely, given the current pandemic of T2DM, which shows no sign of abating. Currently, it is conservatively estimated that more than 400 million people are affected worldwide, and it is predicted that cases will exceed 500 million by 2030. Diabetes increases the risk of secondary complications such as cardiac disease, renal failure, and retinopathy. Impaired microvascular function combined with peripheral neuropathy make diabetes the most common cause of nontraumatic amputation. As a result, it causes a marked increase in both morbidity and mortality. Costs associated with diabetes consume (at least) $322 billion per year, and one in five healthcare dollars is spent caring for people with diabetes (see http://www/diabetes/org/diabetes-basics/statistics/infographics/adv-staggering-cost-of-diabetes.html). This burden is rapidly becoming unaffordable. Thus it is important to identify the cause of diabetes and ways in which it may be ameliorated. This requires a better understanding of the disease etiology.
All forms of diabetes are characterized by elevated blood glucose due to an insufficiency (absolute or relative) of the hormone insulin, which is secreted by the β-cells of the pancreatic islets. It is now clear that although both defective insulin secretion and impaired insulin action in peripheral tissues contribute to T2DM, the principal defect lies in the pancreatic β-cell (reviewed in Refs. 30, 580). Although T2DM is associated with a reduction in β-cell mass, this is too small to account for the reduced insulin secretion. Furthermore, there is evidence that β-cell mass in T2DM may have been underestimated due to equating a reduction in insulin staining/content with a reduction in β-cell number (97, 429, 667). Thus the evidence favors the idea that T2DM is a disease of impaired β-cell function, resulting from a reduction in insulin content, and a failure of the β-cell to respond to glucose stimulation with insulin secretion. Consequently, if we are to grasp what goes wrong with this process in T2DM, it is imperative that we understand how insulin secretion is regulated physiologically.
Insulin plays a major role in glucose homeostasis as it is the only hormone capable of lowering the blood glucose concentration. It is for this reason that defects in insulin secretion produce the severe metabolic disorders associated with uncompensated T2DM. Insulin is secreted by the β-cells of the pancreatic islets of Langerhans in response to elevation of the intracellular Ca2+ concentration ([Ca2+]i). This is produced by an influx of extracellular Ca2+ via voltage-dependent Ca2+ channels, whose activity, in turn, is regulated by the β-cell membrane potential. This means that electrical activity is critically important for insulin release.
This review provides an electrophysiologist’s perspective of insulin secretion. We consider how glucose, via its uptake and metabolism, initiates β-cell electrical activity and insulin exocytosis. We discuss the ion channels involved and their modulation by stimulators and inhibitors of insulin release. We then examine how action potential firing, via an increase in cytoplasmic [Ca2+]i, triggers insulin exocytosis. Finally, we briefly consider what goes awry in T2DM and the nature of the defect(s) responsible for impaired insulin secretion.
It is worth pointing out that diabetes is a disease of Western lifestyle and that elevation of blood glucose is not deleterious provided it is not prolonged. In contrast, it can be fatal if the blood sugar falls too low (<3 mM) for just a few minutes, as the brain is starved of fuel. Homeostatic mechanisms have evolved to ensure this does not happen. This probably explains why insulin is the only hormone that is able to lower the blood sugar, whereas several hormones are capable of increasing it (189). It is only in the last 50 yr or so that food has been plentiful in many societies. Consequently, our bodies are adapted to conserve resources and not to deal with an excess [the thrifty phenotype hypothesis (250)].
Much of our knowledge of insulin secretion comes from work on rodent islets and β-cells. However, the ultimate goal is to understand the human β-cell. Although human and rodent β-cells share many features, it should not be taken for granted that they are identical. Indeed, a number of important differences have been identified. Mice are nocturnal whereas humans are diurnal (with the possible exception of teenagers). Moreover, feeding behaviors differ. Rodents feed continuously while awake whereas humans feed 3–4 times per day, and although both mice and humans are omnivores, it is clear that their diets are quite different. It seems likely that these differences will have an impact on the physiology of insulin secretion and action. Consequently, it is essential that observations in rodents are confirmed in human cells. Here we compare the properties of mouse islets (the most widely used experimental preparation) with those of human islets. We focus principally on studies of primary β-cells. However, it should be remembered that some studies are performed on freshly isolated islets, whereas others use islets that have been cultured overnight. Short-term culture has its advantages (e.g., cells damaged by the isolation procedure may recover), but tissue culture may also lead to changes in gene expression and metabolism.
Much valuable information has been also obtained from global or β-cell-specific genetically engineered mice, but there are a few caveats with the use of such models that should not be forgotten. First, compensatory changes may take place in response to gene deletion. Second, mouse models of disease do not necessarily recapitulate the human phenotype. For example, the SUR1 knockout mouse exhibits a much weaker phenotype than its human counterpart. Whereas loss of SUR1 in humans results in severe hypoglycemia (life-threatening if untreated) (213), SUR1-KO mice are viable and essentially normoglycemic (620). Third, as we discuss below, the complement of ion channels, receptors, transporters, and exocytotic proteins differs in human and mouse β-cells.
II. THE PANCREATIC ISLET AND THE β-CELL
A. The Pancreatic Islet
The pancreatic islets are endocrine microorgans that are embedded in the exocrine parenchyma of the pancreas. They lie scattered throughout the organ, often with a higher density in the tail region. With the use of elegant optical projection tomography, that allows all islets to be visualized and counted, it has been shown that a pancreas from an 8-wk-old mouse contains ~1,100 pancreatic islets, that occupy 10−7 l (8). From these values we estimate that the average islet has a volume of 100 pl, corresponding an islet diameter of 60 μm (assuming spherical geometry). Furthermore, given that a single mouse β-cell has a volume of 1.3 pl,3 this suggests the average mouse islet only contains ~80 cells. This is surprisingly small and considerably less than the ~1,000 cells/islet commonly quoted as ‟typicalˮ for an islet. Interestingly, <10% of the islet number accounts for 50% of the β-cell volume. Presumably this subset of large islets corresponds to those that are isolated when the pancreas is dispersed by collagenase digestion (where the typical yield is 100–200 islets/mouse), and thus to those islets used for experiments. A human pancreas contains ~1 million pancreatic islets with an average diameter of 130 µm (259), corresponding to ~200 β-cells per islet (568).
The mature pancreatic islet consists of several types of endocrine cell (FIGURE 1, A–D). The most important are the insulin-secreting β-cells (which make up 50% of cells in human islets and 75% in the mouse), the glucagon-releasing α-cells (35–40% in human and 15–20% in mice), and the somatostatin-releasing δ-cells (10–15% in human and ~5% in the mouse) (102). There are also a small number of pancreatic polypeptide (PP)-secreting cells. Mouse islets consist of a clearly demarcated core of β-cells surrounded by a mantle of non-β-cells. In contrast, human islets have a more complex architecture in which β-cells are directly juxtaposed to non-β-cells, and non-β-cells are also found within the islet core (82). In both species, the δ-cells have many long processes that ramify throughout the islet and make contact with several α- and β-cells. The complex microanatomy of the islet will determine the extent to which paracrine interactions impinge on β-cell secretion, and this may differ between individual islets, especially in humans.
FIGURE 1.

A and B: immunohistochemistry of mouse (A) and human (B) pancreatic islets (red, insulin; green, glucagon; blue, somatostatin). Images provided by Dr A Clark, Oxford. Scale bars: 20 μm. C and D: schematic of mouse (C) and human (D) islets highlighting differences in blood supply, innervation, and islet cell distribution. The α- (green), β- (red), and δ-cells (blue) are indicated. Also illustrated (C, gray) is a pancreatic ganglion cell (613). E and F: electron micrographs of mouse (E) and human (F) β-cells. Scale bars: 500 nm. In F, the β-cell is surrounded by a δ- and an α-cell (granules indicated by α and δ). Electron micrographs provided by Prof. L. Eliasson, Lund (E), and Dr. A. Clark, Oxford (F). m, Mitochondrion; l, lipfuscin body; sg, secretory granule.
The pancreas receives a rich vascular supply originating from the splenic artery so that the islets are exposed to the systemic, not the portal, glucose concentration. It is drained by the splanchnic veins, which empty into the hepatic portal vein (FIGURE 1, C and D). Although the islets only comprise 1–2% of the pancreas, they receive up to 20% of the pancreatic blood supply (313). The blood flow is 0.5–1 ml·min−1·g pancreas−1 in both humans and mice (108, 304). For a mouse pancreas that weighs 0.2–0.4 g (69), the perfusion rate is thus 0.1–0.4 ml/min. The weight of a human pancreas is 60–100 g (597), from which a perfusion rate of 60–80 ml/min can be estimated (that is, 0.5–1% of the cardiac output).
The pancreatic islets are extensively innervated by cholinergic, adrenergic, and peptidergic nerve branches (405, 556, 683). In mouse islets, these nerve branches extend into the pancreatic islets and make direct contact with the endocrine cells (405). However, human islet cells appear more sparsely innervated, and the little innervation that is observed makes contact with smooth muscle rather than endocrine cells (555). Thus autonomic regulation of human islets may be primarily indirect, via changes in blood flow.
B. The β-Cell
The β-cells are the principal component of the pancreatic islets in all species. They are polygonal cells, with an average diameter of 13–18 μm (221) that possess ~10,000 secretory granules (489), each containing up to 8–9 fg insulin (1.6–1.8 amol insulin). This corresponds to an intragranular insulin concentration of ~100 mM (294, 571). Insulin is stored in crystalline form in the secretory vesicles as a Zn2-insulin6 complex (FIGURE 1, E and F), and accounts for 5–10% of the total protein content of the β-cell, more than any other protein. It is released by regulated exocytosis. Only a small fraction of the secretory granules (<1%/h) undergo exocytosis even at high glucose concentrations (571). Whereas the insulin granules in mouse β-cells typically have a ‟fried eggˮ appearance, the insulin core/crystal is more irregular in human β-cells (FIGURE 1, E and F). Human β-cells also contain lipofuscin bodies (wear-and-tear pigments) that can be used to estimate the age of the β-cell (121).
III. GLUCOSE METABOLISM
Physiologically, insulin is released following food ingestion. However, only a few nutrients are capable of eliciting insulin secretion on their own. Classified as ‟initiatorsˮ of insulin secretion, these include glucose (the most important physiological stimulus), the amino acid leucine, substances that stimulate metabolism of endogenous nutrients, and drugs such as the antidiabetic sulfonylureas. Many other nutrients are capable of enhancing insulin release but because they require the presence of an initiator to be effective, they are referred to as ‟potentiatorsˮ or ‟amplifiersˮ of insulin secretion (31, 276). These include most amino acids (270), fatty acids (31), hormones, and neurotransmitters. The mechanisms by which they act are considered in section VIIB, 1 and 2.
It is worth noting that in vivo the primary signal for insulin secretion is not usually glucose, but neurotransmitters released in response to the sight or smell of food (the cephalic phase of release) or incretins, peptides released from the gut due to the presence of food in the gut lumen. This is because circulating glucose concentrations at rest (4–5 mM in human) are sufficient to enable the action of these potentiators of release. Together, these mechanisms prepare the body for the subsequent increase in plasma glucose and prevent blood glucose levels from rising too high after a meal. It also explains why insulin secretion is greater in response to an oral glucose challenge than an intravenous one. Indeed, increasing glucose from 5 to 7.5 mM only produces a 30% stimulation of insulin release from isolated human islets (717), whereas it produces an up to 500% increase in plasma insulin in vivo (712).
In human islets, insulin secretion is detectable at glucose concentrations as low as 3 mM (89, 150, 266), which is significantly lower than the secretion threshold in mouse islets (~5 mM; Ref. 257). This correlates with the lower fasting plasma glucose concentration in humans (4–5 mM vs. 7–10 mM in mice) (189, 389, 540) (FIGURE 2A). It probably reflects differences in intrinsic β-cell properties as when human islets are transplanted into diabetic mice they control plasma glucose at the normal nonfasting glucose concentration of humans (551). In both mouse and human islets, insulin secretion is half-maximal at 10–12 mM glucose and saturates at glucose concentrations above 20 mM.
FIGURE 2.

A: relationship between glucose concentration and insulin secretion in static incubations of isolated mouse (red) and human (black). Secretory responses have been normalized to secretion at 20 mM glucose. [Modified from Walker et al. (717).] B: relationship between glucose concentration and oxidative metabolism of the sugar (measured as 14CO2 production radiolabeled glucose) in isolated mouse islets (75% β-cells). Glucose oxidation is half-maximal at ~6 mM glucose (arrow). [Modified from Ashcroft et al. (33).] C: relationship between glucose concentration and ATP content (red) and β-cell whole-cell KATP channel conductance (GK,ATP: normalized to conductance at 0 mM glucose) in isolated mouse islets. Effects of glucose on both parameters are half-maximal at ~3 mM glucose (arrowed). [Modified from Ashcroft et al. (34) and Zhang et al. (762).]
As will be discussed at length in section IX, glucose-induced insulin secretion (GIIS) follows a biphasic time course in both mouse and human islets. A step increase in glucose produces an initial transient response, referred to as 1st phase secretion, that is subsequently followed by a slower and more gradual rise in secretion (2nd phase release) (473).
A. Glucose Uptake and Metabolism
It is well established that GIIS requires the metabolic degradation of the sugar and an increase in intracellular ATP. This is a vast topic that has received a considerable amount of attention, and it is not our aim to give a comprehensive account. Rather, we provide a brief overview of β-cell metabolism as an introduction to the metabolic regulation of β-cell electrical activity and insulin secretion (for further details of glucose metabolism, see reviews in Refs. 315, 416, 476, 520). It is likely that this differs in T2DM, and indeed changes in metabolism are likely to contribute to the etiology of the disease.
Glucose metabolism by the β-cell has several unique properties that enable it to act as a glucose sensor and adjust insulin secretion to the plasma glucose level. These ensure that glucose metabolism is controlled by substrate availability rather than energy demand.
1. Glucose uptake
Glucose is transported into the β-cell by facilitated diffusion. In mice, this is mediated by Glut2 (Slc2a2), which has a low affinity for glucose (i.e., has a high Km) and a high transport capacity (682). This enables fast equilibration of intracellular glucose with that in the extracellular medium and ensures that glucose uptake is not rate limiting for glycolytic flux. GLUT1 (SLC2A1) constitutes the primary glucose transporters in human β-cells (258, 437) (see Supplemental Figure 1B, available online; see footnote 1). This transporter has a lower Km (6) than GLUT2 (SLC2A2; 11 mM) (437), which may explain why insulin secretion is initiated at lower glucose concentrations in human islets (3 mM) than in mouse islets (6 mM). However, GLUT2 is not only expressed in human β-cells but is likely to play a functional role, because patients with homozygous loss-of-function mutations in GLUT2 develop transient neonatal diabetes (589). Furthermore, even in human β-cells, glucose uptake is not rate limiting for glucose metabolism (385).
Gene expression (RNAseq) data suggest human β-cells also express the Na+-dependent glucose transporters SGLT1 and 2 (SLC5A1 and SLC5A2), but at levels considerably lower than found for GLUT1 (68). In contrast, neither SGLT is expressed in significant amounts in mouse β-cells (56) (see Supplemental Figure 1B).
2. Glucose metabolism
Glucose metabolism is initiated by the phosphorylation of glucose, a reaction catalyzed by the enzyme glucokinase (GCK; also known as hexokinase IV) (80). Unlike hexokinase, GCK is not subject to product inhibition by glucose-6-phosphate (435). It also has a far lower affinity for glucose, GCK activity being half-maximal at 8 and 4 mM in mouse (395) and human (150) islets, respectively (hexokinase is half-maximal at 1 mM glucose). Phosphorylation of glucose is the rate-limiting step in insulin secretion, and loss-of-function mutations in GCK impair GIIS, with heterozygous mutations leading to maturity-onset-diabetes-of-the-young (MODY) and homozygous mutations to neonatal diabetes (214). Similarly, knockout of glucokinase in mice produces hyperglycemia and is eventually fatal (679). The critical role of GCK in GIIS is also demonstrated by the effects of inhibitors and activators of the enzyme. For example, mannoheptulose, an inhibitor of GCK, strongly suppresses GIIS (536) and, conversely, glucokinase activators stimulate GIIS (319). Indeed, the GCK activator piragliatin restores GIIS in islets isolated from T2DM donors to levels comparable to those seen in islets from nondiabetic donors (150).
The concerted effects of Glut2 and Gck result in a steep concentration-dependent acceleration of glucose metabolism in β-cells with increasing glucose, such that elevation of glucose from 1 to 10 mM produces a >10-fold rise in the rate of glucose metabolism in rodent β-cells (FIGURE 2B) (33).
An important aspect of glucose metabolism in the β-cell is that almost all the glucose entering glycolysis proceeds into the Krebs cycle (520, 602). Under normal conditions, glycogen synthesis accounts for <7% of glucose uptake, the conversion of glucose to lipids and amino acids is of minor quantitative significance, and the sorbitol and pentose phosphate pathways are relatively unimportant. Little or no glucose is metabolized to lactate because expression of both lactate dehydrogenase (LDH1) and the monocarboxylate transporter MCT1 (Slc16a1) is suppressed (684). The absence of these ‟disallowedˮ genes is necessary to prevent triggering of insulin secretion during exercise by circulating pyruvate or lactate; indeed, expression of MCT1 in human β-cells (due to a mutation in its promoter) leads to exercise-induced hypoglycemia (495). The close coupling between glycolysis and mitochondrial oxidation explains why β-cells lack the Warburg effect (in which glycolysis accelerates under conditions in which ATP is lowered).
β-Cells are unusual in that an increase in blood glucose elevation leads to elevation of intracellular ATP. This inhibits KATP channel activity, so triggering membrane depolarization, electrical activity, and insulin release. The glucose-induced increase in intracellular ATP is half-maximal at 3 mM glucose and saturates at ~20 mM (FIGURE 2C). This glucose dependence is comparable to that of the glucose-induced closure of the KATP channels (FIGURE 2C), but it is much lower than that of insulin secretion (FIGURE 2A).
Mitochondrially generated ATP is crucial for glucose-stimulated electrical activity and insulin secretion as both are prevented by mitochondrial inhibitors such as azide (144). Similarly, β-cell lines in which mitochondria have been removed (rho-0 cells) fail to release insulin in response to glucose stimulation, and reintroduction of normal mitochondria restores normal insulin secretion (418). Inactivation of the von Hippel-Lindau (vHL) protein, which results in HIF1-α stabilization and the diversion of glucose from oxidative metabolism towards glycolysis, also leads to strong suppression of ATP production, β-cell electrical activity, and GIIS (106, 756). Conversely, agents that are metabolized entirely within the mitochondria (e.g., ketoisocaproate, methyl pyruvate4) are able to stimulate insulin secretion (375, 500; but see Refs. 86, 158).
An important aspect of ATP production is that it leads, via KATP channel closure and initiation of β-cell electrical activity, to an increase in [Ca2+]i. This in turn produces elevation of mitochondrial (matrix) Ca2+ ([Ca2+]m) (677), which stimulates oxidative mitochondrial metabolism (417). Ca2+ import into the mitochondria is mediated by the mitochondrial Ca2+ uniporter (MCU), and silencing MCU in β-cells reduces the increase in intracellular ATP/ADP produced by glucose (676).
Mitochondrial metabolism not only leads to ATP production, it also generates essential coupling factors that amplify insulin secretion (416). For example, glutamate is generated from α-ketoglutarate (419). It can also be derived from the malate-aspartate shuttle (205). The loss of these Krebs cycle reactants (cataplerosis) is compensated for by their continual replenishment (anapleurosis). Other mitochondrially derived potential coupling factors include ATP and NADPH (416) (see sect. IXD, 3 and 7a).
Most of what is known about β-cell metabolism derives from rodent studies, and there is a dearth of data on human islets, and especially for T2DM β-cells. In islets from nondiabetic donors, the rate of glucose oxidation increases threefold when glucose is raised from 1 to 6 mM, and there is only a small additional (25%) acceleration when glucose is further increased to 12 mM or beyond (150). Surprisingly, it seems that (unlike mouse β-cells) only a fraction of glucose entering glycolysis proceeds into the Krebs cycle (150). The significance of this finding is not understood. Nevertheless, mitochondrial oxidative metabolism is essential for GIIS, and the mitochondrial uncoupler FCCP promptly lowers insulin secretion to basal levels (266). The importance of mitochondrially generated ATP for GIIS also accounts for the fact that mitochondrial disease can lead to diabetes, as in maternally inherited diabetes with deafness (MIDD), an extremely rare genetic disorder (390, 408, 418). Interestingly, a common variant of the transcription factor B1 mitochondrial (TFB1M; also known as dimethyladenosine transferase 1 mitochondrial) predicts future risk of T2DM, and its lowered expression in mouse β-cells leads to reduced nutrient-induced ATP generation and GIIS (353).
Oxidative glucose metabolism (measured as oxygen consumption rate) is reduced in islets from T2DM donors (141), and the dose-response relationship is shifted towards slightly higher concentrations; the half-maximal rate of oxygen production occurred at 4.4 and 5.4 mM glucose in control and T2DM islets, respectively (150). The ability of glucose to elevate ATP is also lacking, suggesting oxidative metabolism is reduced (18). These observations suggest that glucose metabolism is impaired in T2DM. This idea is supported by the fact that like diabetic mouse β-cells (97), T2DM human islets accumulate large amounts of glycogen (97, 685). Chronic hyperglycemia is also associated with marked changes in expression of metabolic genes in mouse β-cells (97), and similar changes are observed in islets from individuals with T2DM (409, 430, 606).
IV. β-CELL ELECTRICAL ACTIVITY
Although of endodermal origin, β-cells share many features with nerve cells including electrical excitability. Changes in membrane potential couple variations in the blood glucose concentration to insulin secretion (154, 273). There is a close correlation between the glucose dependence of insulin release and the percentage of time the cell spends firing action potentials (442). This is because Ca2+ influx through voltage-gated Ca2+ channels is essential for insulin exocytosis (169). Here, we first consider the different methods of recording electrical activity and their relative advantages and disadvantages. We then describe the effects of glucose on electrical activity in mouse and human β-cells.
A. Methodological Considerations
Membrane potential recordings were originally conducted in microdissected pancreatic islets using sharp intracellular microelectrodes. With this technique it is possible to record changes in membrane potential and electrical activity (40), even in vivo (587), and the input resistance of the cell can be estimated by injecting a current pulse and monitoring the associated voltage change. Microelectrodes have also been used to record the voltage-gated currents that underlie action potential firing (439, 560). However, they suffer from the disadvantage that a large leak current is produced by insertion of the electrode into the cell, and the high resistance of the electrode means that it is not possible to inject current sufficiently fast to reliably analyze the rapid activation kinetics of the membrane currents involved in action potential firing.
More recently, techniques have been developed that allow the extracellular recording of β-cell electrical activity using microelectrode arrays (373, 512). This methology has been applied to both mouse and human β-cells. It has the advantage that it is complely noninvasive and recordings can be be maintained for many hours/days. However, it does not provide as much detailed information as intracellular membrane potential recordings.
Most voltage-clamp measurements have been performed on isolated β-cells maintained in tissue culture using the patch-clamp technique (31, 32, 251). Important aspects of this method are summarized in FIGURE 3.
FIGURE 3.

Patch-clamp techniques. A: the experiments start with establishment of the cell-attached configuration. In this recording mode, a patch electrode is tightly sealed to the surface of an intact cell, allowing channel activity in the patch of membrane under the electrode tip to be studied under physiological conditions. For example, changes in channel activity in response to glucose metabolism can be measured by adding glucose to the bath solution. The seal between the electrode and the membrane is mechanically very stable, which enables additional configurations to be obtained. B: upon withdrawal of the electrode, the piece of membrane spanning the electrode tip is ripped off, forming an excised membrane patch that has its intracellular surface exposed to the bath solution (an inside-out patch). This is used for testing the effects of cytosolic constituents, such as ATP, on channel function. C: the plasma membrane outside the recording electrode can be permeabilized using detergents [like digitonin or saponin (162)] or the pore-forming peptide α-toxin [from Staphyloccus aureus (674)] to allow exchange of small molecules with a diameter of <1.5 nm (such as ATP) but not larger molecules (like enzymes). This recording configuration is referred to as the open cell-attached. D: the membrane beneath the electrode tip can be destroyed by suction, providing electrical access to the cell interior. This is known as the standard whole-cell configuration as it measures the summed activity of all ion channels in the cell membrane. It allows dialysis of the cell contents with the pipette solution. For example, the intracellular ion concentrations and cytosolic constituents (like ATP) can be manipulated by this route. The whole-cell configuration can also be used to preload the cells with biologically inert precursors of intracellular regulators that can then be photoliberated by a flash of ultraviolet light (‟caged compoundsˮ). E: withdrawal of the pipette from the standard whole-cell configuration produces an outside-out patch, in which the external membrane surface faces the bath solution. This is used to test the effects of extracellular ligands on channel activity. It can also be used as a ‟snifferˮ patch to probe the release of substances from the β-cell, if the membrane patch contains receptors to the compound of interest. F: the perforated patch whole-cell configuration allows measurement of electrical activity or whole-cell currents from a metabolically intact cell (291). In this variant of the whole-cell configuration, a pore-forming antibiotic [such as amphotericin (531)] is incorporated into the membrane below the pipette tip, thereby establishing electrical access to the cell while leaving cellular metabolism and intracellular second messenger systems intact.
All patch-clamp configurations can also be applied to freshly isolated intact islets. In this case, the islet is held in place using a wide-bore pipette and the patch-clamp electrode is applied to a cell at the opposite pole of the islet (221). Although this technique is restricted to cells in the periphery of the islet (FIGURE 1, A and C), there are a sufficient number of β-cells to allow in situ electrophysiological analyses (224). Alternatively, voltage-clamp measurements can be performed in pancreatic tissue slices (640) using an adaptation of the original brain slice technique (167).
There are a number of caveats to be born in mind when extrapolating data obtained in patch-clamp studies to the physiological situtation. First, the different patch-clamp configurations may influence the activity of the ion channels being studied, because of washout of channel regulators in excised patches and standard whole-cell recordings. Second, there are potential pitfalls in extrapolating data obtained in dispersed single β-cells to the situation in whole islets, as cell dispersion and overnight culture may affect the ion channel properties, or channel density. Third, electrophysiological experiments are sometimes carried out with unphysiological ion concentrations or with nonphysiological ions. For example, glucose-induced electrical activity is often recorded at 2.6 mM rather than 1.3 mM (which is the free Ca2+ concentration in plasma), although it appears that this has a relatively minor impact on electrical activity in mouse β-cells (38). It should also be noted that the extracellular concentations of Ca2+ and K+ in the islet interstium are not stable, but oscillate in parallel with electrical activity, as can be seen using ion-selective microelectrodes (506, 507).
The use of different permeant ions can also influence the biophysical properties of a given channel. For example, Ca2+-dependent Ca2+ inactivation is not seen if Ba2+ is substituted as a charge carrier for Ca2+, and extracellular divalent cations may shift the channel’s voltage dependence. Similarly, drugs used to facilitate the ability to record an ion channel may influence its properties (e.g., BAY K 8644) and many ion channel blockers are not as selective as is often claimed.
Finally, given that the different islet cell types possess distinct ion channel complements, it is important to know from which cell type the data derive. Islet cells can be identified functionally by their electrophysiological fingerprint (their ion channel complement) (98) or electrical response to glucose (β-cells starts firing action potentials at high glucose whereas the α-cells are active at low glucose; Ref. 761). Alternatively, cells can be identified before the electrophysiological measurements are made by expressing fluorescent proteins in a cell-specific fashion, or subsequently, by immunocytochemical identification of cells marked by infusion of the tracer biocytin into the cell from the pipette (760).
B. The Importance of Membrane Resistance
The membrane current represents the summed activity of all ion channels in the membrane: the greater their activity, the larger the membrane current. Its relationship to the membrane conductance is given by Ohm’s law, which states that conductance (G) is current (I) divided by potential (V), i.e., G = I/V. Resistance (R) is the reciprocal of conductance. Thus, for the same current magnitude, the change in membrane potential it produces will be determined by the membrane conductance, being greater when the conductance is small, and less when the conductance is low. This explains why a small background inward current is unable to depolarize the β-cell when KATP channels are open, but can do so when they are mostly closed (FIGURE 4).
FIGURE 4.

A: at low glucose, high KATP channel activity (thick black arrow) keeps the β-cell membrane potential negative, and depolarizing conductances (narrow red arrow) are too small to have a major impact. B: at high glucose, KATP channel activity is strongly reduced, and depolarizing currents (even small ones) will exert a stronger effect on the membrane potential. C: the input resistance (R) of the β-cell membrane determines the ease with which electrical activity can be initiated. When KATP channel activity is high, R is low. Conversely, when the KATP channels are shut, R is high. From Ohm's Law (), it is evident that the same magnitude of current (I) will produce a much greater change in membrane potential (ΔV) when R is high (red trace) than when it is low (black trace). At high glucose, a small current may depolarize the β-cell sufficiently to trigger action potential firing (dotted red line). The ‟tug-of-warˮ between repolarizing and depolarizing membrane currents explains why potentiators of insulin secretion such as acetylcholine and arginine, which activate small depolarizing currents, are ineffective in the absence of glucose, when the activity of the KATP channels is high (i.e., R is low), but are able to stimulate electrical activity and insulin secretion at glucose concentrations that shut most KATP channels (i.e., R is high).
C. Electrical Activity
1. Mouse β-cells
At nonstimulatory glucose concentrations, the mouse β-cell is hyperpolarized and electrically silent. The membrane potential can be as negative as −80 mV, which is close to that predicted for a pure K+ conductance. Increasing glucose to 5 mM depolarizes the β-cell to about −60 mV. However, electrical activity is not elicited until the glucose concentration exceeds 6 mM, which depolarizes the membrane to between −60 and −50 mV (the threshold for electrical activity).
Mouse β-cells have a highly distinctive glucose-dependent pattern of electrical activity. At glucose concentrations between 6 and 20 mM, this consists of short-lived bursts of action potentials, superimposed on a depolarized plateau and separated by repolarized electrically silent intervals. This pattern of activity is sometimes referred to as slow wave activity (FIGURE 5, A and B). At 10 mM glucose, the active and silent phases last 5–10 s and 10–20 s, respectively, and there are 2–4 bursts/min (154, 273, 570). The peak of the action potential usually does not exceed 0 mV (154, 761). Its width is somewhat variable but, at 20 mM glucose, the time spent above −20 mV (the voltage where insulin granule exocytosis first becomes detectable; Ref. 222) is ~30 ms (288). Increasing glucose concentrations produce a progressive increase in burst duration at the expense of the repolarized intervals until firing finally becomes continuous, usually above ~20 mM glucose (154, 273).
FIGURE 5.

A: glucose-induced electrical activity recorded from a β-cell in an acutely isolated intact mouse islet when the glucose concentration was increased from 1 to 10 or 20 mM (as indicated by horizontal bars). Note that 10 mM glucose evokes a biphasic response and that continuous action potential firing is replaced by oscillatory (bursting) electrical activity after the initial 3–4 min (red horizontal line beneath the membrane potential recording). At 20 mM glucose, electrical activity is continuous and there is a time-dependent ~15 mV reduction in the amplitude of the action potentials (recording provided by Dr. Q. Zhang, Oxford). B: bursts of action potentials in a β-cell in a freshly isolated mouse islet exposed to 10 mM glucose shown on an expanded time base. C: changes in cytoplasmic free Ca2+ ([Ca2+]i) in response to a step increase in glucose from 1 to 15 mM glucose. Note the triphasic response to glucose: an initial lowering below basal [Ca2+]i (red dashed line) (1), a rapid increase to a peak (2) followed by a decline to an elevated plateau on which small oscillations are superimposed (3). D: the consensus model of GIIS. Glut2, glucose transporter; KATP channels, ATP-sensitive K+ channels; Ψ, membrane potential; SG, secretory granules. The + and – signs denote stimulation and inhibition, respectively, whereas the arrows (↑,↓) indicate an increase or decrease of the indicated parameter. The red arrow connecting insulin and glucose indicates feedback regulation of insulin secretion via changes in plasma glucose.
Electrical activity is accompanied by changes in cytoplasmic free Ca2+ ([Ca2+]i) (701) (FIGURE 5C), which in turn drive pulsatile insulin secretion (210). Glucose induces a triphasic response in [Ca2+]i consisting of an initial small fall in [Ca2+]i, followed by an initial peak and then a sustained lower plateau on which small oscillations in [Ca2+]i are superimposed. As β-cells are electrically coupled, electrical activity and changes in [Ca2+]i spread across the islet in a synchronous fashion with the ‟waveˮ being initiated in one part of the islet and then propagating to other parts with a slight delay (165, 440, 591). At glucose concentrations just above the threshold for electrical activity (~6 mM), only 7% of the β-cells exhibit [Ca2+]i oscillations (761), but as the glucose concentration is increased, the fraction of active β-cells increases. Thus, although every β-cell has the capacity to respond to glucose in a graded fashion, the islet’s response to glucose is also due to recruitment of an increasing number of β-cells (283, 402).
The mechanisms by which glucose initiates electrical activity and stimulates insulin secretion are well understood. The consensus model is summarized in FIGURE 5D. Although a gross simplification, it provides a useful introductory background. It posits that ATP-sensitive K+ channels (KATP channels) are spontaneously active at low glucose and that efflux of positively charged K+ through these channels generates the negative membrane potential of the unstimulated β-cell. Glucose enters the β-cell via the Glut2 transporter, and the ensuing metabolic breakdown of the sugar leads to the generation of ATP at the expense of ADP. This results in closure of the KATP channels, membrane depolarization, and initiation of electrical activity. Action potential firing is dependent on the opening of voltage-gated Ca2+ channels, and the resulting increase in [Ca2+]i then triggers exocytosis of insulin granules. Insulin travels via the bloodstream to its target organs when it activates glucose uptake and thereby lowers plasma glucose. As a result, glucose uptake by the β-cell is reduced, decreasing glucose metabolism, reactivating KATP channels and switching off insulin secretion. This mechanism provides feedback regulation of insulin secretion via changes in plasma glucose.
Electrical activity is stimulated by glucose, amino acids (like arginine, leucine, alanine, lysine, and glycine) (269, 270, 744), pharmacological agents (like sulfonylureas and glinides) (272, 442), neurotransmitters (like acetylcholine) (81, 126, 277), and hormones (including GLP-1) (381). However, only glucose, leucine (and its deamination product α-ketoisocaproic acid), and sulfonylureas are capable of initiating electrical activity on their own. All other agents are only effective in the presence of glucose concentrations that are close to the threshold for insulin secretion, when the KATP channels are inhibited and the input resistance is so high that a small current can exert a big effect on the membrane potential (FIGURE 4).
A number of hormones and neurotransmitters (e.g., somatostatin and epinephrine) transiently repolarize the β-cell and inhibit action potential firing and insulin secretion (154, 543). Their effects on β-cell electrical activity and the underlying mechanisms are considered in section VIIB, 1 and 2.
The properties of electrical activity described above refer to β-cells within freshly isolated intact mouse islets. Interestingly, electrical activity changes following short-term tissue culture, with bursts of action potentials and their associated [Ca2+]i responses becoming longer (208). These changes are even more dramatic if the islets are dispersed into single cells or small clusters (223, 273, 631). Why this happens is unresolved, but it may be related to the fact that isolated cells are invariably cultured before study.
2. Electrical activity: human β-cells
Much less is known about the properties of glucose-induced electrical activity in human β-cells. There are only few recordings and (because of the logistics of isolation) studies are restricted to cells/islets maintained in tissue culture.
Like their mouse counterparts, human β-cells are hyperpolarized (−70 mV) and electrically silent at low glucose (210, 452). Glucose produces a concentration-dependent depolarization, and once the membrane potential exceeds −60 mV, this initiates electrical activity and insulin secretion (FIGURE 6, A and B). As in mouse islets, the action potentials are frequently grouped in bursts, but these are shorter (~2 s) and more frequent than those of mouse islets. The action potentials of human β-cells are also triggered from a more negative membrane potential in human than in mouse β-cells (−60 mV rather than −50 to −40 mV) and often overshoot, peaking at potentials above 0 mV (568).
FIGURE 6.

A: glucose-induced electrical activity in a β-cell in an intact human pancreatic islet in response to increasing glucose concentrations, as indicated by the staircase above the membrane potential recording. Note that it takes >40 min for β-cell to repolarize following exposure to glucose. B: response to 6 mM glucose (indicated by red rectangle in A) shown on an expanded time base. The action potentials undergo complex time-dependent changes in amplitude and peak voltage (see also FIGURE 13C). At 6 mM glucose, bursts of 3 or 4 action potentials are sometimes observed (red horizontal line under recording: see FIGURE 13B). Recordings in A and B were performed by Dr. E. Rebelato, Oxford. C: changes in [Ca2+]i in response to increasing glucose from 1 to 6 mM in a cell (presumably a β-cell) in an intact human islet. [Modified from Rorsman et al. (569).]
As the glucose concentration is increased, the β-cell becomes progressively more depolarized, action potential amplitude gradually declines, and (at 11 mM and above) firing becomes continuous. A distinctive feature of electrical activity in human β-cells is that, while it is rapidly initiated, it is only slowly reversible on return to low glucose, taking as long as 30 min following exposure to 22 mM glucose (FIGURE 6A). As the β-cell starts repolarizing, the amplitude of the action potential also increases. This correlates with a paradoxical and transient stimulation of insulin secretion following exposure to high (>20 mM) glucose concentrations (265). The underlying mechanism is not known, but may reflect mobilization of glycogen stored within the β-cell during high-glucose stimulation (148).
Although glucose-induced [Ca2+]i oscillations are observed in human β-cells, and are probably driven by oscillatory electrical activity (529) (FIGURE 6C), they are less well synchronized across human islets than mouse islets (102). This suggests that electrical coupling between individual human β-cells may be weaker, perhaps because of the different architectures of mouse and human islets. Whereas mouse islets contain a central core of electrically coupled β-cells that functions as a syncytium, β-cells in human islets are organized into several subdomains, with β-cells in one domain being electrically insulated from β-cells in other domains by strands of non- β-cells (82, 102) (FIGURE 1, B and D). Within the subdomains, synchronized glucose-induced [Ca2+]i oscillations can be recorded (529).
As in mouse islets, glucose and sulfonylureas (618) are true initiators of electrical activity in human β-cells. Amino acids (arginine and glycine) (744), neurotransmitters (like adenine nucleotides, GABA, and acetylcholine) (89, 92, 347, 568) and hormones (618) can also stimulate electrical activity and insulin release but only in the presence of a ‟permissiveˮ concentration of glucose. Epinephrine and somatostatin lead to transient repolarization and suppression of glucose-induced electrical activity (327, 568).
3. Other species
Surprisingly, it remains unclear if bursting electrical activity is found in all species. It was initially reported that rat β-cells do not show oscillatory electrical activity (20), but more recent data indicate that in fact they generate oscillatory electrical activity very similar to that of mouse β-cells (424). Membrane potential recordings have also been performed on canine and porcine β-cells (454, 455). These revealed no obvious oscillatory electrical activity, but as these measurements were made in dispersed β-cells, it remains unclear whether this reflects a true species difference or if it is simply a consequence of cell isolation (as in the mouse).
V. ION CHANNELS
In both mouse and human β-cells, electrical activity results from a complex interplay between voltage-dependent and voltage-independent conductances. Here we summarize, in turn, the biophysical and pharmacological properties of the ion currents involved, and their underlying channels and transporters in mouse and human β-cells. We start with the ion channels that are active at rest (that is, at low glucose), then consider those channels that might underlie the glucose-dependent depolarization, and finally discuss the voltage-gated ion channels involved in action potential firing and bursting.
A. The ATP-Sensitive K+ Channel
The ATP-sensitive K+ channel (KATP channel) is the predominant ion channel open at rest in β-cells of all species. As a consequence, the β-cell is hyperpolarized in the absence of glucose, and the membrane potential approaches the reversal potential of the KATP current, which lies close to the K+ equilibrium potential. Glucose metabolism closes the KATP channel, thereby depolarizing the β-cell and initiating β-cell electrical activity, Ca2+ influx, and insulin secretion (27, 573). Metabolic regulation of the channel is mediated by changes in the intracellular concentrations of ATP and MgADP, which inhibit and stimulate the channel, respectively (127, 163, 328). The name of the channel derives from this characteristic nucleotide regulation. As described below, mutations in KATP channel genes lead to either hypersecretion or hyposecretion of insulin. The KATP channel is also the molecular target of sulfonylurea drugs, which have been used for more than 60 yr to treat T2DM (262). Consequently, it is of critical importance for insulin secretion.
Because of its key role in insulin release, the KATP channel has been extensively studied. Here, we summarize the most pertinent findings. For a more comprehensive review, see References 4, 25, 29, 30, 32, 477, 479, 644.
1. Structure of the channel
The successful cloning and molecular characterization of the KATP channel revealed it is an octameric complex of four pore-forming Kir6.2s and four regulatory SUR1 subunits (4, 302, 583). Their membrane topology is shown in FIGURE 7A. Kir6.2 (Kcnj11/KCNJ11) is a member of the inward rectifier (KIR) family, although it shows only weak rectification. Binding of ATP (or ADP) to Kir6.2 causes channel inhibition (698). SUR1 (ABCC8) belongs to the ATP-binding cassette transporter family (5), but unlike most other ABC transporters, it has no known transporter function. Instead, it regulates the activity of the Kir6.2 pore, endowing it with sensitivity to inhibitory sulfonylurea drugs (hence its name), to K+ channel openers such as diazoxide, and to the stimulatory effects of Mg-nucleotides (5, 232, 621, 698). Metabolic regulation of the channel thus involves interactions of adenine nucleotides with both Kir6.2 (4 sites) and SUR1 (8 sites). Both subunits are also required for correct trafficking of the channel to the plasma membrane (757).
FIGURE 7.

A: topology of Kir6.2 and SUR1, showing two (of 4) Kir6.2 and two (of 4) SUR1 subunits. Kir6.2 has two transmembrane domains and cytosolic NH2 and COOH termini. SUR1 has 17 transmembrane domains arranged as groups of 5, 6, and 6 (TMD0, TMD1, and TMD2) and 2 nucleotide-binding domains (NBD1 and NBD2) that associate to form 2 nucleotide-binding sites at their interface. Binding of ATP (or ADP) to Kir6.2 inhibits channel activity. Binding of MgADP/MgATP to SUR1 stimulates activity. B and C: the KATP channel complex viewed from the side (B) and bottom (C). The Kir6.2 tetramer is surrounded by 4 SUR1 subunits. In B, the front subunit has been removed for clarity. Blue: TMD1, TMD2 of SUR1. Pink: TMD0 of SUR1. Green: NBDs of SUR1. Gray: Kir6.2. Brown: 3rd cytosolic loop of SUR1. The plasma membrane (yellow) is shown behind the channel in B. Figure provided by Dr. M. Puljung, Oxford.
The structure of the KATP channel complex reveals the SUR1 subunits are arranged around a central tetrameric Kir6.2 pore (393, 432, 450) (FIGURE 7, B and C). Each SUR1 is anchored to a Kir6.2 via its NH2-terminal transmembrane domain (TMD0), which interacts with the first transmembrane domain of Kir6.2. As is the case for other ABC proteins, SUR1 contains two cytoplasmic nucleotide-binding domains (NBDs), arranged in a head-to-tail dimer, which associate to form two nucleotide-binding sites (NBS1 and NBS2) at the interface (FIGURE 7A). Occupancy of NBS2 of SUR1 by MgADP is believed to increase the channel open probability.
In the 6 Å cryo-EM structure of the KATP channel complex, the NBDs of SUR1 lie far apart (393, 432, 432a), presumably because the structure was determined in the presence of the sulfonylurea glibenclamide, which inhibits the channel and prevents Mg-nucleotide interaction with the NBS of SUR1 (524). In other ABC proteins, MgATP binding closes the cleft between the NBDs, resulting in a conformational change in the transmembrane domains (TMDs) from an inward-facing to an outward-facing configuration. It is possible that binding of MgATP or MgADP to SUR1 causes a similar conformational change that results in opening of the Kir6.2 pore. The core of SUR1 (TMD1 and TMD1) lies in the inward-facing configuration (393, 432) and by analogy with other ABC proteins it can be hypothesized that this reorientates to an outward-facing conformation upon Mg-nucleotide binding to the NBDs.
Extensive mutagenesis, coupled with molecular modeling, identified a putative ATP-binding site of Kir6.2 (19). This was supported by the discovery of many disease-causing mutations lining the binding pocket and subsequently confirmed when the 6 Å structure was obtained (393, 432). It sits at the interface between the NH2 terminus of one subunit and the COOH terminus of the other, with the β-phosphate of ATP interacting with residue K185.
In both mouse and human, the Kir6.2 and SUR1 genes are expressed side-by-side on chromosomes 7 and 11, respectively. Numerous proteins have been suggested to interact with the KATP channel, including 14-3-3 proteins (280), syntaxin (474), EPAC2 (616), actin (85), and many others (344), but in most cases the physiological importance of these interactions remains unclear. It is notable that Abcc8/ABCC8 is expressed at 5- to 20-fold higher levels than Kcnj11/KCNJ11 (see Supplemental Figure 7B). Each Kir6.2 subunit requires an SUR1 partner to traffic to the plasma membrane (and vice versa) (757). Thus the level of KCNJ11 will dictate KATP channel density and the increased level of SUR transcripts may simply be to ensure that all Kir6.2 subunits have a partner. Alternatively, SUR1 may have functions other than as a KATP channel subunit. For example, large amounts of the protein are found in the secretory vesicles, where their role remains to be defined (204).
2. Biophysical properties
The single KATP channel conductance is 13 pS when measured in cell-attached recordings using quasi-physiological ion gradients at voltages close to the resting potential (21, 695). Under these conditions, the channel shows weak inward rectification due to a voltage-dependent block of outward currents by intracellular Mg2+ and spermine ions (400). When measured in symmetrical (140 mM) extra- and intracellular [K+], the single-channel conductance is ~50 pS (27, 695). In ATP-free medium, the open probability in inside-out patches is 0.06 (366). The whole-cell conductance measured under comparable conditions is ~10 nS (573). These values of single-channel open probability and conductance provide an estimate of at least 12,000 KATP channels per β-cell. However, the open probability varies considerably according to how it is measured: in cell-attached patches it varies with the intracellular ATP concentration, while in inside-out patches it diminishes with time after patch excision (rundown) (526). Accordingly, the number of KATP channels per β-cell is not known with certainty.
KATP channel activity at potentials negative to the K+ equilibrium potential (i.e., where the current is inward) shows complex kinetics consisting of bursts of brief openings and closures with time constants of 0.3 and 2 ms, respectively (573), that are separated by longer closed intervals. ATP acts by decreasing the duration of the channel openings and increasing the lifetimes of the long closed states (132, 173, 691). Conversely, MgADP increases burst duration and reduces the interburst intervals (328).
3. Metabolic regulation
In the intact cell, KATP channel activity is determined by the balance between the stimulatory and inhibitory effects of nucleotides (25). Intracellular ATP (or ADP) inhibits the KATP channel by binding to Kir6.2 in a reaction that does not require Mg2+ (234, 698). In excised inside-out patches, ATP inhibition is half-maximal at ~10 μM in the absence of Mg2+ (127), but this increases to 20–30 µM in the presence of Mg2+ due to the simultaneous presence of Mg-nucleotide stimulation via SUR1 (234). This nucleotide sensitivity would suggest that the KATP channel should be permanently closed at resting ATP concentrations within the β-cell (0.5–6 mM; Ref. 480), which is not the case (27, 573). Resolution of this puzzle is provided by the fact that the MgATP sensitivity of the channel is much lower when measured in the whole-cell configuration; inhibition is half-maximal at 0.4–0.8 mM rather than 0.01 mM as in excised patches (641). Similarly, it is lower in β-cells in which the plasma membrane has been permeabilized with α-toxin (FIGURE 3C); under these conditions, the IC50 is 156 µM and channel activity is 16% of maximal at 1 mM MgATP (674). This is due to Mg-nucleotide stimulation at the NBDs of SUR1, which shifts the ATP concentration-inhibition curve into a range of intracellular ATP concentrations over which glucose-induced changes in [ATP]i occur. Metabolic modulation of the KATP channel may therefore involve changes in ATP, MgADP, or both.
As intracellular ATP levels never fall below 1 mM in β-cells, even in the absence of glucose (143, 422), KATP channels are largely closed, even at resting glucose concentrations. At 5 mM glucose, the conductance is 7% of maximal, and this falls to 3% of maximal when glucose is increased to 10 mM (674). Nevertheless, this tiny change in conductance can cause a marked change in membrane potential because of the high-input resistance of the β-cell membrane at these glucose concentrations. The fact that glucose regulation is mediated by minute changes in the open probability of many channels is advantageous as it prevents the random opening of a single KATP channel from hyperpolarizing the membrane at high glucose (when the input resistance is high) and so inhibiting insulin secretion (129).
A pertinent question is what submembrane ATP concentration is seen by the KATP channel? This is difficult to answer. Ideally, one would use a submembrane ATP sensor to measure it directly, but while this has been applied to other cell types (228), it has not been attempted in β-cells. However, measurements of channel activity in cell-attached patches before and after permeabilization of the plasma membrane with α-toxin, followed by dialysis with different ATP concentrations, yield values of ~1 mM in glucose-free solution and ~3 mM in 11 mM glucose (674). These values are in broad agreement with biochemical measurements of [ATP]i in purified rat β-cells which suggest ATP is ~2 mM under basal conditions and rises to 4 mM when glucose is increased to 10 mM (142).5
There is considerable evidence that the lower ATP sensitivity of the KATP channel in the intact cell is due to the presence of intracellular MgADP, as in this configuration KATP channel activity persists at intracellular ATP concentrations normally associated with nearly complete inhibition of channel activity (72, 163, 328, 366). This suggests that glucose acts by changing the relative concentrations of ATP and ADP. Indeed, parallel measurements of the cytosolic ATP/ADP ratio and KATP channel activity have provided direct evidence for an inverse correlation between these two parameters (675).
It is difficult to determine the EC50 for channel activation by MgADP (or MgATP) at SUR1, because both ATP and ADP (IC50 = 60 µM) also block the channel at Kir6.2. However, it can be estimated by coexpressing SUR1 with Kir6.2 carrying a mutation that renders it ATP insensitive. This yields EC50 of 8 µM for MgADP and 112 µM for MgATP (523); it is believed that the lower affinity of MgATP reflects the fact that it must be hydrolyzed to MgADP before it can stimulate channel opening. It is also worth noting that the nucleotide affinity of the NBDs, together with the intracellular nucleotide concentrations, means that the NBDs will usually be occupied by MgATP (or its hydrolytic product), even at low glucose. Thus the enhanced MgADP occupancy of the NBDs expected on glucose elevation presumably reflects changes in the nucleotide off-rate following MgATP hydrolysis, rather than a direct effect of MgADP binding.
KATP channels are also regulated by lipids such as phosphatidylinositol bisphosphate (PIP2) and long-chain acyl CoAs (e.g., oleoyl CoA), which stimulate channel opening and reduce its ATP sensitivity (229, 622). The predicted PIP2 binding site (645) lies close to the ATP-binding site and may influence binding allosterically. The increase in unliganded open probability induced by PIP2 indirectly also reduces the channel ATP sensitivity. PIP2 and long-chain acyl CoAs probably set the level of KATP channel activity in a given cell, but it is not clear if they participate in its metabolic regulation. Loss of PIP2 is believed to underlie the rundown of channel activity that occurs upon membrane excision (526, 622).
4. Pharmacological regulation
KATP channels are blocked by sulfonylureas (such as gliclazide, glibenclamide, and tolbutamide), by glinides (like repaglinide and nateglinide), and by benzamido derivatives (e.g., meglitinide), and they are activated by K+ channel openers like diazoxide (230). It is important to remember that, in vivo, more than 90% of most sulfonylureas are bound to plasma proteins. With this proviso, there is a good correlation between drug binding to β-cell membranes, KATP channel inhibition, and the free concentration of the drug in the plasma of T2DM patients (773).
Most pharmacological regulation of the KATP channel is mediated via SUR1. A high-affinity binding site on SUR1 mediates the therapeutic effects of sulfonylureas (233). Mutagenesis studies first showed that a key residue (S1238) in the eighth cytosolic loop of SUR1 contributes to SU binding (34a), and a recent cryo-EM structure (432a) of the KATP channel with bound glibenclamide and ATP reveals that this residue makes direct contact with one end of the glibenclamide molecule. Binding is stabilized by additional interactions with transmembrane domains 6, 7, 8, and 16. Although a low-affinity sulfonylurea site also exists on Kir6.2, this is of little therapeutic relevance (233).
Sulfonylureas act as partial antagonists at SUR1, as in excised patches they only inhibit the channel by a maximum of ~60%. In the whole-cell configuration, however, they fully block the KATP current. This discrepancy is due to the fact that sulfonylureas also prevent MgADP activation of the β-cell KATP channel (233, 524). Because channel activity is normally a balance between ATP block (at Kir6.2) and MgADP activation (at SUR1), loss of MgADP activation enhances channel inhibition and enables sulfonylureas to block the channel completely. The KATP channel activator diazoxide also mediates its effect by binding to SUR1 (232). Its binding site is still unknown. Interestingly, diazoxide action requires the presence of intact nucleotide-binding domains and of MgADP (or MgATP) (232, 621). Thus it appears that KATP openers promote nucleotide activation of the channel, whereas sulfonylureas have the opposite effect.
A possible synthesis of these data is that Mg-nucleotides promote dimerization of the NBDs, as they do in other ABC proteins, and that this conformational change is associated with KATP channel activation. K+ channel openers likely stabilize the dimer, explaining why they require Mg-nucleotides to be effective and why they slow the off-rate of MgADP (231). Conversely, sulfonylurea binding induces dissociation of the NBDs dimer, which leads to MgADP unbinding and explains why these drugs prevent the stimulatory effect of Mg-nucleotides (233).
5. KATP current magnitude and glucose-induced electrical activity
The KATP current is largely responsible for the resting conductance of the β-cell at low glucose which explains the negative membrane potential of the β-cell (about −70 to −80 mV), close to the K+ equilibrium potential (EK).
The whole-cell KATP conductance is often expressed relative to cell capacitance, which is proportional to cell size. In whole-cell recordings, the maximum KATP conductance, measured when ATP has washed out of the cell, is ~2 nS/pF (573). When measured in the perforated patch configuration (where cytosolic ATP levels are undisturbed), it is ~1 nS/pF in single β-cells exposed to glucose-free solution (584, 631). In β-cells within freshly isolated pancreatic islets, it is significantly lower and ranges between 0.25 and 0.4 nS/pF (762). In both preparations, glucose produces a concentration-dependent reduction of the whole-cell conductance which, when expressed as a fraction of that in the absence of glucose, is half-maximal at 3–4 mM glucose (26, 212, 584, 674, 762). Notably, significant KATP channel activity remains at glucose concentrations ≥5 mM in β-cells in intact islets and whole-cell KATP channel activity falls from ~0.1 nS/pF to <0.02 nS/pF when glucose is varied between 7 and 15 mM (762) (FIGURE 2C). Similarly, the KATP current of dispersed β-cells is not fully inhibited until glucose reaches ~20 mM (212, 674) (FIGURE 2C). These data indicate, consistent with conclusions based on membrane potential measurements (261, 335), that glucose and tolbutamide influence KATP-channel activity also at suprathreshold glucose concentrations. As pointed out in section IIIA2, the glucose dependence of KATP channel activity is in good agreement with the glucose-induced increase in intracellular ATP (FIGURE 2C).
The KATP channel is not only involved in the membrane depolarization induced by glucose, it also regulates the frequency of action potentials (and thereby insulin secretion) at higher glucose concentrations, as discussed in more detail in section VIIIA6. In addition, it influences the burst duration. This arises because KATP channel activity can be enhanced by activation of plasmalemmal (PMCA) and sarco/endoplasmic reticulum (sER) Ca2+-ATPases (SERCA), which lowers intracellular ATP (196).
Electrical activity in human β-cells is also controlled by KATP channels with properties very similar to those of mouse β-cells (90, 452, 453). However, the whole-cell KATP conductance in the absence of glucose is <10% of that found in mouse β-cells, being around 60 pS/pF (535). As there is no major difference in single-channel conductance or channel density (28), the difference is likely to be due to a lower open probability. Furthermore, because the ATP sensitivity of human and mouse KATP channels is comparable, the difference in open probability probably reflects differences in β-cell metabolism.
6. KATP channels and disease
Mutations in either the Kir6.2 or SUR1 subunit cause human disease. As this has been reviewed extensively elsewhere (25, 29, 32, 644, 681), we provide only a brief summary here.
Loss-of-function mutations cause congenital hyperinsulinism because they result in permanent membrane depolarization and persistent insulin release (479, 644, 681). In most cases, the disease is recessively inherited. Some patients have a focal lesion due to uniparental disomy and they can be treated by surgical excision of the affected part of the pancreas, which results in a cure. Most patients with diffuse disease require total or near-total pancreatectomy. However, patients with dominant disease, which is usually less severe, can often be treated by diet and/or diazoxide; for unknown reasons, some of these patients may later progress to T2DM (485, 538).
Interestingly, neither Kir6.2 nor SUR1 knockout mice are a good model of human hyperinsulinism as they do not exhibit lower blood glucose levels (except very briefly after birth) and they gradually develop glucose intolerance (295, 451, 541, 607). Somewhat surprisingly, the effects of ablating on Kir6.2 or SUR1 on insulin secretion are marginal; insulin secretion at nonstimulatory glucose concentrations is slightly increased, whereas it is actually reduced at high glucose (170, 411, 620). In part, this may reflect a decrease in insulin content (619). It is also possible that mice cope with loss of functional KATP channels because they are more extensively innervated than human islets (555). For example, increased sympathetic tone, via activation of adrenoreceptors in β- and α-cells, will inhibit insulin secretion (α2-receptors) and stimulate glucagon secretion (β-receptors). The combination of these effects may be sufficient to prevent severe hypoglycemia.
Gain-of-function mutations in either Kir6.2 or SUR1 result in neonatal diabetes (ND), which usually presents within the first six months of life and is associated with a low birth weight (25, 29, 32, 43, 215, 255). All mutations result in a reduced ability of MgATP to block the channel, thereby preventing membrane depolarization and insulin secretion in response to glucose metabolism. Sulfonylurea drugs, which bypass metabolism and block the channel directly, provide an effective therapy in >90% of patients (44, 504). The mechanism of action of ND mutations includes loss of ATP binding, failure of ATP binding to induce channel closure, an increased open probability (which indirectly reduces ATP inhibition), and enhanced MgATP activation via SUR1 (25, 29). About 20% of ND patients also experience neurological symptoms due to the effect of the mutant KATP channel on brain function (255); some, but not all, of these effects are ameliorated by sulfonylurea therapy.
Finally, a common variant in Kir6.2 (E23K) is associated with a slightly increased risk of T2DM (216, 586). While its effects on KATP channel activity (604, 605) and insulin secretion (696) are very small (585) and controversial (552), it is important to note that even a tiny increase in channel activity can lead to ND (705). Thus it will be hard to experimentally demonstrate a functional change associated with a mutation causing an increased risk of T2DM that only achieves significance in large population studies.
B. The Depolarizing Inward Current
Closure of KATP channels alone is not sufficient to cause membrane depolarization. In the absence of other ion channels, even a minute amount of K+ channel activity is sufficient to maintain the membrane potential close to the K+-equilibrium potential. Accordingly, the β-cell must be equipped with an inward current, with an equilibrium potential positive to the threshold for regenerative electrical activity (−60 mV), that is too small to affect the membrane potential when KATP channel activity is high but is large enough to depolarize the membrane when KATP channel activity is low. Given the high input resistance of the glucose-stimulated β-cell (5–10 GΩ), a current as tiny as 1–2 pA would suffice to produce the 10 mV depolarization required to reach threshold (FIGURE 4). It is important to note that it is not necessary for this current to be activated by glucose. It may be tonically active but simply so small that it only affects the membrane potential once KATP channels are almost fully closed. This would explain how a variety of nonmetabolic stimuli [including sulfonylureas (272) and quinine (37)] can evoke electrical activity even in the complete absence of glucose.
A tiny inward current, which is spontaneously active around the resting potential, can be recorded when KATP channel activity is blocked. The identity of this ‟background current,ˮ which underlies the glucose-induced depolarization, remains an enigma. The fact that removal of extracellular Na+ does not prevent glucose-induced depolarization (549, 618) argues that the current cannot be Na+-dependent. Ca2+ might carry the current and Cl− is also a possibility as lowering the extracellular Cl− concentration from 145 to 12 mM depolarized glucose-stimulated β-cells by 5–10 mV (608). In this section, we consider possible candidates for the background inward current.
1. Chloride channels
Pancreatic β-cells have an intracellular chloride concentration ([Cl−]i) as high as 35 mM (93). This predicts a Cl− reversal potential (ECl) of −35 mV and argues that at more negative potentials opening of Cl− channels will result in Cl− influx, and thus membrane depolarization. A number of different Cl−-permeable channels have been reported in β-cells, including the cystic fibrosis transmembrane conductance regulator (CFTR), volume-regulated anion channels (VRAC), Ca2+-activated Cl− channels, and the H/Cl− cotransporter ClC3.
a) cftr.
As many as 30–50% of cystic fibrosis (CF) patients develop CF-related diabetes (244). It has been proposed that this reflects an important role of the CFTR in glucose-induced membrane depolarization. However, at least two lines of argument suggest this is unlikely.
First, CFTR is not expressed at all (mouse) or expressed only at very low levels (human) in β-cells (see Supplemental Figure 2A). Although it has been reported at 10-fold higher levels in some preparations (68), this may reflect contamination of isolated islets with exocrine acinar and ductal cells, where CFTR is highly expressed (606).
Second, it has been proposed that glucose activates CFTR. However, quantitative considerations question this idea. The reported (244) current-voltage relationship of the glucose-activated CFTR current in metabolically intact mouse β-cells corresponds to a whole-cell conductance of 20 nS. This is 5-fold larger than the resting conductance recorded from metabolically intact β-cells in the absence of glucose (221), and 10-fold larger than the peak voltage-gated Ca2+ current (see legend to FIGURE 9) (221). With CFTR currents as large as these, the β-cell membrane potential would be clamped at the Cl− equilibrium potential (ECl; −35 mV) in the presence of glucose, and voltage-gated currents would be unable to elicit action potential firing as they are (relatively) too small. The fact that this is not the case suggests the importance of CFTR in β-cell electrical activity may have been overstated.
FIGURE 9.
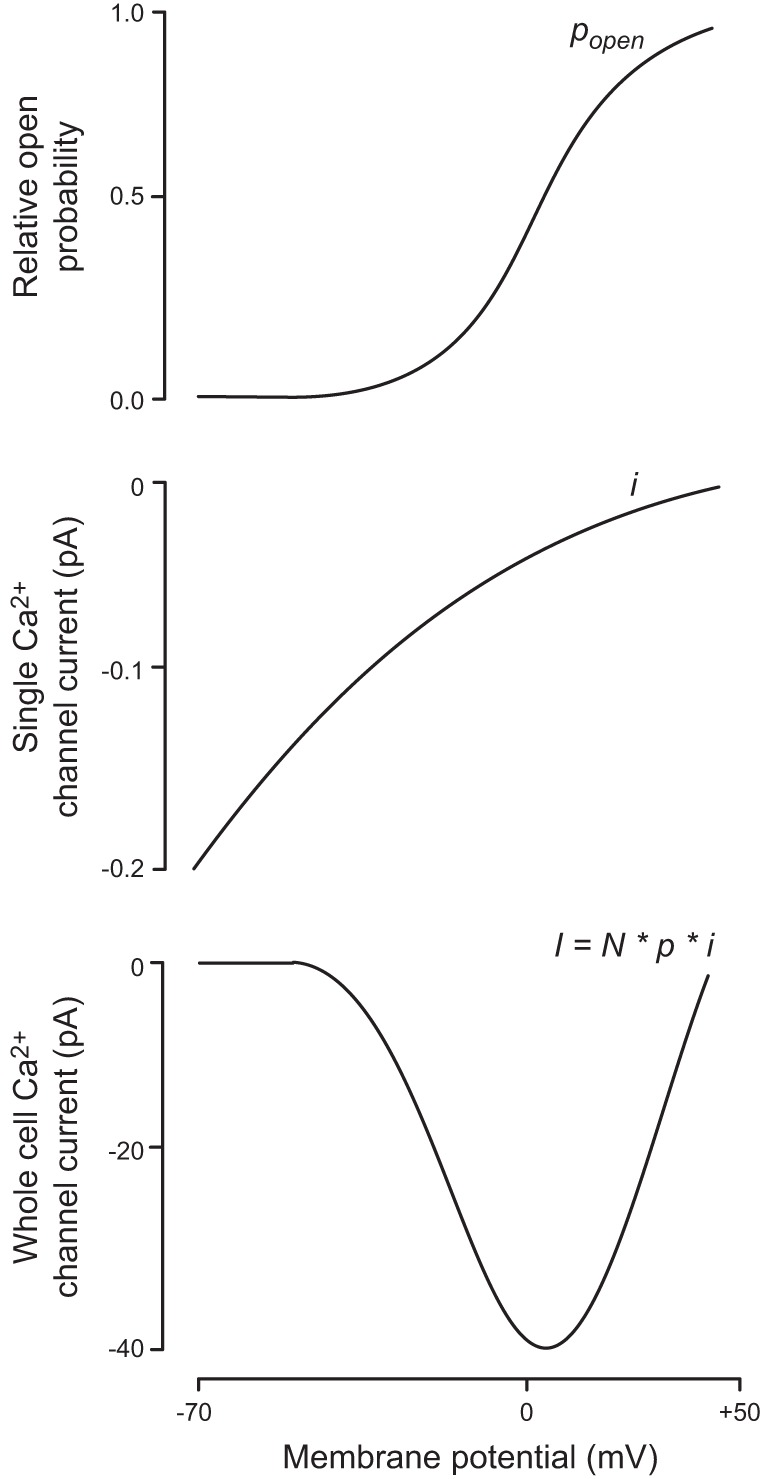
Relationship between membrane potential and the open probability (Popen), the single Ca2+ channel current (i), and the whole-cell Ca2+ current (I; i.e., the product of ) Note the U-shaped current-voltage relationship for the whole-cell current. The whole-cell slope conductance (G = I/V) between 0 and +50 mV (where the Ca2+ channels are maximally active) is ~1 nS (i.e., 50 pA/50 mV). [Modified from Larsson-Nyrén et al. (369).]
It has also been suggested that CFTR may play a role in the response to elevated intracellular cAMP, such as that elicited by the incretin hormone GLP-1 (166). It was proposed that cAMP-dependent activation of CFTR, by mechanisms that remain poorly understood, activates the Cl− channel Ano1 (see below) and that the associated Cl− influx facilitates priming of insulin granules for release. However, this mechanism appears to operate only in the presence of high intracellular cAMP, such as that induced by the adenylate cyclase activator forskolin. Under these conditions, CFTR-inhibitors (like GlyH-101) reduced insulin secretion by 50%, but no inhibitory effect was observed when insulin secretion was stimulated by high glucose alone. Thus the cAMP-dependent Cl− current does not appear to contribute to the background inward current that underlies membrane depolarization in response to glucose.
Taken together, the available data suggest it is unlikely that CFTR plays a role in GIIS. Thus, although lack of CFTR undeniably often leads to diabetes in humans, this is likely to be a secondary consequence of blockage of the pancreatic ducts, which leads to degeneration of both pancreatic acinar cells and the pancreatic islets, rather than reflecting a critical role of CFTR in β-cell electrophysiology. This conclusion is in line with the finding that a mouse model globally expressing CFTR-ΔF508 (the most common CFTR mutation) exhibits only subtle effects on β-cell function (188).
b) vrac.
There is evidence that glucose may activate the volume-regulated anion (VRAC) Cl− channel (65, 349). VRAC was recently identified as the ‟leucine-rich repeats containing 8Aˮ protein (Lrrc8a/LRRC8A) (527, 713), which is expressed at low but detectable levels in both mouse and human β-cells (3, 68, 146, 475). Its physiological importance in β-cells remains to be determined, but it is of interest that a niflumic acid-blockable and volume-sensitive Cl− current (that may correspond to VRAC) has been reported to contribute to the glucose- and cAMP-dependent membrane depolarization in mouse β-cells (191).
c) Ca2+-activated Cl− channels.
Recent data implicate Ca2+ release, via NAADP-induced activation of Tpc1 and/or Tpc2 Ca2+ release channels in near-plasma-membrane acidic (lysosomal) Ca2+ stores, as modulating β-cell electrical activity (22, 104). The increase in [Ca2+]i below the plasma membrane may open Ca2+-activated Cl− channels such as Ano8, Ano10, and Best3, all of which are expressed in mouse and human β-cells (FIGURE 8A). Ano6 is also expressed in mouse β-cells, at relatively at high levels, but it is found at a much lower level in human β-cells. Conversely, ANO5 is expressed in human β-cells but not detected in mouse β-cells. Recently, it was reported that T-A16AInh-AO1 (T-AO1), a selective blocker of Ano1 channels, elicited a slight membrane repolarization, reduced action potential firing in glucose-stimulated mouse β-cells, and inhibited GIIS in isolated rat islets (134). However, as the expression data indicate Ano1 channels are not present either in mouse and human β-cells, it seems more likely that T-AO1 is not selective for Ano1 but also interacts with other types of (Cl−) channel.
FIGURE 8.

A: schematic of voltage-gated Na+ current (INa; red), Ca2+ current (ICa; green), delayed rectifying K+ current (IK; blue), and total membrane current (Itotal; i.e., INa + ICa + IK; black) elicited by a voltage-clamp depolarization (V) from −70 to −10 mV. The dashed line indicates the zero-current level. Downward and upward deflections represent inward (depolarizing) and outward (repolarizing) membrane currents, respectively. B: time-dependent changes in the value of m (dotted red), m3 (continuous red), and h (black) where m and h vary with time after the onset of depolarization (t) according to the expressions and . The parameter values for m and h have been normalized to their maximum. The green trace (below) shows the product m3h, which approximates the activation and inactivation of the whole-cell Na+ current. The curve has been inverted to facilitate comparison with the Na+ current. C: time-dependent changes in values of n and n4 where n varies with time after onset of depolarization (t) according to the expression . This approximates the time course of the whole-cell K+ current. Note that m3 and n4 result in sigmoidal activation kinetics. [Modified from Hille (282).]
d) clc3.
Both mouse and human β-cells express high levels of the H+/Cl− cotransporter ClC3 (Clcn3/CLCN3). In mouse β-cells Clc3 has been implicated in the regulation of exocytosis (389, 427) and is highly expressed in human β-cells, where it may fulfill a similar function. However, the fact that β-cells from Clcn3 knockout mice show normal [Ca2+]i responses to glucose (389) suggests that any ClC3 transporters that reach the plasma membrane do not provide the depolarizing inward current.
2. Transient receptor potential (TRP) channels
Transient receptor potential (Trp) channels represent a large family of cation-selective channels (707) that potentially might contribute to the background inward current of β-cells. Insulin-secreting cells have been reported to express a number of Trp channels, but in mouse β-cells significant expression is restricted to Trpc1, Trpm2, Trpm3, Trpm4, Trpm5, and Trpm7 (56) (see Supplemental Figure 2B). Most TRP channels are nonselective cation channels, but their relative permeability to Ca2+ and Na+ (PCa/PNa) varies considerably (125), with Trpm4 and Trpm5 being impermeable to Ca2+.
A decisive role for Trpm4 and Trpm5 channels in glucose-induced membrane depolarization seems unlikely given that glucose remains capable of evoking electrical activity and insulin secretion in β-cells/islets from mice in which these channels have been genetically ablated (194, 618). However, subtle effects on electrical activity have been reported in β-cells from Trpm5-deficient mice (124). Furthermore, Trpm4 and Trpm5 are critical for the ability of GLP-1 to evoke β-cell electrical activity and stimulate insulin secretion (618). Ablation of Trpm2, however, has been shown to reduce the capacity of glucose to stimulate insulin secretion and elevate [Ca2+]i (754). This suggests that Trpm2 provides at least part of the background inward current in the mouse β-cell. Trpm2 channels are activated by nicotinic acid dinucleotide phosphate (NAADP) (657), which increases in response to glucose stimulation of β-cells (433), and may account for the reported glucose-induced increase in background current (754).
Other Trp channels may also be involved. In both insulin-secreting cell lines and rat β-cells, Trpv1 channels appear to be involved in GIIS (145). However, in vivo genetic ablation of Trpv1 channels affected insulin sensitivity but not insulin secretion in mice (376), which is not unexpected given the very low expression of Trpv1 in mouse β-cells (see Supplemental Figure 2B).
Trpc1 has been proposed to provide the conduit for store-operated Ca2+ entry (in conjunction with Orai1 and STIM-1; see Section VIIB1a) (578), but its contribution to the depolarizing background current has not been explored.
Human β-cells express particularly high levels of TRPC1, TRPM2, TRPM3, TRPM4, TRPM5, TRPM6, and TRPM7 (see Supplemental Figure 8B). It seems likely that, as in rodent β-cells, activation of the TRPM4 channel contributes to the stimulation of electrical activity induced by GLP-1. The finding that electrical activity evoked by GLP-1 (but not glucose) is abolished by removal of extracellular Na+ supports this idea (618).
3. Pumps and transporters
Plasmalemmal pumps and transporters could contribute to the background inward current if they are electrogenic. The most important pump in the β-cell is the Na+-K+-ATPase, which utilizes cytosolic ATP to maintain the transmembrane Na+ and K+ gradients. It is highly expressed and as much as 75% of the energy consumption of the β-cell is due to the operation of this pump (421). A tetrameric complex of two α- and two β-subunits, the Na+-K+-ATPase of both mouse and human β-cells is predominantly composed of α1-, β1-, and β3-subunits (56, 68). For each ATP consumed, the pump extrudes 3 Na+ in exchange for 2 K+. This leads to the net loss of positive charges inside the cell and consequently membrane hyperpolarization. Inhibiting the pump with oubain produces a 5–10 mV depolarization (271), indicating the pump makes a small hyperpolarizing contribution to the membrane potential. Glucose activates the Na-K pump, presumably via increased availability of ATP, an effect that is believed to contribute to the fact that glucose stimulation is associated with reduction of the cytoplasmic Na+ concentration in β-cells (9). In summary, the Na+-K+-ATPase contributes a hyperpolarizing current and thus cannot be the direct source of the depolarizing background inward current.
The Na+ gradients resulting from the activity of the Na+-K+-ATPase indirectly ‟energizeˮ a number of other membrane transporters, such as the Na+/Ca2+ antiporter and the Na+/K+/Cl− transporter. The Na+/Ca2+ antiporter Ncx1 (Slc8a1/SLC8A1) is expressed in both mouse and human β-cells (3, 68, 146, 475). Human β-cells also express Ncx2 (SLC8A2). Both proteins export one Ca2+ in exchange for the import of 3 Na+ and are therefore electrogenic. However, they are typically only activated at high [Ca2+]i (194), which implies they do not contribute to the background inward current or the initiation of electrical activity. In support of this idea, removal of extracellular Na+ does not interfere with glucose-induced membrane depolarization in either mouse or human β-cells (618).
Mouse and human β-cells also express the Na+/K+/Cl− cotransporter 2 (Nkcc2; encoded by the gene Slc12a2) (56, 68). Inhibitors of Nkcc2 (like furosemide) suppress GIIS and cause hyperglycemia (588), but exactly how this occurs remains obscure because the operation of this transporter is electroneutral (1 Na+, 1 K+, and 2 Cl− enter the cell simultaneously). It is therefore unlikely to be linked to membrane depolarization and electrical activity.
4. Summary
In conclusion, the origin of the inward current that mediates depolarization in response to glucose remains an enigma, but an increasing body of genetic and electrophysiological evidence makes it likely that TRP channels are involved. The evidence is particularly strong for Trpm2 in mouse β-cells, but further investigation is needed in human β-cells. However, we should remember that the background current may not be a single entity and that many voltage-independent channels and transporters may each separately make a tiny contribution to the total current. Furthermore, a small contribution from voltage-dependent inward currents (e.g., Na+ and Ca2+ currents, discussed below) also seems possible as these channels have a finite (albeit very small) open probability, even at very negative potentials.
C. Currents Responsible for Regenerative Electrical Activity
Once the membrane has depolarized above a certain threshold level, regenerative bursts of action potentials are initiated (FIGURE 5). Many different voltage-dependent currents contribute to this electrical activity. Confusingly, the currents involved, and their relative importance, may vary between species, between primary and clonal β-cells, between individual cells in the same islet, and under different experimental conditions. For example, activation of TTX-sensitive Na+ channels plays a prominent role in human β-cell action potential firing at low glucose but not at high glucose (90, 521), and in most mouse β-cells, it is not involved in action potential firing at all (761). A further problem is that relative transcript levels do not necessarily correspond to relative current magnitudes, suggesting mRNA levels either do not equate to protein levels, or that trafficking and/or posttranslation modifications influence channel density in the surface membrane or modulate the channel open probability. Differences in experimental methodology, such as whether β-cells/islets were cultured or not, and under what conditions, or differences in mouse strains, might also underlie the observed variability.
Here, we first discuss, in turn, the properties of each of the currents that contribute to β-cell electrical activity in mouse and human β-cells. We then consider how these currents work together to produce the pattern of β-cell firing. The activation and inactivation of the voltage-gated membrane currents are traditionally described using Hodgkin-Huxley terminology, and FIGURE 9 provides a brief background to this method of biophysical characterization of voltage-gated membrane currents.
1. Voltage-gated Ca2+ currents
An increase in the cytoplasmic Ca2+ concentration is essential to trigger insulin release (732). At least in mouse β-cells, the increase in [Ca2+]i responsible for GIIS is almost entirely due to influx of extracellular Ca2+ and intracellular Ca2+ stores contribute marginally (if at all). Voltage-gated Ca2+ channels are the primary conduit for extracellular Ca2+ entry. Their opening plays a critical role in the upstroke of the β-cell action potential, and their blockade leads to inhibition of action potential firing and insulin secretion (570). It is, however, important to recognize that the fact that GIIS principally depends on Ca2+ influx does not exclude the possible involvement of intracellular Ca2+ stores in the shaping of β-cell electrical activity (734). Furthermore, Ca2+ release from intracellular Ca2+ stores contributes to insulin secretion elicited by agents such as acetylcholine (see sect. VIIB1a).
a) mouse beta-cells.
I) Molecular composition. Voltage-gated Ca2+ channels are composed of a pore-forming α1-subunit and two auxiliary subunits (α2δ and β) that influence protein folding and intracellular trafficking (151). The α-subunit comes in various flavors, which have different functional properties. Mouse β-cells express L-type Ca2+ channels (Cacna1c and Cacna1d) and smaller amounts of N- (Cacna1b) and P/Q-type (Cacna1a) Ca2+ channels (211, 222, 603) (see Supplemental Figure 3A). At the transcript level, Cacna1c, Cacna1d, Cacna1b, and Cacna1a contribute 15, 40, 15, and 30%, respectively. There is no evidence of low-threshold T-type Ca2+ currents in mouse β-cells, and T-type transcripts are only expressed at very low levels (although they may be induced following treatment with cytotoxic interleukins; Ref. 771).
The different types of Ca2+ channel can be identified by their biophysical properties and the effects of selective inhibitors. Thus L-type Ca2+ channels are blocked by dihydopyridines (DHPs) like nifedipine and isradipine, R-type by SNX482, N-type by ω-conotoxin, and P/Q-type by ω-agatoxin. A cocktail of isradipine, SNX482, ω-contoxin, and ω-agatoxin almost completely inhibits the β-cell whole-cell Ca2+ current (603). Isradipine blocks 50% of the current, SNX482 blocks 20%, ω-conotoxin blocks <10% inhibition, and ω-agatoxin blocks 20%. This suggests that L-type channels make the dominant contribution to the Ca2+ current of mouse β-cells, with lesser contributions from R-, N-, and P/Q-type channels.
Intriguingly, SNX482 blocks a much greater proportion of the Ca2+ current in mouse β-cells than expected from the relative expression of Cacna1e (317). It is possible that this reflects the fact that different mouse strains were used for the electrophysiological and transcriptome studies. Alternatively, mRNA and protein levels may not be linearly correlated. A similar discrepancy exists with regard to the relative roles of the L-type Ca2+ channels Cacna1c and Cacna1d. Although Cacna1d is expressed at higher transcript levels, genetic ablation of Cacna1d has little, if any, effect on the magnitude of the β-cell Ca2+ current (48) or GIIS, but merely produces a slight shift in the voltage dependence of the Ca2+ current towards more depolarized voltages (470). Conversely, ablation of Cacna1c leads to complete loss of the DHP-sensitive Ca2+ current (603), and mice expressing a DHP-resistant variant of Cacna1c (α-1CDHP−/−) possess no DHP-sensitive Ca2+ currents (625). These findings argue that Cacna1c represents the principal L-type Ca2+ channel in mouse β-cells. Biophysical measurements of exocytosis (capacitance measurements) and secretion studies on β-cell-specific knockouts of L-type Ca2+ channel subunits suggest that Cacna1c (Cav1.2) is particularly important for 1st phase insulin secretion, possibly by physical interaction with release-competent secretory granules (603) (see sect. IXD2). R-type Ca2+ channels (Cacna1e) have been proposed to underlie sustained (2nd phase) insulin secretion, tentatively by promoting mobilization of new granules to the release sites (317).
Mouse β-cells also express Cacnb1, b2, and b3 β-subunits and the Cacna2d1 α2δ-subunit (see Supplemental Figure 3B). Ablation of β3 affects neither the gating nor the amplitude of the β-cell Ca2+ current (57). In neurons, the α2δ-subunit modulates exocytosis by regulating Ca2+ channel density at the synapse and facilitating Ca2+ channel assembly (290). Ablation of α2δ in β-cells is associated with a 50% reduction in Ca2+ current amplitude and a strong reduction of insulin secretion (434), suggesting its role in β-cells may be similar to that in neurons.
II) Biophysical properties. In mouse β-cells, membrane depolarization to −50 mV or above elicits detectable Ca2+ currents (73, 694). The Ca2+ current is maximal at 0 mV and has a peak amplitude of 160 pA in metabolically intact β-cells within intact pancreatic islets when measured at a physiological extracellular Ca2+ concentration (2.6 mM). At more depolarized voltages, the current amplitude is smaller, reflecting the decreased electrochemical gradient. The combination of the sigmoidal voltage dependence of Ca2+ channel activation and the fact that the Ca2+ channel reversal potential occurs at positive potentials (>50 mV) underlies the U-shaped voltage dependence of the whole-cell Ca2+ current (FIGURE 9). There is a suggestion that the current magnitude may be reduced by islet dissociation and subsequent culture of single β-cells, as the maximal Ca2+ current amplitude was only 50–80 pA in isolated β-cells, ~50% of that measured in intact islets (223, 369).
The time course of Ca2+current activation can be fitted with m2 kinetics (see FIGURE 9). The time constant of activation (τm) decreases with depolarization, from ~1 ms at −40 mV to <0.5 ms at 0 mV (221), i.e., Ca2+ current activation becomes more rapid with depolarization. The voltage dependence of activation has been estimated by tail current analysis to be half-maximal at −20 mV at extracellular Ca2+ concentrations of 1.3–2.6 mM (73, 369, 630). However, at supraphysiological Ca2+ concentrations, activation is shifted towards more depolarized membrane potentials; for example, there is a 25-mV shift when [Ca2+]o is increased from 2.6 to 10.2 mM (565, 572, 694), a concentration often used for studies of the β-cell Ca2+-current. This is because divalent cations increase the membrane surface potential, and so affect the voltage seen by channel’s voltage sensor. It is important to remember this when extrapolating to the physiological situation.
During membrane depolarization, β-cell Ca2+ currents undergo both Ca2+- and voltage-dependent inactivation (515). When measured in β-cells within intact islets using the perforated patch whole-cell configuration, inactivation is slow during depolarizations up to −30 mV (which evoke small Ca2+ currents) but becomes faster and more prominent at stronger depolarizations that evoke larger Ca2+ currents. At −10 mV, Ca2+ current inactivation is biphasic and consists of an initial rapid component with a time constant of 2 ms followed by a second slower process with a time constant of >100 ms. The rapid and slow components account for >80 and <20% of the total Ca2+ current, respectively. The rapid component was abolished when Ba2+ was used as the charge carrier. This is characteristic of Ca2+-dependent inactivation, which is mediated by calmodulin and insensitive to Ba2+ (511). Ca2+-dependent inactivation is also influenced by intracellular Ca2+ buffers and in whole-cell recordings its rate depends on the concentration and avidity of the Ca2+ buffer used to dialyse the cell cytoplasm. The slow component of inactivation persists in the presence of Ba2+ and probably reflects voltage-dependent inactivation (570).
The rapid inactivation of the Ca2+ current during depolarizations to −10 mV suggests that the current may undergo significant inactivation during a single action potential, which typically peaks at −10 mV and lasts 30 ms (288). Recovery from rapid inactivation proceeds with a time constant of ~140 ms. With an action potential frequency of 5 Hz, the interval between successive action potentials is 170 ms. This is insufficient for the Ca2+ current to recover fully and leads to progressive inactivation (350). Recovery from slow inactivation is a much slower process, with a time constant of ~5 s (570). The possible impact of Ca2+ channel inactivation on β-cell electrical activity is considered in section VIIIA, 6 and 7.
Single Ca2+ channel currents have been recorded from cell-attached patches. As these were studied in the presence of the L-type Ca2+channel activator BAY K 8644, they are likely to be L-type Ca2+ channels. With 100 mM Ba2+ as the charge carrier, the single-channel conductance is 24 pS (565). By comparing the whole-cell Ca2+ current amplitude recorded at 100 mM Ba2+ with that measured at 2.6 mM Ca2+, and assuming that neither the open probability nor the voltage dependence is significantly affected by the permeant cation, a single-channel conductance of 2–3 pS and a single-channel amplitude of 0.1 pA at 0 mV (which approximates the peak voltage of the β-cell action potential) can be estimated. This is in reasonable agreement with the 6 pS and 0.15 pA recorded at 10 mM extracellular Ca2+ (630). A single-channel current of 0.1 pA corresponds to an influx of 800,000 Ca2+ ions every second. As the mean open time of the β-cell Ca2+ channel is only ~2 ms (48), this suggests that, on average, opening of a single Ca2+ channel results in the influx of >1,500 Ca2+ ions into the β-cell.
The number of voltage-gated Ca2+ channels per β-cell has been estimated as ~500 by nonstationary fluctuation analysis of whole-cell Ca2+ currents in isolated β-cells maintained in tissue culture (48).
III) Modulation by metabolism and protein phosphorylation. There is evidence that Ca2+ channel activity is regulated by glucose metabolism. Thus the glucose metabolite glyceraldehyde increases Ca2+ channel activity by a protein kinase C (PKC)-dependent mechanism in RINm5Fcells (706). Furthermore, in mouse β-cells, glucose increases the whole-cell Ca2+ current and stimulates Ca2+ channel activity (635). These effects are reversed by mannoheptulose (which blocks glucose phosphorylation) and inhibitors of mitochondrial ATP production (including oligomycin) (635). However, the effects of glucose on Ca2+ channels are not consistently observed. It is possible this variability may reflect metabolic heterogeneity between β-cells. For example, metabolically compromised β-cells may be unable to maintain Ca2+ channels in a functional state at low glucose, an idea supported by the fact that Ca2+ channel activity quickly declines in isolated patches (565). Similarly, Ca2+ currents in rat β-cells are upregulated by acute elevation of glucose (341). Interestingly, this was not the case for β-cells from hyperglycaemic diabetic GK rats, suggesting that Ca2+ channel activity may already have been maximally upregulated due to the prevailing hyperglycemia in vivo (and that this effect persisted after cell isolation). The underlying causal mechanism remains to be elucidated.
L-type Ca2+ channels are regulated by protein phosphorylation. In mouse β-cells, forskolin, which elevates cAMP, increases the Ca2+ current by 80% (11), suggesting the channel may be activated by protein kinase A (PKA). The effects of forskolin on the β-cell Ca2+ current are amplified by okadaic acid, an inhibitor of serine-threonine protein phosphatases type 1 (PP1), type 2A (PP2A), and type 3 (PP3) (13). InsP6, the dominant inositol phosphate in insulin-secreting cells, has also been reported to increase the Ca2+ current (367). This may be because InsP6 inhibits PP1, PP2A, and PP3. Glucose produces a 10% increase in InsP6 in insulin-producing cells, and thus it is possible that glucose mediates its stimulatory effect on Ca2+ channel activity by InsP6-induced inhibition of protein phosphatases. The PKC activator PMA also increases the β-cell Ca2+ current (13). In addition, it has been reported that glucose-induced activation of the AMPK-related kinase salt-inducible kinase 2 (SIK2) leads to increased Ca2+ influx and insulin secretion in mouse β-cells (582). Whether this mechanism contributes to the acute regulation of Ca2+ channel activity by glucose remains to be determined.
b) human beta-cells.
I) Molecular composition. The molecular composition of the Ca2+ current in the human β-cell differs significantly from that of the mouse. Human β-cells express L-type (CACNA1C and CACNA1D), P/Q-type (CACNA1A), and T-type (CACNA1H) Ca2+ channels (see Supplemental Figure 3A). N-type (CACNA1B) Ca2+ channels are expressed at lower levels than in mouse β-cells, and CACNA1E is undetectable.
II) Biophysical properties. The T-type Ca2+ channel activates at much more negative membrane potentials than other types of Ca2+ channel. For this reason, it is sometimes referred to as low-voltage-activated (LVA) Ca2+ channel. It gives rise to a transient (rapidly inactivating) current that in human β-cells can be detected at membrane potentials as negative as −60 mV. The whole-cell current is maximal at −40 to −30 mV, where its peak amplitude is ~4 pA/pF, and it is reversibly blocked by the selective T-type Ca2+ channel blocker NNC 55–3096 (90). T-type Ca2+ channels undergo voltage-dependent inactivation, which is half-maximal at −65 mV. At −50 mV, the current inactivates with a time constant of 40 ms and inactivation becomes faster at more depolarized membrane potentials.
By analogy to what has been proposed for other excitable cells (509), it seems likely that T-type Ca2+ channels play a role in pacemaking, facilitating initiation of action potential firing. They are likely to be particularly important at around the threshold potential (−55 mV). Indeed, in membrane potential recordings, application of NNC 55–3096 suppressed action potential firing and insulin secretion elicited by 6 mM glucose. The T-type Ca2+ channels expressed in human β-cells (CACNA1H) are also blocked by micromolar concentrations of zinc (687). Given the high intragranular concentration of Zn2+ in β-cells (30 mM),6 it seems possible that Zn2+ released locally during exocytosis may exert an inhibitory autocrine effect on electrical activity and insulin secretion in human β-cells (92, 347).
At potentials positive to about −40 mV, other Ca2+ channels contribute to the whole-cell Ca2+ current in human β-cells. Ca2+ current amplitude is maximal at 0 mV, where it amounts to 14 pA/pF. L-type and P/Q-type Ca2+ channels contribute 50 and 25%, respectively, of the peak current at 0 mV, with the rest being principally attributable to T-type Ca2+ channels (90). Unlike mouse β-cells, R-type Ca2+ channels contribute marginally (if at all) to the whole-cell Ca2+ current. The L-type (isradipine-sensitive) Ca2+ current becomes detectable at −40 mV and is maximal at −20 mV, whereas the P/Q-type (ω-agatoxin-sensitive) Ca2+ current is small at voltages more negative than −20 mV and maximal at 0 mV. As will be discussed below (see sect. VIIIB6), these differences in voltage dependence have implications for the relative functional importance of L-type and P/Q type channels at different glucose concentrations with the former being more important at high glucose concentrations.
The high-voltage-activated (HVA) component of the human β-cell Ca2+ current flows through L- and P/Q-type Ca2+ channels and activates at more depolarized voltages than the LVA T-type Ca2+ current. It activates very rapidly, the time constant of activation (τm) being 0.4 ms at 0 mV. Inactivation is biphasic: at 0 mV, time constants for the fast component (35% of the total current) and slow component (45% of the current) were 6 and 60 ms, respectively. The decline in current represents inactivation of T- and L-type Ca2+ channels, as P/Q-type Ca2+ channels give rise to a non-inactivating Ca2+ current (90).
III) Ca2+ channels and disease. Expression of CACNA1D is reduced in islets from donors with T2DM, and a polymorphism (rs312480) in the gene influences GIIS, the ‟at-riskˮ C-allele being associated with lower GIIS and insulin content than the T-allele (539). Interestingly, serum from T1DM patients contains a factor that results in a marked increase in L-type Ca2+ channel activity (324). This factor was subsequently identified as apoliprotein CIII (325). Secreted frizzled-related protein 4 (SFRP4), released from β-cells in response to interleukin1β has been shown to decrease β-cell L- and P/Q-type Ca2+ channel activity. SFRP expression is increased in T2DM patients, and increased serum SFRP levels are detectable years before diagnosis and predict the later development of T2DM (420). Collectively, these data suggest that modulation of Ca2+ channel activity may contribute to diabetes risk in some patients.
2. Voltage-gated Na+ channels
In most excitable cells, the depolarizing phase of the action potential reflects activation of both voltage-gated Ca2+ channels and Na+ channels (282). This is also the case for the pancreatic β-cell, although the relative contribution of the Na+ current varies between species.
Na+ channels consist of an α-subunit, which forms the voltage-dependent pore, and an auxiliary β-subunit that modulates gating, protein trafficking, and interaction with the cytoskeleton (109). There are nine different α-subunits (Nav1.1-Nav1.9) and four different β-subunits (β1–4). These are encoded by SCN1A-SCN5A and SCN8A-SCN11A (α-subunits) and SCN1B-SCN4B (β-subunits), respectively.
a) mouse.
I) Molecular composition. Mouse β-cells predominantly express Nav1.7 (Scn9a) with a small contribution of Nav1.3 (Scn3a) and Nav1.6 (Scn8a) (see Supplemental Figure 4). Of the β-subunits, β1 (Scn1b) is expressed at much higher levels than β3 (Scn3b) (761).
II) Biophysical properties. Mouse pancreatic β-cells possess large, tetrodotoxin (TTX)-sensitive voltage-gated Na+ currents (514). In β-cells within intact pancreatic islets, Na+ currents are detectable at depolarizations above −50 mV. The maximum Na+ current is observed at −10 mV and has a peak amplitude of 800–900 pA (120 pA/pF). The voltage dependence of activation is sigmoidal with a half-maximum at −20 mV (761).
Like other voltage-dependent Na+ currents, that of the β-cell activates and inactivates very rapidly. During a depolarization to 0 mV, it reaches its peak within 0.25 ms and is fully inactivated within ~1 ms. Following complete voltage-dependent inactivation, the Na+ current recovers with a time constant of ~20 ms (514). There is a single report of unitary Na+ channel currents in β-cells, which states they have an amplitude of 1 pA at −30 mV (514).
The mouse (and rat) β-cell Na+ channel is highly unusual in the voltage dependence of its inactivation. In most β-cells, it inactivates at such negative membrane potentials that little or no Na+ current remains within the physiological range of membrane potentials (221, 514, 761) (FIGURE 10). Thus Na+ current inactivation has consistently been reported to be half-maximal (Vh) at about −100 mV, and in most β-cell Na+ currents cannot be elicited from the normal resting potential (–70 mV). In these cells, Na+ current cannot contribute to the action potential, which explains why their electrical activity is resistant to the Na+ channel blocker TTX. Not all β-cells conform to this picture, however: in ~30% of β-cells, an additional Na+ current component is present, which accounts for 15–25% of the total Na+ current (i.e., ~150 pA) (709, 761). As its inactivation is half-complete at −40 mV, this Na+ current component persists at physiological membrane potentials and is therefore capable of contributing to the upstroke of the action potential (761). Accordingly, those β-cells in which it is present exhibit rapidly activating, large-amplitude action potentials that peak at potentials above 0 mV. In such cells, TTX reduces action potential amplitude. It is possible that β-cells that generate Na+-dependent action potentials account for the observation that glucose induces [Na+]i oscillations in a subset of β-cells (225).
FIGURE 10.
Steady-state voltage-dependent inactivation of Na+ channels in mouse and human β-cells. This was analyzed by a two-pulse protocol (inset) in which a test pulse to 0 mV (to maximally activate the Na+ channels) was preceded by a conditioning depolarization (50–100 ms) to various membrane potentials. During the conditioning pulse, the Na+ channels undergo voltage-dependent activation: the more depolarized the conditioning voltage, the fewer the Na+ channels that remain to be activated by the test pulse. This gives rise to a sigmoidal relationship between membrane potential and the peak current during the test pulse that describes the voltage dependence of inactivation. In human β-cells, inactivation of the principal Na+ current component is half-maximal at −45 mV (arrowed). The corresponding value in mouse β-cells is 50–60 mV more negative (about −100 mV, arrowed). [Modified from Braun et al. (90) and Göpel et al. (221).]
It is tempting to attribute the biphasic nature of Na+ current inactivation to the activity of distinct types of Na+ channel. Indeed, studies using knockout mice suggest that Nav1.7 channels are responsible for the current that inactivates at very negative membrane potentials and that Nav1.3 contributes to the Na+ current that inactivates at more positive voltages (761).
Why Nav1.7 channels inactivate at such negative membrane potentials in mouse β-cells remains an enigma. In neurons, steady-state inactivation of Nav1.7 is half-maximal at potentials 30–40 mV more positive than those observed in β-cells (131). The same difference is seen when Nav1.7 as well as Nav1.3 and Nav1.6 are expressed in INS1-cells or in HEK cells (own unpublished data). This shift in the voltage dependence of inactivation towards more negative voltages in insulin-secreting cells may reflect differences in membrane lipid composition.
III) Pharmacology. The β-cell Na+ current is inhibited by TTX and, as expected, the toxin inhibits β-cell electrical activity (761). However, in one study (678) it was stimulatory. It is possible that this discrepancy is due to confounding paracrine effects, mediated by changes in somatostatin release from pancreatic δ-cells, which are equipped with voltage-gated Na+ channels (221, 761). A similar explanation may account for the fact that one study reported that the Na+ channel activator veratridine inhibited glucose-induced β-cell electrical activity (678).
IV) Metabolic regulation. Whether β-cell Na+ channels are metabolically regulated has not been widely studied. However, it has been reported that increasing intracellular ATP from 2 to 8 mM produces a 10-mV positive shift in the voltage dependence of inactivation (772). Given that glucose increases intracellular ATP, it is tempting to speculate that glucose may exert a similar effect on inactivation in metabolically intact β-cells and thus that Na+ channels may make a greater contribution to electrical activity at high glucose. However, as β-cells are depolarized at high glucose, which leads to Na+ channel inactivation, the significance of this ATP-dependent effect is not immediately clear.
V) Functional significance. The mouse β-cell Na+ current appears to be particularly important at low glucose concentrations, because the increase in electrical activity, [Ca2+]i, and insulin secretion evoked by 6 mM glucose are all strongly reduced by TTX (761). It seems possible that the subset of β-cells that possess Na+ currents that are activated within the physiological range of membrane potentials, have a slightly lower threshold for firing action potentials. Although most Na+ channels will be completely inactivated at −70 mV, the remaining 10–15% will provide a significant depolarizing current (the peak current at 0 mV is 100–150 pA, which is comparable to, or larger than, the voltage-gated Ca2+ current). Because of electrical coupling between β-cells, this may result in electrical activity (and insulin secretion) across the entire islet.
b) human.
I) Molecular composition. Whereas mouse β-cells mainly express Scn9a (Nav1.7), SCN8A (Nav1.6) accounts for 40% of Na+ channel transcripts in human β-cells (68, 475) with the remainder being SCN9A (Nav1.7), SCN3A (Nav1.3), and SCN2A (Nav1.2) (see Supplemental Figure 4). Of the β-subunits, SCN1B (β1) and SCN3B (β3) each account for 50% of transcripts.
II) Biophysical characteristics and pharmacology. Human β-cells are equipped with large TTX-sensitive Na+ currents. The maximum amplitude of the Na+ current is >40 pA/pF, approximately threefold larger than the Ca2+ current. Na+ currents are elicited by depolarizations above −40 mV (90). They activate rapidly, peak within <1 ms (at −20 mV and above), and are almost completely inactivated within 5 ms. Steady-state inactivation is half-maximal at −40 mV, ~60 mV more positive than the corresponding value in mouse β-cells (221, 514) (FIGURE 10). Why this is the case is unclear. Possibly, it is because human β-cells express a different Na+ channel complement than mouse β-cells (FIGURE 10).
III) Functional significance. The inactivation properties of the human β-cell Na+ current suggest that voltage-gated Na+ channels are functionally more important than in mouse β-cells. Indeed, addition of TTX to electrically active human β-cells reduces the peak height of the action potential and in some cells virtually abolishes regenerative electrical activity. Furthermore, TTX reduces insulin secretion: its effect is particularly strong at glucose concentrations just above the threshold for GIIS (6 mM) but is weaker at higher (10–20 mM) concentrations (52, 90) (see FIGURE 13, C AND D).
FIGURE 13.

A: stimulus-secretion coupling in a human β-cell. Glucose uptake via GLUT1 and GLUT2 leads to accelerated mitochondrial glucose metabolism, increased ATP production, and closure of the KATP channels (consisting of the pore-forming subunit Kir6.2 and the sulfonylurea-binding protein SUR1). Inwardly rectifying Kir5.1 and Kir7.1 channels also contribute to the resting conductance of the human beta-cells, but their contribution is small. The increased membrane resistance (Rm↑) resulting from closure of the KATP channels allows occasional spontaneous opening of T-type Ca2+-channels (Ca[T]) to depolarize the β-cell (Ψ↓), and this leads to regenerative opening of additional T-type Ca2+ channels and a further membrane depolarization that culminates in rapid activation of L-type Ca2+ channels (Ca[L]) and voltage-gated Na+ channels (NaV) during the upstroke of the action potential. At the peak of the action potential, P/Q-type Ca2+ channels (Ca[P/Q]) open and the associated Ca2+ influx triggers exocytosis of insulin-containing secretory granules (SG). Opening of Ca2+-activated high-conductance K+ channels (BK) underlies action potential repolarization. Interspike membrane potential is influenced by TALK-1/TASK-1, small conductance Ca2+-activated K+ channels (SK), and/or delayed rectifying K+. B: burst of overshooting action potentials recorded from a human beta-cell. Rm↑ and Ψ↓ indicate an increase in membrane resistance and membrane depolarization, respectively. The numbers 1–9 highlight different phases of β-cell electrical activity: 1) the initial depolarization, 2) the upstroke of the action potential, 3) the peak of the action potential, 4) action potential repolarization, 5) the afterhyperpolarization, 6) the plateau/interspike potential, 7) the progressive reduction of action potential amplitude during electrical activity, 8) burst termination, and 9) the pacemaker depolarization between two bursts of action potentials that eventually results in the initiation of a new burst of action potetentials (1) (see sect. VIIIB6). C and D: schematics explaining why the Na+ channel blocker TTX has a weaker inhibitory effect on electrical activity (above) and insulin secretion (below) at high glucose than at low glucose concentrations. GIIS is greater at high glucose (D) than at low glucose (C) despite the reduction of action potential height because glucose exerts an amplifying effect on insulin secretion in addition to triggering electrical activity (see sect. IXD7a).
3. Voltage-gated K+ channels
The action potentials of both mouse and human β-cells are short, ranging between 5 ms (human) and 30–40 ms (mouse) (568, 570). Rapid repolarization is mediated by activation of voltage-gated K+ channels. Action potential repolarization still occurs in the absence of K+ channel activation because of inactivation of the voltage-gated Na+ and/or Ca2+ channels, but under such conditions the action potentials can last hundreds of milliseconds (570). Voltage-gated K+ channels carry an outward K+ current that is sometimes known as the delayed outward current, because it activates more slowly than the Na+ current. As it shows outward rectification, the current is also referred to as the delayed rectifier. Although Ca2+-activated K+ channels are also voltage dependent, we consider them separately below.
Whereas voltage-gated Na+ and Ca2+ channels consist of a single α-subunit with four homologous domains, voltage-gated K+ channels consist of four separate α-subunits, each corresponding to a single domain of the Na+ channel. These coassemble to form a tetrameric pore (247). Voltage-gated K+ channels are the largest family of ion channels, and the genome contains ~40 genes encoding α-subunits with diverse functional properties. This diversity is further increased by the fact that heteromeric channels may be formed by the association of two types of closely related α-subunit.
a) mouse
I) Molecular composition. At the transcript level, mouse β-cells express Kcnb1 (Kv2.1), Kcnh1 (Kv10.1), Kcnh2 (Kv11.1), and Kcnh6 (Kv11.2) pore-forming α-subunits at relatively high levels (see Supplemental Figure 5A). Kcna2 (Kv1.2), Kcnb2 (Kv2.2), Kcnc3 (Kv3.3), Kcnd1 (Kv4.1), and Kcnh5 (Kv10.2) are also expressed at detectable levels. However, experiments on Kcnb1-deficient mice make it clear that these channels account for most of the delayed outward current in mouse β-cells (310). Kcnh2 (Kv11.1 or HERG1) and Kcnh6 (Kv11.2 or HERG2) are involved in spike frequency modulation. These channels carry little outward current during the action potential and are thus unlikely to contribute much to action potential repolarization. However, when the β-cell is repolarized, they give rise to long-lasting outward ‟tail currents.ˮ This is predicted to increase the interval between successive spikes, slowing spike frequency. Inhibition of Kcnh2/Kcnh6 channels leads to elevation of [Ca2+]i and stimulation of GIIS (254). Mouse β-cells also express Kcnc3 (Kv3.3), which encodes a rapidly inactivating TEA-sensitive K+ current (224). An early report indicated the presence of a transient TEA-resistant and 4-AP-sensitive A-type K+ current (633). However, mouse β-cells only express classical A-type K+ current transcripts (Kcnd1–4) at low levels (FIGURE 12A), and thus it is likely that the 4-AP-sensitive currents were actually recorded from non-β-cells (some of which express A-type K+ channels; Refs. 2, 146).
FIGURE 12.

A: schematic of the effects of increasing glucose concentrations (indicated at top) on mouse β-cell membrane potential and electrical activity, ATP/ADP ratio, and whole-cell KATP channel conductance (GKATP). The dashed vertical lines separate phase I, II, III, and IV (see main text). Schematic based on Cha and co-workers (112, 113). Schematic courtesy of Dr. C. Cha, Oxford. B and C: bursts of β-cell action potentials recorded from a β-cell exposed to 10 mM glucose. The numbers 1–6 highlight different phases of β-cell electrical activity: 1) the upstroke of the action potential, 2) action potential repolarization, 3) the plateau potential, 4) the progressive decrease in action potential amplitude during the burst, 5) burst termination, and 6) the pacemaker depolarization between two successive bursts (see sect. VIIIA).
Like voltage-gated Ca2+ and Na+ channels, voltage-gated K+ channels also possess auxiliary β- subunits (FIGURE 12B). Kcnip1 is the major β-subunit expressed in mouse β-cells. Its functional role has not been investigated, but silencing of Kcnip1 in mouse insulinoma cells leads to stimulation of GIIS (377).
II) Biophysical properties. Action potential repolarization in mouse β-cells results from activation of a delayed outward K+ current. This current becomes detectable during depolarizations positive to −20 mV (572, 632). Current activation is slower than that of Ca2+ channels and is best described by n4 kinetics (FIGURE 9C). At −10 mV (the peak of the action potential), the time constant of activation (τn) is ~10 ms, and the current amplitude is ~100 pA, which is of similar magnitude to the Ca2+ current. Although their activation is slow compared with that of voltage-gated Na+ and Ca2+ channels, Kv2.1 channels activate with sufficient speed to contribute to the repolarization of the action potential in mouse β-cells. This explains why genetic knockdown of Kv2.1 channels increases both action potential amplitude and duration (310). Deactivation of Kv2.1 channels is fairly slow, and the K+ conductance of the β-cell will therefore be somewhat increased following each action potential; we have proposed that this increase in K+ permeability may contribute to the temporal separation of two successive action potentials (31).
Voltage-gated K+ channels with a single-channel conductance of 8 pS (when using physiological intra- and extracellular K+ concentrations) and 16 pS (at 145 mM extracellular K+) have been recorded (632). These values are close to those expected for Kv2.1 channels exposed to these ionic gradients (690). Openings of K+ channels with properties conforming to those of delayed rectifying K+ channels are also observed during action potential repolarization in cell-attached patches on mouse β-cells (632).
III) Pharmacology. Kv2.1 channels are blocked by the broad-spectrum K+ channel blocker TEA with an IC50 of 1.4 mM (76, 178). They are also inhibited by the Kv2 channel inhibitor stromatoxin, which has the same effect on the delayed outward current as genetic ablation of Kcnb1. The toxin has no effect in Kv2.1 knockout β-cells (310), suggesting its effect is selective and mediated by block of Kv2.1.
IV) Modulation. The magnitude of the voltage-gated K+ current has been reported to increase in response to glucose and other stimulators of β-cell metabolism (including glyceraldehyde and α-ketoisocaproic acid) and is reduced by the mitochondrial inhibitor FCCP or by intracellular dialysis with ATP-free medium or nonhydrolyzable ATP analogs (753). The effect of increasing glucose from 2.8 to 16.7 mM is fairly dramatic: within 25 min the amplitude of the delayed rectifying K current is threefold larger. The magnitude of the effect is similar at all membrane potentials and is apparent even at −30 mV, where K+ currents are small (753). The functional significance of this effect is not immediately evident, as it would be expected to shorten the action potentials at high glucose and thereby restrict Ca2+ influx and reduce GIIS. One possibility is that it may prevent the β-cell from becoming overloaded with Ca2+. This might help prevent Ca2+-induced inactivation of Ca2+ channels and the resulting reduction in insulin secretion. It would also facilitate a faster firing rate.
Protein kinases also regulate voltage-gated K+ channels. For example, cAMP (via PKA activation) reduces the magnitude of the delayed rectifying current (413). It has been proposed that by prolonging the action potential duration, and thus the amount of Ca2+ entry, this effect will contribute to the insulinotropic action of compounds that act via the cAMP/PKA pathway, such as the gut hormones GLP-1 and GIP. Acute exposure of β-cells to the free fatty acid linoleic acid also reduced delayed outward current amplitude (181). This is predicted to prolong action potential duration and potentiate GIIS (491). It is possible that the effect of glucose on the K+ channel is mediated via changes in cAMP (164).
Interestingly, Kv2.1 channels have been shown to interact with a number of exocytotic SNARE proteins (see sect. IXB1). For example, syntaxin-1 and SNAP25 both shift the voltage dependence of inactivation, albeit in opposite directions (syntaxin shifts it negative and SNAP25 positive) (413, 448). Syntaxin-1 also interferes with the trafficking of Kv2.1 to the plasma membrane (386).
b) human.
Human β-cells possess a slowly activating, stromatoxin-sensitive (delayed-rectifier) K+ current (90). The time constant of activation (τm) measured at −20 mV is >10 ms. Thus delayed rectifying K+ channels may not activate much during the human β-cell action potential, which lasts only ~5 ms (568). This explains why stromatoxin has little effect on action potential height in human β-cells. Based on studies in mouse β-cells, it was proposed that pharmacological inhibition of Kv2.2 channels, via increased action potential duration, might represent a means of enhancing GIIS (279). Given the small effects of stromatoxin on glucose-induced electrical activity and insulin secretion in human islets, however, it is not evident that this strategy will be therapeutically meaningful in humans.
In contrast to mouse β-cells, the delayed rectifying K+ current in human β-cells is carried principally by Kv2.2 (KCNB2) and Kv1.6 (KCNA6) channels (see Supplemental Figure 5A), which are expressed at high levels in the human β-cell and encode inactivating TEA-sensitive voltage-gated K+ channels (68, 475). Human β-cells also express fairly high levels of KCNA5, KCNC3, and KCND3.
Interestingly, human β-cells express high levels of KCNQ2 (Kv7.2). In neurons, this channel gives rise to the M-current, so named because its activity is inhibited by muscarinic receptor activation. KCNQ2 is voltage-activated, but it is also active at the resting potential and thereby dampens electrical activity (614). Whether reduced activity of this channel contributes to the stimulatory effect of muscarinic receptor activation on electrical activity in human β-cells (568) has not been investigated.
Like mouse β-cells, human islets express very high levels of KCNH2 (Kv11.1 or HERG1), more than twice as high as KCNB2. HERG-like currents also have been recorded from human β-cells (575). Application of the HERG1-blocker WAY 123–398 produced an approximately twofold increase in action potential frequency and insulin secretion evoked by a combination of glucose and arginine (575), consistent with the idea that these channels are active in the human β-cell and determine the interval between two successive action potentials.
Human β-cells also express a number of auxiliary (β) K+ channel subunits (see Supplemental Figure 5B), particularly KCNB1 and KCNG3. Little is known of the function of these proteins, but in other cell types KCNB1 confers fast inactivation on otherwise noninactivating Kv1 channels (323).
c) kv channels and disease.
Copy number variations (CNVs) are major players in genetic susceptibility to common disease (377). CNVs in KCNIP1 have been identified in Korean T2DM subjects. As described above, deletion of KCNIP1 leads to stimulation of insulin secretion, as expected if the Kv current is reduced. Why this predisposes to T2DM remains unclear. One possibility is that the resulting prolongation of the action potential results in Ca2+ overload and ultimately β-cell death.
Genetic evidence suggests that the KCNQ1 locus is associated with increased T2DM risk (700, 749). However, Kcnq1/KCNQ1 is expressed at very low levels in both mouse and human β-cells (see Supplemental Figure 5A), and mice lacking Kcnq1 show normal GIIS (23). This makes it unlikely that Kcnq1 channels contribute much to the outward current in mouse or human β-cells, and thus that the KCNQ1 locus increases T2DM risk by affecting β-cell channel activity. Nevertheless, patients with long QT-syndrome 1 (LQT1), which is associated with heterozygous loss-of-function mutations in KCNQ1, have increased insulin secretion and are more prone to hypoglycemia than control individuals. There is now evidence that the T2DM risk associated with the KCNQ1 locus is mediated through another gene in the imprinted 11p15.5 region of the chromosome (692). Regional gene expression is regulated by differential methylation at the promoter of KCNQ1 overlapping transcript 1 (KCNQ1OT1), a nontranslated antisense RNA that regulates maternal-specific expression of downstream genes. One possibility is that this leads to increased expression of the cell cycle inhibitor Cdknc1 (23), which reduces β-cell mass and thereby increases T2DM risk. However, it worth pointing out that both KCNJ11 (Kir6.2) and ABCC8 (SUR1) lie in the same region of the chromosome (11p15.1), and gain-of-function variants in these genes enhance T2DM risk (255, 293).
4. Large-conductance Ca2+-activated K+ channels
Although the amplitude of the delayed outward current was initially thought to be largely unaffected by blocking the Ca2+ current (632), more recent data indicate that some β-cells (especially those with large Ca2+ currents) also possess a Ca2+-activated K+ component of outward current (293). This reflects activation of large-conductance Ca2+-activated K+ channels (big K+ channels, or BK channels).
a) mouse.
I) Molecular structure. The BK channel is an octameric complex of four pore-forming α-subunits and four regulatory β-subunits (62). In mouse β-cells, BK channels comprise α1 (encoded by Kcnma1) and β2-subunits (encoded by Kcnmb2) (see Supplemental Figure 6B).
II) Biophysical properties. The BK current is activated by both membrane depolarization and Ca2+, which binds to the Ca2+ bowl in the cytoplasmic COOH-terminal domain of the BK channel and shifts the voltage dependence of activation to more negative membrane potentials (370). The BK current can be isolated by subtracting the K+ current remaining after Ca2+ current inhibition from that recorded under control conditions. It becomes detectable during depolarizations above −30 mV, is maximal at +20 mV, and undergoes a secondary decrease at more positive voltages. This N-shaped current-voltage relationship reflects the fact that the Ca2+ current first increases and then decreases with membrane depolarization (FIGURE 10C).
At −10 mV, BK current activation can be described by a single exponential with a time constant of 5 ms. Thus the BK current activates at least fivefold faster than Kv2.1, making it ideally placed to contribute to spike repolarization. Indeed, blocking BK channels increases spike amplitude by ~20 mV and enhances GIIS (293). BK channels also undergo voltage-dependent inactivation, which at −10 mV (the peak of the action potential) is described by a single exponential with a time constant of 20 ms (293).
In cell-attached patches, single BK channel currents with activation and inactivation kinetics similar to those observed at the whole-cell level can be activated by membrane depolarizations from −70 mV to ~20 mV (632). The single-channel conductance is 70–90 pS for outward currents recorded in cell-attached patches when measured using physiological K+ gradients (632) and 240 pS for inward currents recorded in excised inside-out patches exposed to symmetrical 140 mM K+ (128). In inside-out patches, BK channel activity at physiological membrane potentials (i.e., <0 mV) is barely detectable at [Ca2+]i below 10 μM. In the β-cell, Ca2+ concentrations higher than this will only occur very close to the voltage-gated Ca2+ channels, which predicts that BK channels and voltage-gated Ca2+ channels should colocalize. This is well established in other cell types (464) but remains to be determined in β-cells.
III) Pharmacology. BK channels are blocked by the broad-spectrum K+ channel blocker TEA with an IC50 of 100 µM, which is 15- and 200-fold lower than the corresponding values for the delayed rectifier and KATP channel, respectively (75, 76). They are also blocked by peptide toxins such as charybodotoxin, iberiotoxin, and slotoxin (293, 311) and by the alkaloid paxilline (293).
IV) Functional role. Early experimental and theoretical studies led to the proposal that BK channels play a key role in glucose-dependent regulation of electrical activity (41, 115). Support for this idea came from the finding that BK channels are present at high density in isolated outside-out membrane patches exposed to high (0.2 mM) intracellular Ca2+ (75) and in cell-attached patches on β-cells in which intracellular [Ca2+]i was elevated (via high-[K+]o depolarization) (632). In addition, lowering intracellular pH reduced BK channel activity, suggesting a potential link between glucose metabolism (via cytoplasmic acidification) and BK channel closure (128). However, following the discovery of the central role of the KATP channel in glucose-dependent depolarization, and the finding that charybdotoxin, a blocker of BK channels, did not affect electrical activity, action potential firing (356), or the voltage-gated K+ current of mouse β-cells (632), it was concluded that BK channels contribute little to either glucose-induced membrane depolarization or action potential repolarization in β-cells.
Nevertheless, more recent data have reopened the question of the functional role of BK channels. It was reported that the BK channel blocker paxilline inhibited a rapidly activating and deactivating current in mouse β-cells (293). This suggests BK channel activation potentially might reduce action potential duration, Ca2+ influx, and insulin release. Surprisingly, however, both insulin secretion and glucose tolerance were reduced in BK KO mice (159). Why this is the case is unclear. One possibility is that it reflects a reduction in β-cell mass since BK channels are important for the capacity of the β-cell to handle oxidative stress and BK KO mice showed a threefold increase in the number of apoptotic cells (159).
b) human.
At the molecular level, human β-cells express the same α- and β-subunits as mouse β-cells. Thus KCNMA1 and KCNMB2 are the principal BK channel genes (see Supplemental Figure 6B). KCNMA1 is expressed at fivefold higher levels in human than in mouse β-cells, which may explain why BK channels appear to play a more prominent role in human β-cells.
Like the mouse BK current, that of the human β-cell becomes detectable during depolarizations above −40 mV, is maximal at +20 mV, and undergoes a secondary decrease at more positive voltages, reflecting the decline in the Ca2+ current (FIGURE 9). The peak amplitude of the BK current at +20 mV is 40 pA/pF, comparable to the magnitude of the voltage-gated Na+ current. The current activates with a time constant of 2 ms at −10 mV, which is threefold faster than Kv2.1. This is sufficiently rapid to influence action potential duration. As predicted, inhibition of BK channels with iberiotoxin was associated with an ~10 mV increase in action potential amplitude and stimulation of insulin secretion (90).
5. Small-conductance Ca2+-activated K+ channels
The identity of the channel(s) that regulate the duration of the glucose-induced bursts of action potentials was a mystery for many years. Because burst duration is influenced by the external Ca2+ concentration and the repolarization terminating each burst is associated with a transient increase in membrane conductance (40, 547), a Ca2+-activated K+ conductance was suggested to contribute to burst repolarization. However, the demonstration that BK channel blockers did not affect the slow waves (356) implied a different type of Ca2+-activated K+ channel must be involved. Identification of this K+ channel turned out to be more challenging than expected. This is because when isolated mouse β-cells are maintained in tissue culture they do not exhibit the rapid oscillatory electrical activity observed in β-cells within intact mouse islets (15, 368, 631). The conundrum was finally solved by using the perforated patch whole-cell technique to study β-cells within freshly isolated intact islets and measuring currents induced by simulated action potentials (223). This revealed a time-dependent increase in a K+ current (Kslow) that gradually develops during a burst of action potentials. Kslow is a compound current that includes a contribution from both the KATP channel and the small-conductance Ca2+-activated K+ (SK) channel. Here, we focus on the role of the SK channel. Although it is not a voltage-gated channel, it is nevertheless considered in this section because its activation is intimately linked to β-cell electrical activity.
a) mouse.
I) Molecular composition. Mouse β-cells express SK1, SK2, SK3, and SK4 (Kcnn1–4) channels, but the expression of SK3 (Kcnn3) is ~10-fold higher than that of the other SK channels (see Supplemental Figure 6A). SK2 and SK3 channels are blocked by apamin. An early report that apamin is without effect on either glucose-induced electrical activity (374) or the amplitude of the Kslow current (12, 223) was therefore surprising (although see Ref. 669). Consequently, it was proposed that β-cells express an apamin-insensitive isoform of SK3 (759). Although SK4 channels (Kcnn4) are expressed at low levels (only 5% of Kccn3), β-cells from Kcnn4-deficient mice exhibit increased excitability, enhanced GIIS, and improved glucose tolerance in vivo (156). A possible resolution to both puzzles is if heteromultimeric assembly of different SK channel subunits (i.e., SK3 and SK4) underlies the apamin-insensitive Kslow channel of the mouse β-cell (460).
II) Biophysical properties. The time and voltage dependence of activation of Kslow echoes the changes in [Ca2+]i associated with (simulated) electrical activity (223). Following cessation of electrical activity, the current deactivates with a time course similar to the slow depolarization between two successive bursts of action potentials (223). Thus Kslow likely contributes to the pacemaker current. The single-channel conductance recorded with symmetrical 130 mM K+ solutions is 39 pS (156).
III) Pharmacology. As expected for a Ca2+-activated current, Kslow is inhibited by Ca2+ channel blockers. It is also blocked by TEA with an IC50 of ~5 mM (223) and by UCL 1684. Azimilide appears to be a specific blocker of the SK3 channel in mouse β-cells (759) and increases the duration of the slow waves and potentiates GIIS (759). Kslow is seemingly unaffected by apamin (see above) or the SK4 inhibitor chlotrimazole. However, when apamin is applied to β-cells lacking functional Kv2.1 channels (due to genetic ablation or pharmacological inhibition), a dramatic increase in action potential duration is observed (311). Conversely, pharmacological activation of SK channels using riluzole in mouse β-cells exposed to tolbutamide leads to a prompt membrane repolarization that is reversed by apamin (311). Collectively, this pharmacological profile suggests that SK channels not only are expressed in mouse β-cells but also are functionally active and contribute to action potential and burst repolarization.
IV) Modulation. The Kslow current is reduced by 50% when glucose is increased from 5 to 10 mM (217). This finding is somewhat counterintuitive, given that glucose increases [Ca2+]i. Perhaps it reflects a direct effect of glucose metabolism on the KATP-dependent component of Kslow, or an indirect one mediated by increased Ca2+ buffering, but this has not been determined. It is interesting that the SERCA inhibitor thapsigargin exerts a dual effect on Kslow, transiently increasing and then reducing its amplitude (217). This suggests [Ca2+]i buffering plays an important role in Kslow regulation. The modulation of Kslow by glucose is of functional importance as it may underlie the shortening of the electrically silent interburst intervals that occurs with increasing glucose concentrations.
The existence of a KATP channel-independent component of Kslow (contributed by SK channels) might explain the paradox that slow waves persist in islets of SUR1 knockout mice, which lack functional KATP channels (157, 223). The finding that Kslow is influenced by glucose may also help explain why some SUR1 knockout islets remain capable of responding to glucose with increased electrical activity (157).
It has been reported that Kslow is reduced in dispersed β-cells maintained in tissue culture (223) which may account for the loss of normal oscillatory electrical activity seen in this preparation (but see Ref. 217).
b) human.
As in mouse islets, action potential firing in β-cells in intact human islets is often grouped into bursts (FIGURE 6B), although these are not as distinct as those of mouse β-cells. Their electrophysiological basis remains unclear, but extrapolation from mouse islets implicates small-conductance Ca2+-activated K+ channels. Like their mouse counterparts, human β-cells predominantly express SK3 (KCNN3) with other SKs being expressed at 10-fold lower levels (68, 475). Blocking SK channels with apamin, or the nonselective antagonist NS8593, results in membrane depolarization and action potential firing in human β-cells (311) and stimulates insulin secretion (568).
6. Hyperpolarization-activated cation channels
The hyperpolarization-activated cation current (HCN; also referred to as Ih or If) modulates the resting potential and rhythmic electrical activity of cardiac and neuronal cells (67). Both mouse (3, 146) and human (68, 475) β-cells express Hcn/HCN2–4.
HCN channels are activated by membrane hyperpolarization and are permeable to Na+ and Ca2+, so potentially they could provide a depolarizing inward current. However, in mouse insulinoma cells, there is normally little HCN current at voltages more positive than −70 mV (activation is half-maximal at −88 mV). Thus there is little current at the resting membrane potential. In the presence of cAMP, however, activation of HCN channels is shifted to more positive membrane potentials (half-maximal at −74 mV in the presence of forskolin and IBMX). This shift means a significant depolarizing current will exist at physiologically meaningful membrane potentials; for example, ~20% of the maximal HCN current will be activated at −60 mV (168). HCN channels may therefore contribute to the reported shortening of the interburst interval observed in β-cells (211) of mouse islets exposed to high (50 nM) concentrations of GLP-1 (238). Pharmacologically, the HCN current is blocked by ZD7288 and cilobradine. These blockers do not affect glucose- and forskolin-induced insulin secretion (168).
D. Voltage-Independent Currents
Here we consider ion channels that are not involved in generating electrical activity but whose activity influences action potential firing by providing background conductances.
1. Inwardly rectifying K+ channels (KIR)
KIR channels are widely expressed. Their principal physiological roles are to set the resting membrane potential and regulate the excitability of electrically active cells and to facilitate K+ transport in epithelial cells (281). Their name derives from the fact that they display inward rectification; in other words, in symmetrical K+ concentrations, the current-voltage relation is not linear, but is larger in the inward than in the outward direction. This is also observed at the single-channel level and derives from a voltage-dependent block of outward currents by intracellular cations such as Mg2+ and spermine (478). Differences in cation binding affinity explain why some channels show weak rectification and others are strong inward rectifiers. The apparent activation kinetics of strong inward rectifiers result from a time-dependent unblock of the channel on hyperpolarization, as channel open probability does not vary with membrane potential. The most important KIR channel in the β-cell is Kir6.2 (encoded by Kcnj11/KCNJ11), the pore-forming component of the KATP channel. This is discussed above. Here, we focus on other members of the KIR channel family.
a) molecular composition.
Mouse β-cells express Kcnj12 (Kir2.2), lesser amounts of Kcnj13 (Kir7.1), Kcnj5 (GIRK4), and Kcnj6 (GIRK2) and very low levels of Kcnj1 (Kir1.1), Kcnj3 (Kir3.1), and Kcnj4 (Kir2.3) (see Supplemental Figure 7A). Human β-cells express particularly high levels of KCNJ13 (Kir7.1) and KCNJ16 (Kir5.1). KCNJ2 (Kir2.1), KCNJ3 (Kir3.1), KCNJ8 (Kir6.1), and KCNJ14 (Kir2.4) are also expressed, but at lower levels. Quantitative PCR studies also revealed expression of KCNJ4 (Kir2.3) and KCNJ12 (Kir2.2) in human islets (88), but these transcripts were not detected at significant levels by RNAseq (68, 475). The reason for this discrepancy is unclear. We point out that although a number of KIR genes are expressed in both human and mouse β-cells, most (60–85%) of the transcripts are Kcnj11/KCNJ11.
b) functional properties.
KIR channel properties can be studied in the presence of a sulfonylurea (e.g., tolbutamide) to selectively block KATP currents. In human β-cells, hyperpolarizing voltage steps from a holding potential of −70 mV evoked progressively larger tolbutamide-resistant inward currents (553, 568). These currents were blocked by low concentrations of Ba2+ (100 μM) and Cs+ (1 mM). The Ba2+-sensitive current displayed strongly inward rectification and reversed at about −85 mV, which is close to EK.
Inhibition of inward rectifiers with Ba2+ (0.1 mM) or Cs+ (1 mM) in human β-cells exposed to glucose (6 mM) and tolbutamide (0.1 mM) resulted in membrane depolarization and stimulation of action potential firing (568). It is not clear which of the KIR channels expressed in the human β-cell contribute to this current. Studies of KIR channels in mouse β-cells have mainly focused on GIRK channels, which have been reported to mediate the hyperpolarization of mouse β-cells by epinephrine (308) and somatostatin (566) (see sect. VIIB2a).
c) kIR channels and disease.
The most important KIR channel associated with diabetes is Kir6.2 (KCNJ11), which is discussed above (see sect. VA6). In addition, a polymorphism in KCNJ15 (the gene encoding Kir4.2) increases the risk of T2DM >1.7-fold (522). As KCNJ15 is expressed at low, albeit detectable, levels in human β-cells (see Supplemental Figure 7A), and there is no evidence its expression is increased in diabetic β-cells (606), its effect on T2DM risk is probably mediated via other cell types.
2. Tandem pore channels
The tandem pore domain K+ channels constitute a large family of K+ channels. They are dimers comprising two α-subunits, each of which contains four transmembrane domains and two pore domains. Functionally, they serve as ‟leakˮ channels that give rise to almost instantaneous, noninactivating currents. They should not be confused with the two-pore Tpc channels found in intracellular membranes.
a) mouse.
I) Molecular composition. Mouse β-cells express TALK1 (Knck16) at levels higher than Kcnj11 (Kir6.2) channels (see Supplemental Figure 8B). They also express low levels of TWIK1 (Kcnk1) and TASK1 (Kcnk3) channels (see Supplemental Figure 8A).
II) Functional properties. TASK1 is an outwardly rectifying K+ current that activates around −20 mV and increases with depolarization. As might be predicted from its very low expression, comparison of the membrane currents in wild-type and Knck3 knockout mice indicated its contribution to the β-cell membrane conductance is negligible at physiological membrane potentials, and even at −10 mV (the peak of the action potential), the outward current flowing through these channels is only 1 pA/pF (135). Despite the small size of the current, β-cells from Knck3-deficient mice were slightly more depolarized in the presence of glucose than their wild-type counterparts. Action potential frequency was paradoxically reduced (135), but this may be a consequence of voltage-dependent inactivation of other currents involved in action potential generation.
Kcnk16 (TALK1) gives rise to an outwardly rectifying current that becomes detectable at voltages above −45 mV (708). Despite the high expression, the TALK1-dependent current is small and limited to ~1 pA/pF at 0 mV. With a reversal potential of −50 mV (708), this corresponds to an estimated membrane conductance of 0.02 nS/pF. Thus, under most conditions, the contribution of this current to the resting membrane conductance of the β-cell is dwarfed by that of the KATP channel (~1 nS/pF) (see sect. VA5).
β-Cells in islets from Kcnk16/TALK1-deficient mice are more depolarized than wild-type β-cells, during both the depolarized plateau from which action potentials arise, and the silent interburst intervals (708). Action potential frequency is unaffected, but the average interburst duration is reduced by 60%. GIIS is enhanced, especially at glucose concentrations that are just above the threshold for stimulation of insulin release. This correlates with a moderate improvement of glucose tolerance in vivo (708). Collectively, these observations suggest that TALK-1 stabilizes the interburst intervals. Whether TALK1 channels are regulated by glucose metabolism and thereby directly influence glucose-induced electrical activity, or whether they just provide an additional background K+ conductance, has not been studied.
There have been no functional studies of Kcnk1 (TWIK1) in mouse β cells to date.
b) human.
I) Molecular composition. Human β-cells express KCNK16 (TALK1) at levels more than threefold higher than KCNJ11 (KIR6.2) and much lower (10–20% of KCNK16) levels of KCNK1 (TWIK1), KCNK3 (TASK1), and KCNK17 (TALK2) (see Supplemental Figure 8, A and B).
II) Functional properties. TASK1 activates around −30 mV in human β-cells (135). In 14 mM glucose, blocking the channel with the selective inhibitor A1899 depolarizes the β-cell by ~5 mV and increases action potential frequency. Action potential height is also reduced, presumably due to depolarization-induced inactivation of ion channels involved in action potential firing.
Downregulation of KCNK16 (TALK1) suggests that these channels give rise to a current with outwardly rectifying properties that activates above −20 mV. An increased risk of T2DM has been linked to a coding sequence polymorphism in TALK1 (rws1535500) (708) that increases TALK1 channel activity. This would be expected to cause membrane hyperpolarization, reduced excitability, and a decrease in insulin secretion; however, this has not been studied.
Nothing is known about the functional roles of TWIK1 (KCNK1) and TALK2 (KCNK17) in human β-cells.
E. Intracellular Ion Channels
Pancreatic β-cells are also equipped with multiple types of ion channels in the membranes of intracellular organelles such as secretory vesicles, lysosomes, and sER. These include InsP3 receptor (InsP3R), ryanodine receptor (RyR), and two-pore (TPC) Ca2+ release channels.
Three isoforms of the InsP3 receptor are found in mouse β-cells: Itpr1, Itpr2, and Itpr3 (see Supplemental Figure 9). In human β-cells, ITPR3 is expressed at particularly high levels (~10-fold greater than ITPR1 or ITPR2) (68, 475). Activation of these receptors leads to Ca2+ release and a rise in [Ca2+]i. As outlined in section VIIB1a, the effects of acetylcholine are, in part, mediated by elevation of InsP3, which stimulates sER Ca2+ release via binding to InsP3 receptors.
Ca2+-induced Ca2+ release (CICR) has been proposed to mediate the effects of GLP-1 on [Ca2+]i in β-cells (239, 332). However, Ryrs are only expressed at low levels in mouse β-cells (56). In contrast, Ryr3 is highly expressed in somatostatin-secreting δ-cells (760). Therefore, the reported effects of ryanodine in mouse islets may actually be a consequence of the paracrine action of somatostatin. In human islets, activation of RYR2 by ryanodine stimulates insulin secretion at 3 mM glucose but is without effect at higher glucose concentrations (320). Furthermore, expression of RYR2 is low (<15% of that of ITP3), and no other RYRs are expressed in human β-cells (see Supplemental Figure 9). Collectively, these observations indicate that CICR mediated by RyRs plays a relatively minor role in both mouse and human β-cells.
Recently, it was proposed that Ca2+ release from acidic intracellular stores, via two-pore channels, plays a role in the initiation of electrical activity by activating a depolarizing inward current (22, 104) (see sect. VB2). Both mouse and human β-cells express Tpc1 (Tpcn1) and Tpc2 (Tpcn2), with the former being found at much higher levels. However, GIIS is little affected in Tpcn1/Tpcn2 double knockout mice (618), which suggests two-pore channels may not play an important role in glucose-induced electrical activity, at least in mouse β-cells.
VI. CELL COUPLING
In mouse islets, glucose-induced [Ca2+]i oscillations tend to be synchronized across the entire islet (591), indicative of electrical coupling between β-cells. This has been documented by parallel electrophysiological measurements from two β-cells in the same islet (165, 441). The synchronicity is so strong that [Ca2+]i responses in individual cells are almost superimposable on those seen at the whole-islet level. In contrast, synchronous [Ca2+]i oscillations across the whole islet are not usually observed in human islets (102), although they have been reported for small groups of β-cells (529). This weaker synchronicity may reflect the less organized architecture of the human islet, where β-cells are intermingled with non-β-cells (FIGURE 1B). However, although [Ca2+]i oscillations elicited by high glucose appear to be stochastic in human islets, coordinated oscillations are observed in the presence of the incretin hormones GLP-1 and GIP (284).
The synchronous [Ca2+]i oscillations are driven by electrical coupling between adjacent β-cells. Electrical activity in one β-cell results in the injection of current into neighboring cells, and a membrane depolarization that may be sufficient to trigger their electrical activity. When a β-cell in an intact islet is voltage-clamped at −70 mV, its electrical activity is suppressed but bursting electrical activity in neighboring β-cells can be detected as changes in the holding current that resemble inverted bursts of action potentials (FIGURE 11, A AND B).
FIGURE 11.

A: schematic of electrical coupling between β-cells in an intact islet. The green rectangles indicate gap junctions. Electrically coupled cells are indicated in pink. One β-cell within the islet is voltage-clamped at −70 mV through the recording electrode, so inhibiting electrical activity. Spontaneous electrical activity in neighboring electrically coupled cells (black traces in pink cells) results in an inward current in the voltage-clamped cell that resembles an inverted burst of action potentials. B: membrane potential (top) and membrane current (bottom) recorded from the same β-cell under current- and voltage-clamp conditions, respectively. Assuming that electrical activity recorded in the β-cell connected to the patch electrode before voltage-clamping approximates that of its neighbors, the total gap-junctional conductance (Gj) can be estimated from the equation Gj = ΔI/ΔV, where ΔV (above) and ΔI (below) represent the current and voltage differences between the plateau current/potential and the most repolarized voltage/least negative current. C: an example of bursting electrical activity where low-amplitude action potentials (coming from an adjacent β-cell) precede full-amplitude action potentials (red rectangle). These low-amplitude action potentials probably reflect electrical activity in a neighboring β-cell(s) that ‟leaksˮ into the cell from which the recording is made via gap junctions.
Electrical coupling is mediated by gap junction channels, which are formed from the association of two hemi-channels, one in the membrane of each participating cell. Each hemi-channel is a complex of six subunits (known as connexins), surrounding a central pore. Connexin-36 (Gjd2/GJD2) is the only connexin expressed at significant levels in either mouse or human islets (see Supplemental Figure 10), and in connexin-36 KO mice, synchronized [Ca2+]i oscillations are no longer observed at the whole-islet level (537).
Connexin-36 is distinguished by its low unitary conductance (10–15 pS) (643, 680). The coupling conductance between two β-cells in an islet has been estimated as 180 pS (762), and that between a pair of isolated β-cells as 150–220 pS (17, 508). Comparison of the whole-cell and single-channel junctional conductances suggests that electrical coupling between two β-cells is mediated by 15–20 gap junctions.
The membrane conductance of β-cells in intact islets (measured in perforated patch recordings at 20 mM glucose as outlined in FIGURE 11A) is ~1 nS (221), 10-fold higher than that observed in dispersed single β-cells (631). This difference is primarily thought to reflect electrical coupling between β-cells in the intact islet. In support of this idea, the resting conductance is reduced by >80% (to 0.2 nS) in connexin36 KO islets (639), a value similar to that observed in isolated β-cells (0.1–0.2 nS).
Importantly, the efficiency of electrical coupling depends on the resting conductance of the β-cell: the greater the resting membrane conductance, the weaker the cell-to-cell coupling. This argues that glucose elevation will increase coupling, because KATP closure reduces the plasma membrane conductance. In islets expressing nonfunctional KATP channels in 70% of β-cells, normal KATP channel activity in the remaining 30% of β-cells is sufficient to ensure nearly normal [Ca2+]i oscillations across the entire islet in response to glucose (554). Conversely, increased KATP channel activity, even in a subset of β-cells, will impede the passive spread of the depolarization across the islet by electrically ‟insulatingˮ spiking cells from their neighbors. This would explain why although some β-cells (~40%) in islets from mice expressing a gain-of-function KATP channel mutation displayed glucose-induced [Ca2+]i responses, these were not synchronized across the islet (212).
Electrical coupling between β-cells in mouse islets is sufficient to explain the propagation of [Ca2+]i waves across the islet, which have a velocity of 80 μm/s (762). Synchronization of electrical activity and [Ca2+]i oscillations is chiefly determined by the time it takes to change the membrane potential and initiate an action potential in the adjoining β-cell (~150 ms). Some β-cells appear to act as ‟pacemakersˮ (or hubs) that initiate the [Ca2+]i waves, and there many be several hubs in a single mouse islet (321).
Although it is clear that gap junctions are essential for the synchronicity of the [Ca2+]i oscillations, whether they are functionally important for insulin secretion remains unclear. Little synchronicity is seen in human islets, yet they exhibit excellent GIIS. This echoes observations in connexin36 KO mice, which exhibit an increase in basal [Ca2+]i and in both basal and GIIS. Possibly, the role of cell coupling is not to enhance insulin secretion but rather to ensure that insulin is not oversecreted. Too much insulin, after all, is rapidly fatal.
The above description considers gap junctions as passive mediators of cell coupling but does not address the possible modulation of electrical coupling. Such modulation may occur via changes in expression of Gja2, gating (via PKA, PKC, CaMKII), trafficking, or pharmacological regulation of the connexin-36 channel. This topic has recently been reviewed (177), but its functional significance remains largely unexplored. Another question is whether there is exchange of cytosolic constituents between adjacent β-cells via gap junctions. However, this appears unlikely given the fact there is little dye-coupling (as measured using low-molecular-weight fluorescent probes) in electrically coupled cells (528, 762).
VII. MODULATORS OF β-CELL ELECTRICAL ACTIVITY AND SECRETION
GIIS is modulated by a plethora of agents, which include drugs, circulating nutrients, hormones, neurotransmitters, or paracrine hormones released locally within the islets as well as ions and small molecules released by the β-cell that may exert autocrine effects on insulin release. These may potentiate or inhibit GIIS. With the exception of the amino acid leucine and certain drugs (e.g., sulfonylureas) and NEFA (albeit only weakly), none of these agents is capable of initiating insulin secretion on their own. However, several are capable of triggering electrical activity and insulin secretion in the presence of a substimulatory glucose concentration (3 mM and above). This is because they generate an inward current that is too small to counteract the hyperpolarizing influence of the KATP channel under glucose-free conditions, but which is large enough to do so when KATP channels are largely closed by subthreshold glucose concentrations (FIGURE 4). Many modulators of insulin secretion mediate their main effects by influencing exocytosis rather than electrical activity; however, as exocytosis requires Ca2+ entry, they are again only effective in the presence of depolarizing glucose concentrations.
Hormones and neurotransmitters interact with receptors that are (mainly) located in the plasma membrane, the exception being certain steroid hormone receptors that have intracellular (cytosolic or nuclear) receptors. In terms of their effects on β-cell electrical activity, these receptors can broadly be divided into ionotropic and metabotropic receptors. The ionotropic receptors contain an ion channel, and receptor activation therefore directly modulates ion flux. Metabotropic receptors are mostly G protein-coupled receptors, and receptor activation results in indirect activation of ion channels via G proteins or an intracellular second messenger (such as Ca2+).
In this section, we consider some of the agents that modulate β-cell electrical activity and insulin secretion. This constitutes a large and ever-increasing list. Our discussion makes no claim to be comprehensive or to discuss all papers on a given agent in depth. Instead, we focus on those compounds that are known to alter β-cell electrical activity, or that, in our opinion, are of particular significance. We also refer the reader to the many excellent reviews of the field (6, 189, 248, 559).
A. Nutrients
Following a mixed meal, there is an increase in the circulating levels of amino acids (646) that is likely to contribute to the stimulation of insulin secretion. In contrast, the uptake of dietary fat (mediated by chylomicrons and delivered into the circulation via the lymphatic system) is delayed and takes several hours to reach the circulation (189).
1. Amino acids
Here we consider the effects of those amino acids whose effects on β-cell electrical activity have been studied or can be predicted. With a single exception, amino acids potentiate insulin secretion only in the presence of glucose. This is because although their uptake generates a small inward current it is too small to depolarize the β-cell unless most KATP channels are closed.
a) arginine and lysine.
Arginine and lysine are strong stimulators of β-cell electrical activity and insulin secretion (77, 270, 499), but they are effective only in the presence of glucose. They enter the β-cell via the y+ family of cationic amino acid transporters (636). Mouse and human β-cells both express high levels of cationic amino acid transporters CAT-1 (Slc7a1/SLC7A1) and CAT-2 (Slc7a2/SLC7A2) (see Supplemental Figure 11, A and D).
Electrogenic uptake of positively charged arginine (or lysine) evokes a small depolarizing current (636). Provided KATP channel activity is low (as is the case in the presence of >3 mM glucose), this current is capable of stimulating β-cell electrical activity (278, 499). Because it acts by a rapid biophysical effect that is independent of β-cell metabolism, arginine has been used clinically in vivo to assess insulin secretory capacity (723), in a manner analogous to the use of high extracellular K+ in vitro. The fact that arginine remains an effective secretagogue in T2DM argues that the KATP channels must be largely closed in T2DM β-cells.
b) alanine.
Alanine has been reported to stimulate 45Ca efflux from islets preloaded with the isotope in a Na+-dependent fashion (114). However, although alanine depolarizes the β-cell, the current is so small it is only capable of eliciting electrical activity in the presence of glucose, when the KATP conductance is very low (270) (see FIGURE 4). Alanine uptake may be mediated by either Slc1a4/SLC1A4 (encoding the transporter ASCT1; Supplemental Figure 11, B and D) (329) or Slc38a2/SLC38A2 (encoding the transporter SNAT2; Supplemental Figure 11, C and D) (415). Mouse β-cells also express very high levels of Slc38a4 (SNAT4), another Na+-dependent transporter of neutral amino acids (see Supplemental Figure 11C).
c) glutamate.
Both mouse and human β-cells express several members of the glutamate/neutral amino acid transporter family SLC1 (329). Mouse β-cells express Slc1a5 (encoding the transporter ASCT20) (56), which mediates Na+-dependent uptake of glutamate into the β-cell (see Supplemental Figure 11A). Its operation is therefore potentially electrogenic, but it should be noted that glutamate is negatively charged (pKa ~4) at physiological pH, in which case its uptake would be electroneutral. Glutamate may also be transported by SNAT2 (Slc38A2/SLC38A2), which mediates electrogenic Na+-dependent amino acid uptake and is expressed in both mouse and human β-cells (see Supplemental Figure 11C). Human β-cells also express SLC1A1 (encoding EAAT3) (180), but a recent proteome analysis indicates that mouse islets do not contain significant levels of this transporter (767), in agreement with gene expression data (see Supplemental Figure 11B). The operation of EAAT3 involves the outward transport of one K+ coupled to the inward transport of three Na+ and one protonated glutamate (329).
Taken together, the data suggest that electrogenic uptake of extracellular glutamate gives rise to a depolarizing inward current. It should therefore be considered whether the stimulatory effects of glutamate on electrical activity and insulin secretion may, at least in part, be mediated by electrogenic glutamate uptake rather than activation of ionotropic glutamate receptors (see sect. VIIC5). Conversely, release of metabolically generated glutamate via the reverse operation of EAAT3 would give rise to a hyperpolarizing current and result in reduced action potential firing and impaired insulin secretion. This idea is supported by the observation that pharmacological inhibition of EAATs results in stimulation of GIIS (180) and demonstrates that glutamate release imposes a tonic inhibition on insulin release. Although this mechanism is unlikely to be of any functional significance in mouse β-cells (given the low expression of Slc1a1), it may potentially operate in human β-cells, in which SLC1A1 is expressed (see Supplemental Figure 11B).
d) leucine.
Leucine is the only amino acid capable of serving as an initiator of insulin release and stimulating β-cell electrical activity in the absence of glucose. It is likely to be transported into β-cells via the neutral amino acid transporter Slc7a5, which is highly expressed in both mouse and human β-cells (see Supplemental Figure 11, A and D) and transports amino acids in a Na+-independent (nonelectrogenic) fashion. Leucine stimulates β-cell electrical activity and insulin secretion by serving both as a metabolic fuel and as an allosteric activator of glutamate dehydrogenase (746).
2. Fats
Isolated islets exhibit both stimulatory and inhibitory responses to nonesterified fatty acids (NEFA). Whereas acute exposure to NEFA stimulates insulin secretion in both rodent and human islets (111, 647), exposures for longer than 24 h are inhibitory (488). The functional significance of the acute stimulatory effect of NEFA on insulin secretion is not immediately evident as there is a delay of several hours between the ingestion of a mixed (triglyceride-containing) meal and the appearance of NEFA in circulation (189). Indeed, because of the suppression of lipolysis in response to meal-induced insulin secretion, the plasma NEFA concentration drops to very low levels following a meal (189). Although there is a long delay between ingestion of triglycerides and their appearance in plasma, it is nevertheless possible that NEFA may influence insulin secretion in the short term by an indirect mechanism mediated by stimulation of GLP-1 release from enteroendocrine L-cells (463).
The mechanisms accounting for the acute stimulatory effect of NEFA remain to be elucidated but have been reported to include stimulation of exocytosis and a slight elevation of [Ca2+]i (491, 542). Some of these effects may be secondary to activation of the FFA receptor GPR40 (354) (encoded by FFAR1), which is expressed at high levels in both mouse and human β-cells (see Supplemental Figures 11E and 12A). Activation of FFAR1 elevates [Ca2+]i and stimulates insulin secretion in a glucose-dependent fashion (306). Similarly, the FFAR1 agonist TAK-875 potentiates GIIS (748). The effects on β-cell electrical activity have not been thoroughly investigated, but it has been reported that FFAR1 agonists reduce the amplitude of the delayed outward current (181). This would be expected to broaden the action potential, increase Ca2+ entry, and so stimulate secretion. There is also evidence that FFAR1 might signal via Gαq/Gα11 GTP-binding proteins to reduce KATP channel activity and thereby increase electrical activity (592).
More recently, the GPR40 agonist fasiglifam was shown to activate a small inward current in the presence of glucose and tolbutamide (~1 pA/pF at −70 mV) (743). This was associated with an ~15 mV depolarization and a moderate stimulation of insulin secretion at both 8.3 and 16.7 mM glucose. The stimulatory effect of the fasiglifam could be antagonized by the TRPC3 channel-specific blocker pyrazole-3 (743). However, Trpc3/TRPC3 is expressed at very low levels (if at all) in both mouse and human β-cells (see Supplemental Figure 2B).
Although NEFA stimulate insulin secretion at subthreshold glucose concentrations, and even in the complete absence of glucose, their effect is relatively weak compared with that of glucose. However, this may simply be a consequence of the low NEFA concentrations used. Thus the rate of oxidation of 0.7 mM oleate and palmitate is comparable to that of 3 mM glucose (63).
B. Hormones and Neurotransmitters
In this section we consider the effects of circulating hormones and neurotransmitters released by nerve endings within the islet. We consider in turn the actions of the potentiators and inhibitors of GIIS.
1. Stimulatory agonists
Potentiators of insulin release include (but are not limited to) acetylcholine (ACh), vasoactive intestinal polypeptide (VIP), glucagon-like peptide 1 (GLP-1), gastric inhibitory peptide (GIP), opioids, pituitary adenylate cyclase-activating peptide (PACAP), and bombesin (303).
While there is a variety of ways in which stimulators of release may exert their effects, two main pathways dominate G protein receptor (GPCR)-mediated responses (67a). The first involves G protein-mediated stimulation of adenylyl cyclase and elevation of cAMP. This leads to activation of PKA and EPAC2 (exchange protein directly activated by cAMP). GLP-1, VIP, and PACAP all act by increasing cAMP. The second mechanism is via activation of the phosphoinositide pathway. This involves G protein-mediated activation of phospholipase C (PLC), which cleaves phosphoinositol bisphosphate to generate the signaling molecules inositol trisphoshate (InsP3) and diacylglycerol (DAG). The former binds to InsP3 receptors in the sER to liberate intracellular Ca2+. DAG primarily acts via activation of PKC. Acetylcholine, cholecystokinin and bombesin are examples of that agents that stimulate insulin secretion by activating the PLC-PKC pathway.
a) acetylcholine.
ACh stimulates β-cell electrical activity and insulin secretion in a glucose-dependent fashion. In mouse islets it is released from cholinergic nerve endings, which derive from branches of the vagus nerve. In human islets, it has also been proposed that ACh is released from α-cells (459, 558). Paradoxically, this means ACh should be released under conditions associated with inhibition of glucagon secretion. It will be interesting to determine precisely how the human α-cell can differentially regulate the release of glucagon and ACh.
ACh acts on muscarinic receptors in the β-cell membrane. Both mouse and human islets express M3 receptors (Chrm3/CHRM3) and lower levels of CHRM4 (see Supplemental Figure 12, A and B) that signal via Gq and Gi proteins, respectively. In addition, human (but not mouse) β-cells express the ionotropic nicotinic ACh receptor α-subunits CHRNA3 and CHRNA5 (see Supplemental Figure 12A) and high levels of the β-subunit CHRNB2 (see Supplemental Figure 12C). There is a positive correlation between the expression of CHRNB2 expression and GIIS (199). It has been proposed that the expression of nicotinic receptors in β-cells may contribute to the increased risk of T2DM associated with smoking, for example, via nicotine-induced receptor desensitization and thus reduced responsiveness to endogenous ACh release (199).
When ACh is applied at a substimulatory glucose concentration (3 mM), membrane depolarization is limited to <5 mV. In the presence of 10 mM glucose, however, 1 µM ACh depolarizes mouse β-cells in a Na+-dependent fashion by ~10 mV (267). This increases action potential firing and ‟locksˮ the membrane at the ‟plateauˮ potential, from which action potentials are generated (277, 590). These effects of ACh are mimicked by carbachol and oxotremorine-m (oxo-m), consistent with the idea they are mediated via muscarinic receptors (81). Similarly, in human β-cells exposed to 6 mM glucose, carbachol evoked membrane depolarization (~5 mV) and increased action potential firing (568).
In mouse β-cells, ACh activates a small Na+ current in a concentration-dependent manner (EC50 2.5 μM). At a holding potential of −70 mV, the amplitude of the current elicited by ACh concentrations >10 μM is limited to 0.75 pA/pF (561). The relative smallness of the current explains why ACh has no effect in the absence of glucose; the current is simply too small to overcome the hyperpolarizing action of the KATP current. However, such a small current may still have a significant effect on the membrane potential in the presence of glucose because KATP channel closure increases the input resistance of the cell to >1 GΩ (see FIGURE 4).
The ACh-activated current is mediated by a nonselective Na+ leak channel (Nalcn/NALCN) (659), which is expressed at low but detectable levels in both mouse and human β-cells (3, 68, 146, 475), where it forms a protein complex with the M3 receptor (659). In neurons, Nalcn provides the background leak conductance that keeps the cell sufficiently depolarized to allow spontaneous electrical activity (209). It is unlikely that it fulfils this role in β-cells, however, as the resting conductance is not affected by silencing of Nalcn (561). Furthermore, omission of extracellular Na+ does not interfere with the capacity of glucose to evoke electrical activity (194, 618).
Although the initial response to low concentrations of ACh (1 μM) is an increase in membrane conductance (in keeping with the activation of Nalcn), this is later superseded by a decrease in conductance that is associated with membrane depolarization (590). The reduced conductance may reflect a PKC-dependent inhibition of KATP channels that is secondary to activation of PLC and generation of DAG, similar to the response to GLP-1 (618) (see sect. VIIB1b). In human β-cells it is possible (see sect. VB3b) that inhibition of the M-current (Kcnq2/KCNQ2) also contributes.
The effects of high concentrations (100 μM) of ACh and other muscarinic agonists are biphasic. In addition to the initial depolarization, ACh produces a subsequent transient repolarization that correlates with elevation of [Ca2+]i. This presumably reflects activation of Ca2+-activated K+ channels (SK and BK). In support of this idea, the transient repolarization is reduced by the BK blocker charybdotoxin (81). Repolarization is only transient and is followed by a sustained membrane depolarization, which is postulated to result from activation of a store-operated conductance (SOC) due to InsP3-induced depletion of intracellular Ca2+ stores (64). The store-operated conductance involves a stromal interaction molecule (STIM) that extends into the lumen of the sER and senses the intraluminal Ca2+ concentration. Following InsP3-induced depletion of sER Ca2+, STIM interacts with, and activates, the plasmalemmal store-operated Ca2+ channel (Orai) (763). In both mouse and human β-cells Orai3/ORAI3 predominates, but Orai1/ORAI1 and Orai2/ORAI2 are also expressed. Expression of Stim1/STIM1 and Stim2/STIM2 is equal and comparable in both species (3, 68, 146, 475).
Store-operated Ca2+ entry has been studied by Mn2+ quenching of fura 2 fluorescence (399). Under basal conditions, Mn2+ entry is low, but it is accelerated by high concentrations (100 μM) of carbachol. However, it is debatable whether SOC activation contributes to the stimulatory effects of ACh on β-cell electrical activity as low concentrations of ACh are associated with little mobilization of intracellular Ca2+ (207), yet they still result in a sustained depolarization.
In addition to the effects of ACh described above, which are mediated by M3 receptors and activation of Gq, ACh also inhibits L-type Ca2+ channels (211). This effect may be mediated by activation of M4 receptors, which are coupled to Gi.
Finally, ACh also potentiates depolarization-evoked exocytosis by a CaM kinase II-dependent stimulation of granule mobilization (241) (see sect. IXD7).
b) glp-1, gip, and glucagon.
The hormones glucagon, GLP-1, and GIP stimulate β-cell electrical activity and enhance exocytosis. They do so by interacting with Gs protein-coupled receptors that are coupled to elevation of intracellular cAMP. Other agents that increase intracellular cAMP (like the adenylate cyclase activator forskolin, the phosphodiesterase inhibitor IBMX, and membrane-permeable forms of cAMP such as dibutyryl cAMP) also strongly stimulate insulin secretion (609).
I) GLP-1. The gut hormone GLP-1 is a potent stimulator of GIIS and is of importance in T2DM therapy (286). It is secreted by the enteroendocrine L-cells of the distal small intestine and colon (286) and reaches the pancreas via the circulation. There is some evidence that in human islets GLP-1 is also released from α-cells, possibly due to alternative splicing of the pro-glucagon gene (425). The GLP-1 content of human α-cells appears surprisingly high, ~10% of the glucagon content. In contrast, mouse α-cells contain just 1.5 fg GLP-1 per cell, which is only ~0.05% of the glucagon content (550).
GLP-1 acts by activation of the GLP-1 receptor (Glp1r/GLP1R), which is highly expressed in both mouse and human β-cells (see Supplemental Figure 12A). Although the GLP-1 receptor is regarded as an archetypal Gs-coupled receptor (leading to elevation of cAMP), there is also evidence that at low physiological concentrations it preferentially activates Gq, leading to activation of PKC (via activation of PLC and generation of DAG) (618).
GLP-1 is ineffective at nonstimulatory glucose concentrations, but in the presence of a near-threshold glucose concentration (6–7.5 mM), it elicits intracellular Ca2+ oscillations and insulin secretion (381, 618). In intact mouse islets at glucose concentrations that are themselves stimulatory, the effects of GLP-1 on electrical activity are fairly moderate and limited to an increase in the percentage of time the β-cell spends firing action potentials (238). In isolated β-cells, application of GLP-1 at substimulatory glucose concentrations (5–6 mM) initiates action potential firing in previously inactive cells (618).
The depolarizing effect of GLP-1 on electrical activity is, in part, mediated by closure of the KATP channel (238, 287). The mechanism has not been fully elucidated, but there is evidence that cAMP itself, via the cAMP-sensing protein Epac2 (cAMP-GEFII), inhibits KATP channel activity (331). Downregulation of Epac2/cAMP-GEFII does not affect GIIS, but it reduces the stimulatory effect of both GLP-1 and GIP by 50% (339). It is noteworthy that GLP-1 stimulates β-cell electrical activity at physiological concentrations (1–10 pM) without producing any detectable increase in cAMP. Thus >1 nM GLP-1 is needed to elevate cAMP (EC50 6 nM; Ref. 618), which is ~100-fold higher than the level of the hormone measured in the plasma (286). That argues that the effect of low concentrations of GLP-1 involves a cAMP-independent mechanism. In addition to closing KATP channels, GLP-1 increases a background inward current that is carried by Trpm2, Trpm4, or Trpm5 channels (381, 618, 754). Knockout of Trmp4 prevents GLP-1-mediated depolarization and stimulation of insulin secretion (618), indicating that KATP channel closure alone is not sufficient to mediate the stimulatory effect of GLP-1.
When tested at physiological (pM) concentrations, GLP-1 produces a small PKC-dependent increase (15%) in the perforated patch whole-cell voltage-gated L-type Ca2+ current (618). However, this increase is too small to explain its stimulatory effect on electrical activity and insulin secretion.
In addition to the effects on electrical activity, GLP-1 also exerts a direct stimulatory effect on exocytosis in both mouse and human islets (237, 238). This effect is considered in greater detail in section IXD7.
II) Glucagon. Glucagon, released locally by the α-cells within the islet, stimulates insulin secretion by a cAMP-dependent mechanism. Glucagon receptors (Gcgr/CCGR) are expressed at comparable levels in mouse and human β-cells, but at lower/much lower levels than those found for the GLP-1 receptor (see Supplemental Figure 12A). Glucagon inhibits KATP channel activity and thereby stimulates β-cell electrical activity (256). Like GLP-1, the effects of glucagon on β-cell electrical activity are subtle and manifest as a slight increase in burst duration at the expense of the silent intervals (301). Glucagon also stimulates insulin secretion by a direct effect on exocytosis (238). In the clinic, a glucagon test is often used to test for β-cell function (598).
III) GIP. GIP stimulates insulin secretion via activation of the GIP receptor (GIPR, encoded by Gipr/GIPR), which is expressed at high levels in both mouse and human β-cells (see Supplemental Figure 12A). It is released by the enteroendocrine K-cells of the proximal small intestine (658) and probably accounts for most of the incretin effect in vivo (286). The stimulatory effect of GIP on insulin secretion is diminished in T2DM (471), possibly due to a hyperglycemia-induced reduction of GIPR expression (740). Consistent with a key role for GIP in the incretin response, Gipr knockout mice show impaired GIIS and glucose tolerance during an oral glucose tolerance test (456). GIP is also a strong stimulator of GIIS in isolated islets (469).
Despite its physiological significance, there are surprisingly few studies exploring the mechanism of action of GIP on β-cell function. However, the available evidence suggests that GIP affects β-cell electrical activity in a similar way to GLP-1. In mouse pancreatic islets exposed to 11 mM glucose, GIP increases the fractional active phase (715). In human β-cells exposed to 5 mM glucose, GIP also initiates electrical activity via a reduction in the resting membrane K+ conductance, probably reflecting closure of KATP channels (237). In both mouse and human β-cells, GIP also stimulates exocytosis (147, 237).
c) estrogens.
Both 17β-estradiol and xenoestrogens stimulate insulin secretion in pancreatic β-cells. They do so by interacting with a plasmalemmal receptor (466), distinct from the classical intracellular receptor, possibly GPR30 (Gper1) (431). Binding of 17β-estradiol at physiological concentrations to this receptor increases cGMP and activates protein kinase G: this phosphorylates the KATP channel, which causes its closure (467, 562). As a consequence the β-cell membrane depolarizes, stimulating voltage-gated Ca2+ entry and insulin secretion.
2. Inhibitory agonists
Every minute of our lives, the body requires an adequate supply of insulin to function properly. Too much insulin causes hypoglycemia, and it is therefore important that there are mechanisms to quickly and effectively shut off insulin secretion. The paracrine hormone somatostatin, the neurotransmitter epinephrine, and the peptide galanin inhibit GIIS (481). In all cases their inhibitory effects are mediated via GPCRs and are prevented by pretreatment with pertussis toxin, suggesting the involvement of an inhibitory G protein (Gi). In mouse islets, epinephrine, galanin and somatostatin inhibit glucose-induced electrical activity, with epinephrine producing the strongest, and somatostatin the weakest, effect (152, 153, 543, 567). Inhibition of electrical activity is mediated by activation of low-conductance K+ channels distinct from KATP channels (566). In addition, depolarization-evoked exocytosis is inhibited (327), but whereas the effect on electrical activity is transient, that on exocytosis is maintained as long as the agent is present. As all three agents are believed to act in the same way, we focus our discussion on somatostatin.
a) somatostatin.
Somatostatin is a powerful inhibitor of insulin secretion. It has both paracrine and systemic effects, being released locally (as somatostatin-14) within the islet by δ-cells and also reaching the islet via the circulation from the gut (as somatostatin-28) (174). At the receptor level, human β-cells express SSTR1, SSTR2, SSTR3, and SSTR5 mRNAs (see Supplemental Figure 12, A and E). In mouse islets, Sstr3 accounts for almost all (>95%) somatostatin receptor mRNAs, and other subtypes are expressed at very low levels (see Supplemental Figure 12, A and E). In contrast, secretion experiments employing ‟selectiveˮ receptor antagonists and agonists have led to the suggestion that somatostatin inhibits insulin secretion by activation of Sstr5 (87). This discrepancy is unresolved. It may reflect differences in mouse strains or illustrate the fact that antagonists are not as selective as claimed. Human β-cells express particularly high levels of SSTR2 but SSTR1, 3, and 5 are also present (see Supplemental Figure 12E).
In human β-cells, application of somatostatin (10–30 nM) hyperpolarizes the β-cell by ~10 mV by activation of SSTR2 (327). The effect persists in the presence of a KATP channel blocker, indicating the involvement of another type of K+ channel, most likely a G protein-coupled KIR channel (GIRK channel) (327). Indeed, the repolarizing effect of somatostatin was prevented by tertiapin Q, an antagonist of GIRK channels. Of the GIRK channels, GIRK2 (Kcnj6/KCNJ6) is expressed at particularly high levels in both mouse and human β-cells (see Supplemental Figure 7A). It is possible that somatostatin, in addition to activating a repolarizing current, also inhibits an inward depolarizing current, as shown for epinephrine and ghrelin (359).
In addition to its effect on electrical activity, somatostatin also suppresses insulin secretion by a distal effect exerted at the level of exocytosis. This effect is quantitatively the more important (327) (see sect. IXD7).
In human (but not mouse) β-cells, somatostatin reduces the voltage-gated Ca2+ current (327). However, this small (<20%) effect contributes little to suppression of exocytosis, and when the duration of the depolarization is increased to compensate for the reduction of Ca2+ entry, exocytosis remains strongly (>80%) inhibited.
b) epinephrine.
Adrenoreceptors (ADRs) are expressed at levels comparable to the SSTRs in mouse β-cells. In human β-cells, expression of ADRs is ~30% of the SSTRs (see Supplemental Figure 12A). α2a ADRs (Adra2a) are the only adrenergic receptors expressed in mouse β-cells (see Supplemental Figure 12D). In human β-cells, α1b (ADRA1B) and β2 (ADRB2) receptors are also expressed, but at much lower levels than ADRA2A (see Supplemental Figure 12D). This may explain why epinephrine exerts both stimulatory (β2) and inhibitory (α2) effects in human β-cells (361).
Epinephrine inhibits electrical activity and exocytosis in mouse and human β-cells, by a mechanism resembling that of somatostatin (327). In the presence of high extracellular K+ (140 mM), epinephrine activated a tolbutamide-resistant but tertiapin Q-sensitive current with an amplitude of 100 pA at −90 mV. This current exhibited strong inward rectification (308), consistent with it being mediated by GIRK channels. When recorded with normal ionic gradients, the α2-adrenergic agonist clonidine activated a K+ current with a whole-cell conductance of 0.15 nS (566). Recently, it was also reported that low concentrations of epinephrine (1 nM) impair insulin secretion and β-cell electrical activity by inhibiting cAMP-activated Trpm2 channels (which may contribute to the background inward current) (305). In contrast to these studies, which implicate ion channels in the response to epinephrine, others have reported that epinephrine inhibits insulin secretion (evoked by arginine) without affecting action potential firing (140). This likely reflects the inhibitory effect of epinephrine on exocytosis (see sect. IXD7).
A common polymorphism located in the 3′ untranslated region of ADRA2A (rs553668), which is found in 30% of the population, is associated with an increased risk of T2DM (577). In vitro human islet data suggest a causal relationship between rs553668, ADRA2A overexpression, and defective insulin secretion, which is mediated by inhibition of β-cell electrical activity and exocytosis. In T2DM patients who carry the high-risk ADRA2A variant, treatment with the α2 adrenoreceptor antagonist yohimbine improved insulin secretion (672).
c) galanin.
In mouse β-cells, galanin receptors (GALRs) are expressed at levels as high as the ADRs and SSTRs (see Supplemental Figure 12A). In contrast, galanin receptors are absent in human β-cells. Two types of galanin receptors are expressed in mouse β-cells: Galr1 and Galr3 (see Supplemental Figure 12E). Galanin inhibits glucose-induced electrical activity in mouse β-cells (543). The effects of galanin on electrical activity in human β-cells have not been investigated, but given the low expression of galanin receptors in human β-cells, its effects (if any) are likely to be weak.
d) ghrelin.
In addition to being produced by the stomach, ghrelin is also produced in the pancreatic islets, especially in fetal and neonatal islets (729). Ghrelin exerts an insulinostatic effect (728). Its effects are mediated by activation of Gi and culminate in inhibition of Trpm2 channels (359), the same ‟backgroundˮ channels that are activated by glucose. This leads to suppression of glucose-induced electrical activity. When considering the functional role of ghrelin signaling, it should be borne in mind that adult mouse and human β-cells express the ghrelin receptor (Ghsr/GHSR) at very low levels (see Supplemental Figure 12, A and E). In contrast, δ-cells express very high levels of Ghsr/GHSR (3, 146). It is therefore likely that the systemic effects of ghrelin are mediated by stimulation of somatostatin secretion, which then inhibits insulin secretion by a paracrine effect. As expected if this were the case, the inhibitory effect of ghrelin on insulin secretion was abolished in the presence of SSTR2/3 antagonists (3).
e) leptin.
Adipose tissue has an important endocrine role, in addition to its role in energy homeostasis and storage (345). Two key adipocyte-derived hormones have reciprocal effects on pancreatic β-cells: leptin and adiponectin. To date, only the effects of leptin on β-cell electrical activity have been studied.
Leptin is secreted from white adipocytes (648) and acts on leptin receptors. Its effects on β-cell function have been reviewed elsewhere (379). Leptin-deficient (ob/ob) mice are obese and hyperinsulinemic and showed increased electrical activity (relative to that of wild-type C57Bl6 mice; Ref. 574). This effect was attributed to reduced K+ channel activity. It is therefore of interest that at 11 mM glucose, leptin promotes AMPK-dependent KATP channel trafficking to the plasma membrane, increases the KATP conductance, and lowers intracellular Ca2+ (35, 501). Treatment of ob/ob mice with leptin reverses hyperinsulinemia (612). Leptin receptors (Lepr/LEPR) are expressed at low levels in both mouse and human β-cells (see Supplemental Figure 12, A and E). Despite the low expression, mice lacking leptin receptors (ObR, db/db mice) are hyperexcitable (443), have reduced K+ permeability (measured as 86Rb flux) (59), and progress to diabetes in an age-dependent fashion (58).
3. Summary
Common to the effects of many (if not all) hormones and neurotransmitters is that they influence insulin secretion by modulating electrical activity, exocytosis, or more usually, a combination of both. With regard to the effects on electrical activity, potentiators act by reducing the repolarizing background KATP conductance and activating an additional depolarizing current (Trpm2 or Trpm4). Conversely, inhibitors may act by increasing the resting K+ channel activity (usually GIRK) and inhibiting the depolarizing inward current (possibly Trpm2).
C. Autocrine Regulators
In addition to insulin, a large number of other substances are present in the β-cell secretory granules and are co-released with insulin in response to glucose. These include islet amyloid polypeptide (IAPP or amylin), ATP, zinc, and the neurotransmitters GABA and glutamate. There is therefore the potential for these agents to act back on the β-cell to enhance or inhibit insulin secretion in an autocrine fashion. We next consider these substances, the mechanism of their release, their receptors on the β-cell, and their effects on β-cell electrical activity and insulin secretion.
1. Insulin
Given that an insulin granule contains 1.6 amol insulin (294) and that the interstitial space of the medium-sized islet used experimentally is ~0.2 nl (10% of 2 nl; Refs. 33, 138), exocytosis of a single insulin granule is sufficient to increase the interstitial insulin concentration to 8 nM. This is an enormous concentration and circulating levels are 100–1,000 times lower (340).
The β-cell expresses two isoforms of the insulin receptor (A and B), and autocrine insulin signaling may influence β-cell function both acutely and in the longer term (383). There is evidence that the different isoforms produce slightly different functional effects (382). The long-term effects of insulin may be mediated by changes in gene transcription, cell proliferation, and apoptosis (383). The acute effects of insulin on stimulus-secretion coupling are less clear. Experiments on β-cell-specific insulin receptor knockout mice (BIRKO mice) suggest that insulin signaling is necessary to maintain β-cell function (357; but see Ref. 626). Moreover, pancreas-specific deletion of insulin receptor substrate 2 (IRS2) leads to impaired glucose tolerance and reduced GIIS (105).
Studies in isolated rodent islets or β-cells have reported inhibitory, stimulatory, no, or mixed responses to exogenous insulin or C-peptide (reviewed in Ref. 92). Acute application of insulin to mouse β-cells was found to produce membrane hyperpolarization and suppression of glucose-induced electrical activity, due to activation of KATP channels (346). The effect of insulin on the membrane potential was dose-dependent and half-maximal at ~25 nM. It was proposed that this inhibitory effect of insulin enables the β-cell to match its energy requirements to its energy production. It is therefore of interest that BIRKO mice are glucose-intolerant and exhibit impaired GIIS (characterized by the loss of 1st phase insulin secretion) but retain normal arginine-induced insulin secretion (357). This phenotype recapitulates the insulin secretion defects seen in T2DM.
The effects of insulin in human islets are less well characterized. In isolated human islets, insulin stimulates C-peptide release at both 1 and 6 mM glucose (92). A similar small but transient stimulation of C-peptide release in response to exogenous insulin (0.2 and 200 nM) was also observed in isolated human islets perifused with 3 mM glucose (404).
In addition, exocytosis of the insulin granule may exert a stimulatory effect on β-cell function and insulin secretion via incorporation of vesicle proteins (e.g., phogrin) in the plasma membrane (465, 688, 689).
Because insulin is stored as a Zn2-insulin6 complex, GIIS will be associated with Zn2+ release (394). Extracellular Zn2+ at concentrations >50 μM inhibits glucose-induced electrical activity and insulin secretion (185), probably by blocking voltage-gated L-type Ca2+ channels (249). In addition, Zn2+ produces a small increase in KATP channel activity, which also been postulated to contribute to autocrine inhibition in mouse islets (628). As discussed in section VC1bII), T-type Ca2+ channels in human β-cells also are blocked by micromolar concentrations of Zn2+ (687). Given the high intragranular Zn2+ concentration, insulin secretion might be predicted to result in inhibition of its own release, due to T-type Ca2+ channel inhibition and suppression of electrical activity. However, a reduction in temperature from +37°C to +27°C, which leads to almost complete suppression of insulin exocytosis (544), has minimal effects on electrical activity (39). This suggests that locally released Zn2+ (and insulin) does not function as an inhibitory autocrine signal.
Zn2+ uptake into the insulin granules is mediated by the ZnT8 (Slc30a8/SLC30A8) transporter (117). A common polymorphism in SLC30A8 (rs13266634) affects T2DM risk (384, 627). This variant leads to a tryptophan-to-arginine switch at position 325 in the protein's intracellular carboxy-terminal domain that is thought to reduce Zn2+ transport into the secretory granules. Although it was originally predicted that this variant increases T2DM risk, data from patients with truncating gene variants suggest that reduced activity of ZnT8 is actually protective (187). In mice lacking ZnT8, the insulin granule core is less condensed, but this is not associated with any gross changes in insulin secretion, either at the level of the isolated islet or the animal (384). It has been argued that the diabetogenic effect of the variant is mediated by regulation of hepatic insulin clearance (increased in the presence of Zn2+) (668).7
2. ATP and ADP
Secretory granules in many cell types contain both ATP and ADP. In β-cells, ATP and ADP co-released with insulin may be expected to result in autocrine activation of β-cell purinergic receptors. Indeed, GIIS is reduced in both human and mouse β-cells when purinergic receptor activation is prevented (202, 312, 347, 737).
Nucleotide uptake into insulin secretory vesicles is mediated by the vesicular nucleotide transporter (vNUT, encoded by Slc17a9/SLC17A9) (596), which is expressed in mouse β-cells (3, 146). The intragranular ATP concentration has been estimated to be 3 mM, comparable to that of the cytosol (195). Thus ATP is not actively accumulated in secretory granules. Although SLC17A9 expression in human β-cells is almost undetectable (68, 475), stimulation of insulin secretion is still associated with ATP release (347). This suggests that either the low expression of vNUT in human β-cells is sufficient to maintain intragranular ATP levels, or alternative uptake mechanisms exist. Nevertheless, it is clear that ATP will be released into the interstitium together with insulin and may thus subserve auto- and paracrine functions. In addition, ATP may be released from intra-islet nerve endings, but the functional effects of this have not been studied.
ATP acts on purinergic receptors, which may be ionotropic (i.e., P2X) or metabotropic (i.e., P2Y). These vary in their structure, relative sensitivity to adenine nucleotides, and the mechanism by which they modulate electrical activity (direct for P2X, indirect for P2Y). Mouse β-cells express P2X4 (P2rx4), P2Y1 (P2ry1), and P2Y6 (P2ry6) (see Supplemental Figure 13A). Extracellular ATP closes KATP channels, stimulates electrical activity, and mobilizes Ca2+ from intracellular stores (516). However, it also inhibits exocytosis via P2Y1 receptors (516). As the latter effect dominates, the result is an inhibition of insulin secretion. The stimulation of electrical activity is associated with the activation of an inward cationic current with an amplitude of 20 pA/pF, measured ~1 s after application of ATP or α,β-methyleneadenosine-5-triphosphate (623), which is broadly consistent with the reported agonist sensitivity of P2X4 receptors (482, 623).
In human β-cells, both P2X and P2Y receptor agonists stimulate GIIS (184). Human β-cells express P2X2, P2X4, P2RY1, and P2RY11 at high/fairly high levels (see Supplemental Figure 13A). It has been reported that ATP acts principally by activating ionotropic P2X3 receptors (312), but this conclusion is difficult to reconcile with the low P2RX3 expression in human β-cells (68) and the observation that the effects of ATP are mimicked by ADP (which is a poor P2X3 agonist) and by the P2Y1 agonist MRS-2365. Furthermore, the stimulatory effects of ATP were abolished by the P2Y1 antagonist MRS-2279 (347). These data instead suggest that the effects of ATP are mediated by P2Y1 receptors (P2RY1). Human β-cells also express P2RX4 (encoding P2X4) at greater levels than P2RY1, but the fact that ADP mimics the effect of ATP again militates against major involvement of such channels in the stimulation of electrical activity in human β-cells.
Stimulation of human β-cells by ATP is associated with activation of a small Na+-dependent inward current with an amplitude of 0.2 pA/pF (184). This current need not necessarily reflect activation of an ionotropic purinergic receptor, as activation of P2Y1 receptors leads to activation of PLC, which may culminate in opening of Trpm4 or 5 channels by a mechanism similar to that proposed for GLP-1 (618) (sect. VIIB2bI).
3. GABA
Pancreatic β-cells express the enzyme glutamic acid decarboxylase, which catalyzes the decarboxylation of glutamate to GABA. Mouse islets express Gad1 (GAD65), whereas GAD2 (GAD67) is found in human β-cells (348). GAD65 and GAD67 are major auto-antigens in type 1 diabetes (343, 599).
GABA is an important inhibitory neurotransmitter in the nervous system. Its presence at high levels in pancreatic β-cells (approaching those of the CNS; Ref. 673) has led to the proposal that GABA may function as an autocrine or paracrine signal within the islet (566).
There are two types of GABA receptor on β-cells: ionotropic GABAA and metabotropic GABAB receptors. GABAA receptors are pentameric complexes of two α-subunits and two β-subunits and a fifth (variable) subunit. Human β-cells express low but detectable levels of the GABAA receptor α-subunits (see Supplemental Figure 13B; almost exclusively GABRA2). Expression of the β-subunits is much higher (see Supplemental Figure 13B) and is entirely accounted for by GABRB3 (68, 475). Activation of GABAA receptors in human β-cells leads to a transient increase in Cl− permeability. The effect is transient because GABAA receptors rapidly desensitize. Because pancreatic β-cells have a high intracellular Cl− concentration (89), an increased Cl− conductance will depolarize the β-cells towards the Cl− equilibrium potential (ECl), which is around −35 mV (89). Indeed, GABA-induced action potential firing has been observed in human β-cells and application of GABA stimulates insulin secretion, effects that can be antagonized by the GABAA receptor antagonist SR95531. Expression of GABAA receptor α-subunits in mouse β-cells is very low (see Supplemental Figure 13B), and application of GABA does not affect GIIS (206). Despite the absence of GABAA receptor subunits, expression of the β-subunits (almost entirely Gabrb3) is as high as in human β-cells (see Supplemental Figure 13B).
GABAB receptors are metabotropic G protein-coupled receptors. Human β-cells only express GABAB receptor 1 (Gabbr1/GABBR1) (see Supplemental Figure 13B). It is also present in mouse β-cells but the GABAB receptor 2 (Gabbr2/GABBR2) is expressed at 10-fold higher levels (see Supplemental Figure 13B). Activation of GABAB receptors (using the specific agonist baclofen) leads to inhibition of insulin secretion in mice (78; but see Ref. 206) via a pertussis toxin-sensitive mechanism (94). GABAB antagonists such as CGP55485 stimulate insulin secretion in human islets (670). Thus, in human islets, GABA appears to exert both stimulatory (GABAA) and inhibitory (GABAB) effects, and which effect prevails will depend on the concentration of GABA and the duration of exposure.
The mechanisms by which GABA is released from the β-cells have only partially been elucidated. However, it is now clear that in both rodent and human β-cells, GABA is released both by a vesicular route (via exocytosis of the insulin-containing granules) and a nonvesicular route (89, 95). The mechanism of nonvesicular release is unknown but may involve bestrophin-1 Cl− channels (as shown in glia cells; Ref. 378). BEST1 is highly expressed in human β-cells, whereas mouse β-cells express Best3 (see Supplemental Figure 2A). The nonvesicular mode of release is remarkably effective, as the rate of release of GABA into the culture medium suggests that the GABA stores in β-cells turn over 30 times during 24 h (629). In human islets, spontaneous activity of endogenous GABAA receptor Cl− channels is observed (670), possibly reflecting the presence of an ambient GABA concentration sufficient to result in receptor activation. In the CNS, GABA is removed from the synaptic cleft by the GABA transporters GAT1–3 (encoded by SLC6A1, SLC6A13, and SLC6A11, respectively). However, these are only expressed at low levels in mouse and human β-cells alike (3, 68, 146, 475). Expression in α- and δ-cells is also low.
Fascinatingly, GABA induces conversion of mouse α-cells to β-cells in vivo, and treatment of transplanted human islets with GABA results (via activation of GABAA receptors in α-cells) in an increased percentage of β-cells and a reduced percentage of α-cells, suggesting a similar conversion may occur in humans (55). However, it should be noted that the expression of functional GABAA receptors is very low in human α-cells (89), an observation confirmed at the mRNA level (68, 475).
4. Glutamate
Glutamate acts as an excitatory neurotransmitter in the pancreas and stimulates electrical activity and insulin secretion in rat islets (220). It is stored together with glucagon in the secretory granules of α-cells and co-released with glucagon (103, 462). Similarly, Vglut2 (SLC17A6) is expressed in human β-cells (68), and glutamate may therefore be co-released with insulin from β-cells (205). As discussed in section VIIA1c, a slow release of glutamate from human β-cells can also occur by reverse operation of the plasmalemmal glutamate transporter EAAT3 (Slc1a1/SLC1A1).
There are multiple types of (excitatory) ionotropic glutamate receptors (693). Traditionally, these are classified according to their agonist specificity as NMDA (encoded by the Grin/GRIN genes), kainate (Grik/GRIK), and AMPA receptors (Gria/GRIA). All receptors are expressed in both mouse and human β-cells, but the levels are higher for human β-cells (see Supplemental Figure 14A). Three different classes of NMDA receptor subunits exist: GluN1 (Grin1/GRIN1 gene), GluN2 (Grin2/GRIN2), and GluN3 (Grin3/GRIN3). GluNs assemble as tetramers, with the GluN1 subunit being obligatory for the formation of a functional NMDAR. Grin1 is expressed in mouse β-cells (see Supplemental Figure 14B), and NMDA-activated currents have been recorded in mouse insulinoma cells (MIN6) (220). Surprisingly, application of the NMDAR antagonist MK 801 or ablation of Grin1 leads to improved glucose tolerance in vivo and amplifies glucose-induced [Ca2+]i oscillations and electrical activity in β-cells in vitro (428). The underlying mechanisms remain undetermined. GRIN1 is expressed at very low levels in human β-cells (see Supplemental Figure 14B), making it uncertain whether functional NMDA receptors are present at all in human β-cells. Nevertheless, a small clinical study suggested insulin secretion and glucose tolerance were improved in T2DM individuals treated with an NMDAR antagonist (428).
AMPA receptors (encoded by Gria2 and Gria3) are expressed in mouse β-cells. Large AMPA- and glutamate-activated CNQX-sensitive inward currents have also been recorded in mouse β-cells within pancreatic slices (736; but see Ref. 118). In human β-cells, GRIA2, GRIA3, and GRIA4 (but not GRIA1) are expressed and (in absolute terms) at levels ~10-fold higher than those found in mouse β-cells (see Supplemental Figure 14A). Kainate receptors are expressed at comparable levels in human (GRIK2 and 5) and mouse (Grik5) β-cells (see Supplemental Figure 14, A and B).
In addition to these ionotropic receptors, rat β-cells have also been reported to express the metabotropic receptors mGluR4, mGluR5, and mGluR8 (99). Based on RNAseq data, expression of metabotropic receptors (GRMs) in mouse β-cells is very low (see Supplemental Figure 14C). However, human β-cells express high levels of mGluR4 (GRM4), which account for ~80% of the GRM mRNA transcripts (see Supplemental Figure 14D).
5. Glycine
Glycine is stored within the secretory granules and coreleased with insulin from β-cells, providing a mechanism for autocrine stimulation of insulin secretion similar to that observed for GABA (744). Both human and mouse β-cells also express Slc6a17/SLC6a17 (3, 68, 146, 475) that encodes a synaptic vesicle glycine transporter (502).
Ionotropic glycine receptors comprise five subunits [four pore-forming α-subunits and one β-subunit (406)] that form a glycine-activated Cl− selective channel. Human β-cells express high levels of the glycine receptor α1-subunits (GLRA1) and β-subunits (GLRB) (see Supplemental Figure 13, B and D). GLRA3 is also expressed in human β-cells but at much lower levels than GLRA1 (see Supplemental Figure 13D). Expression of glycine receptor α-subunits in mouse β-cells is very low, in agreement with the small amplitude of glycine-activated Cl− currents in mouse β-cells (744). Nevertheless, expression of the β-subunit (Glrb) is (if anything) higher than in human β-cells. The functional significance of this observation remains unclear.
In human β-cells, activation of glycine receptors gives rise to sustained inward Cl− currents with an amplitude of 20–25 pA/pF (744). These become detectable at ~30 μM, are half-maximal at 100 μM, and are maximal between 0.3 and 1 mM glycine. This dose-response curve suggests that circulating glycine levels (0.1–0.5 mM) should produce significant activation of GlyRs. Activation of GlyR Cl− channels results in membrane depolarization because of the high intracellular Cl− (see section VIIC4) and thereby increases action potential firing in human β-cells exposed to 6 mM glucose (744). As expected, GIIS in human islets is reduced by the glycine receptor antagonist strychnine.
Following its release, glycine is cleared from the interstitium by the plasmalemmal glycine transporter GlyT1 (SLC6A9) (68, 475), which cotransports Na+. This explains why removal of extracellular Na+ significantly slows glycine uptake. It has been argued that these transporters might be so efficient that, despite the high plasma levels, the interstitial concentration of glycine is kept low enough to prevent tonic activation of the GlyRs (744).
6. IAPP
IAPP (amylin) is expressed at levels 1 and 10% of that of insulin in human (68, 475) and mouse β-cells, respectively (see Supplemental Figure 1A). IAPP is co-secreted with insulin from β-cells in response to glucose stimulation at an approximate >1:20 stoichiometry (330). In humans, IAPP has a tendency to form amyloid plaques within the islets that sometimes may lead to β-cell destruction (727). In addition, IAPP has been proposed to function as an autocrine/paracrine factor. IAPP-deficient male mice exhibit increased GIIS and improved glucose tolerance (201), suggesting that IAPP may be involved in feedback control of insulin secretion. Its mechanism of action on the β-cell includes activation of KATP channels (714). IAPP is believed to interact with the calcitonin receptor (Calcr/CALCR) (84), but the expression of Calcr/CALCR expression is negligible in both mouse and human β-cells (see Supplemental Figure 12A).
VIII. ION CHANNELS AND β-CELL ELECTRICAL ACTIVITY
In earlier sections, we have summarized the properties of β-cell ion channels, transporters, and receptors. We now consider how these membrane conductances contribute to β-cell electrical activity. We discuss, in turn, the ion channels that are responsible for 1) the negative resting membrane potential of the β-cell, 2) the depolarization from the resting potential to the threshold potential, 3) the upstroke of the action potential, 4) action potential repolarization, 5) the oscillations in membrane potential (bursts) observed at intermediate glucose concentrations, and 6) the conversion of oscillatory electrical activity into continuous action potential firing at high glucose. We consider human and mouse β-cells separately because (as described above) there are considerable differences in their ion channel complements.
A. A Model for Mouse Pancreatic β-Cell Electrical Activity
1. The resting membrane potential
In the absence of glucose, the resting potential is around −70 to −80 mV mV, due to the high resting KATP channel activity resulting from the low intracellular ATP/ADP ratio (FIGURE 12A, phase I). The membrane potential is slightly more positive than expected if it was determined by the KATP channel activity alone, reflecting the tonic activity of a depolarizing background conductance.
2. The depolarization to the threshold potential for electrical activity
Elevation of extracellular glucose leads to stimulated metabolic degradation of the sugar, an increase in the intracellular ATP/ADP ratio, and reduction of KATP channel activity (675) (FIGURE 12A, phase II). As a consequence, the influence of the depolarizing inward current becomes more prominent (FIGURE 4), and the β-cell depolarizes. This explains why KATP channel blockers like tolbutamide produce membrane depolarization and induce glucose-like electrical activity. As discussed above, the identity of the depolarizing (inward) current has not been unequivocally established (see sect. VB). The fact that glucose is capable of initiating electrical activity in the absence of extracellular Na+ suggests the inward current may be Ca2+ permeable (194, 549), consistent with the reported role of Trpm2 channels (754). The background inward current must depolarize the membrane to approximately −50 mV, which is the threshold for electrical activity and the membrane potential at which voltage-gated Ca2+ channels start to open (221) (FIGURE 9).
3. The upstroke of the action potential
Regenerative activation of voltage-gated L-type Ca2+ channels explains the initial depolarization and the upstroke of the action potential (FIGURE 12B). In 30% of β-cells, voltage-gated Na+ channels also contribute to action potential firing (761). Action potential firing in this subset of β-cells may trigger electrical activity in neighboring cells that lack Na+ channels by spread of depolarization via gap junctions (554). This explains the paradox that the Na+ channel blocker TTX has a stronger inhibitory effect on GIIS than expected, given that Na+ channels are functionally inactivated in most β-cells.
The important role of the voltage-gated Ca2+ channels in action potential generation is illustrated by the effects of inorganic Ca2+ channel blockers (such as Co2+), which block all types of Ca2+ channel (548). This results in rapid membrane hyperpolarization and suppression of electrical activity. During sustained application of these blockers, a paradoxical membrane depolarization is observed (548), which correlates with a decrease in the resting β-cell K+ conductance (221), and presumably reflects KATP channel inhibition. Organic L-type Ca2+ channel antagonists (like verapamil and nifedipine) also transiently suppress electrical activity, but low-amplitude action potential activity then resumes (704). This is in agreement with membrane potential recordings from L-type Ca2+ channel knockout mice (603). The L-type Ca2+ channel-independent action potentials presumably reflect opening of P/Q- and N-type Ca2+ channels, with a possible contribution of voltage-gated Na+ channels in some β-cells.
4. Action potential repolarization
Rapid Ca2+-dependent inactivation of Ca2+ channels and voltage-dependent inactivation of Na+ channels during the action potential, combined with activation of Kv2.1 and large-conductance (BK) Ca2+-activated K+ channels, underlies action potential repolarization (293). Although delayed rectifying (Kv2.1) channels open during the action potential, their opening is so slow that the membrane potential will have largely returned to the plateau potential before they have activated significantly (293, 310, 632). Thus they primarily contribute to the late phase of action potential repolarization.
Inhibition of large-conductance Ca2+-activated (BK) channels increases action potential height (263, 293), consistent with the idea that the BK channels start activating during the upstroke of the action potential. The paradox that blocking BK channels does not affect the downstroke velocity of the action potential can be explained by greater activation of Kv2.1 channels at this time (293).
5. Plateau potential
Following the first action potential, the membrane does not fully repolarize, but returns to a level between −50 and −40 mV (the plateau potential). Several channels potentially influence the plateau potential. Their role is discussed below.
a) delayed rectifying k+ channels.
The deactivation of delayed rectifying K+ current is slow (572). This means that those channels that activated on depolarization will remain open for some time, even after the membrane has repolarized. The maintained K+ conductance may help stabilize the membrane at the plateau potential and prolong the time before the next spike is fired (31). Pancreatic β-cells from mice lacking Kv2.1 channels show complex action potentials with signs of ‟reentry,ˮ i.e., a new action potential is triggered during the downstroke of the previous one (311). This is consistent with the idea that Kv2.1 channels contribute to the temporal separation of successive action potentials.
Time-dependent deactivation of Kv2.1 and Kv11.1/11.2 channels (156, 311) and reactivation of Ca2+ channels (570) during the ~200 ms interspike interval prepares the β-cell for a new action potential, which is initiated when the depolarizing influence of the Ca2+ current exceeds the repolarizing action of the K+ channels.
b) two-pore k+ channels.
The two-pore K+ channels TASK-1 and TALK-1 may also influence the plateau potential. A role for these outwardly rectifying channels is suggested by the fact that their genetic ablation results in β-cell membrane depolarization (135, 708). Their biophysical properties suggest that they might only be functionally important at membrane potentials above −40 mV, which roughly corresponds to the plateau membrane potential.
c) Ca2+-activated K+ channels.
Accumulation of Ca2+ during action potential firing leads to activation of small-conductance Ca2+-activated SK3/SK4 channels. This underlies the brief “afterhyperpolarizations” that sometimes separate groups of 3–5 action potentials that sit superimposed on the plateau potential.
6. Progressive decrease in action potential amplitude and frequency
Cumulative Ca2+-dependent (and voltage-dependent) inactivation of the Ca2+ channels (515, 570, 593) may account for the time-dependent decrease in action potential height and frequency that is often observed during the burst.
7. Burst termination
Numerous hypotheses have been advanced to explain how the bursts are generated. These include (but are not limited to) activation of large-conductance (BK) Ca2+-activated K+ channels by Ca2+ influx during β-cell electrical activity (41), opening of Ca2+-activated K+ channels by mobilization of intracellular Ca2+ stores (15), opening of a depolarizing store-operated conductance (734), inactivation of the voltage-gated Ca2+current (594), and cyclical variations in KATP channel activity due to oscillations in the intracellular ATP/ADP-ratio driven by metabolism (368).
Patch-clamp experiments on β-cells in intact islets (in which bursting electrical activity is maintained) have helped to resolve the mechanisms involved. It appears that termination of the burst is due to activation of Kslow which comprises both KATP and SK3/SK4 channels. Ca2+ entry and the resultant elevation of [Ca2+]i directly activates Kslow (mediated by Ca2+-activated SK3/SK4 channels). In addition, Ca2+ entry during electrical activity increases the KATP conductance by an indirect effect. This is mediated as follows. Ca2+ entering the cell during electrical activity is removed by the activity of the Ca2+-ATPases in the plasma membrane and the sER. This consumes energy, utilizing ATP and generating ADP. Eventually, after 25–30 action potentials, the fall in the cytoplasmic/submembrane ATP/ADP ratio results in sufficient activation of KATP channels to terminate the burst and fully repolarize the β-cell to −70 mV (143, 675) (FIGURE 12, A and B).
Each action potential is associated with the entry of ~107 Ca2+ ions (16 amol). Given that the Ca2+-ATPase transports 1 Ca2+ for each ATP, it can be calculated that Ca2+ entry during a burst of 30 action potentials will generate 0.5 fmol ADP during its removal. In a cell with a volume of 1 pl, this would correspond to an average ADP concentration of 0.5 mM, sufficient to activate the KATP channel even in the presence of mM ATP (328). Thus the KATP channels provide a background conductance that varies in amplitude throughout the burst. It is important to remember that only a tiny increase in KATP current is needed to hyperpolarize the β-cell.
Interestingly, KATP channel activity sometimes continues to increase even after electrical activity has ceased (570). Thus KATP channel activity might not precisely echo [Ca2+]i. This may explain why the β-cell membrane potential often hovers around the plateau potential for a while, following cessation of action potential firing, before eventually repolarizing to the interburst potential (FIGURE 12B). A slight increase in KATP channel activity might suffice to suppress action potential firing but not to repolarize the cell completely. A further increase in KATP channel activity would produce a larger repolarization. Once the β-cell has repolarized to membrane potentials approaching the threshold for voltage-gated Ca2+ channel activation, the regenerative closure of these channels can account for the rapid final repolarization to the interburst membrane potential.
8. Pacemaker depolarization between two successive bursts
Following burst repolarization and the cessation of action potential firing, [Ca2+]i is reduced and the intracellular ATP/ADP ratio gradually increases, resulting in a time-dependent decrease in Kslow and KATP channel activity (FIGURE 12, A, phase III, and C ). This explains the pacemaker depolarization between successive bursts. During the electrically silent and repolarized interval, reactivation of voltage-gated Na+ and Ca2+ channels also occurs. A new burst of action potentials is induced when the β-cell has depolarized to the membrane potential where Ca2+ and Na+ channels start activating.
9. Graded response to increasing glucose concentrations
One of the most fascinating properties of β-cell electrical activity is its capacity to respond to increasing glucose concentrations in a graded fashion. Although increasing glucose recruits quiescent β-cells (402, 761), there is also a concentration-dependent increase in the electrical activity of individual β-cells. This manifests as a glucose-dependent increase in burst duration and frequency at the expense of the interburst silent intervals (273). Eventually, at glucose concentrations of 20–30 mM, continuous action potential firing emerges and the membrane potential never repolarizes from the plateau level. Although it is questionable whether such high concentrations are attained physiologically, it is notable that in vivo membrane potential recordings also demonstrate the transition from oscillatory electrical activity to continuous action potential firing, with the latter occurring at glucose concentrations as low as 10 mM (587).
We postulate that this change in excitability arises because the increased metabolic rate of the β-cell at higher glucose levels is associated with enhanced ATP production (FIGURE 12A, phase IV). Ca2+ entering the cell during electrical activity is removed from the cytosol by activation of the Ca2+-ATPases SERCA and PMCA, resulting in increased ATP consumption. However, because of the increased metabolic rate at high glucose, the β-cell has a greater capacity to buffer ATP. Thus electrical activity will not lower the ATP/ADP ratio as much as at low glucose levels. As a consequence, there will be little or no KATP channel reactivation and the β-cell will remain permanently depolarized.
Recent measurements of the cytoplasmic ATP/ADP ratio support this idea. They show that initiation of electrical activity is associated with a transient drop in the cytoplasmic ATP/ADP ratio (676, 677). Subsequently, this is superseded by increased ATP production, secondary to an increase in the intramitochondrial Ca2+ concentration (417). As the glucose concentration is increased, there is a progressive increase in the cytoplasmic ATP/ADP ratio. This hypothesis also explains why sulfonylureas (like high glucose) produce continuous electrical activity (261, 335); it is because they prevent the increase in KATP channel activity that results from the fall in cytoplasmic ATP/ADP ratio (due to Ca2+-ATPase activity).
B. A Model for Human β-Cell Electrical Activity
Although work on mouse β-cells is very helpful for defining mechanisms that confer glucose regulation of electrical activity, the ultimate goal is to understand what happens in human β-cells. However, our knowledge of the electrophysiology of human β-cells remains fragmentary compared with that of the mouse β-cell. Given the close relationship between glucose-induced electrical activity and GIIS, further work on human β-cells (and in particular those from T2DM donors) is needed to establish whether changes in β-cell electrical activity contribute to the etiology of T2DM. FIGURE 13, A and B, summarizes current understanding of the stimulus-secretion coupling in the human β-cell.
1. The resting membrane potential
At nonstimulatory glucose concentrations (<3 mM) (89), KATP channel activity maintains a negative membrane potential (−70 to −60 mV) (FIGURES 6A and 13A). Inwardly rectifying K+ channels (Kir5.1 and Kir7.1) are also active but their contribution to the resting conductance is small (10–15% of that provided by the KATP channels) (535, 553).
2. The depolarization to the threshold potential for electrical activity
In human islets, insulin secretion is triggered at glucose concentrations as low as 3 mM, whereas 6 mM is required in mouse islets (89). Two features of human β-cell electrical activity account for this difference. First, KATP channel activity in human β-cells, even at low (1 mM) glucose concentrations, is only ~10% of that in mouse β-cells (535). Second, human β-cells (unlike mouse β-cells) are equipped with T-type Ca2+ channels that activate at membrane potentials as negative as −60 mV (52, 90). The combination of these two effects means that the human β-cell is much closer to the threshold for initiation of electrical activity. Thus a small decrease in KATP channel activity may be sufficient to take the membrane potential into the range where T-type Ca2+ channels activate and initiate regenerative electrical activity. As discussed above for mouse β-cells, agents that activate small currents will only influence β-cell electrical activity at glucose concentrations associated with near-maximal closure of the KATP channels. This also applies to human β-cells. However, these currents may have a greater impact on both membrane potential and electrical activity in human β-cells because of its lower resting K+ conductance and the presence of T-type Ca2+ channels.
3. The upstroke of the action potential
Eventually, the depolarization resulting from the opening of T-type Ca2+ channels is large enough to activate TTX-sensitive Na+ channels and L-type Ca2+ channels. Na+ channels play a particularly important role at glucose concentrations just above the threshold for triggering electrical activity (52, 90). Likewise, the important role played by the L-type Ca2+ channels in the initiation and upstroke of the action potential explains the inhibitory effects of isradipine on insulin secretion. At the peak of the action potential (−20 mV and above), P/Q-type Ca2+ channels open and trigger exocytosis of insulin granules (51, 90).
4. Action potential repolarization
The elevation of [Ca2+]i resulting from the opening of the Ca2+ channels leads to activation of BK channels and initiates action potential repolarization (51, 90). BK channel activity also contributes to the rapid afterhyperpolarization following each action potential. Stromatoxin-sensitive delayed rectifying K+ channels and (possibly) HERG channels also open during the action potential but because their activation is slow they contribute little to spike repolarization (311, 575). However, their slow deactivation contributes to the pacemaker depolarization between two action potentials.
5. Bursts of action potentials
The increase in Ca2+ during a burst of action potentials is sufficient to activate Ca2+-activated SK3 channels. Apamin (a blocker of SK1, SK2, and SK3 channels) stimulates action potential firing in human β-cells and reduces action potential height (311). The decrease in action potential amplitude may be explained by the associated membrane depolarization; this produces voltage-dependent inactivation of Na+ channels involved in the upstroke of the β-cell action potential. Support for the role of SK channels comes from the fact that the SK1–3 antagonist NS8593 produced a 10-mV depolarization, increased action potential firing, and strongly potentiated insulin secretion evoked by 10 mM glucose in human islets (311).
Activation of SK3 channels may explain why action potentials in human β-cells often come in groups of five or six action potentials separated by slightly repolarized electrically silent intervals. These resemble the bursts of action potentials in mouse β-cells, but they are shorter, the action potentials usually originate from a more negative membrane potential, and there is no well-defined plateau potential. It is tempting to attribute the silent (slightly repolarized) intervals separating groups of action potentials to Ca2+-dependent activation of SK3 channels. Activation of SK channels may also explain why the membrane potential becomes more repolarized after a period of electrical activity human β-cells (see FIGURES 6B and 13C). As in mouse β-cells, the plateau potential may be stabilized by the activity of TALK-1 and TASK-1 channels, which provide a small voltage-independent background K+ permeability. In accordance with this idea, pharmacological blockade of TASK-1 channels increases action potential firing (135, 708). Once electrical activity has stopped, buffering/lowering of [Ca2+]i reduces SK, explaining the pacemaker depolarization between two successive bursts. When the SK current is sufficiently reduced, the β-cell depolarizes with ensuing activation of T-type Ca2+ channels. Cumulative voltage-dependent inactivation of the Na+ channels and Ca2+-dependent inactivation of the L-type Ca2+ channels contributes to the decline in action potential height during the burst (768).
6. Response to increasing glucose concentrations
Increasing glucose from 1 to 6 mM and then to 20 mM glucose results in a progressive increase in the intracellular ATP/ADP ratio in human β-cells and a concentration-dependent inhibition of the KATP channels. Channel activity is reduced by 90% at 6 mM glucose and by >95% at 10 mM glucose (453). This reduction of KATP channel activity results in further membrane depolarization. This may be seen from the fact that the membrane potential during the interspike intervals may be as negative as −60 mV at 6 mM glucose (89), but is between −50 and −40 mV at 10 mM glucose or above (52) (FIGURE 6 and schematic in FIGURE 13, C and D ).
As the β-cell depolarizes, the T-type Ca2+ channels and the TTX-sensitive Na+ channels undergo progressive voltage-dependent steady-state inactivation leading to a reduced action potential amplitude in the presence of high glucose (FIGURES 6A and 13, C and D). When the Na+ channels are in the inactivated state, they no longer contribute to β-cell electrical activity, and blocking them with TTX will exert no additional effect. This explains why TTX is a weaker inhibitor of GIIS at high glucose than at low glucose (FIGURE 13, C and D).
The peak amplitude of the action potential directly correlates with the activation of voltage-gated P/Q-type Ca2+ channels, which is maximal at 0 mV and diminishes steeply at more negative voltages. Accordingly, the low-amplitude action potentials generated at high glucose (once Na+ channel inactivation has occurred) will principally be associated with Ca2+ entry via L-type Ca2+ channels as these activate at more negative voltages than the P/Q-type Ca2+ channels (90). This accounts for the switch in the Ca2+ channel dependence of GIIS.
IX. INSULIN SECRETION
The importance of β-cell electrical activity is that it generates the increase in [Ca2+]i that is required to trigger exocytosis of insulin-containing secretory granules. FIGURE 14A shows insulin secretion elicited by an increase in glucose from 1 to 20 mM. After a delay of a few minutes, glucose elicits a biphasic stimulation of insulin secretion. This consists of an initial transient response, lasting <10 min (1st phase secretion), followed by a 2nd (sustained) phase, lower than peak 1st phase insulin release but still 10-fold higher than basal secretion. In many cases, insulin secretion gradually increases during the 2nd phase (473). Application of the KATP channel blocker glibenclamide in the presence of 1 mM glucose triggers insulin secretion without a delay and the secretory response consists only of ‟1st phaseˮ insulin secretion with little sign of a sustained ‟2nd phaseˮ despite the continued presence of glibenclamide (FIGURE 14B). When glucose is subsequently elevated to 20 mM in the continued presence of glibenclamide, insulin secretion increases to a level equivalent to steady-state 2nd phase GIIS.
FIGURE 14.
A: insulin secretion measured in the perfused mouse pancreas preparation when glucose was elevated from 1 to 20 mM (as indicated by horizontal bar). B: same as in A but secretion was elicited by 2 µM glibenclamide (a KATP channel blocker) (horizontal bar). Glucose was subsequently elevated to 20 mM glucose (horizontal bar) in the continued presence of glibenclamide. The dotted line illustrates the insulin response to glucose shown in A. In A and B, data points indicate mean values and the shaded areas the SE (shaded areas). Data provided by N. Rorsman, Oxford. C: geometric mean of plasma insulin concentrations during a hyperglycemic clamp at 15 mM glucose (horizontal bar) in nondiabetic subjects (ND) and in patients with type 2 diabetes mellitus treated by diet (T2D) or the sulfonylurea gliclizide (T2D + SU). [Modified from Hosker et al. (292).]
It is worth pointing out that an abrupt (step) increase in glucose from a low to a high concentration is very far from physiological. Following a meal, plasma glucose increases from a basal level of ~5 to 8 mM over a ~30 min period (an increase of 0.1 mM/min) (189). In both mouse and human islets, when the glucose concentration is ramped from a nonstimulatory glucose concentration (<2 mM) to a high concentration (>25 mM) insulin secretion is essentially monophasic with little sign of a 1st phase (150, 494). Nevertheless, understanding the cell biology of biphasic insulin secretion is important because T2DM is associated with the almost complete loss of 1st phase GIIS and a reduction of 2nd phase insulin secretion (FIGURE 14C) (242, 292).
In this section we consider the properties of insulin secretion (both qualitatively and quantitatively) and the molecular machinery of insulin granule exocytosis.
A. Quantitative Considerations: In Vivo Versus In Vitro
In the perfused mouse pancreas, insulin release increases from a basal rate of ~0.2 ng to 5–10 ng insulin/min during 1st phase insulin secretion and 2 ng/min during 2nd phase secretion when stimulated with 20 mM glucose (FIGURE 15A). A mouse pancreas contains 50 μg insulin (see legend to FIGURE 15). The above rates therefore convert to fractional release rates of 0.002, 0.01–0.02, and 0.005% per min. As a mouse β-cell contains ~10,000 secretory granules (650), these values correspond to release rates of 0.02, 1–2, and 0.5 granules/min (FIGURE 15A). These calculations assume that all β-cells respond uniformly to glucose, which is not the case. Imaging of exocytosis in intact mouse islets indicates that in fact the secretory capacity varies considerably between β-cells (402). Although these differences may in part be due to ‟sectioning biasˮ (because exocytosis occurred in a part of the cell not included in the imaged segment), it is clear that the release rates calculated above should be regarded as an average.
FIGURE 15.

A: estimated rates of insulin secretion in the perfused mouse pancreas based on the experiment shown in FIGURE 14A and a total pancreatic insulin content of 50 μg [based on 0.008 g pancreas/g body weight, 0.25 μg insulin/g pancreas and a body weight of ~25 g (69)]. B: rates of insulin secretion in vivo estimated by solving Equation 1 using plasma insulin levels reported by (275) and a granule insulin content of 1.6 amol (294)(equivalent to ~8 fg of insulin). C: pulsatile insulin secretion in humans. The red and black traces show a 3-min moving average of the plasma insulin and glucose concentrations of a normal subject upon glucose infusion. Note that increasing glucose by ~0.5 mM more than doubles plasma insulin levels during the first oscillation. [Data modified from Lang et al. (363).] D: pulsatile insulin secretion measured using the perfused mouse pancreas preparation and a perfusion rate of ~0.3 ml/min. Data provided by N. Rorsman, Oxford.
It is interesting, nevertheless, to compare the release estimated above to those observed in vivo. To our knowledge, insulin secretion in vivo in mice has not been analyzed by the deconvolution methods that have been applied to humans (see Ref. 340). In the absence of such data we have estimated the rate of insulin secretion (dInsulin) during a glucose tolerance tests by solving the differential equation
| 1 |
where Vplasma is the volume of the plasma (0.55 ml in the mouse), Content corresponds to the insulin content of a single insulin granule (1.6 amol) (294), and Rate is the number of granules released per minute. In fed mice, the steady-state plasma insulin level at a plasma glucose of ~9 mM is 120 pM, and this increases to a steady-state concentration of 300 pM at glucose concentrations >16.7 mM (275). Solving the above equation suggests 20,000 and 50,000 granules/min are released at 8.9 and 16.7 mM glucose, respectively. Given that a mouse pancreas consists of 1,000 islets that contain on average ~80 β-cells (see sect. IIA), these release rates correspond to 0.25 and 0.62 granules·β-cell−1·min−1. During 1st phase secretion, plasma insulin levels are four- to fivefold higher, corresponding to a release rate of ~2 granules·β-cell−1·min−1. The latter values are in fair agreement with 1st and 2nd phase GIIS derived from perfused pancreas measurements (FIGURE 15B). Thus there appears to be a good correlation between the rates of GIIS in vivo and in vitro. As we have previously reported (569), there is also a good agreement between GIIS in vitro and in vivo in humans.
With these low release rates, the 500–600 secretory granules that are docked below the plasma membrane in mouse (489) and human β-cells (447) should be adequate to maintain blood glucose within the normal range for at least 10–20 h. This argues that the small (25%) reduction in β-cell/islet insulin content associated with T2DM (576) should not be of major pathophysiological importance and that other functional defects (for example, impaired initiation of electrical activity) are more significant drivers of disease.
When blood samples are taken at high frequency (every min), it becomes clear that elevation of plasma glucose (by intravenous glucose infusion) triggers oscillations in plasma insulin with a period of ~10 min (363) (FIGURE 15C). These oscillations are less apparent in patients with T2DM (285, 483). Interestingly, oscillatory insulin secretion is also seen occasionally in the perfused mouse pancreas (FIGURE 15D), especially when using a physiological perfusion rate (0.3 ml/min; see sect. IIA). Precisely how insulin secretion in the 1,000 islets of the mouse pancreas is synchronized remains an open question. Although the oscillations can be suppressed by application of TTX in some pancreases [which would be consistent with a role for intrapancreatic ganglion cells (613)], in other experiments they persist in the presence of the Na+ channel blocker (own unpublished).
B. Molecular Machinery of Exocytosis
The molecular machinery of insulin granule exocytosis bears a strong resemblance to that of synaptic vesicle release at the neuronal synapse (364). Here we briefly summarize the current view of the molecular regulation of exocytosis (FIGURE 16, A and B). This is provided only as a background to a description of insulin granule exocytosis and is not intended to be comprehensive. We refer the reader to the many excellent reviews of the topic elsewhere for a more detailed account (193, 336, 652).
FIGURE 16.

Schematic of the molecular machinery mediating Ca2+-dependent synaptic vesicle release. A: the core fusion machinery comprises the SNARE/SM protein complex. It consists of the vesicular (v-)SNARE protein VAMP-2 (red), the plasma membrane (target, t) SNARE proteins syntaxin-1 (yellow), and SNAP-25 (green), Munc18–1 (gray), and the Ca2+-sensor synaptotagmin-1 (blue, on vesicle). Tethering of the synaptic vesicle to the active zone involves a plasmalemmal voltage-gated Ca2+ channel (VGCC) and an active zone protein complex consisting of RIM (violet), Munc13 (brown), and RIM-BP (purple). RIM binds to the vesicular rab proteins Rab3 and Rab27 (sea green). B: five stages of exocytosis are illustrated: i) tethering; ii) docking and assembly of a loose trans-SNARE complex; iii) the formation of a tight 4-helix or ternary SNARE complex, with one helix coming from syntaxin, two helices from SNAP-25 and one helix from VAMP-2. This process is stabilized by complexin; iv) Ca2+ binding to synaptotagmins results in displacement of complexin, completion of zippering, and fusion pore opening; and v) expansion of the fusion pore and release of the cargo (a dense core vesicle is shown rather than a clear synaptic vesicle). After fusion, the resulting cis-SNARE complexes (cis, in the same membrane; trans, in opposite membrane) are disassembled by the NSF/SNAP ATPases and recycled. Modulators of β-cell exocytosis and where they act are given in green (see sect. IX, A and B). [Modified from Südhof (652).]
1. The SNARE model of synaptic vesicle release
Regulated exocytosis is a highly controlled process that involves the docking and tethering of the secretory vesicles to the plasma membrane, their fusion with the plasma membrane and the emptying of their cargo into the extracellular space. It involves distinct sets of proteins in the vesicular/granular and plasma membranes (651), which are known as v-SNAREs and t-SNAREs, respectively. According to the SNARE model of exocytosis, SNAP-25 and syntaxin-1 in the plasma membrane interact with VAMP2 in the secretory vesicle membrane to form the SNARE complex. This process is orchestrated by the proteins munc18, munc13, and the active zone protein RIM (652–654).
RIM is a binding partner of the vesicular protein Rab3 (336) and plays a key role in vesicle docking. In keeping with this idea, deletion of RIM leads to a decrease in the number of vesicles available for release (252). Munc13 contains a RIM-binding domain and RIM may therefore recruit munc13 to the release sites. Both these proteins interact with RIM-binding proteins (RIM-BP) in the synaptic terminal (652).
Prior to exocytosis, the SNARE proteins undergo a series of conformational changes that culminate in the fusion of the granule with the plasma membrane. Initially, munc18 binds to a ‟closedˮ form of syntaxin-1, and thereby prevents SNARE complex formation. The NH2-terminal ends of syntaxin and VAMP-2 then become aligned (FIGURE 16B, step i). Munc13 opens syntaxin-1 (407), allowing the trans-zippering of the SNAREs in the plasma and granular membranes from the NH2 to the COOH terminals (which are anchored to the membrane). This leads to the formation of a four-helix trans-complex, also referred to as the ‟ternary complexˮ (100) (FIGURE 16B, step ii). At this stage, binding of munc18 to the SNARE complex stabilizes it and prevents its premature disassembly. Thus munc18 plays a dual role in SNARE complex formation.
Once SNARE complexes are partially assembled, complexin associates to the complex. Complexin stabilizes the partially zippered SNARE complex, and its negatively charged accessory helix prevents COOH-terminal zippering (FIGURE 16B, step iii). Following an increase in cytosolic Ca2+, the Ca2+ sensor synaptotagmin displaces complexin, allowing full zippering to take place. As a result, the granule and plasma membranes are brought into very close proximity and, via mechanisms that remain poorly understood, leads to disruption of the lipid membrane enclosed by the SNARE complexes, resulting in the opening of the fusion pore (FIGURE 16B, step iv). The surface tension produced by the curvature of the granule then leads to further expansion of the fusion pore (FIGURE 16B, step v). Some granules may even fully collapse into the plasma membrane, allowing efficient delivery of the granule cargo (309). Finally, the SNARE complex is disassembled in an ATP-dependent reaction catalyzed by NSF, allowing recycling of the SNARE proteins (398).
2. Molecular machinery of β-cell exocytosis
All the essential proteins required for synaptic vesicle release are also expressed in the β-cell and presumably fulfill the same function(s) (193, 336, 364). Here, we consider their roles in insulin exocytosis from mouse and human β-cells. We discuss the proteins in the order that they (or the protein families to which they belong) are likely to participate in the control of insulin secretion, as deduced from the model of synaptic vesicle exocytosis outlined above (FIGURE 16, A and B). There are a number of caveats associated with extrapolating the ‟classicalˮ picture of exocytosis to the pancreatic β-cell that should be borne in mind. First, the subcellular localization of many exocytotic proteins in β-cells is often not clearly determined, especially at the electron microscope level. Second, protein-protein interactions are determined in detergent homogenates and thus may differ from the living β-cell. Third, manipulating any part of the vesicle trafficking machinery (from granule biogenesis to endocytosis) may have unexpected effects. For example, ablation of the dynamin-2, a protein involved in endocytosis, results in glucose intolerance and almost complete loss of 2nd phase insulin secretion (176). This effect is mediated via changes in clathrin-mediated endocytosis and actin remodeling. Thus the picture presented here should not be regarded as the definitive story but rather as a ‟work in progress.ˮ
a) rab3 and rab27.
The GTP-binding proteins Rab3 and Rab27 play key roles in the docking of synaptic vesicles at the active zone in neurons (652). There are four Rab3 isoforms that are encoded by separate genes (Rab3a-d/RAB3A-D). In mouse β-cells, only Rab3a, b, and d are expressed at comparable levels in human β-cells; RAB3B is expressed at particularly high levels (see Supplemental Figure 15A).
As for other GTPases, the activities of Rab3 and Rab27 reflect their cycling between an inactive GDP-bound (cytosolic form) and an active GTP-bound (membrane-associated) form. These transitions are mediated by guanine nucleotide exchange factors (GEFs) that result in GTP binding and by GTPase activating proteins (GAPs) that accelerate the intrinsic Rab-GTPase activity (54).
Detailed physiological studies of Rab3A knockout mice have revealed that Rab3a is required for maintenance of the readily releasable pool (RRP) of granules (444). Thus its deletion is associated with glucose intolerance and reduced GIIS both in vivo and in vitro (742). The role of the other Rab3s has not been investigated in as much detail, but studies employing overexpression of nonfunctional (GTPase-deficient) Rab3 mutants suggest that, with the exception of Rab3c, they are all required for Ca2+-dependent secretion in insulin-secreting cells (298).
Rab27 exists in two isoforms (Rab27a-b/RAB27A-B). In mouse and human β-cells, only Rab27a/RAB27A is expressed (see Supplemental Figure 15B). Rab27A is associated with the insulin secretory granules (362, 724, 766), and the data suggest it is involved in granule docking with the plasma membrane (338), influences insulin exocytosis (444), and is essential for the replenishment and release of an immediately releasable pool of secretory granules (110).
Pancreatic β-cells express many other Rab proteins in addition to Rab3 and Rab27 (3, 68, 146, 475). The roles of these small GTPases have (to the best of our knowledge) not been extensively studied. However, both Rab11 and its effector Rip11 participate in cAMP-induced (but not glucose-induced) insulin secretion, possibly by regulating the recycling of the proteins involved in the exocytotic process back to immature granules (655).
b) rim.
The RIM protein family has seven identified members (RIM1α, RIM1β, RIM2α, RIM2β, RIM2γ, RIM3γ, and RIM4γ) encoded by four genes (Rims1–4/RIMS1–4). In nerve terminals, RIM1 and RIM2 serve as key factors determining Ca2+ channel density and synaptic vesicle docking at the active zone (252) and are involved in the control of neurotransmitter release (600). Little is known about the role of RIM3 and RIM4.
Mouse and human β-cells express Rims/RIMS1, and Rim2 (Rims/RIMS2) at low and high levels, respectively (see Supplemental Figure 15C). Like Rim1, Rim2 interacts with Rab3A (496) and Munc13–1 (160, 750). It also interacts with the cAMP-sensing protein cAMP-GEFII (also known as Epac2) (496, 615).
Ablation of Rims2 leads to impaired glucose tolerance and inhibition GIIS or high K+-induced insulin secretion (750). This is associated with a 70% reduction in the number of docked granules. In β-cells lacking Rim2, exocytosis proceeds almost entirely by release of granules that have newly arrived at the plasma membrane (newcomer granules; see sect. IXD5 and FIGURE 23A) (750).
FIGURE 23.

A: different modes of insulin granule fusion. Release of previously ‟dockedˮ granules upon stimulation (left), release of granules not originally docked with the plasma membrane but that are recruited to the plasma membrane and undergo exocytosis following a variable period of being docked (‟newcomer,ˮ middle) and granules that arrive at the plasma membrane and are instantly released without being docked (‟crash fusion,ˮ right). [Modified from Shibasaki et al. (617).] B: imaging of secretory granules by TIRF microscopy. Red area indicates the evanescent field. Illumination leads to time-dependent bleaching of fluorescently labeled secretory granules (bright green → dark green). Granules that were initially bright (at t = 0) might thus appear to vanish within a few minutes (t = 2 min) because of loss of fluorescence. When such granules subsequently undergo exocytosis, the dramatic increase in fluorescence as the fluorophore moves further into the evanescent field will result in the reappearance of fluorescence giving the impression that granules released by glucose (at t = 2 min) were not docked with the plasma membrane. With agents that act more quickly (like K+), exocytosis will occur (at t = 0.5 min) before granules have faded enough to make them invisible. C: insulin secretion elicited by sequential stimulation with high extracellular K+ ([K+]o) and glucose. Note that high [K+]o transiently stimulates insulin secretion and that release rates subsequently decline back towards baseline values and that subsequent addition of glucose leads to a slowly developing increase in insulin release without any sign of a 1st phase (compare FIGURE 21A). Values are means (red circles) + SE (gray shaded area). Experiment performed by M. Söderström, Oxford.
Both mouse and human β-cells express high levels of the RIM-binding proteins Rimbp2/RIMBP2 and Erc1/ERC1 (3, 68, 146, 475). These may contribute to the tethering of the insulin granules to the plasma membrane (see FIGURE 16A).
c) syntaxin.
Mouse β-cells express syntaxin-1a (Stx1a), -2 (Stx2), -3 (Stx3), and -4 (Stx4a) (see Supplemental Figure 16A). Syntaxin-5, -7, -12, -16, and -18 (Stx5, 7, 12, 16, and 18) are also highly expressed, but these are believed to mediate intracellular membrane trafficking (i.e., membrane fusion in the endoplasmic reticulum and the Golgi network) and are not thought to participate in exocytosis. Intriguingly, syntaxin-1 also modulates the activity of the KATP and voltage-gated delayed rectifying K+ channels in β-cells (387), and its function may therefore not be limited to the regulation of exocytosis. Variable results have been obtained when Stx1a is knocked down. In one study where TIRF microscopy was used to monitor granule exocytosis, Stx1a deficiency resulted in a marked reduction of docked granules and strong suppression of 1st phase insulin secretion (487). However, 2nd phase secretion was unaffected. In contrast, a second study found only a slight decrease in the number of docked granules, but both 1st and 2nd phase insulin secretion (measured using perifused islets) were strongly reduced (718). Given the redundancy of syntaxin gene expression in mouse β-cells, it is perhaps not unexpected that deletion of Stx1a does not abolish exocytosis as syntaxin-3 and -4 can substitute for syntaxin-1 in the release process.
As remarked above, mouse β-cells also express high levels of syntaxin-3 (769). Genetic silencing of Stx3 inhibits GIIS, suggesting that syntaxin-3 participates in exocytosis. Unlike syntaxin-1, syntaxin-3 localizes to the secretory granules. It is involved in the release of newcomer granules (769). Syntaxin-3 has also been proposed to be involved in compound exocytosis, a process in which the secretory granules fuse with one another within the cell before fusing with the plasma membrane (193) (see sect. IXD9).
Islets from heterozygous Stx4+/− mice exhibit reduced 1st and 2nd phase GIIS (642). Syntaxin-4 interacts with the submembrane F-actin network (490), and it has been proposed that glucose-induced remodeling of the submembrane actin network induces a conformational change in syntaxin-4 that enables it to interact with VAMP-2. Thus the actin network not only acts as a physical barrier, it also prevents insulin secretion by blocking SNARE complex formation (316). This may explain why agents that disrupt the actin network (like cytochalasin) stimulate insulin secretion (571).
In human β-cells, syntaxin-1 (STX1A) accounts for close to 50% of all syntaxin transcripts and is expressed at levels 10–20 times higher than STX2–4 (FIGURE 16A). Expression of STX1A is also 30-fold higher in human than in mouse β-cells (FIGURE 16B), possibly suggesting that it plays a relatively more significant role in insulin secretion in humans. Indeed, there is a positive correlation between STX1A expression and GIIS in isolated islets and a negative correlation between STX1A expression and HbA1C (a surrogate measure of long-term glucose control). Islets from T2DM donors have reduced expression of STX1A (30% less) compared with islets from nondiabetic individuals (16).
d) snap-25.
Both mouse and human β-cells express SNAP-25 at high levels (Snap25/SNAP25) (see Supplemental Figure 17A). Alternative splicing of exon 5 of SNAP-25 gives rise to two isoforms, SNAP-25a and SNAP-25b (50), which differ by nine amino acids. In adrenal chromaffin cells, expression of Snap25b in Snap25-deficient cells results in a larger pool of primed vesicles than that seen when SNAP-25a is reintroduced (637). Mouse β-cells coexpress SNAP25b and SNAP25a. The blind-drunk (Bdr) mouse has a mutation in Snap-25b (314), and islets from heterozygous (+/Bdr) mice exhibit reduced glucose-stimulated insulin secretion (314). SNAP25 expression is reduced in islets from T2DM donors (16, 493). Furthermore, there is negative relationship between SNAP25 expression and HbA1C in vivo, and a positive relationship between SNAP25 expression and GIIS in vitro (16). Thus SNAP-25 is necessary for insulin release in both mouse and human β-cells.
Like SNAP-25, SNAP-23 (Snap23/SNAP23) has been reported to participate in β-cell exocytosis (579), but its expression is only 10–25% of SNAP25 in both mouse and human β-cells. Surprisingly, insulin secretion is increased in Snap23 knockout mice. It was proposed that this effect reflects formation of fusion-incompetent SNAP-23/syntaxin-1A/VAMP-2 complexes. As a consequence, syntaxin-1 and VAMP-2 are depleted, leading to reduced formation of fusion-competent SNAP-25/syntaxin-1A/VAMP-2 complexes (358).
e) vamp.
Both mouse and human β-cells express high levels of VAMP-2 and 3 (Vamp2–3/VAMP2–3) (see Supplemental Figure 17B). There is no β-cell-specific mouse knockout of VAMP-2, and its global deletion is neonatally lethal (601). There is a negative correlation between VAMP2 expression in human islets and HbA1C in vivo (16). VAMP-3-deficient mice are viable and exhibit normal glucose and insulin tolerance (745). Mouse (but not human) β-cells also express high levels of Vamp4. There are no functional studies of the role of VAMP-4 in β-cells, but it has been proposed to play a role in synaptic vesicle biogenesis/recycling in neurons (355).
Both mouse and human β-cells also express VAMP-8 (Vamp8/VAMP8). First identified as an endosomal SNARE (named endobrevin), overexpression of VAMP-8 has been reported to inhibit GIIS in mouse insulinoma cells by interfering with endocytosis (468). More recently, VAMP-8 has been postulated to play a role in the release of newcomer granules (770). In mice lacking Vamp8, GIIS is not reduced, but the response to GLP-1 is attenuated (770).
f) munc18 and other syntaxin-binding proteins (stxbps).
The interaction of Munc18 with syntaxin plays a key role in priming insulin granules for release (196). Munc18 belongs to a protein family collectively referred to as Sec1/Munc18 (SM) proteins. They include Munc18–1, −2, and −3 (Stxbp1–3), all of which are expressed in mouse β-cells. In human islets, STXBP1 is expressed at 5- to 10-fold higher levels than STXBP2 and STXBP3 (see Supplemental Figure 17C).
I) Munc18–1. Munc18–1 (Stxbp1/STXBP1) comes in two different splice variants (-a and -b). Munc18–1a has been reported to control 1st phase insulin secretion (155). Munc18–1b interacts with syntaxin-3 and VAMP-8 (770) and accordingly may be involved in exocytosis of newcomer granules and compound exocytosis (770). In humans, there is a negative correlation between the expression of STXBP1 and HbA1C and a positive correlation with GIIS (16).
II) Munc18–2. Whereas munc18–1 is recruited to the plasma membrane in a glucose-dependent fashion, munc18–2 (Stxbp2/STXBP2) is principally cytosolic (423). It was proposed that munc18–2 controls refilling of the docked pool of granules. Munc18–2 may also mediate the release of newcomer granules that undergo release without prior docking. Indeed, munc18–2 interacts with syntaxin-3 (193), which would be in agreement with a role in newcomer release. In humans, there is a positive correlation between STXBP2 expression and HbA1C (16), indicating that increased levels of munc18–2 may paradoxically inhibit GIIS.
III) Munc18–3. Munc18–3 (Stxbp3/STXBP3) knockout mice are not viable, but heterozygous knockouts (Stxbp3+/−) are glucose-intolerant, an effect that is at least partially due to reduced GIIS (486).
IV) Syntaxin-interacting protein. Syntaxin-interacting protein (Synip, encoded by Stxbp4/STXBP4) binds specifically to syntaxin-4. Overexpression of Synip inhibits both 1st and 2nd phase insulin secretion in clonal mouse insulin-secreting βH9 cells (581). Stxbp4/ STXBP4 is expressed at low levels in primary mouse β-cells (~5% of Stxbp1) and human islets (<2% of STXBP1) (see Supplemental Figure 17C) (68, 475).
V) Tomosyn. Tomosyn (Stxbp5/STXBP5) is another syntaxin-binding protein (tomo is Japanese for ‟friendˮ; Ref. 192). It is expressed at levels only 15–20% of those found for Stxbp1 in mouse and human β-cells (see Supplemental Figure 17C). There are three different isoforms of tomosyn: b (big), m (medium), and s (small). In mouse β-cells, overexpression and silencing of tomosyn have been reported to inhibit and stimulate insulin exocytosis/secretion, respectively (764). In rat insulinoma cells, in contrast, silencing of tomosyn inhibits insulin granule exocytosis elicited by a combination of high [K+]o, IBMX, and glucose (116).
VI) Amisyn. Amisyn (Stxbp6/STXBP6) is a sixth member of the family of syntaxin-binding proteins (ami is French for friend). Its role in kiss-and-run exocytosis is discussed below (see sect. IXD8). In mouse and human β-cells, Stxbp6/STXBP6 is expressed at levels 10–25% of Stxbp1/STXBP1 (Munc18–1) (see Supplemental Figure 17C). Despite its relatively low expression, there is a positive correlation between the expression of SYTBP6 and HbA1C and a negative correlation with GIIS (46, 122).
g) munc13.
Munc13 is involved in the final priming of the secretory vesicles for release. There are four homologs of Munc13 (munc13–1, −2, −3, and −4) that are encoded by four separate genes (Unc13a-d/UNC13A-D). Both mouse and human islets express munc13–1 and munc13–2 (unc13a/UNC13A and unc13b/UNC13B), whereas expression of unc13c and unc13d is very low (see Supplemental Figure 17D). The role of munc13–2 in the β-cell has not been studied, but ablation of munc13–1 selectively suppresses 2nd phase insulin secretion (333). Islets from mice expressing a variant of munc13–1 lacking the DAG-binding site are refractory to stimulation with the phorbol ester PMA. Thus munc13–1 may represent an important target of glucose-induced and/or cholinergic and incretin-induced DAG signaling in pancreatic β-cells.
It has been reported that munc13–1 levels are reduced in islets from T2DM donors (493). However, a more recent report failed to detect such an effect, although there was a tendency towards a correlation between increased munc13–1 (UNC13A) expression and a reduced HbA1C or increased GIIS (16). It has also been reported that RIM2 (see sect. IXB2a) regulates munc13–1 activity and that this effect may contribute to the strong suppression of GIIS in Rim2 knockout mice (750).
h) complexin.
Complexin-2 (Cplx2/CPLX2) is expressed at high levels in mouse and human β-cells (see Supplemental Figure 17E). Human β-cells also express fairly high levels of complexin-1 (CPLX1), but the levels are still only <15% of those of CPLX2 (see Supplemental Figure 17E). Silencing complexin-1 in mouse βTC3 cells leads to a moderate reduction of glucose- and leucine-induced insulin secretion (1). However, overexpression of complexin-1 also inhibited insulin secretion. Clearly, the interactions of complexin-1 with the exocytotic machinery in β-cells are complex (as befits a protein with this name) (see also sect. IXB5a).
i) synaptotagmin.
Like synaptic vesicle release, insulin secretion is a Ca2+-dependent process. This explains the necessity for β-cell electrical activity, as it generates the Ca2+ signal required to trigger insulin release. Synaptotagmins represent the Ca2+ sensors for synaptic vesicle release (652). The identity of the β-cell Ca2+ sensor has only partially been elucidated, but the available evidence strongly implicates a member of the synaptotagmin family. There are 17 synaptotagmin isoforms. Of the Ca2+-dependent synaptotagmins (Syt1, 2, 3, 5, 6, 7, 9 and 10), only Syt5 and 7 are expressed at significant levels in mouse and human pancreatic β-cells. Mouse (but not human) β-cells also express low levels of Syt2, Syt3, and Syt9 (see Supplemental Figure 18A). Although SYT1 is not detectably expressed in human β-cells, there is a positive correlation between SYT1 expression and GIIS in human islets. There is also evidence that expression is reduced in islets from donors with T2DM (16). Given the low expression of SYT1 in β-cells, it is likely that this correlation reflects a paracrine/systemic effect of synaptotagmin-1 rather than an intrinsic β-cell effect.
Both high- and low-affinity Ca2+ sensors (operating at low and high [Ca2+]i, respectively) appear to be involved in insulin secretion from pancreatic β-cells. In permeabilized β-cells, an increase in [Ca2+]i has been reported to stimulate insulin secretion with an EC50 as low as ~2 μM (733). A similar Ca2+ dependence of exocytosis has been documented by capacitance measurements of insulin granule exocytosis evoked by dialysis with intracellular Ca2+ (525, 747). In addition, there is evidence for a low-affinity component of Ca2+-dependent exocytosis that underlies the large and rapid exocytotic responses elicited by membrane depolarization and that operates at [Ca2+]i levels as high as 20 μM (48, 663). Such high [Ca2+]i levels are only likely to occur in close proximity to the Ca2+ channels.
Both mouse and human β-cells also express Syt5 at high/fairly high levels (Supplemental Figure 18A). Reduced Syt5 expression correlates with decreased GIIS following long-term exposure to elevated glucose (697). Downregulation of Syt5 has also been reported to reduce GIIS in INS1-cells (299). Given the high expression of Syt5 in β-cells, it is possible that Syt5 contributes to the high-affinity component of Ca2+-dependent exocytosis. Further functional analyses of Syt5-deficient β-cells should be considered.
Knockout studies suggest that synaptotagmin-7 (Syt7) represents one of the Ca2+ sensors involved in exocytosis in mouse β-cells (245). Genetic ablation of Syt7 in mice results in mild glucose intolerance during an IPGTT and a modest 50% reduction of GIIS both in vivo and in vitro. However, the finding that GIIS and depolarization-evoked exocytosis persist in Syt7 KO mice/β-cells suggests the existence of additional Ca2+ sensors. Syt7 is also important in human β-cells for the Ca2+-dependent mobilization of secretory granules (149), which largely corresponds to 2nd phase insulin secretion.
Although these data implicate Syt7 as an exocytotic Ca2+ sensor, it should be noted that Syt7 is not present in the insulin secretory granules but instead is found in association with components of the endosomal/lysosomal pathway and at the plasma membrane (461). Recently, Syt7 was implicated in intracellular cholesterol transport (120). Furthermore, depleting cholesterol in the plasma membrane leads to a ~50% reduction of 1st phase and 2nd phase insulin secretion (710). This is similar to the effects of genetic ablation of Syt7, raising the possibility that ablation of Syt7 mediates its effects on secretion indirectly, via changes in plasmalemmal levels of cholesterol.
The identity of the low-affinity Ca2+ sensor of exocytosis that mediates the effect of high [Ca2+]i remains an enigma. Syt3, Syt5, and Syt7 are all high-affinity Ca2+ sensors (656)8 and therefore unlikely to explain the rapid component of exocytosis triggered by high [Ca2+]i. In neurons, Syt1, Syt2, and Syt9 serve as low-affinity Ca2+ sensors (365, 741). It was previously thought that Syt9 might represent the low-affinity Ca2+ sensor for β-cell exocytosis (297, 299), but studies on Syt9-deficient islets/mice suggest this is not the case (246). As pointed out above, mouse β-cells do in fact express low levels of Syt2, and its role as a low-affinity Ca2+ sensor should therefore be considered.
Of the Ca2+-independent synaptotagmins, particularly high levels of synaptotagmin 4, 11, and 13 are found in human β-cells, whereas synaptotagmins 4 and 13 (Syt4 and 13) predominate in mouse β-cells (see Supplemental Figure 18B). Silencing of either Syt4 or Syt13 inhibits GIIS in rat insulinoma cells, and their expression is reduced in islets from T2DM donors (16). In peptidergic nerve endings, knockdown of Syt4 has been shown to decrease exocytosis in response to large Ca2+ increases, such as those that are likely to occur close to the mouth of voltage-gated Ca2+ channels (765). It is possible that the Ca2+-independent synaptotagmins (Syt4 and Syt13) exert a similar (indirect) modulatory effect (see Ref. 458) on exocytosis in insulin secreting cells, but it is perhaps equally possible that the effect is due to interference with granule recycling and/or granule generation (see sect. IXB2).
Finally, the cytoskeletal matrix protein piccolo (Pclo/PCLO) binds to the cAMP sensor cAMP-GEFII (Epac2) and to Rim2 (two proteins that mediate the stimulatory effect of cAMP on exocytosis; see below) in a Ca2+-dependent fashion, with a Ca2+ affinity as low as 1.5 mM (190). This concentration is ~100-fold higher than the Ca2+ dependence of rapid exocytosis in β-cells (17–25 μM). It is unlikely that such high Ca2+ concentrations are attained anywhere in the β-cell. Nevertheless, downregulation of Pclo in mouse islet inhibits insulin secretion (190). In mouse β-cells, Pclo/PCLO is expressed at levels 30–40% of Syt7/SYT7 in mouse and human β-cells (3, 68, 146, 475).
2. Molecular modulators of β-cell exocytosis
Insulin secretion presents some interesting variations on the basic theme of exocytosis outlined above, that may represent an adaptation to the β-cell’s role as a fuel sensor, and that involves proteins in addition to the canonical exocytotic proteins discussed above. These proteins are considered next and their sites of action are indicated in the schematic in FIGURE 16.
a) sur1 and epac2.
Interestingly, the KATP channel subunit SUR1 also appears to be required for insulin exocytosis. A large body of biochemical evidence indicates that the secretory granules contain SUR1 (204, 243, 497), and it has been reported to interact with the SNARE protein syntaxin-1 (503) (see sect. VA1). A role for SUR1 in exocytosis is also suggested by the finding of impaired glucose- and incretin-induced insulin secretion in SUR1 knockout mice (170). At the single-cell level, β-cells from these mice exhibit complete loss of an ultrafast cAMP-dependent component of exocytosis. The underlying mechanism is not clear, but there is evidence for a cross-talk between SUR1 and cAMP signaling. Using Sur1 as the bait in a yeast-two-hybrid screen for SUR1-binding partners led to the identification of Epac2 (Rapgef4/RAPGEF4) (496). Epac2 mediates the PKA-independent effects of cAMP on exocytosis by interacting with the Rab3 effector protein RIM2, which is critical for the docking and priming of insulin granules (see above). Interestingly, mouse β-cells express Epac2 at ~10-fold higher levels than their human counterparts (56).
Epac2 has also been identified as a sulfonylurea-binding protein (758), binding glibenclamide and tolbutamide (but not gliclazide). This suggests Epac2 mediates the reported effects of sulfonylureas on β-cell exocytosis (170, 171; but see Ref. 426). It was subsequently found that sulfonylureas and cAMP interact cooperatively to activate Epac2 (666) and thereby enhance insulin secretion (660). However, it should not be forgotten that KATP channel inhibition is essential for triggering sulfonylurea-stimulated insulin secretion, to elevate [Ca2+]i.
It is striking that most of the cytosolic agents that regulate the activity of the KATP channel also influence secretory granule exocytosis in β-cells. Thus it is tempting to speculate that SUR1 integrates metabolic signals not only to regulate KATP channel activity but also insulin granule exocytosis.
An acidic intragranular pH is important both for correct processing and maturation of insulin, and for insulin secretion (47, 649). Acidification depends on a vesicular proton pump (v-ATPase). In β-cells lacking the SUR1, no cAMP-induced acidification was observed, and exocytosis was reduced (170). Exactly how SUR1 influences intragranular pH and how, in turn, this modulates β-cell exocytosis has not been elucidated. Regardless of the exact mechanism, it is clear that H+ pumping requires a counter-ion, and this may account for the reported involvement of ClC3 transporters (389) and granular glutamate uptake in glucose- and incretin-stimulated insulin secretion (205, 419).
b) granuphilin.
Granuphilin (Sytl4; synaptotagmin-like 4) associates with the secretory granules and interacts with syntaxin-1a (686), Munc18–1 (130), and the small GTPase Rab27a (751). It appears to play a critical role in the docking of insulin granules at the plasma membrane, and genetic ablation of Sytl4 leads to a marked reduction in the number of docked granules (219, 457). Paradoxically, this is associated with stimulation of insulin secretion in response to high glucose or high extracellular K+, indicating that docking is not a prerequisite for exocytosis to occur (337) (see also sect. IXB2c).
Long-term (48 h) exposure of pancreatic islets to the nonesterified free fatty acid (NEFA) palmitate has been reported to increase Sytl4 expression and inhibit insulin secretion (342). Knockdown of granuphilin was found to partially restore glucose-induced secretion in palmitate-treated islets. These findings raise the interesting possibility that granuphilin may mediate the suppression of insulin secretion produced by chronic exposure to NEFA. SYTL4 expression in human β-cells is very low, yet long-term exposure to palmitate suppresses GIIS (288). It is possible that SYTL1, which is highly expressed, mediates the effect of NEFA in human β-cells (see Supplemental Figure 17, F and G).
c) rabphilin-3a and noc2.
Rabphilin-3 (encoded by Rph3a/RPH3A) is a secretory vesicle protein and a potential Rab3a effector (137). Downregulation of Rph3a reduces GIIS, suggesting that rabphilin-3a stimulates insulin release, an effect that does not require high-affinity Rab3 binding (318). However, although RPH3A is expressed at detectable levels in human β-cells, its expression in mouse β-cells is very low (see Supplemental Figure 15F). In contrast, Noc2 (Rph3al/RPH3AL; ‟rabphilin3a-likeˮ) is highly expressed in both human and mouse β-cells (see Supplemental Figure 15F). Isolated islets from Rph3al-deficient mice have dimished glucose- and depolarization-evoked secretion, effects that are rescued by pretreatent with pertussis toxin (436).
d) doc2β.
Double C2 domain (DOC2) proteins are soluble cytosolic proteins that are positive regulators of exocytosis (236). There are at least two mouse isoforms: Doc2A and Doc2B. These are both expressed at low levels in mouse and human β-cells (3, 68, 146, 475). In neurons, Doc2a and b translocate to the plasma membrane when [Ca2+]i is elevated (235) and promote vesicle fusion (534). A transgenic mouse overexpressing Doc2b has improved glucose tolerance and increased GIIS (mainly due to increased 2nd phase secretion). In contrast, ablation of Doc2a, Doc2b, or both Doc2a and Doc2b leads to deterioration of glucose tolerance. However, the effect of ablating either Doc2a or Doc2b alone on glucose-induced insulin release is marginal, both in vivo and in vitro, and even when both are ablated the reduction in secretion is limited to 20% (391).
4. Other proteins influencing exocytosis
a) caps.
Ca2+-dependent activator protein for secretion (CAPS) was initially discovered as a regulator of Ca2+-triggered large dense core vesicle fusion in PC12-cells (716). There are two forms of CAPS, CAPS-1 and CAPS-2, which are encoded by two separate genes (Cadps/CADPS and Cadps2/CADPS2). There is evidence that CAPS is involved in the ATP-dependent priming of the insulin granule (492).
Mouse and human β-cells express CAPS1 (Cadps/CADPS) and low amounts of CAPS2 (Cadps2/CADPS2) (3, 68, 146, 475). In Cadps2 knockout mice, 1st phase insulin secretion is unaffected but 2nd phase secretion is strongly reduced, consistent with the idea that the protein plays a role in granule priming (638). CAPS proteins are not required for insulin secretion and depolarization-evoked exocytosis proceeds even in their complete absence (i.e., in Cadps and Cadps2 double knockout mice) (638). In addition to playing a role in granule priming, CAPS proteins also influence insulin secretion by more indirect mechanisms. For example, CAPS proteins affect granule stability, and their ablation leads to increased crinophagy (638).
b) synapsin.
The synapsins were the first synaptic vesicle proteins to be identified (226). There are three synapsins, encoded by the genes Syn1–3/SYN1–3. They are thought to influence the availability of synaptic vesicles for release by controlling their trafficking and interaction with the cytoskeleton (101). In human β-cells, only SYN1 is expressed, whereas mouse β-cells express both Syn1 and Syn2 (3, 68, 146, 475). GIIS in Syn1*Syn2 double knockout mice is not different from that of wild-type mice, but insulin secretion evoked by high [K+]o is slightly reduced (726).
5. MicroRNAs
MicroRNAs (miRs) are small noncoding RNAs that regulate the translation of specific target proteins (200). Currently, >2500 miRs have been identified in humans of which >140 are present in human β-cells (352). A particular feature of miRs is that each one can influence the regulation of multiple genes. Because of this ‟promiscuity,ˮ miRs collectively influence the expression of at least 75% of β-cell genes. However, their precise mode of action is often not immediately evident and a combination of multiple experimental approaches is needed to dissect it.
Several miRs have been reported to affect insulin secretion and to target components of the secretory machinery. These include miRs 7, 9, 30, 33, 96, 124, 145, and 375 (reviews in Refs. 175, 186, 498). It is therefore appropriate to consider them in this section. This topic remains a work in progress, and it is likely that the list of miRs affecting exocytosis will expand as our understanding of their effects on secretion and exocytosis improves. It should be emphasized that in addition to the direct effects on exocytosis considered below, miRs may also affect insulin release indirectly via changes in electrical activity (due to altered expression of ion channels), insulin biosynthesis, or glucose metabolism.
a) miR-7.
MiR-7 is the second most abundant miR in human β-cells. GIIS is enhanced in miR-7a knockout mice (371), despite there being no obvious effect on [Ca2+]i or the number of docked granules. It was concluded that miR7 acts by influencing the release competence of docked granules. Interestingly, overexpression of miR7a resulted in reduced expression of the exocytotic protein complexin-1. This would be consistent with a role for complexin-1 in priming of the secretory granules.
b) miR-9.
MiR-9 is almost exclusively expressed in the brain and pancreatic islets. Overexpression of miR9 inhibits insulin secretion, an effect attributed to its ability to enhance expression of granuphilin (513).
c) miR-29.
MiR-29 is increased in β-cells exposed to elevated glucose, and it was proposed that this mediates the reduction of glucose-stimulated insulin secretion caused by long-term hypoglycemia via reduced expression of syntaxin-1 (186, 218).
d) miR-96.
Overexpression of miR-96 also inhibits insulin secretion. This effect may be due to increased expression of granuphilin (cf. effect of miR-9) (401).
e) miR-124.
In mouse insulinoma cells, increased expression of miR-124 stimulated basal insulin secretion and reduced GIIS. This was mediated by increased expression of SNAP-25 and Rab3a, and reduced expression of Noc-2 (401). Stimulation of basal secretion may be due to an increase in [Ca2+]i resulting from decreased KATP channel activity and stimulation of electrical activity (53). It has also been proposed that the increased expression of SNAP25 inactivates Kv2.1 voltage-dependent K+ channels (414), slowing action potential repolarization and increasing Ca2+ entry. However, the latter effect will not be important unless the β-cell is firing action potentials and would therefore not explain the increase in basal insulin secretion.
f) miR-375.
MiR-375 is the miRNA with the highest expression in islets and the first to be studied in detail (517). Increased expression of miR-375 inhibits insulin exocytosis by targeting myotrophin, a regulator of actin depolymerization, and (possibly as a consequence) exocytosis (see sect. IXB2c). miR-375 is also involved in adaptive β-cell expansion in response to insulin resistance (518).
C. Methods of Measuring Exocytosis
Traditional biochemical assays of insulin secretion allow (at best) measurements from individual islets with a temporal resolution as high as 20 s (61). However, elucidation of the cell biology of islet hormone secretion requires an even higher temporal resolution and ideally should be performed at the single-cell level.
There are now several methods that permit high-resolution studies of β-cell exocytosis, the secretion of insulin and other granule constituents, the events that precede exocytosis, and the fate of the granule following exocytosis. Here, we first briefly summarize the strengths and limitations of the various techniques and then discuss some of the findings and controversies that have emanated from these studies.
1. Single-cell and single-vesicle measurements of exocytosis
a) capacitance measurements.
Measurements of membrane capacitance report the increase in cell surface area that occurs when the secretory granule fuses with the plasma membrane (334, 505) (FIGURE 17A). This is because the electrical property of capacitance (the capacity to separate and store electric charge) is proportional to the area of the capacitor. Biological membranes act as capacitors and have a specific membrane capacitance of ~10 fF/μm2. A β-cell with a diameter of 15 µm will therefore have a capacitance of ~7 pF, close to that measured experimentally (221). Similarly, a secretory granule with a diameter of 0.35 μm can be estimated (assuming spherical geometry) to add ~4 fF to the cell capacitance upon fusion with the plasma membrane. The strengths of capacitance measurements include their high time resolution (~1 ms), the fact that cells are studied under voltage-clamp conditions, and the ability to load the cytosol with putative regulators of exocytosis, or Ca2+ indicators, via the recording pipette (see FIGURE 3D).
FIGURE 17.

A: capacitance measurements of exocytosis. Ca2+ influx triggered by a brief depolarization (ΔV) leads to the fusion of (five) secretory granules with the plasma membrane (gray). The resultant increase in membrane area can be detected as an increase in cell capacitance (ΔC) because cell capacitance (C) is proportionally related to cell surface area (A) [i.e., C = ε*A, where ε is the specific membrane capacitance (10 fF/μm2)]. For technical reasons, the recording is usually interrupted during the depolarization (illustrated schematically by the red trace). The net increase in cell capacitance (ΔC) that occurred during the pulse is shown by the black trace (334). B: schematic of on-cell (cell-attached) single-granule capacitance measurements and the equivalent circuit. Orange, green, and gray lines correspond to the plasma membrane, the granule membrane, and the walls of the recording pipette, respectively (not to scale). Gp, Cv, and Cp are fusion pore conductance, granule capacitance, and patch capacitance, respectively. [Modified from Lindau (397).] C: carbon fiber amperometry. A carbon fiber connected to an amplifier is placed in the vicinity of the cell. Exocytosis can be detected as amperometric current spikes (right) that develop when the substance released (e.g., serotonin) is oxidized by the high voltage (e.g., 0.65 V) applied to the carbon fiber giving rise to a rapid current transient (right). D: electrophysiological detection of ATP release. ATP is co-released with insulin and activates P2X2 receptors (P2X2Rs) in β-cells engineered to express such receptors. ATP release and activation of P2X2Rs result in rapid current transients.
Capacitance measurements of exocytosis are mostly conducted at the whole-cell level and the background noise is usually too high to resolve the small capacitance steps that result from fusion of individual secretory granules. However, by recording from small patches of membrane in the cell-attached mode, the signal-to-noise ratio increases sufficiently to allow single-vesicle exocytotic events to be resolved. Studies on human and mouse β-cells (253) have revealed that the 80–90% of capacitance steps belong to a single class of events with a mean magnitude of 3–5 fF, as expected for granules with a diameter of 0.3–0.4 μm. Cell-attached single-vesicle capacitance measurements have the additional benefit that they can provide information about the fusion pore, which connects the granule lumen with the extracellular space (FIGURE 17B).
Capacitance measurements also constitute a means to study endocytosis. This interesting topic is beyond the scope of this review. Suffice it to say that the bulk of endocytosis occurs by retrieval of small (endocytotic) vesicles with a size only 5–10% of the exocytotic events (200 aF vs. 2–4 pF) (412).
b) electrochemical detection of β-cell exocytosis.
A limitation of capacitance measurements of exocytosis is that they only report that an increase in cell surface area has occurred. It is therefore important that they are complemented by other means of monitoring exocytosis.
Carbon fiber amperometry affords the possibility of studying the kinetics of the emptying of the granule lumen (FIGURE 17C). It relies on the fact that oxidation of the compound at the tip of the carbon fiber electrode can be detected as an electric current. Insulin is not easily oxidized and is difficult to detect by this method, but both catecholamines and serotonin can be detected electrochemically (119). This technique has been successfully applied to rodent β-cells in which the granules have preloaded with serotonin (36, 74, 634, 663), and modified carbon fibers have been used to detect the release of insulin itself (294).
c) exocytosis monitored by ionotropic membrane receptors.
Exocytosis in β-cells not only results in insulin release. The secretory granules also contain a number of other substances, such as adenine nucleotides, glycine, glutamate, and GABA. The release of these compounds can be studied by ‟engineeringˮ β-cells to express ionotropic receptors (ligand-gated ion channels) in their plasma membrane. For example, β-cells have been genetically engineered for detection of ATP, GABA, or glycine release by adenoviral infection with ATP-activated P2X2 receptors (P2X2R) (484), GABAA receptors (GABAAR) (93), or glycine receptors (GlyR) (744). During exocytosis, these compounds are released into the extracellular space, diffuse over the cell surface, and bind to their receptors (FIGURE 17D). Activation of a large number of receptors situated close to the granule release site gives rise to a transient current similar to the postsynaptic currents of neurons. In this way, the β-cell acts as its own ‟sniffer cellˮ (195).
d) real-time measurements with a cell-based bioassay.
The release of granule contents can also be monitored using biosensor cells expressing metabotropic receptors coupled to intracellular Ca2+ mobilization, with an increase in [Ca2+]i indicating exocytosis. This method has been used to detect ATP release from β-cells (312), ACh and glutamate release from α-cells, and somatostatin release from δ-cells (557).
Insulin secretion has also been detected in real time using the phosphoinositide biosensor GFP4-Grp1 in conjunction with TIRF microscopy, to measure an increase in the plasma membrane level of phosphatidylinositol 1,3,5-trisphosphate (PIP3) (164). Following insulin secretion, insulin receptor activation in the β-cell membrane leads to PIP3 formation and thereby a transient increase in GFP4–GRP1 fluorescence.
e) imaging of exocytosis using tagged proteins.
Despite their improved spatial and temporal resolution, all the methods discussed above share the weakness of traditional biochemical assays of secretion in that they report only that exocytosis has occurred. They provide no information about the pre- and postexocytotic events. However, such information can be obtained from experiments in which the secretory granule membrane, and/or the granule ‟cargoˮ (the peptide content of the granules), is labeled with different variants of green fluorescent protein (GFP). Confocal imaging or TIRF microscopy is then used to measure changes in fluorescence (FIGURE 18, A and B). For example, by tagging the peptide cargo with the pH-insensitive fluorescent protein Emerald, granules inside the cell can be observed before exocytosis by their Emerald fluorescence, and cargo release can be detected as an abrupt loss of Emerald fluorescence (484) (FIGURE 18C). Likewise, by tagging the granule membrane protein VAMP2 with the pH-sensitive fluorescent protein pHluorin, opening of the fusion pore can be visualized by the resulting intragranular alkalinization (secretory granules normally have an acidic pH; Ref. 47), which increases pHluorin fluorescence (449).
FIGURE 18.

A: imaging of granules in a β-cell using conventional and two-photon confocal microscopy. Note that the multiphoton microscopy allows imaging within a thinner slice of the cell (because two photons must simultaneously excite the fluorophore, which is unlikely to occur outside the focal plane). Insulin or other proteins of interest are visualized by expression of GFP-tagged proteins. B: evanescent wave (TIRF) microscopy. Illumination of the specimen is restricted to a layer above the coverslip, only a few hundred nanometers thick, thus facilitating the study of events taking place in the immediate vicinity of the plasma membrane. C: TIRF imaging of granule exocytosis. Images were captured at 20 Hz (50-ms intervals) in a voltage-clamped β-cell held at −70 mV and depolarized to 0 mV as indicated above the images. Granules close to the membrane are seen as bright spots at −70 mV. Exocytosis on depolarization to 0 mV is seen as a brief flash of light followed by the rapid dissipation of fluorescence as the tagged protein diffuses away from the release site. D: imaging of exocytosis using a fluorescent fluid phase marker (e.g., sulforhodamine; abbreviated SRB). SRB occupies the thin space between adjacent cells (top). Exocytosis results in the fusion of granules with the plasma membrane, enabling SRB to enter the granule lumen, producing fluorescent invaginations that can be visualized by multiphoton confocal imaging (bottom). E: three examples (red arrows) of granules labeled with SRB that have undergone exocytosis in response to high glucose (20 mM). Scale bar: 3 μm. [From Hoppa et al. (289).] F: monitoring fusion pore expansion and exocytosis by using extracellular fluid space markers (e.g., 10 kDa Alexa-conjugated dextran; red dots) with the membrane label FM1–43 (green). Upon membrane fusion, FM1–43 (which has prelabeled the outer leaflet of the plasma membrane; dashed green line) labels the granule membrane via lateral diffusion. Entry of fluorescent dextran will only occur once the fusion pore has expanded sufficiently to accommodate dextran. G: schematic of parallel recordings of FM1–43 fluorescence (green trace) and dextran fluorescence (red trace). Note that FM1–43, measured by two-photon confocal microscopy within a rectangular square (superimposed on the leftmost granule), increases promptly upon exocytosis. Uptake of dextran is delayed relative to FM1–43 uptake and the fluorescence signal (measured within the rectangle) for both FM1–43 and dextran decrease when the granule membrane collapses into the plasma membrane (see Ref. 664).
f) imaging of exocytosis using polar fluid-phase tracers.
A potential problem with GFP-based methods of monitoring exocytosis and granule dynamics is that they involve the attachment of a large and bulky protein (22 kDa and 4–5 nm) to proteins involved in, or released by, exocytosis. There is some evidence that this may influence the kinetics of secretion (49, 445). These caveats must be borne in mind when interpreting the data. Use of the extracellular and polar fluid-phase tracers sulforhodamine B (SRB) or Alexa594, combined with two-photon microscopy (method illustrated in FIGURE 18, D and E), avoids many of these problems. In this assay, the extracellular space is labeled with the chosen tracer. As the space between two adjacent β-cells is very narrow (20–40 nm), the fluorescence signal is normally faint. Upon fusion of the secretory granule and opening of the fusion pore, the fluid phase tracer enters the granule space and labels the entire Ω-shaped structure, which has a diameter of 300–400 nm. This is detected as a fluorescent spot within ~5 s of fusion (FIGURE 18, D and E). When the granule membrane subsequently collapses into the plasma membrane, the fluorescence gradually returns towards baseline with a variable time course (ranging from a few seconds to 20–30 s). By using extracellular space markers of different molecular weights or dimensions, in conjunction with the membrane probe FM1–43 that inserts into the outer leaflet of the membrane, this technique can also be used to estimate the diameter of the fusion pore connecting the granule lumen to the extracellular space and to assay the kinetics of cargo release (289, 664) (FIGURE 18, F and G).
g) insulin secretion monitored as zn2+ release.
Insulin secretion can also be monitored as an increase in extracellular Zn2+, which parallels insulin secretion. Insulin is stored as a Zn2-insulin6 crystal, whose stability depends on the pH (36). In this method, the β-cell plasma membrane is first labeled with the Zn2+ indicator ZIMIR. Zn2+ co-released with insulin binds to ZIMIR and thereby increases its fluorescence (388). This technique permits spatially resolved measurements of release but does not currently have sufficient sensitivity to detect exocytosis of individual granules.
D. Cell Physiology of Insulin Exocytosis
The application of the battery of the high-resolution techniques described above (FIGURES 17 and 18) has resulted in a wealth of cell physiological data. Here we review these findings and attempt to correlate them with the whole-body, whole-islet, and gene expression data summarized in the preceding sections.
1. Kinetics of β-cell exocytosis
Most measurements of β-cell exocytosis (whether by amperometry, optical imaging or recordings of membrane capacitance) have been performed in isolated β-cells, whereas insulin secretion itself is normally measured in intact islets. In this section we therefore consider to what extent studies of the properties of exocytosis in isolated β-cells maintained in tissue culture can be extended to β-cells in the intact and acutely isolated pancreatic islet.
a) isolated β-cells versus β-cells in intact islets.
In capacitance measurements, depolarization-evoked exocytosis results in step increases in membrane capacitance (FIGURE 19A AND B). Exocytosis is triggered by depolarizations above −30 mV, is maximal around 0 mV, and then undergoes a secondary decrease at more positive voltages (FIGURE 19C). This ∩-shaped voltage dependence mirrors that of depolarization-evoked Ca2+ entry (see FIGURE 9) and the associated elevation of [Ca2+]i (14). In isolated β-cells, peak rates of exocytosis obtained from capacitance measurements are in excess of >0.5 pF/s (i.e., 50 fF in 100 ms; corresponding to >100 granules per second), and responses plateau within 100–200 ms (FIGURE 19C). When measured in β-cells within intact islets, exocytosis becomes detectable during a 40-ms depolarization, does not plateau, and operates at a maximum rate of 50 fF/s (222), only ~10% of that seen in isolated β-cells. The rate of capacitance increase observed for β-cells in intact islets agrees well with that of insulin secretion measured biochemically (222). Thus measurements of cell capacitance in β-cells within intact islets might be more physiologically relevant than those from isolated β-cells.
FIGURE 19.

A: relationship between pulse duration and exocytotic response (measured as depolarization-evoked increases in cell capacitance) in isolated β-cells (red trace) and in β-cells within intact acutely isolated islets (black trace). Measurements were performed in the presence of 0.1 mM intracellular cAMP to potentiate depolarization-evoked exocytosis. [Modified from Eliasson et al. (170) and Göpel et al. (222).] B: example of capacitance increase elicited by a 300-ms depolarization from −70 mV to 0 mV in a β-cell within an intact islet. Note that the capacitance increase is restricted to the depolarization and that there is little sign of exocytosis continuing beyond the depolarization. [Modified from Göpel et al. (222).] C: voltage dependence of exocytosis in β-cells in intact islets. The ∩-shaped voltage dependence mirrors that of the voltage-gated Ca2+ current (cf. FIGURE 9). The gray rectangle indicates the approximate voltage range of the β-cell action potential. Note that exocytosis is steeply voltage-dependent between −20 and 0 mV. D: ‟active zoneˮ of elevated [Ca2+]i produced by Ca2+ channel opening is restricted to the vicinity of the channel. In the resting (hyperpolarized) state, the Ca2+ channels are closed, submembrane [Ca2+]i is low, and exocytosis of secretory granules (SG) cannot proceed. Upon membrane depolarization, Ca2+ channels activate, [Ca2+]i increases to very high levels close to the inner mouth of the Ca2+ channels (red zone), and exocytosis is initiated. When the membrane potential is subsequently repolarized, Ca2+ channels instantly close, the active zone quickly collapses and [Ca2+]i rapidly falls below that required to trigger exocytosis, so insulin release ceases.
It is unlikely that the low rate of β-cell secretion in intact islets is due to paracrine inhibition (for example, exerted by somatostatin released from neighboring δ-cells) as pretreatment with pertussis toxin (which blocks inhibitory G proteins) only marginally increased the rate of exocytosis (222). There is also no evidence to suggest that the difference arises because β-cells in intact islets contain fewer near-membrane (i.e., docked) granules (see sect. IXD5). The reduced exocytotic capacity of β-cells in intact islets is likewise not the consequence of reduced Ca2+ entry: if anything, the Ca2+ channel density is greater in β-cells in intact islets (223). Thus the reason why exocytosis in isolated β-cells proceeds at much higher rates than observed in the intact tissue remains a mystery. Nevertheless, the discrepancy may have important implications for understanding the molecular regulation of insulin granule exocytosis (see discussion in sect. IXD6).
2. The Ca2+ channels are tethered to the secretory granules
Depolarization-evoked exocytosis in mouse β-cells is restricted to the duration of the depolarization (when the Ca2+ channels are open) (FIGURE 19B). Although [Ca2+]i remains elevated for several seconds following repolarization, exocytosis stops immediately. This behavior is expected if exocytosis is regulated by the high [Ca2+]i close to the Ca2+ channels. The immediate cessation of exocytosis upon repolarization probably reflects the rapid collapse of active zones of very high [Ca2+]i when the Ca2+ channels close (FIGURE 19D).
Genetic ablation of Cav1.2 L-type Ca2+ channels selectively abolished 1st phase GIIS while 2nd phase release remained intact. The kinetics of depolarization-evoked exocytosis suggests that insulin secretion depends on the close physical proximity of the secretory granules to the Ca2+ channels. There is evidence that Ca2+ channels are physically anchored to the secretory granules (FIGURE 20). This interaction is mediated by residues 753–893 of the Cav1.2 Ca2+ channel, which correspond to the intracellular loop between the second and the third homologous domain of the α1-subunit (731). This region of the channel is termed the synprint peptide (synaptic protein interaction peptide). It interacts with syntaxin-1, SNAP25 and synaptotagmin. Based on the length of the synprint peptide, it has been estimated that the secretory granules must reside within 10 nm from the inner mouth of the Ca2+ channel (48) (FIGURE 20A).
FIGURE 20.

Tethering of voltage-gated Ca2+ channels to secretory granules. A: SNAREs bind to the II-III loop of the L-type Ca2+ channel, the synaptic protein interaction (synprint peptide), and thereby tether the granule close to the inner mouth of the channel. Upon Ca2+ channel activation, the exocytotic machinery becomes exposed to a localized increase in [Ca2+]i. These transients are too rapid to be buffered by slow Ca2+ chelators like EGTA, explaining why depolarization-evoked exocytosis is resistant to this Ca2+ buffer. B: following the addition of a large excess of the ‟synprint peptide,ˮ the endogenous synprint peptide (which is part of the Ca2+ channel) is competitively displaced, leading to the disassembly of the granule-channel complex. Although Ca2+ channel activity is unperturbed, the secretory granule is no longer sufficiently close to the inner mouth of the Ca2+ channel (where [Ca2+]i exists at exocytotic levels), leading to the suppression of insulin release. C: the rapid Ca2+ chelator BAPTA binds Ca2+ so quickly that even granules tethered to the inner mouth of the Ca2+ channels are not exposed to Ca2+ concentrations high enough to evoke secretion (right). Figure courtesy of Professor E. Renström, Lund.
The importance of the interaction between the Ca2+ channels and secretory granules for rapid exocytosis is illustrated by the selective loss of rapid exocytosis following infusion of an excess of recombinant synprint peptide into the β-cell cytosol (731) (FIGURE 20B). This competes for endogenous synprint, preventing interaction of the Ca2+ channel with the secretory vesicle. Exocytosis evoked by a train of depolarizations (48) or photorelease of caged Ca2+ (731), two conditions that can be assumed to produce a high [Ca2+]i throughout the β-cell, was unaffected by the synprint peptide.
Regulation of rapid insulin granule exocytosis by [Ca2+]i close to the Ca2+ channel would be consistent with the observed low Ca2+ sensitivity of exocytosis (tens of micromolar) and the finding that 1st phase (but not 2nd phase) insulin secretion is resistant to introduction of the slow Ca2+ chelator EGTA but is inhibited by the fast chelator BAPTA (510) (illustrated schematically in FIGURE 20C).
In isolated human β-cells, exocytosis proceeds beyond the end of the depolarization (90, 91) (FIGURE 21B), and exocytotic responses elicited by short depolarizations are much smaller than in mouse β-cells (FIGURE 21A). The kinetics of ‟asynchronousˮ exocytosis in human β-cells largely mirrors the depolarization-evoked [Ca2+]i transients (FIGURE 21B). These differences might indicate differences in Ca2+-dependent exocytosis between mouse and human β-cells. Whereas human β-cells appear to rely exclusively on high-affinity Ca2+ sensors, low-affinity Ca2+ sensors may play a more prominent role in mouse β-cells. It is perhaps relevant in this context that mouse, but not human, β-cells express the low-affinity synaptotagmins Syt2 and Syt9, albeit at much low levels than Syt7 and Syt5 (see Supplemental Figure 18A).
FIGURE 21.

A: relationship between exocytosis (measured as the depolarization-evoked increase in membrane capacitance) in isolated human β-cells (solid black line) and isolated mouse β-cells (dashed red line). Responses have been normalized to exocytosis elicited by a 500-ms depolarization. Note that depolarizations <100 ms evoke only small increases in capacitance in human β-cells. In β-cells within intact human islets, there is an additional, rapid component of exocytosis (568). Measurements were performed in the presence of 0.1 mM cAMP. [Modified from Braun et al. (90) and Eliasson et al. (170).] B: example of capacitance increase (ΔCm, red trace) elicited by a 500-ms depolarization in an isolated human β-cell. Note that in human β-cells, unlike mouse β-cells (FIGURE 19B), exocytosis continues for ~500 ms beyond the end of the pulse (gray rectangle). The dashed red line represents a linear extrapolation of the postdepolarization exocytosis to the start of the depolarization. The blue trace shows the decay of depolarization-evoked [Ca2+]i recorded in a β-cell following membrane repolarization. The dashed line superimposed on the capacitance trace is the inverted [Ca2+]i signal. This type of “asynchronous” exocytosis suggests that exocytosis in human β-cells is determined by the bulk rather than the local (near-membrane) [Ca2+]i. [Modified from Braun et al. (90) and Rorsman et al. (563).]
3. The readily releasable pool, the reserve pool, and granule priming/depriming
Secretory granules in β-cells belong to (at least) two functionally distinct pools: the readily releasable pool (RRP) and the reserve pool (RP) (472). The RRP includes granules that are biochemically prepared for release (i.e., ‟primedˮ granules). Granules belonging to the reserve pool have not yet acquired release competence and must undergo a series of ATP-, Ca2+- and temperature-dependent reactions (‟mobilizationˮ or ‟primingˮ) before exocytosis (571). This may involve physical translocation of the granules within the cell to the plasma membrane but could also result from priming of granules already in place below the plasma membrane.
A pancreatic β-cell contains ~10,000 secretory granules, but only a fraction (1–2%, i.e., 100–200 per β-cell) of these are immediately available for release. These granules are defined as the RRP (489). When islets are depolarized with glibenclamide (FIGURE 14B) or high [K+]o (FIGURE 23C), insulin secretion declines to basal within 5 min. Interestingly, high-[K+]o depolarization was associated with a 30% reduction in the number of near-plasma membrane (docked) granules, from 650 to 450 per β-cell (489). The reduction in the number of docked granules (~200) is similar in size to the RRP. Importantly, no depletion of docked granules occurred when islets were depolarized in the presence of glucose. Rather than rapidly declining to basal levels, insulin secretion elicited by the combination of high-[K+]o depolarization and glucose exhibited a sustained 2nd phase insulin secretion (268). Indeed, when glucose is added to islets stimulated by glibenclamide or high [K+]o after insulin secretion has almost declined to basal levels, a secondary slowly developing acceleration of insulin secretion is observed (FIGURES 14B and 23C). Collectively, these observations suggest that 1) depolarization-induced insulin secretion involves granules situated close to the plasma membrane and 2) glucose stimulates the replenishment of the docked pool and the RRP.
The mechanisms by which [Ca2+]i and glucose/ATP stimulate exocytosis have been studied using capacitance measurements and photorelease of caged Ca2+. A step increase in [Ca2+]i in the presence of intracellular ATP elicits a biphasic response: an initial rapid increase in capacitance increase being followed by a sustained slower increase (172). The initial component was attributed to the release of the RRP, i.e., granules that have already acquired release competence by ATP-dependent priming and that can be released without further ATP consumption. The slower component of capacitance increase reflects the time- and ATP-dependent mobilization of granules.
ATP stimulates insulin granule exocytosis in a concentration-dependent fashion, with half-maximal efficacy (EC50) at 0.5 mM (492). A similar ATP dependence of the rapid component of β-cell exocytosis has been detected in experiments involving photorelease of caged Ca2+ and amperometric detection of granule release (662). This ATP dependence probably means that, in vivo, ATP is already present at concentrations high enough to sustain exocytosis even at low glucose levels, and it is therefore unlikely that ATP itself exerts any modulatory effect on exocytosis. However, exocytosis is reduced by increasing concentrations of ADP with an IC50 of ~0.3 mM. At ADP concentrations >1 mM, depolarization-evoked exocytosis in the presence of 3 mM ATP was reduced by 80%. ADP does not affect the release of RRP granules but interferes with granule mobilization/priming (45). Thus changes in the cytoplasmic near-membrane ATP/ADP ratio can be expected to influence insulin granule exocytosis and GIIS. This may explain the absence of 2nd phase insulin secretion in islets stimulated with high [K+]o or sulfonylureas at low glucose.
ATP-dependent priming occurs with a latency as short as <400 ms (172). Given that granule mobility is 1.5 μm/s (49, 702), this suggests that an increase in the intracellular ATP/ADP ratio increases exocytosis principally by priming of granules that are already situated close to the plasma membrane (within ~0.5 μm). Exactly how ATP and ADP influence exocytosis is not known, but there is evidence that they act by modulating phosphatidylinositol 4-kinase (PI 4-kinase), PI 3-kinase, and CAPS (492).
4. Ca2+ channel clustering and domain overlap
The maximal Ca2+ current amplitude in isolated mouse β-cells is ~100 pA, and the number of voltage-gated Ca2+ channels per β-cell has been estimated as ~500 by nonstationary fluctuation analysis (48). Based on a specific membrane capacitance of 10 fF/μm2 and a whole-cell capacitance of ~5 pF, this corresponds to a Ca2+ channel density of 1 Ca2+ channel/μm2 (182). This is only 5–10% of that found in adrenal chromaffin cells, where the Ca2+ channel density is as high as 20 channels/μm2 (45). It is therefore perhaps surprising that depolarization-evoked exocytosis (at least in isolated β-cells; see sect. IXD1a) operates at speeds comparable to those seen in other endocrine cells. Since exocytosis is triggered with a latency of 10 ms and the mean open time of the Ca2+ channel is only 2 ms (45), the likelihood that opening of a single channel would be sufficient to increase [Ca2+]i high enough to elicit exocytosis would seem to be low. However, it turns out that the Ca2+ channels are not evenly distributed in the β-cells and that they aggregate into clusters that associate with the secretory granules: single-channel recordings revealed that whereas most membrane patches contain no active Ca2+ channels, other patches contained three or more active channels, suggesting that their distribution is nonstochastic (48). Clustering of Ca2+ channels has also been documented by recording near-membrane depolarization-evoked [Ca2+]i transients (FIGURE 22A) (288).
FIGURE 22.

A: TIRF imaging of near-membrane [Ca2+]i increases in a single voltage-clamped β-cell stimulated by a single 50-ms depolarization from −70 to 0 mV. The cell was infused with EGTA (10 mM) to restrict intracellular diffusion of Ca2+. Changes in [Ca2+]i are displayed in pseudocolors with black/blue and yellow/red corresponding to very low and high concentrations, respectively. Scale bar: 2 μm. Note that Ca2+ entry is not uniform but restricted to many ‟hotspotsˮ (red). [Modified from Hoppa et al. (288).] B: schematic showing how spatial [Ca2+]i domains overlap. Resting: the membrane contains four Ca2+ channels (green). Three of these channels sit beneath a secretory granule (the outline of which is indicated by dashed circle). Depolarization leads to localized Ca2+ entry and [Ca2+]i increases within spatially restricted domains (red). If the Ca2+ channels sit close to each other, these domains overlap. C: spatiotemporal domain overlap. Changes in [Ca2+]i at four different locations, as indicated in B. During the depolarization (gray area), the individual Ca2+ channels open and close stochastically and [Ca2+]i (traces 1–3) echoes Ca2+ channel activity. Individually, the [Ca2+]i are too brief to evoke exocytosis, but a sufficiently long elevation is generated by spatiotemporal domain overlap (location/trace 4).
Clustering of Ca2+ channels has, due to spatiotemporal [Ca2+]i domain overlap (FIGURE 22, B and C), dramatic effects on the likelihood of a granule being exposed to elevated Ca2+ for long enough (10 ms) to evoke exocytosis: from 0.5% for individual Ca2+ channels to ~20% for a triplet of Ca2+ channels (48).
Ca2+ channel clustering has also been visualized by attaching fluorescent tags to Ca2+ channel subunits (e.g., the α1-subunit) and performing single-molecule tracking (198). Such studies suggest that clusters of ~10 Ca2+ channels are found near secretory granules about to undergo exocytosis. These clusters disappear in cells overexpressing the synprint peptide, suggesting that their formation is mediated by interaction with the exocytotic machinery (198). Interestingly, Ca2+ channel clustering is absent following chronic exposure to palmitate (a diabetogenic condition) or in glucose-intolerant high-fat-fed mice (123). In contrast, acute exposure to palmitate did not prevent clustering (198), and instead led to a stimulation of exocytosis, via an increase in the readily releasable pool of granules (491). T2DM is also associated with dispersion of Ca2+ channel clusters, and the resultant loss of efficient domain overlap may thus contribute to the reduction of insulin secretion (198).
Protein aggregates are usually found in cholesterol-rich membrane regions (lipid rafts). It is possible that the membrane lipid composition is altered in T2DM, obesity, or following chronic elevation of circulating free fatty acids and that this leads to the disruption of lipid rafts. Indeed, depletion of membrane cholesterol using methyl-β-cyclodextrin (MBCD), or inhibition of cholesterol biosynthesis, leads to reduction of voltage-gated Ca2+-currents, depolarization-evoked exocytosis, and GIIS (710, 738).
5. Docked granules versus newcomer granules
Whereas the docked pool is defined morphologically and ultrastructurally as the granules that are situated immediately beneath the plasma membrane, the RRP is defined functionally as the subset of granules that are release-competent and immediately available for release (see sect. XD3). As discussed above, ultrastructural studies indicate that β-cells contain ~600 docked granules (489), corresponding to a granule density of ~1 granule/μm2. Similar values for granule density have been obtained by live-cell imaging using fluorescently tagged granules: 0.8 granule/μm2 in mouse β-cells (288) and 0.7 granules/μm2 in human β-cells (447). In the latter studies, most of these near-membrane granules were immobile, thus confirming they were functionally as well as morphologically docked (49, 288).
There is general agreement that many docked granules are released in response to membrane depolarization produced by sulfonylureas or high [K+]o (288, 446, 487, 617). There is also agreement that 2nd phase GIIS largely reflects the release of ‟newcomerˮ granules (FIGURE 23A) rather than granules that are initially docked below the plasma membrane (610). However, there is considerable controversy over what type of granules are released during 1st phase GIIS. It has variably been attributed to release of either docked (487), newcomer (617), or even granules that undergo exocytosis immediately upon arrival at the plasma membrane without docking at all [termed ‟restless newcomersˮ (610) or ‟crash fusionˮ (661)] (FIGURE 23A).
It is not immediately clear how the docked insulin granules in mouse β-cells discriminate between Ca2+ entry induced by membrane depolarization evoked by high-[K+]o or by high glucose. One possible explanation is that they do not and that the difference simply reflects the different time courses by which these two stimulation paradigms initiate insulin secretion. Whereas high [K+]o stimulates insulin secretion instantly, glucose only stimulates insulin secretion after a delay of several minutes (FIGURE 14B). A problem inherent with the use of fluorescent proteins is that their fluorescence fades upon illumination. In TIRF microscopy, the energy of the excitation light decays exponentially from the surface of the coverslip on which the cells are mounted. It is possible that the fluorescence of the docked granules (i.e., those immediately adjacent to the coverslip) bleaches so much between the time of glucose addition and the onset of secretion that they are effectively invisible at the time exocytosis occurs (FIGURE 23B) (197). In contrast, when secretion is triggered by high-[K+]o depolarization, the latency between application of the stimulus and the onset of secretion is so short that the docked granules remain visible. In both cases, once the granules have fused and the fluorescent cargo is released into the space between the cell membrane and the coverslip, the fluorescence will increase dramatically and become observable even for granules in which the pre-exocytotic fluorescence may have decayed below the detection level (FIGURE 23B).
One way of testing whether glucose and high-[K+]o stimulate release of the same, or different, subsets of granules would be to measure GIIS with or without prior stimulation with high-[K+]o. In the perfused mouse pancreas, increasing glucose from 1 to 20 mM glucose triggers biphasic insulin secretion (FIGURE 14A). With the use of the same preparation, high-[K+]o stimulation (35 mM) only provokes a rapid transient stimulation of insulin secretion equivalent to 1st phase release (FIGURE 23C). The transitory nature of high-[K+]o evoked secretion presumably reflects depletion of a rapidly releasable/docked pool of granules that is not replenished. If high glucose (20 mM) is subsequently applied in the continued presence of high [K+]o, the response merely consists of a slowly developing increase in insulin secretion. This is presumably because it is dependent on glucose metabolism. The slow increase induced by subsequent glucose application likely reflects the time-dependent priming or physical translocation of secretory granules.
In contrast to what has been reported for mouse β-cells (617), there is agreement that 1st and 2nd phase exocytosis in human β-cells reflects the release of docked and newcomer granules, respectively (197, 447). This agrees with the fact that high glucose does not evoke biphasic insulin secretion when applied subsequent to high-[K+]o depolarization (266).
6. SNARE-independent granule docking?
It would be a mistake to equate granule docking with priming of the SNARE complex. Indeed, advanced imaging data from β-cells within intact pancreatic islets suggest the two processes are distinct. Using a fluorescence resonance energy transfer (FRET) probe that can report the conformational state of the t-SNARE SNAP25, it was shown that the SNARE complex exists in two different conformations: a tight (high-FRET) and a loose (low-FRET) conformation (661). The conformational state predicts the granules’ readiness to undergo exocytosis. The high-FRET state was associated with rapid exocytosis (visualized by Alexa 594 uptake into the granule lumen; see FIGURE 18, D and E). Conversely, in the low-FRET state, there was a significant delay (of up to 10 s) between elevation of [Ca2+]i and exocytosis, and an increase in FRET preceded fusion indicating the complex underwent a conformational change. In a subsequent study, the interaction between plasmalemmal t-SNAREs (syntaxin-1a or SNAP25) and the granular v-SNARE VAMP2 was studied using a similar FRET-based approach. It was found that FRET increased, indicating assembly of the SNARE complex, 2–10 s before exocytosis. Thus, in β-cells, the SNAREs are unassembled in the resting state and assemble only shortly before exocytosis (665). In contrast, in neurons the SNARE complex is preassembled in the resting state, enabling ultrafast exocytosis. The lack of a preassembled SNARE complex in β-cells in intact islets is consistent with the relatively slow time course of exocytosis in acutely isolated intact islets (222) (see sect. IXD1a).
Assembly of the SNARE complex has also been studied in INS1-cells (196). In these cells, granule docking involves the clustering of the syntaxin-1 and munc18 from a pool of freely diffusible molecules. Additional proteins required for exocytosis (including munc13 and SNAP25) are then recruited to the incipient release site (196). Thus it is the arrival of the secretory granule at the plasma membrane that initiates and orchestrates the building of the release site. Only 30% of docking attempts are successful. The granules that successfully dock are decorated with Rab3a, whereas those that do not dock lack this protein (196). Once the granules have successfully docked, Ca2+ channels are recruited to the release sites (198). However, this often occurs with a long delay. Following exocytosis, the exocytotic machinery is quickly disassembled, and its components may be recycled.
Taken together, these data suggest that there are no preexisting docking sites in β-cells (198); rather, they are assembled on demand. This is consistent with the lack of obvious active zones like those found in neurons, where Ca2+ channels and secretory granules congregate at high density. Moving the Ca2+ channels to the secretory granules rather than the reverse seems energetically favorable for the cell. Although both SNARE proteins and Ca2+ channels are fairly large (50–250 kDa), they are dwarfed by size of the insulin secretory granule (300 nm, containing 1.6 amol or 8 fg insulin, equivalent to 5,000 MDa).
Clearly, even if the RRP of insulin granules is not molecularly defined by the presence of a preassembled SNARE complex (665), it remains possible that the release sites have another molecular ‟signature.ˮ There is evidence that insulin granule exocytosis is targeted towards the capillaries and that the ‟scaffoldingˮ proteins RIM2 and piccolo (but not the SNARE proteins) localize to these regions (403). As discussed in section IXB2b, these proteins interact with Rab3a. This finding, taken together with the observation that only those granules decorated with Rab3a successfully dock, suggests that a Rab3a/RIM2 interaction plays a key role in the tethering of the secretory granules to the plasma membrane.
7. Modulation of exocytosis by glucose, hormones, and neurotransmitters
Glucose, hormones, and neurotransmitters modulate exocytosis by a distal effect on exocytosis exerted at a level beyond electrical activity and the increase in [Ca2+]i. These effects are rapid and are therefore likely to be mediated by a direct effect on the release competence of granules already situated close to the plasma membrane. Regulation of insulin secretion at a late step of the stimulus-secretion pathway seems teleologically rational, as it enables exocytosis to rapidly change when glucose demand precipitously rises (as during exercise) or increases (during feeding).
In this section, we consider the modulation of exocytosis by glucose, hormones and neurotransmitters.
a) amplifying effect of glucose.
Glucose has been shown to amplify Ca2+-induced insulin secretion by a direct effect on the secretory machinery (203, 264, 595). The identity of the amplifying factor(s) has been a matter of much debate. Changes in the intracellular ATP/ADP ratio may be involved, but it seems unlikely that this changes sufficiently under physiological conditions to be functionally relevant and a high ATP/ADP ratio may therefore perhaps be better regarded as a permissive factor. Instead, accumulating data point to an important role for NADPH (307). In both mouse and human β-cells there is evidence that this modulation involves the reversible attachment of small ubiquitin-like modifier protein 1 (SUMO-1) to synaptotagmin-7. Glucose increases exocytosis by NADPH-dependent activation of the deSUMOylating enzyme SENP-1 (136). Indeed, mice lacking SENP-1 are hyperglycemic, have impaired GIIS in vivo and in vitro, and have a 50% reduction of the glucose-induced amplification of insulin secretion (183). NADPH derived from the activity of cytosolic isocitrate dehydrogenase (Idh1) plays a key role in the process. Intriguingly, defective insulin secretion in islets from T2DM islets could be restored by introduction of isocitrate, NADPH, or GSH (183). Notably, the amplifying effect of glucose (via desumoylation of synaptotagmin-7) can occur without physical translocation of secretory granules (136).
b) effects of potentiators and inhibitors.
Many potentiators of insulin secretion exert a direct effect on late steps of exocytosis in addition to stimulating β-cell electrical activity. For example, GLP-1 stimulates exocytosis via Epac2 (170) and by cAMP-dependent activation of PKA, which phosphorylates synaptotagmin-7 at serine-103 (735). In addition to this late effect on exocytosis, cAMP/PKA also acts upstream to accelerate granule mobilization (see sect. IXD8a).
Inhibitory agonists (like somatostatin and adrenaline) also act on late steps of exocytosis to reduce insulin release, for example, by depriming of granules (240, 543). Somatostatin (as well as many other inhibitory agonists) inhibits adenylate cyclase, and it has been proposed that its inhibitory effects on exocytosis may, in part, be mediated via a reduction of intracellular cAMP (438). However, somatostatin remains capable of inhibiting exocytosis and insulin secretion even when intracellular cAMP levels are clamped at 0.1 mM or when the extracellular medium is supplemented with dibutyryl cAMP (139). Thus a reduction of intracellular cAMP is not an obligatory component of the inhibitory mechanism. There is some evidence that somatostatin’s inhibitory effect on exocytosis is mediated by activation of the protein phosphatase calcineurin (543), possibly by dephosphorylation of the same serine residue in Syt7 that is phosphorylated in the presence of GLP-1 (735).
8. Mobilization and refilling of the RRP
The RRP amounts to 100–200 granules. It represents just a subset (~20%) of the 500–600 docked granules and only 1–2% of the total number of secretory granules in the β-cell. Once the RRP has been depleted, exocytosis stops unless the RRP is replenished by mobilization of granules from the reserve pool. In this section we consider how quickly the RRP can be replenished following depletion, the mechanisms involved, and the contributions of the RRP and granule mobilization to biphasic GIIS.
Capacitance measurements have revealed that following depletion of the RRP, as evidenced by the cessation of exocytosis despite continued stimulation, refilling of the RRP follows an exponential time course with a time constant of ~30 s (241). Granule mobilization is accelerated by cAMP (via activation of PKA) (545) and by Ca2+ (via activation of CAMKII) (241). Importantly, mobilization may expand the RRP beyond its original size. When β-cells were exposed to forskolin (to increase cAMP and activate both PKA and Epac2) or to ACh (to mobilize intracellular Ca2+ and activate CAMKII), the RRP increased three- to fourfold relative that seen under control conditions (241, 545). This enhanced 1st phase insulin secretion in response to a subsequent challenge with glucose, tolbutamide, or high [K+]o (274), as expected if the size of the RRP determines 1st phase insulin secretion.
Although the granules situated just below the plasma membrane are sufficient to sustain insulin secretion for many hours (see sect. IXA), ultimately new granules must be physically translocated to the plasma membrane. Granule mobility is very low in the absence of ATP, but increases with a rise in the intracellular ATP concentration (57, 702). In rodent β-cells, granule translocation occurs along microtubules in a process propelled by kinesins and controlled by the cytoplasmic ATP/ADP ratio (703). The final approach towards the plasma membrane is influenced by the submembrane actin web (721). This normally restricts exocytosis, as evident from the observation that treatment of islets with cytochalasin B potentiates GIIS. Glucose-induced remodeling of the submembrane actin network is required for the full insulin secretory response. It depends on glucose metabolism and is controlled by small GTPases such as Cdc42 (24, 720). Activation of Cdc42 leads to activation of ras-related C3 botulinum toxin substrate 1 (Rac1), an effect that is mediated by p21-activated kinase 1 (PAK1) (720). Rac1 is a small Rho family GTPase involved in the regulation of cytoskeletal reorganization and vesicular traffic (83). Genetic knockdown of PAK1 and Rac1 leads to impaired 2nd phase insulin secretion (24, 720). Interestingly, there is evidence that PAK1 is reduced by 80% in islets from patients with T2DM (719). Glucose stimulation also promotes the interaction of Cdc42 and N-WASP (neuronal Wiskott-Aldrich Syndrome protein). Active N-WASP promotes actin polymerization (G actin → F-actin), and this in turn facilitates the approach of the secretory granules to the plasma membrane (699).
Although the concept that the secretory granules are delivered to specialized release sites and are guided there by cytoskeletal tracks is an attractive idea, recent observations in PC12 neuroendocrine cells suggest a different interpretation: that the path the vesicles follow to the release sites is determined by random (Brownian) movements between clusters of nonreactive and reactive (ready to sustain docking/exocytosis) SNARE proteins in the plasma membrane. This gives rise to the impression of directed/organized vesicle navigation (161).
FIGURE 24, A and B, depicts schematically the relationship between granule pools, mobilization, and biphasic insulin secretion. We favor the idea that 1st phase insulin secretion involves release of RRP granules, many of which are situated at the plasma membrane. Depletion of this pool combined with reduced Ca2+ channel activity, either because of Ca2+-dependent inactivation or reduced action potential height, underlies the cessation of 1st phase insulin secretion. Once this has occurred, new granules must be ‟mobilizedˮ for release. This may involve ‟primingˮ of granules already in place but also active (directed by the cytoskeleton) or passive (due to Brownian movement) translocation of granules towards the plasma membrane. In addition, Ca2+ channel redistribution occurs. This provides a mechanism by which granules that have attained release competence can undergo release even if they are (initially) not situated next to a voltage-gated Ca2+ channel.
FIGURE 24.

A: schematic of glucose-induced biphasic insulin secretion. The numbers 1, 2, and 3 indicate the onset of 1st phase secretion, the end of 1st phase secretion, and steady-state 2nd phase insulin secretion, respectively. The green and red areas correspond to the release of docked granules and mobilization of granules, respectively. B: at the onset of 1st phase insulin secretion (1), exocytosis principally involves docked granules situated close to voltage-gated Ca2+ channels. The depletion of this granule pool, together with inactivation of voltage-gated Ca2+ channels (from 3 to 1 active channels in the schematic, indicated by the lack of arrows through Ca2+ channels), account for the transient nature of 1st phase insulin secretion (2). The 2nd phase insulin secretion (3) involves the priming of granules already docked with the plasma membrane but that had previously not acquired release competence (going from red to green: i), physical translocation of granules from within the β-cell (ii), and redistribution of the voltage-gated Ca2+ channels within the plasma membrane (iii).
9. Insulin release, the fusion pore, and kiss-and-run exocytosis
Whereas we know a lot about how insulin secretion is initiated and the processes that precede exocytosis, we know far less about the final steps, i.e., how insulin is delivered to the extracellular space. Within the secretory granule, insulin is usually stored as a crystal of insulin hexamers stabilized by two Zn2+ and the acidic pH of the secretory granule. The insulin molecule has a diameter of 4 nm in its monomeric form (49) and 11 nm in its hexameric form (296). Clearly, membrane fusion must generate a pathway <4 nm in order that insulin can exit.
On-cell capacitance measurements (FIGURE 17B) indicate that the fusion pore is initially narrow but then expands rapidly to a diameter of ~10 nm (7). Expansion of the fusion pore has also been monitored by optical methods that compare the granular uptake of fluid phase tracers of different molecular weights/sizes with the membrane tracer FM1–43 (FIGURE 18, D–G). Interestingly, there was a delay of up to 10 s between the opening of the fusion pore (measured by FM1–43 labeling) and the uptake of 10–70 kDa dextran, which has a diameter of ~6–12 nm, similar to that of hexameric insulin (11 nm) (664). It was hypothesized this long delay reflects the slow solvation of the insulin crystal, which retards the granule swelling that drives the expansion of the fusion pore. Several pieces of evidence support this idea. First, the kinetics of granule release is much faster in guinea pig β-cells that contain Mn2+ instead of Zn2+ (leading to a less stable insulin crystal) (664). Second, recent data obtained using the Zn2+ dye ZIMIR confirmed there was an ~2.5 s delay between membrane fusion (as detected by a capacitance increase) and the extracellular appearance of Zn2+ (388). Finally, imaging of the solvation of Zn2+-insulin crystals (using fluorescent Zn2+ indicators) revealed they fall into three distinct classes: ultrafast (<1 s), intermediate (1–7 s), and slow (>30 s) events (446). The fast events may correspond to the rapid insulin release events detected by amperometric detection of insulin (294). Clearly, the insulin cargo leaves the secretory granule at widely varying rates.
On-cell capacitance measurements have also allowed the demonstration of kiss-and-run exocytosis in mouse and human β-cells (253, 410). In kiss-and-run exocytosis, opening of the fusion pore connecting the granule lumen to the exterior is only transient (compare FIGURE 25, A and B with C and D). As the fusion pore has a diameter of only 1–2 nm (7, 410), it has been argued that it acts as a molecular sieve permitting the exit of small (<1 nm) low-molecular-weight granule constituents (like ATP and GABA) while retaining larger molecules (like insulin; diameter 3–4 nm) (95). Indeed, parallel measurements of ATP release (by electrophysiological techniques) and peptide cargo release (by optical methods) indicate that the two are not obligatorily associated with one another (484). In amperometic measurements of serotonin release, as well as recordings of ATP release in β-cells expressing purinergic ATP receptors (see FIGURE 17, C and D), the current spike that represents full fusion and the rapid loss of granule contents is sometimes preceded by a ‟pedestalˮ that reflects slower release via the narrow fusion pore (FIGURE 25A, arrow). In some cases, stand-alone pedestals are observed. These activate more slowly and are much smaller than the large current spikes that represent full fusion (FIGURE 25C). They represent the loss of ATP and other low-molecular-weight constituents via the fusion pore (which is not large enough to allow the exit of insulin). Dissociation of the release of low- and high-molecular-weight compounds allows the former to exert paracrine/autocrine functions within the islet independent of insulin. Modeling of the fusion pore dynamics suggests that a sizeable fraction of the ATP and GABA content of the secretory granules can be released via the fusion pore (410).
FIGURE 25.

A: single-vesicle capacitance increases (Cm, black trace), fusion pore conductance changes (G, red trace), and patch-clamp measurements of ATP release (green trace). Exocytosis leads to a step increase in membrane capacitance of 4–8 fF and a transient increase in G that reflects the initial opening of the fusion pore. In measurements of ATP release, activation of the purinergic receptors results in a membrane current that can be recorded using the whole-cell patch-clamp technique (see FIGURE 17D). For display, the current responses have been inverted. Arrow indicates the pedestal. B: schematic of full fusion. The fusion pore is initially large enough to accommodate ATP but not bulkier molecules (like insulin). Release of ATP via the fusion pore gives rise to a pedestal (arrow in A), whereas the main spike reflects full fusion and emptying of the entire cargo. ATP and insulin are shown as space-filling models and are inserted to scale. The width of the fusion pore is also to scale but not the dimensions of the rest of the granule. C: as in A but measurements obtained during ‟kiss-and-runˮ exocytosis. In kiss-and-run exocytosis, the increase in capacitance is only transient and is associated with a maintained (for the duration of the capacitance increase) increase in fusion pore conductance. The persistence of the increased fusion pore conductance indicates the failure of the fusion pore to expand. During kiss-and-run exocytosis, a more sustained but lower amplitude ATP-activated current is observed. D: as in B but showing kiss-and-run exocytosis. Slow release of ATP via the fusion pore results in the sustained but smaller ATP-activated current in C. Insulin remains trapped inside the granule lumen. Thus the fusion pore may function as a molecular sieve.
Imaging experiments using extracellular polar fluid phase markers of different molecular weights/dimensions suggest that the opening of the fusion pore is reversible as long as its diameter is <4 nm, but that opening becomes irreversible once this diameter is exceeded (664). In normal β-cells, the vast majority (~90%) of release events are of the full-fusion type and only 10% are kiss-and-run events (122). An increase in kiss-and-run exocytosis at the expense of full fusion (with less efficient delivery of insulin into the extracellular space) might contribute to the diminution of insulin secretion in T2DM. Indeed, there is evidence that the increased T2DM risk associated with the CDKAL1 locus might be mediated by impaired fusion pore expansion (122). This is mediated by increased expression of the transcription factor Sox4, which in turn leads to increased expression of the syntaxin-binding protein amisyn (Stxbp6) (532).
10. Single-vesicle and compound secretion
In on-cell capacitance measurements, most exocytotic events are normally distributed, with a mean value close that expected from the granule diameter (4–5 fF, see sect. IXC1a). However, 5–10% of the events are much bigger (>30 fF) (253), suggesting several granules fuse simultaneously. Such compound (multivesicular) exocytosis occurs when the granules prefuse inside the cell and then exocytose as a single unit (193).
Optical imaging shows that whereas multivesicular exocytosis is normally a fairly rare occurrence (<5%), its likelihood increases dramatically (up to 20%) in the presence of GLP-1 (360) or other agents that increase intracellular cAMP levels (634), in response to ACh (289), or under conditions that lead to a uniform increase in [Ca2+]i. These multivesicular release events involve on average five secretory granules. It has been estimated that the transient potentiation of insulin secretion produced by 20 μM of the muscarinic agonist carbachol is almost entirely due to increased occurrence of multivesicular exocytosis (289). The mechanisms by which multivesicular exocytosis occurs is not entirely clear, but it may be related to the presence of both v-SNAREs (including VAMP-8) and t-SNAREs (like syntaxin-3) on the insulin granules (193) (see sect. IXB2c). After overexpression of syntaxin-3, the number of granules fused with another secretory granule was doubled in mouse β-cells (769).
X. WHAT CAUSES T2DM?
Although this review focuses on the physiology of insulin secretion, it would be disappointing if a detailed understanding of β-cell physiology did not provide some illumination as to the causes of T2DM. Although much valuable information has been gleaned from animal models of diabetes (351), in this section we restrict ourselves to the human disease.
A key question is, of course, what initiates the sequence of events that culminates in overt diabetes. The simplest explanation is that the T2DM occurs when the amount of insulin required exceeds that which the β-cell is able to supply. However, the β-cell is normally able to adjust its output to compensate for huge variations in insulin demand (326, 610) and the amount of insulin released in an entire day represents only a fraction of the total β-cell insulin content. Furthermore, most studies agree that insulin content and β-cell mass are only reduced by a maximum of 30% in diabetic patients (533). Thus the main deficit appears to be in glucose-induced stimulus-secretion coupling (possibly at the level of the initiation of β-cell electrical activity).
T2DM results from the cross-talk between the environmental factors (including diet and body weight) and the genetic predisposition. There is a strong correlation between obesity (expressed as body mass index) (107) and the risk of developing T2DM. A similar correlation exists for age (133) and diabetes risk. Thus, when considering the molecular and cellular causes of T2DM, it is instructive to first consider how these parameters may affect β-cell function.
Genome-wide scan association studies (GWAS) have identified >120 gene variants that result in an increased risk of T2DM (79, 519). Genes associated with T2DM risk currently fall into two groups: rare mutations/variants that cause monogenic diabetes, such as those that lead to neonatal diabetes and MODY, and common gene variants that have only a small effect on T2DM risk individually and which act in concert with other genes. It should also be borne in mind that the >120 genes identified to date account for only 10–20% of the heritability of diabetes, suggesting many more remain to be identified. Importantly, heritability only accounts for 25% of overall diabetes risk; the rest is due to lifestyle and age (30). The significance of diet/lifestyle is eminently illustrated by the dramatic (>80%) decrease in diabetes incidence during World War II (30). Arguably, much of the current T2DM epidemic could be reversed simply by lifestyle changes.
There is a steep relationship between the incidence of T2DM and age with an approximate doubling of diabetes incidence for every decade of life. Above 70 yr of age the incidence of diabetes is ~30% (133). It has been proposed that aging is associated with an accumulation of mitochondrial DNA mutations that culminates in impaired ATP production and β-cell function (624). Indeed, donor age has been found to correlate with a progressive loss of glucose-dependent NADH utilization that may underlie the age-dependent deterioration of GIIS reported in some (227, 300), but not all (10), studies on human islets. This variability is not understood, but it is worthy of note that both a negative (392) and positive (10, 42, 227, 260) correlation between age and insulin secretion has been reported in mice. The improved β-cell function was found to correlate with reduced KATP channel activity (227), possibly representing a compensatory mechanism to maintain insulin secretion in the face of compromised mitochondrial function.
Whereas the risk of T2DM is low in lean individuals, it increases dramatically with weight: in individuals with a body mass index (BMI) above 30, diabetes risk is 10- to 15-fold higher than in lean individuals (BMI <22). It is undeniable that obesity results in insulin resistance (71) but insulin resistance per se does not obligatorily result in T2DM and the β-cells are (at least initially) able to compensate by increased insulin secretion (60). However, in some individuals insulin release ultimately becomes insufficient to match the increase insulin demand.
It has been proposed that an elevation of plasma NEFA might explain the link between obesity and T2DM (70, 730). Indeed, chronic exposure (>24h) of islets to NEFA leads to diminished GIIS (647). However, the problem with this idea is that the plasma NEFA concentration undergoes huge diurnal variations, and there is little evidence that obesity is associated with high circulating NEFA levels (66). This argues that elevation of plasma NEFA levels does not cause T2DM. However, once T2DM has developed, the accompanying increase in plasma NEFA levels may contribute to the progressive exacerbation of insulin secretion and glycemia via suppression of insulin release (FIGURE 26). Another idea is that obesity results in ectopic pancreatic fat which has a detrimental metabolic on β-cell function. Intriguingly, T2DM can be reversed by 8 weeks on a low-calorie diet (600 kcal/day) (396). This was paralleled by a 1.8% reduction in pancreatic triglyceride content (1–2 g of intrapancreatic fat) (396). However, it was also accompanied by normalization of plasma glucose levels (which fell from 9 to 6 mM), and it is possible that the reduction in hyperglycemia contributed to the restoration of β-cell function.
FIGURE 26.
Vicious cycle of hyperglycemia and GIIS initiating another vicious cycle of increased lipolysis/elevated NEFA and reduced GIIS. The combination of these two vicious cycles may account for the progression of T2DM. The slight initial increase in plasma glucose that initiates the vicious cycles may be caused by age-dependent insulin resistance or reduced β-cell metabolism and ATP production that culminates in T2DM (see sect. XA3).
It is well-established that chronic elevation of plasma glucose has deleterious effects on insulin secretion. Culture of human islets at 8 mM glucose for as little as 1–2 days almost completely abolishes GIIS (265). Hyperglycemia evoked by induction of the mutant gain-of-function KATP channels (that are not closed by intracellular ATP) in mice leads to a rapid reduction of islet insulin content, a decreased number of insulin-positive cells, and an increase in islet glucagon content as well as the number of glucagon-positive cells (96, 722). These effects are (almost) fully reversed by restoration of normoglycemia using insulin or the KATP channel blocker glibenclamide. Similar data were found in pancreatectomized rats (372). Thus a small increase (perhaps precipitated by a slight age-dependent elevation of plasma glucose caused by a combination of reduced insulin secretion and insulin resistance) might initiate a vicious cycle of progressive β-cell decompensation and hyperglycemia (725) (FIGURE 26).
A. Understanding the Insulin Secretion Defects of T2DM
In nondiabetic individuals, circulating insulin oscillates with a period of 10 min in healthy individuals (483) (FIGURE 22C). This periodicity is impaired in T2DM (363). Furthermore, T2DM is associated with the loss of biphasic GIIS: 1st phase insulin release is absent and only a slowly developing insulin response, resembling 2nd phase secretion, remains (242, 292) (FIGURE 21C). It is tempting to speculate that the loss of 1st phase and oscillatory insulin secretion reflects the same cellular defect within the β-cell. The ability of arginine and tolbutamide to elicit biphasic insulin secretion argues that the loss of 1st phase secretion is not attributable to a selective loss of, for example, the RRP. If this were the case, the response to all secretagogues should be similarly affected in T2DM. We therefore favor that the defect results from impaired β-cell electrical activity.
Unfortunately, there is only limited information about the electrophysiological properties of human diabetic β-cells. The few recordings that are available indicate that β-cells from diabetic donors have a reduced resting K+ conductance (low KATP channel activity), that they are slightly depolarized, and as a result generate the occasional action potential at low glucose (which is not seen in β-cells from non-diabetic donors) (FIGURE 27A). Elevation of glucose has little effect on the β-cell membrane potential and action potential firing is either much reduced compared with what is seen in non-diabetic cells (FIGURE 27A) or not evoked at all (FIGURE 27B). This will lead to decreased Ca2+ influx and impaired GIIS. These observations echo reports that glucose does not cause an increase in ATP/ADP ratio in islets from T2DM donors (141). Interestingly, the ATP/ADP ratio at glucose concentrations below the threshold for GIIS is, if anything, higher in islets from T2DM donors than nondiabetic donors. This may explain why β-cells from diabetic donors are partially depolarized and fire occasional spontaneous action potentials at basal glucose levels.
FIGURE 27.

A: effects of increasing glucose concentrations (top) on electrical activity recorded from a β-cell from an organ donor diagnosed with T2DM (male, 51 yr of age, BMI 31 kg/m2, newly diagnosed, HbA1C 6.3). Note the action potential firing at 1 mM glucose (arrow), weak depolarizing effect of 10–20 mM glucose, and low action potential frequency even at the highest glucose concentration (compare FIGURE 6A). B: as in A but the experiment concluded by the addition of the sulfonylurea tolbutamide, which produced strong membrane depolarization, stimulation of action potential firing, and a time-dependent decrease in action potential height. In this cell, 6–10 mM glucose (if anything) appeared to hyperpolarize the β-cell. Data in A and B provided by Dr. M. Shigeto, Oxford, as described in Ref. 618. Data are representative of a total of 9 experiments on β-cells from one donor. C and D: schematics showing KATP channel activity (top), membrane potential (middle), and insulin secretion (bottom) in a healthy β-cell (C) and a T2DM β-cell (D). In the nondiabetic β-cell (C), KATP channel activity is high at low glucose, keeping the membrane hyperpolarized and inhibiting insulin secretion (1). When glucose is elevated to an insulin-releasing concentration, KATP channels close, the β-cell depolarizes and starts firing action potentials, which leads to stimulation of insulin secretion (2). At steady state, KATP channel activity remains low, electrical activity continues, but the amplitude of the action potentials has declined because of membrane potential-dependent inactivation of voltage-gated Na+ channels, resulting in a reduction of insulin secretion. In the diabetic β-cell (D), KATP channel activity in low glucose is less than that in the nondiabetic β-cell, leading to a slight depolarization and firing of the occasional action potential (1). This may underlie the elevated basal insulin secretion seen in T2DM. Increasing glucose has only a small effect on KATP channel activity so depolarization is limited and the stimulation of action potential firing and insulin secretion is marginal (2).There is no diminution of action potential height and thus no biphasic insulin secretion. Upon addition of sulfonylureas (abbreviated SU; such as glibenclamide), KATP channel activity is strongly reduced, strong membrane depolarization, stimulation of action potential firing, and a time-dependent decrease in action potential height follows, leading to a biphasic stimulation of insulin secretion (3).
It is important to note that although the stimulatory effect of glucose on insulin secretion and electrical activity is reduced in islets from T2DM donors, they are not completely refractory to glucose (576). Nevertheless, the effect is smaller and requires higher glucose concentrations than found for β-cells from nondiabetic donors. The fact that T2DM islets retain some GIIS is not simply an artifact of islet isolation and subsequent culture at normal glucose concentrations as it is also observed in vivo (723).
Although glucose fails to evoke the normal electrophysiological response in T2DM β-cells, the effects of agents that bypass metabolism and depolarize the β-cell by a direct (nonmetabolic) effect (such as sulfonylureas or arginine) remain intact (FIGURE 27B). This explains the capacity of these agents to elicit biphasic insulin secretion (179, 292, 723).
FIGURE 27, C and D, compares schematically KATP channel activity, membrane potential, and insulin secretion in nondiabetic and T2DM β-cells. We propose the resting KATP channel activity is lower in T2DM β-cells than in nondiabetic β-cells, which accounts for the slightly depolarized resting membrane potential and occasional action potential firing seen at low glucose. This accounts for the elevated basal insulin secretion. Increasing glucose has less effect on KATP channel activity in T2DM than in nondiabetic β-cells, reflecting impaired ATP production. Consequently, membrane depolarization is smaller, action potential firing is more limited, and insulin secretion is only marginally stimulated. In contrast to nondiabetic β-cells, there is no diminution of action potential height (because of slight membrane depolarization and low action potential frequency) and thus no biphasic insulin secretion. Upon addition of sulfonylureas, KATP channel activity is strongly inhibited, producing action potential firing and a time-dependent decrease in action potential height that leads to biphasic stimulation of insulin release in T2DM β-cells.
The exact magnitude of the secretory response in T2DM will also be influenced by factors other than the KATP current, including the size of the RRP, altered expression of exocytotic proteins, and reduced insulin content (576). This may explain why the amplitude of arginine-induced insulin secretion is reduced in T2DM patients (although it remains biphasic) (723). However, the principal cause of the loss of biphasic GIIS is the sluggish electrophysiological response manifested as failure of the sugar to fully depolarize the β-cell and reduced action potential firing. It is therefore imperative that we understand the metabolic changes that accompany T2DM.
In many ways, the electrophysiological and secretory properties of human diabetic β-cells resemble those previously reported for fetal rat islets (564): basal KATP channel activity is reduced, glucose fails to evoke electrical activity, 1st phase GIIS is reduced, and the response to high-[K+]o depolarization is maintained. It is therefore tempting to speculate that the β-cells revert to a less mature state in T2DM (322). Indeed, T2DM is associated with changes in gene expression suggestive of β-cell dedifferentiation (671).
X. CONCLUSIONS: ‟SEEING WONDERFUL THINGSˮ
It is clear that the last 30 yr have seen dramatic advances in our knowledge of the physiology of the pancreatic β-cell and an improved understanding of how this is impaired in disease.
What have been the most surprising developments? The ion channel diversity is much higher than previously appreciated, and many more types of ion channel are found in the β-cell than initially envisaged. Another, perhaps, is the many differences in the ion channel complement of β-cells from different species. As this review makes clear, although mouse and human β-cells share many features, they are far from identical. Are these differences important? The answer is equivocal. Clearly, it does not make much difference to the β-cell whether Ca2+ enters via Cav1.2 or Cav2.1 Ca2+ channels. However, it does matter when it comes to the interpretation of genetic data; a gene variant in a Ca2+ channel that is not expressed in human β-cells cannot underlie impaired insulin secretion, and any increase in risk must be mediated via another cell type.
A major development during the past few years has been the complete analysis of the β-cell transcriptome of both mouse and humans. We now know all the genes expressed in the β-cell, even at the single-cell level, and how these vary between mouse strains and between different species, and we are beginning to recognize the changes caused by insults such as hyperglycemia and T2DM (606, 739). The challenge now is to understand what all these genes do. While some are obvious, others are simply a mystery.
The discovery that activating mutations in KATP channel genes cause neonatal diabetes has led to a new approach to therapy for affected patients, and shown that, despite many years of diabetes, most patients retain sufficient β-cells to control glycemia when the open KATP channels are closed with sulfonylureas (255). This suggests that β-cells may be similarly quiescent in T2DM; we just need to find the means to ‟reawakenˮ them. The use of sulfonylureas to treat KATP channel-associated neonatal diabetes is now considered a classic example of personalized medicine, and one of the first to be implemented.
Although it was recognized over 25 yr ago that diabetes is a polygenic disorder, few would have predicted exactly how polygenic it is (79). Identifying how all these variants predispose to T2DM will be a major challenge, not least because many of them lie in noncoding regions of the genome and may influence expression of genes some distance away.
Clearly, a major problem is the lack of experimental studies on β-cells from donors with T2DM. As a consequence, we still only have a fragmentary picture of what ails the T2DM β-cell. This must, and should, be a focus for future research. A detailed understanding of the metabolism of the T2DM β-cell will be essential. It is also now apparent that dysregulation of glucagon secretion from the pancreatic α-cell plays an important role not only in normal glucose homeostasis and also in T2DM (380). Indeed, from an islet-centric viewpoint, T2DM is a bihormonal disease, and possibly even a trihormonal disease (somatostatin secretion is also dysregulated; Ref. 755). Clearly, research on other islet cell types and their role in T2DM is also a priority for the future.
When we (rashly) accepted to write this review, we believed it would be fairly straightforward and that we had a good overview of the field. However, it quickly turned out to be far more challenging than we had anticipated. Nevertheless, we think it is appropriate to conclude this review with Lord Carnarvon’s response to the question posed by his assistant Howard Carter, when he first peeped through the hole into Tutankhamen’s tomb. ‟What do you see?ˮ Carter asked. ‟I see wonderful thingsˮ was the reply. Our view is the same; during the past 25 years, the field has seen many wonderful new things, and they have certainly not ‟been largely wasted.ˮ It will be exciting to see what the next 25 years bring.
GRANTS
Work in our own laboratories has been supported by the Wellcome Trust Grants 095551, 089795, and 084655; the European Research Council Grant 322620; Royal Society; Diabetes UK; Medical Research Council, Knut and Alice Wallenberg Foundation (Wallenberg Scholar Programme); and Swedish Research Council. F. M. Ashcroft holds a Royal Society/Wolfson merit award.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Supplemental Data
Supplemental Figures S1-S18 - .pdf (1.08 MB)
ACKNOWLEDGMENTS
We thank all our colleagues in the field, whose hard work has led to a dramatic expansion of our knowledge about the control of β-cell electrical activity and insulin secretion. We are particularly indebted to the many students and postdoctoral fellows who have worked with us over the years.
We are grateful to Dr. Sebastian Barg for valuable discussions on the mechanisms of exocytosis in pancreatic β-cells. We are also indebted to the two anonymous reviewers who provided very valuable feedback on an initial version of the review.
Although we have tried to be comprehensive, we will inevitably have overlooked important contributions, and the failure to cite these should not be regarded as an assessment of their validity or significance.
Address for reprint requests and other correspondence: P. Rorsman, Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Oxford OX3 7LJ, UK (e-mail: patrik.rorsman@drl.ox.ac.uk).
Footnotes
Expression data from mouse and human β-cells are presented in Supplemental Figures 1–18, available online.
Type 1 diabetes (T1DM) results from an autoimmune attack on the pancreatic β-cells which leads to their destruction. These patients require life-long insulin therapy or islet transplanation. This review will therefore not consider this form of diabetes and instead focus on T2DM.
Based on the assumption that the β-cell is an ellipsoid with long (a) and short (b) diameters of 16.5 and 12.1 μm. The volume of an ellipsoid is given by the equation V = πab2/6.
Pyruvate itself does not stimulate insulin secretion as it is unable to enter the β-cell due to lack of MCT1 (611).
Calculated from measured ATP concentrations of 2 pmol/103 cells in the absence of glucose and 4 pmol/103 cells at 10 mM glucose (142), and assuming a β-cell volume of 1 pl.
Based on an intragranular insulin concentration of ~100 mM and a 3:1 insulin:Zn2+ stoichiometry.
A problem with this hypothesis is whether there is enough Zn2+ to account for the effects on insulin clearance. Even if the entire pancreatic islet Zn2+ depot (30 pmol/islet, 300 islets) were released, the resulting increase in plasma Zn2+ would be limited to <20 nM (assuming a plasma volume of 0.5 ml in the mouse). This is equivalent to 0.2% of the background plasma Zn2+ concentration. Even if the portal concentration was an order of magnitude higher, these considerations argue that physiological release of insulin will not significantly affect plasma Zn2+ levels seen by the liver.
Note that the exact Ca2+ affinity may vary depending on the phospholipid composition of the plasma membrane (530).
REFERENCES
- 1.Abderrahmani A, Niederhauser G, Plaisance V, Roehrich ME, Lenain V, Coppola T, Regazzi R, Waeber G. Complexin I regulates glucose-induced secretion in pancreatic beta-cells. J Cell Sci 117: 2239–2247, 2004. doi: 10.1242/jcs.01041. [DOI] [PubMed] [Google Scholar]
- 2.Adriaenssens A, Lam BY, Billing L, Skeffington K, Sewing S, Reimann F, Gribble F. A Transcriptome-Led Exploration of Molecular Mechanisms Regulating Somatostatin-Producing D-Cells in the Gastric Epithelium. Endocrinology 156: 3924–3936, 2015. doi: 10.1210/en.2015-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adriaenssens AE, Svendsen B, Lam BY, Yeo GS, Holst JJ, Reimann F, Gribble FM. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59: 2156–2165, 2016. doi: 10.1007/s00125-016-4033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilar-Bryan L, Clement JP IV, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol Rev 78: 227–245, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP IV, Boyd AE III, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268: 423–426, 1995. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 6.Ahrén B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia 43: 393–410, 2000. doi: 10.1007/s001250051322. [DOI] [PubMed] [Google Scholar]
- 7.Alabi AA, Tsien RW. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol 75: 393–422, 2013. doi: 10.1146/annurev-physiol-020911-153305. [DOI] [PubMed] [Google Scholar]
- 8.Alanentalo T, Asayesh A, Morrison H, Lorén CE, Holmberg D, Sharpe J, Ahlgren U. Tomographic molecular imaging and 3D quantification within adult mouse organs. Nat Methods 4: 31–33, 2007. doi: 10.1038/nmeth985. [DOI] [PubMed] [Google Scholar]
- 9.Ali L, Grapengiesser E, Gylfe E, Hellman B, Lund PE. Free and bound sodium in pancreatic beta-cells exposed to glucose and tolbutamide. Biochem Biophys Res Commun 164: 212–218, 1989. doi: 10.1016/0006-291X(89)91704-X. [DOI] [PubMed] [Google Scholar]
- 10.Almaça J, Molina J, Arrojo E Drigo R, Abdulreda MH, Jeon WB, Berggren PO, Caicedo A, Nam HG. Young capillary vessels rejuvenate aged pancreatic islets. Proc Natl Acad Sci USA 111: 17612–17617, 2014. doi: 10.1073/pnas.1414053111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ammälä C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulin release by cyclic AMP in single beta-cells. Nature 363: 356–358, 1993. doi: 10.1038/363356a0. [DOI] [PubMed] [Google Scholar]
- 12.Ammälä C, Bokvist K, Larsson O, Berggren PO, Rorsman P. Demonstration of a novel apamin-insensitive calcium-activated K+ channel in mouse pancreatic B cells. Pflugers Arch 422: 443–448, 1993. doi: 10.1007/BF00375069. [DOI] [PubMed] [Google Scholar]
- 13.Ammälä C, Eliasson L, Bokvist K, Berggren PO, Honkanen RE, Sjöholm A, Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc Natl Acad Sci USA 91: 4343–4347, 1994. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ammälä C, Eliasson L, Bokvist K, Larsson O, Ashcroft FM, Rorsman P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J Physiol 472: 665–688, 1993. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ammälä C, Larsson O, Berggren PO, Bokvist K, Juntti-Berggren L, Kindmark H, Rorsman P. Inositol trisphosphate-dependent periodic activation of a Ca2+-activated K+ conductance in glucose-stimulated pancreatic beta-cells. Nature 353: 849–852, 1991. doi: 10.1038/353849a0. [DOI] [PubMed] [Google Scholar]
- 16.Andersson SA, Olsson AH, Esguerra JL, Heimann E, Ladenvall C, Edlund A, Salehi A, Taneera J, Degerman E, Groop L, Ling C, Eliasson L. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol Cell Endocrinol 364: 36–45, 2012. doi: 10.1016/j.mce.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Andreu E, Soria B, Sanchez-Andres JV. Oscillation of gap junction electrical coupling in the mouse pancreatic islets of Langerhans. J Physiol 498: 753–761, 1997. doi: 10.1113/jphysiol.1997.sp021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, Del Prato S, Rabuazzo AM, Purrello F, Marchetti P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48: 282–289, 2005. doi: 10.1007/s00125-004-1627-9. [DOI] [PubMed] [Google Scholar]
- 19.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24: 229–239, 2005. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antunes CM, Salgado AP, Rosário LM, Santos RM. Differential patterns of glucose-induced electrical activity and intracellular calcium responses in single mouse and rat pancreatic islets. Diabetes 49: 2028–2038, 2000. doi: 10.2337/diabetes.49.12.2028. [DOI] [PubMed] [Google Scholar]
- 21.Arkhammar P, Nilsson T, Rorsman P, Berggren PO. Inhibition of ATP-regulated K+ channels precedes depolarization-induced increase in cytoplasmic free Ca2+ concentration in pancreatic beta-cells. J Biol Chem 262: 5448–5454, 1987. [PubMed] [Google Scholar]
- 22.Arredouani A, Ruas M, Collins SC, Parkesh R, Clough F, Pillinger T, Coltart G, Rietdorf K, Royle A, Johnson P, Braun M, Zhang Q, Sones W, Shimomura K, Morgan AJ, Lewis AM, Chuang KT, Tunn R, Gadea J, Teboul L, Heister PM, Tynan PW, Bellomo EA, Rutter GA, Rorsman P, Churchill GC, Parrington J, Galione A. NAADP and endolysosomal two-pore channels modulate membrane excitability and stimulus-secretion coupling in mouse pancreatic beta cells. J Biol Chem 290: 21376–21392, 2015. doi: 10.1074/jbc.M115.671248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, Kawada Y, Bartolome A, Hashimoto N, Matsuda T, Koyanagi-Kimura M, Kanno A, Hirota Y, Hosooka T, Nagashima K, Nishimura W, Inoue H, Matsumoto M, Higgins MJ, Yasuda K, Inagaki N, Seino S, Kasuga M, Kido Y. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic β-cell mass by epigenetic modification of Cdkn1c. Proc Natl Acad Sci USA 112: 8332–8337, 2015. doi: 10.1073/pnas.1422104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, Matsuda T, Kimura-Koyanagi M, Hashimoto N, Sakahara M, Fujimoto W, Takahashi H, Ueda S, Hosooka T, Satoh T, Inoue H, Matsumoto M, Aiba A, Kasuga M, Kido Y. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 56: 1088–1097, 2013. doi: 10.1007/s00125-013-2849-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashcroft FM. The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab 293: E880–E889, 2007. doi: 10.1152/ajpendo.00348.2007. [DOI] [PubMed] [Google Scholar]
- 26.Ashcroft FM, Ashcroft SJ, Harrison DE. Properties of single potassium channels modulated by glucose in rat pancreatic beta-cells. J Physiol 400: 501–527, 1988. doi: 10.1113/jphysiol.1988.sp017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312: 446–448, 1984. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 28.Ashcroft FM, Kakei M, Gibson JS, Gray DW, Sutton R. The ATP- and tolbutamide-sensitivity of the ATP-sensitive K-channel from human pancreatic B cells. Diabetologia 32: 591–598, 1989. doi: 10.1007/BF00285333. [DOI] [PubMed] [Google Scholar]
- 29.Ashcroft FM, Puljung MC, Vedovato N. Neonatal diabetes and the KATP channel: from mutation to therapy. Trends Endocrinol Metab 28: 377–387, 2017. doi: 10.1016/j.tem.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell 148: 1160–1171, 2012. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 54: 87–143, 1989. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 32.Ashcroft FM, Rorsman P. K(ATP) channels and islet hormone secretion: new insights and controversies. Nat Rev Endocrinol 9: 660–669, 2013. doi: 10.1038/nrendo.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashcroft SJ, Hedeskov CJ, Randle PJ. Glucose metabolism in mouse pancreatic islets. Biochem J 118: 143–154, 1970. doi: 10.1042/bj1180143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashcroft SJ, Weerasinghe LC, Randle PJ. Interrelationship of islet metabolism, adenosine triphosphate content and insulin release. Biochem J 132: 223–231, 1973. doi: 10.1042/bj1320223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.Ashfield R, Gribble FM, Ashcroft SJH, Ashcroft FM. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes 48: 1341–1347, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Ashford ML, Harvey J. Leptin activation of K-ATP channels is mimicked by tyrosine kinase inhibitors in CRI-G1 insulin secreting cells. Br J Pharmacol 122: 56, 1997. [Google Scholar]
- 36.Aspinwall CA, Brooks SA, Kennedy RT, Lakey JR. Effects of intravesicular H+ and extracellular H+ and Zn2+ on insulin secretion in pancreatic beta cells. J Biol Chem 272: 31308–31314, 1997. doi: 10.1074/jbc.272.50.31308. [DOI] [PubMed] [Google Scholar]
- 37.Atwater I, Dawson CM, Ribalet B, Rojas E. Potassium permeability activated by intracellular calcium ion concentration in the pancreatic beta-cell. J Physiol 288: 575–588, 1979. [PMC free article] [PubMed] [Google Scholar]
- 38.Atwater I, Frankel BJ, Rojas E, Grodsky GM. Beta cell membrane potential and insulin release: role of calcium and calcium:magnesium ratio. Q J Exp Physiol 68: 233–245, 1983. doi: 10.1113/expphysiol.1983.sp002715. [DOI] [PubMed] [Google Scholar]
- 39.Atwater I, Goncalves A, Herchuelz A, Lebrun P, Malaisse WJ, Rojas E, Scott A. Cooling dissociates glucose-induced insulin release from electrical activity and cation fluxes in rodent pancreatic islets. J Physiol 348: 615–627, 1984. doi: 10.1113/jphysiol.1984.sp015129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atwater I, Ribalet B, Rojas E. Cyclic changes in potential and resistance of the beta-cell membrane induced by glucose in islets of Langerhans from mouse. J Physiol 278: 117–139, 1978. doi: 10.1113/jphysiol.1978.sp012296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atwater I, Rosario L, Rojas E. Properties of the Ca-activated K+ channel in pancreatic beta-cells. Cell Calcium 4: 451–461, 1983. doi: 10.1016/0143-4160(83)90021-0. [DOI] [PubMed] [Google Scholar]
- 42.Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schübeler D, Glaser B, Kaestner KH. Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved β Cell Function. Cell Metab 22: 619–632, 2015. doi: 10.1016/j.cmet.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babenko AP, Polak M, Cavé H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med 355: 456–466, 2006. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 44.Babiker T, Vedovato N, Patel K, Thomas N, Finn R, Männikkö R, Chakera AJ, Flanagan SE, Shepherd MH, Ellard S, Ashcroft FM, Hattersley AT. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia 59: 1162–1166, 2016. doi: 10.1007/s00125-016-3921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barg S, Eliasson L, Renström E, Rorsman P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes 51, Suppl 1: S74–S82, 2002. doi: 10.2337/diabetes.51.2007.S74. [DOI] [PubMed] [Google Scholar]
- 46.Barg S, Guček A. How Kiss-and-Run Can Make Us Sick: SOX4 Puts a Break on the Pore. Diabetes 65: 1791–1793, 2016. doi: 10.2337/dbi16-0019. [DOI] [PubMed] [Google Scholar]
- 47.Barg S, Huang P, Eliasson L, Nelson DJ, Obermüller S, Rorsman P, Thévenod F, Renström E. Priming of insulin granules for exocytosis by granular Cl− uptake and acidification. J Cell Sci 114: 2145–2154, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Barg S, Ma X, Eliasson L, Galvanovskis J, Göpel SO, Obermüller S, Platzer J, Renström E, Trus M, Atlas D, Striessnig J, Rorsman P. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophys J 81: 3308–3323, 2001. doi: 10.1016/S0006-3495(01)75964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barg S, Olofsson CS, Schriever-Abeln J, Wendt A, Gebre-Medhin S, Renström E, Rorsman P. Delay between fusion pore opening and peptide release from large dense-core vesicles in neuroendocrine cells. Neuron 33: 287–299, 2002. doi: 10.1016/S0896-6273(02)00563-9. [DOI] [PubMed] [Google Scholar]
- 50.Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC. Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci 24: 8796–8805, 2004. doi: 10.1523/JNEUROSCI.1940-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnett DW, Misler S. Coupling of exocytosis to depolarization in rat pancreatic islet beta-cells: effects of Ca2+, Sr2+ and Ba2+-containing extracellular solutions. Pflugers Arch 430: 593–595, 1995. doi: 10.1007/BF00373898. [DOI] [PubMed] [Google Scholar]
- 52.Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet beta-cells: evidence for roles in the generation of action potentials and insulin secretion. Pflugers Arch 431: 272–282, 1995. doi: 10.1007/BF00410201. [DOI] [PubMed] [Google Scholar]
- 53.Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, Rutter GA, Van Obberghen E. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem 282: 19575–19588, 2007. doi: 10.1074/jbc.M611841200. [DOI] [PubMed] [Google Scholar]
- 54.Barr F, Lambright DG. Rab GEFs and GAPs. Curr Opin Cell Biol 22: 461–470, 2010. doi: 10.1016/j.ceb.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, Hadzic B, Druelle N, Napolitano T, Navarro-Sanz S, Silvano S, Al-Hasani K, Pfeifer A, Lacas-Gervais S, Leuckx G, Marroquí L, Thévenet J, Madsen OD, Eizirik DL, Heimberg H, Kerr-Conte J, Pattou F, Mansouri A, Collombat P. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 168: 73–85.e11, 2017. doi: 10.1016/j.cell.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Benner C, van der Meulen T, Cacéres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 15: 620, 2014. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berggren PO, Yang SN, Murakami M, Efanov AM, Uhles S, Köhler M, Moede T, Fernström A, Appelskog IB, Aspinwall CA, Zaitsev SV, Larsson O, de Vargas LM, Fecher-Trost C, Weissgerber P, Ludwig A, Leibiger B, Juntti-Berggren L, Barker CJ, Gromada J, Freichel M, Leibiger IB, Flockerzi V. Removal of Ca2+ channel beta3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell 119: 273–284, 2004. doi: 10.1016/j.cell.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 58.Berglund O, Frankel BJ, Hellman B. Development of the insulin secretory defect in genetically diabetic (db/db) mouse. Acta Endocrinol (Copenh) 87: 543–551, 1978. [DOI] [PubMed] [Google Scholar]
- 59.Berglund O, Sehlin J, Täljedal IB. Influence of the murine diabetes gene on rubidium ion efflux from perifused islets. Diabetologia 19: 45–49, 1980. doi: 10.1007/BF00258310. [DOI] [PubMed] [Google Scholar]
- 60.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68: 1456–1467, 1981. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergsten P, Hellman B. Glucose-induced amplitude regulation of pulsatile insulin secretion from individual pancreatic islets. Diabetes 42: 670–674, 1993. doi: 10.2337/diab.42.5.670. [DOI] [PubMed] [Google Scholar]
- 62.Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 90: 1437–1459, 2010. doi: 10.1152/physrev.00049.2009. [DOI] [PubMed] [Google Scholar]
- 63.Berne C. The metabolism of lipids in mouse pancreatic islets. The oxidation of fatty acids and ketone bodies. Biochem J 152: 661–666, 1975. doi: 10.1042/bj1520661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertram R, Smolen P, Sherman A, Mears D, Atwater I, Martin F, Soria B. A role for calcium release-activated current (CRAC) in cholinergic modulation of electrical activity in pancreatic beta-cells. Biophys J 68: 2323–2332, 1995. doi: 10.1016/S0006-3495(95)80414-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Best L, Benington S. Effects of sulphonylureas on the volume-sensitive anion channel in rat pancreatic beta-cells. Br J Pharmacol 125: 874–878, 1998. doi: 10.1038/sj.bjp.0702148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bickerton AS, Roberts R, Fielding BA, Tornqvist H, Blaak EE, Wagenmakers AJ, Gilbert M, Humphreys SM, Karpe F, Frayn KN. Adipose tissue fatty acid metabolism in insulin-resistant men. Diabetologia 51: 1466–1474, 2008. doi: 10.1007/s00125-008-1040-x. [DOI] [PubMed] [Google Scholar]
- 67.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89: 847–885, 2009. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- 67a.Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov 11: 603–619, 2012. doi: 10.1038/nrd3777. [DOI] [PubMed] [Google Scholar]
- 68.Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, Greiner DL, Garber MG, Harlan DM, diIorio P. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes 64: 3172–3181, 2015. doi: 10.2337/db15-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bock T, Pakkenberg B, Buschard K. Genetic background determines the size and structure of the endocrine pancreas. Diabetes 54: 133–137, 2005. doi: 10.2337/diabetes.54.1.133. [DOI] [PubMed] [Google Scholar]
- 70.Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am 37: 635–646, 2008. doi: 10.1016/j.ecl.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 18: 139–143, 2011. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bokvist K, Ammälä C, Ashcroft FM, Berggren PO, Larsson O, Rorsman P. Separate processes mediate nucleotide-induced inhibition and stimulation of the ATP-regulated K+-channels in mouse pancreatic beta-cells. Proc Biol Sci 243: 139–144, 1991. doi: 10.1098/rspb.1991.0022. [DOI] [PubMed] [Google Scholar]
- 73.Bokvist K, Ammälä C, Berggren PO, Rorsman P, Wåhlander K. Alpha 2-adrenoreceptor stimulation does not inhibit L-type calcium channels in mouse pancreatic beta-cells. Biosci Rep 11: 147–157, 1991. doi: 10.1007/BF01182483. [DOI] [PubMed] [Google Scholar]
- 74.Bokvist K, Holmqvist M, Gromada J, Rorsman P. Compound exocytosis in voltage-clamped mouse pancreatic beta-cells revealed by carbon fibre amperometry. Pflugers Arch 439: 634–645, 2000. [DOI] [PubMed] [Google Scholar]
- 75.Bokvist K, Rorsman P, Smith PA. Block of ATP-regulated and Ca2+-activated K+ channels in mouse pancreatic beta-cells by external tetraethylammonium and quinine. J Physiol 423: 327–342, 1990. doi: 10.1113/jphysiol.1990.sp018025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bokvist K, Rorsman P, Smith PA. Effects of external tetraethylammonium ions and quinine on delayed rectifying K+ channels in mouse pancreatic beta-cells. J Physiol 423: 311–325, 1990. doi: 10.1113/jphysiol.1990.sp018024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bolea S, Pertusa JA, Martín F, Sanchez-Andrés JV, Soria B. Regulation of pancreatic beta-cell electrical activity and insulin release by physiological amino acid concentrations. Pflugers Arch 433: 699–704, 1997. doi: 10.1007/s004240050334. [DOI] [PubMed] [Google Scholar]
- 78.Bonaventura MM, Crivello M, Ferreira ML, Repetto M, Cymeryng C, Libertun C, Lux-Lantos VA. Effects of GABAB receptor agonists and antagonists on glycemia regulation in mice. Eur J Pharmacol 677: 188–196, 2012. doi: 10.1016/j.ejphar.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 79.Bonnefond A, Froguel P. Rare and common genetic events in type 2 diabetes: what should biologists know? Cell Metab 21: 357–368, 2015. doi: 10.1016/j.cmet.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 80.Bonner C, Kerr-Conte J, Gmyr V, Queniat G, Moerman E, Thévenet J, Beaucamps C, Delalleau N, Popescu I, Malaisse WJ, Sener A, Deprez B, Abderrahmani A, Staels B, Pattou F. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 21: 512–517, 2015. doi: 10.1038/nm.3828. [DOI] [PubMed] [Google Scholar]
- 81.Bordin S, Boschero AC, Carneiro EM, Atwater I. Ionic mechanisms involved in the regulation of insulin secretion by muscarinic agonists. J Membr Biol 148: 177–184, 1995. doi: 10.1007/BF00207273. [DOI] [PubMed] [Google Scholar]
- 82.Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 59: 1202–1210, 2010. doi: 10.2337/db09-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci 66: 370–374, 2009. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bower RL, Hay DL. Amylin structure-function relationships and receptor pharmacology: implications for amylin mimetic drug development. Br J Pharmacol 173: 1883–1898, 2016. doi: 10.1111/bph.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brady PA, Alekseev AE, Aleksandrova LA, Gomez LA, Terzic A. A disrupter of actin microfilaments impairs sulfonylurea-inhibitory gating of cardiac KATP channels. Am J Physiol Heart Circ Physiol 271: H2710–H2716, 1996. [DOI] [PubMed] [Google Scholar]
- 86.Bränström R, Efendić S, Berggren PO, Larsson O. Direct inhibition of the pancreatic beta-cell ATP-regulated potassium channel by alpha-ketoisocaproate. J Biol Chem 273: 14113–14118, 1998. doi: 10.1074/jbc.273.23.14113. [DOI] [PubMed] [Google Scholar]
- 87.Braun M. The somatostatin receptor in human pancreatic β-cells. Vitam Horm 95: 165–193, 2014. doi: 10.1016/B978-0-12-800174-5.00007-7. [DOI] [PubMed] [Google Scholar]
- 88.Braun M, Ramracheya R, Amisten S, Bengtsson M, Moritoh Y, Zhang Q, Johnson PR, Rorsman P. Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic delta cells. Diabetologia 52: 1566–1578, 2009. doi: 10.1007/s00125-009-1382-z. [DOI] [PubMed] [Google Scholar]
- 89.Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, Rorsman P. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59: 1694–1701, 2010. doi: 10.2337/db09-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 57: 1618–1628, 2008. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- 91.Braun M, Ramracheya R, Johnson PR, Rorsman P. Exocytotic properties of human pancreatic beta-cells. Ann N Y Acad Sci 1152: 187–193, 2009. doi: 10.1111/j.1749-6632.2008.03992.x. [DOI] [PubMed] [Google Scholar]
- 92.Braun M, Ramracheya R, Rorsman P. Autocrine regulation of insulin secretion. Diabetes Obes Metab 14, Suppl 3: 143–151, 2012. doi: 10.1111/j.1463-1326.2012.01642.x. [DOI] [PubMed] [Google Scholar]
- 93.Braun M, Wendt A, Birnir B, Broman J, Eliasson L, Galvanovskis J, Gromada J, Mulder H, Rorsman P. Regulated exocytosis of GABA-containing synaptic-like microvesicles in pancreatic beta-cells. J Gen Physiol 123: 191–204, 2004. doi: 10.1085/jgp.200308966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Braun M, Wendt A, Buschard K, Salehi A, Sewing S, Gromada J, Rorsman P. GABAB receptor activation inhibits exocytosis in rat pancreatic beta-cells by G-protein-dependent activation of calcineurin. J Physiol 559: 397–409, 2004. doi: 10.1113/jphysiol.2004.066563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Braun M, Wendt A, Karanauskaite J, Galvanovskis J, Clark A, MacDonald PE, Rorsman P. Corelease and differential exit via the fusion pore of GABA, serotonin, and ATP from LDCV in rat pancreatic beta cells. J Gen Physiol 129: 221–231, 2007. doi: 10.1085/jgp.200609658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, Gribble FM, Reimann F, Clark A, Rorsman P, Ashcroft FM. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun 5: 4639, 2014. doi: 10.1038/ncomms5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brereton MF, Rohm M, Shimomura K, Holland C, Tornovsky-Babeay S, Dadon D, Iberl M, Chibalina MV, Lee S, Glaser B, Dor Y, Rorsman P, Clark A, Ashcroft FM. Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells. Nat Commun 7: 13496, 2016. doi: 10.1038/ncomms13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Briant LJ, Zhang Q, Vergari E, Kellard JA, Rodriguez B, Ashcroft FM, Rorsman P. Functional identification of islet cell types by electrophysiological fingerprinting. J R Soc Interface 14: 20160999, 2017. doi: 10.1098/rsif.2016.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brice NL, Varadi A, Ashcroft SJ, Molnar E. Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 45: 242–252, 2002. doi: 10.1007/s00125-001-0750-0. [DOI] [PubMed] [Google Scholar]
- 100.Brunger AT, Weninger K, Bowen M, Chu S. Single-molecule studies of the neuronal SNARE fusion machinery. Annu Rev Biochem 78: 903–928, 2009. doi: 10.1146/annurev.biochem.77.070306.103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bykhovskaia M. Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol 22: 387–392, 2011. doi: 10.1016/j.semcdb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 102.Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA 103: 2334–2339, 2006. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cabrera O, Jacques-Silva MC, Speier S, Yang SN, Köhler M, Fachado A, Vieira E, Zierath JR, Kibbey R, Berman DM, Kenyon NS, Ricordi C, Caicedo A, Berggren PO. Glutamate is a positive autocrine signal for glucagon release. Cell Metab 7: 545–554, 2008. doi: 10.1016/j.cmet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459: 596–600, 2009. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cantley J, Choudhury AI, Asare-Anane H, Selman C, Lingard S, Heffron H, Herrera P, Persaud SJ, Withers DJ. Pancreatic deletion of insulin receptor substrate 2 reduces beta and alpha cell mass and impairs glucose homeostasis in mice. Diabetologia 50: 1248–1256, 2007. doi: 10.1007/s00125-007-0637-9. [DOI] [PubMed] [Google Scholar]
- 106.Cantley J, Selman C, Shukla D, Abramov AY, Forstreuter F, Esteban MA, Claret M, Lingard SJ, Clements M, Harten SK, Asare-Anane H, Batterham RL, Herrera PL, Persaud SJ, Duchen MR, Maxwell PH, Withers DJ. Deletion of the von Hippel-Lindau gene in pancreatic beta cells impairs glucose homeostasis in mice. J Clin Invest 119: 125–135, 2009. doi: 10.1172/JCI26934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carey VJ, Walters EE, Colditz GA, Solomon CG, Willett WC, Rosner BA, Speizer FE, Manson JE. Body fat distribution and risk of non-insulin-dependent diabetes mellitus in women. The Nurses’ Health Study. Am J Epidemiol 145: 614–619, 1997. doi: 10.1093/oxfordjournals.aje.a009158. [DOI] [PubMed] [Google Scholar]
- 108.Carlsson PO, Andersson A, Jansson L. Pancreatic islet blood flow in normal and obese-hyperglycemic (ob/ob) mice. Am J Physiol 271: E990–E995, 1996. [DOI] [PubMed] [Google Scholar]
- 109.Catterall WA. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol 54: 317–338, 2014. doi: 10.1146/annurev-pharmtox-011112-140232. [DOI] [PubMed] [Google Scholar]
- 110.Cazares VA, Subramani A, Saldate JJ, Hoerauf W, Stuenkel EL. Distinct actions of Rab3 and Rab27 GTPases on late stages of exocytosis of insulin. Traffic 15: 997–1015, 2014. doi: 10.1111/tra.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cen J, Sargsyan E, Bergsten P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr Metab (Lond) 13: 59, 2016. doi: 10.1186/s12986-016-0119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cha CY, Nakamura Y, Himeno Y, Wang J, Fujimoto S, Inagaki N, Earm YE, Noma A. Ionic mechanisms and Ca2+ dynamics underlying the glucose response of pancreatic β cells: a simulation study. J Gen Physiol 138: 21–37, 2011. doi: 10.1085/jgp.201110611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cha CY, Santos E, Amano A, Shimayoshi T, Noma A. Time-dependent changes in membrane excitability during glucose-induced bursting activity in pancreatic β cells. J Gen Physiol 138: 39–47, 2011. doi: 10.1085/jgp.201110612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Charles S, Henquin JC. Distinct effects of various amino acids on 45Ca2+ fluxes in rat pancreatic islets. Biochem J 214: 899–907, 1983. doi: 10.1042/bj2140899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chay TR, Keizer J. Minimal model for membrane oscillations in the pancreatic beta-cell. Biophys J 42: 181–190, 1983. doi: 10.1016/S0006-3495(83)84384-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheviet S, Bezzi P, Ivarsson R, Renström E, Viertl D, Kasas S, Catsicas S, Regazzi R. Tomosyn-1 is involved in a post-docking event required for pancreatic beta-cell exocytosis. J Cell Sci 119: 2912–2920, 2006. doi: 10.1242/jcs.03037. [DOI] [PubMed] [Google Scholar]
- 117.Chimienti F, Devergnas S, Pattou F, Schuit F, Garcia-Cuenca R, Vandewalle B, Kerr-Conte J, Van Lommel L, Grunwald D, Favier A, Seve M. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J Cell Sci 119: 4199–4206, 2006. doi: 10.1242/jcs.03164. [DOI] [PubMed] [Google Scholar]
- 118.Cho JH, Chen L, Kim MH, Chow RH, Hille B, Koh DS. Characteristics and functions of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors expressed in mouse pancreatic alpha-cells. Endocrinology 151: 1541–1550, 2010. doi: 10.1210/en.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chow RH, von Rüden L, Neher E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 356: 60–63, 1992. doi: 10.1038/356060a0. [DOI] [PubMed] [Google Scholar]
- 120.Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, Song BL. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161: 291–306, 2015. doi: 10.1016/j.cell.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 121.Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, Gunter JH, de Koning EJ, Walls GV, Gray DW, Johnson PR, Hansen BC, Morris JF, Pipeleers-Marichal M, Cnop I, Clark A. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 53: 321–330, 2010. doi: 10.1007/s00125-009-1562-x. [DOI] [PubMed] [Google Scholar]
- 122.Collins SC, Do HW, Hastoy B, Hugill A, Adam J, Chibalina MV, Galvanovskis J, Godazgar M, Lee S, Goldsworthy M, Salehi A, Tarasov AI, Rosengren AH, Cox R, Rorsman P. Increased Expression of the Diabetes Gene SOX4 Reduces Insulin Secretion by Impaired Fusion Pore Expansion. Diabetes 65: 1952–1961, 2016. doi: 10.2337/db15-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkader F, Bengtsson M, Fearnside J, Ramracheya R, Toye AA, Zhang Q, Clark A, Gauguier D, Rorsman P. Progression of diet-induced diabetes in C57BL6J mice involves functional dissociation of Ca2+ channels from secretory vesicles. Diabetes 59: 1192–1201, 2010. doi: 10.2337/db09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Colsoul B, Schraenen A, Lemaire K, Quintens R, Van Lommel L, Segal A, Owsianik G, Talavera K, Voets T, Margolskee RF, Kokrashvili Z, Gilon P, Nilius B, Schuit FC, Vennekens R. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5-/- mice. Proc Natl Acad Sci USA 107: 5208–5213, 2010. doi: 10.1073/pnas.0913107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Colsoul B, Vennekens R, Nilius B. Transient receptor potential cation channels in pancreatic β cells. Rev Physiol Biochem Pharmacol 161: 87–110, 2011. doi: 10.1007/112_2011_2. [DOI] [PubMed] [Google Scholar]
- 126.Cook DL, Crill WE, Porte D Jr. Glucose and acetylcholine have different effects on the plateau pacemaker of pancreatic islet cells. Diabetes 30: 558–561, 1981. doi: 10.2337/diab.30.7.558. [DOI] [PubMed] [Google Scholar]
- 127.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311: 271–273, 1984. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 128.Cook DL, Ikeuchi M, Fujimoto WY. Lowering of pHi inhibits Ca2+-activated K+ channels in pancreatic B-cells. Nature 311: 269–271, 1984. doi: 10.1038/311269a0. [DOI] [PubMed] [Google Scholar]
- 129.Cook DL, Satin LS, Ashford ML, Hales CN. ATP-sensitive K+ channels in pancreatic beta-cells. Spare-channel hypothesis. Diabetes 37: 495–498, 1988. doi: 10.2337/diab.37.5.495. [DOI] [PubMed] [Google Scholar]
- 130.Coppola T, Frantz C, Perret-Menoud V, Gattesco S, Hirling H, Regazzi R. Pancreatic beta-cell protein granuphilin binds Rab3 and Munc-18 and controls exocytosis. Mol Biol Cell 13: 1906–1915, 2002. doi: 10.1091/mbc.02-02-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898, 2006. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Craig TJ, Ashcroft FM, Proks P. How ATP inhibits the open K(ATP) channel. J Gen Physiol 132: 131–144, 2008. doi: 10.1085/jgp.200709874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Creatore MI, Moineddin R, Booth G, Manuel DH, DesMeules M, McDermott S, Glazier RH. Age- and sex-related prevalence of diabetes mellitus among immigrants to Ontario, Canada. CMAJ 182: 781–789, 2010. doi: 10.1503/cmaj.091551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Crutzen R, Virreira M, Markadieu N, Shlyonsky V, Sener A, Malaisse WJ, Beauwens R, Boom A, Golstein PE. Anoctamin 1 (Ano1) is required for glucose-induced membrane potential oscillations and insulin secretion by murine β-cells. Pflugers Arch 468: 573–591, 2016. doi: 10.1007/s00424-015-1758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dadi PK, Luo B, Vierra NC, Jacobson DA. TASK-1 Potassium Channels Limit Pancreatic α-Cell Calcium Influx and Glucagon Secretion. Mol Endocrinol 29: 777–787, 2015. doi: 10.1210/me.2014-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dai XQ, Plummer G, Casimir M, Kang Y, Hajmrle C, Gaisano HY, Manning Fox JE, MacDonald PE. SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes 60: 838–847, 2011. doi: 10.2337/db10-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Deák F, Shin OH, Tang J, Hanson P, Ubach J, Jahn R, Rizo J, Kavalali ET, Südhof TC. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J 25: 2856–2866, 2006. doi: 10.1038/sj.emboj.7601165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dean PM. Ultrastructural morphometry of the pancreatic β-cell. Diabetologia 9: 115–119, 1973. doi: 10.1007/BF01230690. [DOI] [PubMed] [Google Scholar]
- 139.Debuyser A, Drews G, Henquin JC. Adrenaline inhibition of insulin release: role of cyclic AMP. Mol Cell Endocrinol 78: 179–186, 1991. doi: 10.1016/0303-7207(91)90121-8. [DOI] [PubMed] [Google Scholar]
- 140.Debuyser A, Drews G, Henquin JC. Adrenaline inhibition of insulin release: role of the repolarization of the B cell membrane. Pflugers Arch 419: 131–137, 1991. doi: 10.1007/BF00372998. [DOI] [PubMed] [Google Scholar]
- 141.Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S, Marchetti P. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54: 727–735, 2005. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 142.Detimary P, Dejonghe S, Ling Z, Pipeleers D, Schuit F, Henquin JC. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J Biol Chem 273: 33905–33908, 1998. doi: 10.1074/jbc.273.51.33905. [DOI] [PubMed] [Google Scholar]
- 143.Detimary P, Gilon P, Henquin JC. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem J 333: 269–274, 1998. doi: 10.1042/bj3330269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Detimary P, Gilon P, Nenquin M, Henquin JC. Two sites of glucose control of insulin release with distinct dependence on the energy state in pancreatic B-cells. Biochem J 297: 455–461, 1994. doi: 10.1042/bj2970455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Diaz-Garcia CM, Morales-Lázaro SL, Sánchez-Soto C, Velasco M, Rosenbaum T, Hiriart M. Role for the TRPV1 channel in insulin secretion from pancreatic beta cells. J Membr Biol 247: 479–491, 2014. doi: 10.1007/s00232-014-9658-8. [DOI] [PubMed] [Google Scholar]
- 146.DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, Huising MO. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab 5: 449–458, 2016. doi: 10.1016/j.molmet.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ding WG, Gromada J. Protein kinase A-dependent stimulation of exocytosis in mouse pancreatic beta-cells by glucose-dependent insulinotropic polypeptide. Diabetes 46: 615–621, 1997. doi: 10.2337/diab.46.4.615. [DOI] [PubMed] [Google Scholar]
- 148.Doherty M, Malaisse WJ. Glycogen accumulation in rat pancreatic islets: in vitro experiments. Endocrine 14: 303–309, 2001. doi: 10.1385/ENDO:14:3:303. [DOI] [PubMed] [Google Scholar]
- 149.Dolai S, Xie L, Zhu D, Liang T, Qin T, Xie H, Kang Y, Chapman ER, Gaisano HY. Synaptotagmin-7 Functions to Replenish Insulin Granules for Exocytosis in Human Islet β-Cells. Diabetes 65: 1962–1976, 2016. doi: 10.2337/db15-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Doliba NM, Qin W, Najafi H, Liu C, Buettger CW, Sotiris J, Collins HW, Li C, Stanley CA, Wilson DF, Grimsby J, Sarabu R, Naji A, Matschinsky FM. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am J Physiol Endocrinol Metab 302: E87–E102, 2012. doi: 10.1152/ajpendo.00218.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594: 5369–5390, 2016. doi: 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Drews G, Debuyser A, Nenquin M, Henquin JC. Galanin and epinephrine act on distinct receptors to inhibit insulin release by the same mechanisms including an increase in K+ permeability of the B-cell membrane. Endocrinology 126: 1646–1653, 1990. doi: 10.1210/endo-126-3-1646. [DOI] [PubMed] [Google Scholar]
- 153.Drews G, Detimary P, Henquin JC. Non-additivity of adrenaline and galanin effects on 86Rb efflux and membrane potential in mouse B-cells suggests sharing of common targets. Biochim Biophys Acta 1175: 214–218, 1993. doi: 10.1016/0167-4889(93)90025-K. [DOI] [PubMed] [Google Scholar]
- 154.Drews G, Krippeit-Drews P, Düfer M. Electrophysiology of islet cells. Adv Exp Med Biol 654: 115–163, 2010. doi: 10.1007/978-90-481-3271-3_7. [DOI] [PubMed] [Google Scholar]
- 155.Dubois M, Vacher P, Roger B, Huyghe D, Vandewalle B, Kerr-Conte J, Pattou F, Moustaïd-Moussa N, Lang J. Glucotoxicity inhibits late steps of insulin exocytosis. Endocrinology 148: 1605–1614, 2007. doi: 10.1210/en.2006-1022. [DOI] [PubMed] [Google Scholar]
- 156.Düfer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic beta-cells. Diabetes 58: 1835–1843, 2009. doi: 10.2337/db08-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Düfer M, Haspel D, Krippeit-Drews P, Aguilar-Bryan L, Bryan J, Drews G. Oscillations of membrane potential and cytosolic Ca2+ concentration in SUR1(-/-) beta cells. Diabetologia 47: 488–498, 2004. doi: 10.1007/s00125-004-1348-0. [DOI] [PubMed] [Google Scholar]
- 158.Düfer M, Krippeit-Drews P, Buntinas L, Siemen D, Drews G. Methyl pyruvate stimulates pancreatic beta-cells by a direct effect on KATP channels, and not as a mitochondrial substrate. Biochem J 368: 817–825, 2002. doi: 10.1042/bj20020657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Düfer M, Neye Y, Hörth K, Krippeit-Drews P, Hennige A, Widmer H, McClafferty H, Shipston MJ, Häring HU, Ruth P, Drews G. BK channels affect glucose homeostasis and cell viability of murine pancreatic beta cells. Diabetologia 54: 423–432, 2011. doi: 10.1007/s00125-010-1936-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24: 2839–2850, 2005. doi: 10.1038/sj.emboj.7600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dun AR, Lord GJ, Wilson RS, Kavanagh DM, Cialowicz KI, Sugita S, Park S, Yang L, Smyth AM, Papadopulos A, Rickman C, Duncan RR. Navigation through the Plasma Membrane Molecular Landscape Shapes Random Organelle Movement. Curr Biol 27: 408–414, 2017. doi: 10.1016/j.cub.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dunne MJ, Findlay I, Petersen OH, Wollheim CB. ATP-sensitive K+ channels in an insulin-secreting cell line are inhibited by d-glyceraldehyde and activated by membrane permeabilization. J Membr Biol 93: 271–279, 1986. doi: 10.1007/BF01871181. [DOI] [PubMed] [Google Scholar]
- 163.Dunne MJ, Petersen OH. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett 208: 59–62, 1986. doi: 10.1016/0014-5793(86)81532-0. [DOI] [PubMed] [Google Scholar]
- 164.Dyachok O, Idevall-Hagren O, Sågetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjärvi G, Gylfe E, Tengholm A. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 8: 26–37, 2008. doi: 10.1016/j.cmet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 165.Eddlestone GT, Gonçalves A, Bangham JA, Rojas E. Electrical coupling between cells in islets of Langerhans from mouse. J Membr Biol 77: 1–14, 1984. doi: 10.1007/BF01871095. [DOI] [PubMed] [Google Scholar]
- 166.Edlund A, Esguerra JL, Wendt A, Flodström-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med 12: 87, 2014. doi: 10.1186/1741-7015-12-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflugers Arch 414: 600–612, 1989. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 168.El-Kholy W, MacDonald PE, Fox JM, Bhattacharjee A, Xue T, Gao X, Zhang Y, Stieber J, Li RA, Tsushima RG, Wheeler MB. Hyperpolarization-activated cyclic nucleotide-gated channels in pancreatic beta-cells. Mol Endocrinol 21: 753–764, 2007. doi: 10.1210/me.2006-0258. [DOI] [PubMed] [Google Scholar]
- 169.Eliasson L, Abdulkader F, Braun M, Galvanovskis J, Hoppa MB, Rorsman P. Novel aspects of the molecular mechanisms controlling insulin secretion. J Physiol 586: 3313–3324, 2008. doi: 10.1113/jphysiol.2008.155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eliasson L, Ma X, Renström E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol 121: 181–197, 2003. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Eliasson L, Renström E, Ammälä C, Berggren PO, Bertorello AM, Bokvist K, Chibalin A, Deeney JT, Flatt PR, Gäbel J, Gromada J, Larsson O, Lindström P, Rhodes CJ, Rorsman P. PKC-dependent stimulation of exocytosis by sulfonylureas in pancreatic beta cells. Science 271: 813–815, 1996. doi: 10.1126/science.271.5250.813. [DOI] [PubMed] [Google Scholar]
- 172.Eliasson L, Renström E, Ding WG, Proks P, Rorsman P. Rapid ATP-dependent priming of secretory granules precedes Ca(2+)-induced exocytosis in mouse pancreatic B-cells. J Physiol 503: 399–412, 1997. doi: 10.1111/j.1469-7793.1997.399bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Enkvetchakul D, Loussouarn G, Makhina E, Nichols CG. ATP interaction with the open state of the K(ATP) channel. Biophys J 80: 719–728, 2001. doi: 10.1016/S0006-3495(01)76051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ensinck JW, Laschansky EC, Vogel RE, Simonowitz DA, Roos BA, Francis BH. Circulating prosomatostatin-derived peptides. Differential responses to food ingestion. J Clin Invest 83: 1580–1589, 1989. doi: 10.1172/JCI114055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Esguerra JL, Mollet IG, Salunkhe VA, Wendt A, Eliasson L. Regulation of Pancreatic Beta Cell Stimulus-Secretion Coupling by microRNAs. Genes (Basel) 5: 1018–1031, 2014. doi: 10.3390/genes5041018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fan F, Ji C, Wu Y, Ferguson SM, Tamarina N, Philipson LH, Lou X. Dynamin 2 regulates biphasic insulin secretion and plasma glucose homeostasis. J Clin Invest 125: 4026–4041, 2015. doi: 10.1172/JCI80652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Farnsworth NL, Benninger RK. New insights into the role of connexins in pancreatic islet function and diabetes. FEBS Lett 588: 1278–1287, 2014. doi: 10.1016/j.febslet.2014.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fatherazi S, Cook DL. Specificity of tetraethylammonium and quinine for three K channels in insulin-secreting cells. J Membr Biol 120: 105–114, 1991. doi: 10.1007/BF01872393. [DOI] [PubMed] [Google Scholar]
- 179.Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, Fineman MS, Kim DD, Nauck MA. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 90: 5991–5997, 2005. doi: 10.1210/jc.2005-1093. [DOI] [PubMed] [Google Scholar]
- 180.Feldmann N, del Rio RM, Gjinovci A, Tamarit-Rodriguez J, Wollheim CB, Wiederkehr A. Reduction of plasma membrane glutamate transport potentiates insulin but not glucagon secretion in pancreatic islet cells. Mol Cell Endocrinol 338: 46–57, 2011. doi: 10.1016/j.mce.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 181.Feng DD, Luo Z, Roh SG, Hernandez M, Tawadros N, Keating DJ, Chen C. Reduction in voltage-gated K+ currents in primary cultured rat pancreatic beta-cells by linoleic acids. Endocrinology 147: 674–682, 2006. doi: 10.1210/en.2005-0225. [DOI] [PubMed] [Google Scholar]
- 182.Fenwick EM, Marty A, Neher E. Sodium and calcium channels in bovine chromaffin cells. J Physiol 331: 599–635, 1982. doi: 10.1113/jphysiol.1982.sp014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, Spigelman AF, Wright RC, Plummer G, Suzuki K, Mackay JP, van de Bunt M, Gloyn AL, Ryan TE, Norquay LD, Brosnan MJ, Trimmer JK, Rolph TP, Kibbey RG, Manning Fox JE, Colmers WF, Shirihai OS, Neufer PD, Yeh ET, Newgard CB, MacDonald PE. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J Clin Invest 125: 3847–3860, 2015. doi: 10.1172/JCI82498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fernandez-Alvarez J, Hillaire-Buys D, Loubatières-Mariani MM, Gomis R, Petit P. P2 receptor agonists stimulate insulin release from human pancreatic islets. Pancreas 22: 69–71, 2001. doi: 10.1097/00006676-200101000-00012. [DOI] [PubMed] [Google Scholar]
- 185.Ferrer R, Soria B, Dawson CM, Atwater I, Rojas E. Effects of Zn2+ on glucose-induced electrical activity and insulin release from mouse pancreatic islets. Am J Physiol Cell Physiol 246: C520–C527, 1984. [DOI] [PubMed] [Google Scholar]
- 186.Filios SR, Shalev A. β-Cell MicroRNAs: Small but Powerful. Diabetes 64: 3631–3644, 2015. doi: 10.2337/db15-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP, Mahajan A, Fuchsberger C, Atzmon G, Benediktsson R, Blangero J, Bowden DW, Brandslund I, Brosnan J, Burslem F, Chambers J, Cho YS, Christensen C, Douglas DA, Duggirala R, Dymek Z, Farjoun Y, Fennell T, Fontanillas P, Forsén T, Gabriel S, Glaser B, Gudbjartsson DF, Hanis C, Hansen T, Hreidarsson AB, Hveem K, Ingelsson E, Isomaa B, Johansson S, Jørgensen T, Jørgensen ME, Kathiresan S, Kong A, Kooner J, Kravic J, Laakso M, Lee JY, Lind L, Lindgren CM, Linneberg A, Masson G, Meitinger T, Mohlke KL, Molven A, Morris AP, Potluri S, Rauramaa R, Ribel-Madsen R, Richard AM, Rolph T, Salomaa V, Segrè AV, Skärstrand H, Steinthorsdottir V, Stringham HM, Sulem P, Tai ES, Teo YY, Teslovich T, Thorsteinsdottir U, Trimmer JK, Tuomi T, Tuomilehto J, Vaziri-Sani F, Voight BF, Wilson JG, Boehnke M, McCarthy MI, Njølstad PR, Pedersen O, Groop L, Cox DR, Stefansson K, Altshuler D; Go-T2D Consortium; T2D-GENES Consortium . Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46: 357–363, 2014. doi: 10.1038/ng.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fontés G, Ghislain J, Benterki I, Zarrouki B, Trudel D, Berthiaume Y, Poitout V. The ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Is Associated With Progressive Insulin Resistance and Decreased Functional β-Cell Mass in Mice. Diabetes 64: 4112–4122, 2015. doi: 10.2337/db14-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Frayn KN. Metabolic Regulation: A Human Perspective. Oxford, UK: Wiley-Blackwell, 2010, p. 384. [Google Scholar]
- 190.Fujimoto K, Shibasaki T, Yokoi N, Kashima Y, Matsumoto M, Sasaki T, Tajima N, Iwanaga T, Seino S. Piccolo, a Ca2+ sensor in pancreatic beta-cells. Involvement of cAMP-GEFII.Rim2. Piccolo complex in cAMP-dependent exocytosis. J Biol Chem 277: 50497–50502, 2002. doi: 10.1074/jbc.M210146200. [DOI] [PubMed] [Google Scholar]
- 191.Fujimoto W, Miki T, Ogura T, Zhang M, Seino Y, Satin LS, Nakaya H, Seino S. Niflumic acid-sensitive ion channels play an important role in the induction of glucose-stimulated insulin secretion by cyclic AMP in mice. Diabetologia 52: 863–872, 2009. doi: 10.1007/s00125-009-1306-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama S, Nishioka H, Matsuura Y, Mizoguchi A, Scheller RH, Takai Y. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20: 905–915, 1998. doi: 10.1016/S0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- 193.Gaisano HY. Here come the newcomer granules, better late than never. Trends Endocrinol Metab 25: 381–388, 2014. doi: 10.1016/j.tem.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 194.Gall D, Gromada J, Susa I, Rorsman P, Herchuelz A, Bokvist K. Significance of Na/Ca exchange for Ca2+ buffering and electrical activity in mouse pancreatic beta-cells. Biophys J 76: 2018–2028, 1999. doi: 10.1016/S0006-3495(99)77359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Galvanovskis J, Braun M, Rorsman P. Exocytosis from pancreatic β-cells: mathematical modelling of the exit of low-molecular-weight granule content. Interface Focus 1: 143–152, 2011. doi: 10.1098/rsfs.2010.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gandasi NR, Barg S. Contact-induced clustering of syntaxin and munc18 docks secretory granules at the exocytosis site. Nat Commun 5: 3914, 2014. doi: 10.1038/ncomms4914. [DOI] [PubMed] [Google Scholar]
- 197.Gandasi NR, Barg S, Yin P, Omar-Hmeadi M, Ottosson Laakso E, Vikman P. Glucose dependent granule docking limits insulin secretion and is decreased in human type-2 diabetes. Cell Metab. In press. [DOI] [PubMed] [Google Scholar]
- 198.Gandasi NR, Yin P, Riz M, Chibalina MV, Cortese G, Lund P-E, Matveev V, Rorsman P, Sherman A, Pedersen MG, Barg S. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J Clin Invest 127: 2353–2364, 2017. doi: 10.1172/JCI88491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ganic E, Singh T, Luan C, Fadista J, Johansson JK, Cyphert HA, Bennet H, Storm P, Prost G, Ahlenius H, Renström E, Stein R, Groop L, Fex M, Artner I. MafA-Controlled Nicotinic Receptor Expression Is Essential for Insulin Secretion and Is Impaired in Patients with Type 2 Diabetes. Cell Reports 14: 1991–2002, 2016. doi: 10.1016/j.celrep.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gauthier BR, Wollheim CB. MicroRNAs: ‘ribo-regulators’ of glucose homeostasis. Nat Med 12: 36–38, 2006. doi: 10.1038/nm0106-36. [DOI] [PubMed] [Google Scholar]
- 201.Gebre-Medhin S, Mulder H, Pekny M, Westermark G, Törnell J, Westermark P, Sundler F, Ahrén B, Betsholtz C. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem Biophys Res Commun 250: 271–277, 1998. doi: 10.1006/bbrc.1998.9308. [DOI] [PubMed] [Google Scholar]
- 202.Geisler JC, Corbin KL, Li Q, Feranchak AP, Nunemaker CS, Li C. Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. Endocrinology 154: 675–684, 2013. doi: 10.1210/en.2012-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gembal M, Detimary P, Gilon P, Gao ZY, Henquin JC. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J Clin Invest 91: 871–880, 1993. doi: 10.1172/JCI116308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Geng X, Li L, Watkins S, Robbins PD, Drain P. The insulin secretory granule is the major site of K(ATP) channels of the endocrine pancreas. Diabetes 52: 767–776, 2003. doi: 10.2337/diabetes.52.3.767. [DOI] [PubMed] [Google Scholar]
- 205.Gheni G, Ogura M, Iwasaki M, Yokoi N, Minami K, Nakayama Y, Harada K, Hastoy B, Wu X, Takahashi H, Kimura K, Matsubara T, Hoshikawa R, Hatano N, Sugawara K, Shibasaki T, Inagaki N, Bamba T, Mizoguchi A, Fukusaki E, Rorsman P, Seino S. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Reports 9: 661–673, 2014. doi: 10.1016/j.celrep.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gilon P, Bertrand G, Loubatières-Mariani MM, Remacle C, Henquin JC. The influence of gamma-aminobutyric acid on hormone release by the mouse and rat endocrine pancreas. Endocrinology 129: 2521–2529, 1991. doi: 10.1210/endo-129-5-2521. [DOI] [PubMed] [Google Scholar]
- 207.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev 22: 565–604, 2001. [DOI] [PubMed] [Google Scholar]
- 208.Gilon P, Jonas JC, Henquin JC. Culture duration and conditions affect the oscillations of cytoplasmic calcium concentration induced by glucose in mouse pancreatic islets. Diabetologia 37: 1007–1014, 1994. doi: 10.1007/BF00400464. [DOI] [PubMed] [Google Scholar]
- 209.Gilon P, Rorsman P. NALCN: a regulated leak channel. EMBO Rep 10: 963–964, 2009. doi: 10.1038/embor.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gilon P, Shepherd RM, Henquin JC. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J Biol Chem 268: 22265–22268, 1993. [PubMed] [Google Scholar]
- 211.Gilon P, Yakel J, Gromada J, Zhu Y, Henquin JC, Rorsman P. G protein-dependent inhibition of L-type Ca2+ currents by acetylcholine in mouse pancreatic B-cells. J Physiol 499: 65–76, 1997. doi: 10.1113/jphysiol.1997.sp021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Girard CA, Wunderlich FT, Shimomura K, Collins S, Kaizik S, Proks P, Abdulkader F, Clark A, Ball V, Zubcevic L, Bentley L, Clark R, Church C, Hugill A, Galvanovskis J, Cox R, Rorsman P, Brüning JC, Ashcroft FM. Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes. J Clin Invest 119: 80–90, 2009. doi: 10.1172/JCI35772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Glaser B. Familial hyperinsulinism. In: Gene Reviews, edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K. Seattle, WA: Univ. of Washington, 1993. [Google Scholar]
- 214.Gloyn AL, Odili S, Zelent D, Buettger C, Castleden HA, Steele AM, Stride A, Shiota C, Magnuson MA, Lorini R, d’Annunzio G, Stanley CA, Kwagh J, van Schaftingen E, Veiga-da-Cunha M, Barbetti F, Dunten P, Han Y, Grimsby J, Taub R, Ellard S, Hattersley AT, Matschinsky FM. Insights into the structure and regulation of glucokinase from a novel mutation (V62M), which causes maturity-onset diabetes of the young. J Biol Chem 280: 14105–14113, 2005. doi: 10.1074/jbc.M413146200. [DOI] [PubMed] [Google Scholar]
- 215.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350: 1838–1849, 2004. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 216.Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52: 568–572, 2003. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 217.Goforth PB, Bertram R, Khan FA, Zhang M, Sherman A, Satin LS. Calcium-activated K+ channels of mouse beta-cells are controlled by both store and cytoplasmic Ca2+: experimental and theoretical studies. J Gen Physiol 120: 307–322, 2002. doi: 10.1085/jgp.20028581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gomes PR, Graciano MF, Pantaleão LC, Rennó AL, Rodrigues SC, Velloso LA, Latorraca MQ, Carpinelli AR, Anhê GF, Bordin S. Long-term disruption of maternal glucose homeostasis induced by prenatal glucocorticoid treatment correlates with miR-29 upregulation. Am J Physiol Endocrinol Metab 306: E109–E120, 2014. doi: 10.1152/ajpendo.00364.2013. [DOI] [PubMed] [Google Scholar]
- 219.Gomi H, Mizutani S, Kasai K, Itohara S, Izumi T. Granuphilin molecularly docks insulin granules to the fusion machinery. J Cell Biol 171: 99–109, 2005. doi: 10.1083/jcb.200505179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gonoi T, Mizuno N, Inagaki N, Kuromi H, Seino Y, Miyazaki J, Seino S. Functional neuronal ionotropic glutamate receptors are expressed in the non-neuronal cell line MIN6. J Biol Chem 269: 16989–16992, 1994. [PubMed] [Google Scholar]
- 221.Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 521: 717–728, 1999. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Göpel S, Zhang Q, Eliasson L, Ma XS, Galvanovskis J, Kanno T, Salehi A, Rorsman P. Capacitance measurements of exocytosis in mouse pancreatic alpha-, beta- and delta-cells within intact islets of Langerhans. J Physiol 556: 711–726, 2004. doi: 10.1113/jphysiol.2003.059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, Rorsman P. Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol 114: 759–770, 1999. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Göpel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterisation of somatostatin-secreting δ-cells in intact mouse pancreatic islets. J Physiol 528: 497–507, 2000. doi: 10.1111/j.1469-7793.2000.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Grapengiesser E. Glucose induces cytoplasmic Na+ oscillations in pancreatic beta-cells. Biochem Biophys Res Commun 226: 830–835, 1996. doi: 10.1006/bbrc.1996.1436. [DOI] [PubMed] [Google Scholar]
- 226.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259: 780–785, 1993. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 227.Gregg T, Poudel C, Schmidt BA, Dhillon RS, Sdao SM, Truchan NA, Baar EL, Fernandez LA, Denu JM, Eliceiri KW, Rogers JD, Kimple ME, Lamming DW, Merrins MJ. Pancreatic β-Cells From Mice Offset Age-Associated Mitochondrial Deficiency With Reduced KATP Channel Activity. Diabetes 65: 2700–2710, 2016. doi: 10.2337/db16-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM. A novel method for measurement of submembrane ATP concentration. J Biol Chem 275: 30046–30049, 2000. doi: 10.1074/jbc.M001010200. [DOI] [PubMed] [Google Scholar]
- 229.Gribble FM, Proks P, Corkey BE, Ashcroft FM. Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J Biol Chem 273: 26383–26387, 1998. doi: 10.1074/jbc.273.41.26383. [DOI] [PubMed] [Google Scholar]
- 230.Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia 46: 875–891, 2003. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 231.Gribble FM, Reimann F, Ashfield R, Ashcroft FM. Nucleotide modulation of pinacidil stimulation of the cloned K(ATP) channel Kir6.2/SUR2A. Mol Pharmacol 57: 1256–1261, 2000. [PubMed] [Google Scholar]
- 232.Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J 16: 1145–1152, 1997. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol 504: 35–45, 1997. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA 95: 7185–7190, 1998. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J Neurochem 97: 818–833, 2006. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- 236.Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327: 1614–1618, 2010. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic beta-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes 47: 57–65, 1998. doi: 10.2337/diab.47.1.57. [DOI] [PubMed] [Google Scholar]
- 238.Gromada J, Ding WG, Barg S, Renström E, Rorsman P. Multisite regulation of insulin secretion by cAMP-increasing agonists: evidence that glucagon-like peptide 1 and glucagon act via distinct receptors. Pflugers Arch 434: 515–524, 1997. doi: 10.1007/s004240050431. [DOI] [PubMed] [Google Scholar]
- 239.Gromada J, Dissing S, Bokvist K, Renström E, Frøkjaer-Jensen J, Wulff BS, Rorsman P. Glucagon-like peptide I increases cytoplasmic calcium in insulin-secreting beta TC3-cells by enhancement of intracellular calcium mobilization. Diabetes 44: 767–774, 1995. doi: 10.2337/diab.44.7.767. [DOI] [PubMed] [Google Scholar]
- 240.Gromada J, Høy M, Buschard K, Salehi A, Rorsman P. Somatostatin inhibits exocytosis in rat pancreatic alpha-cells by G(i2)-dependent activation of calcineurin and depriming of secretory granules. J Physiol 535: 519–532, 2001. doi: 10.1111/j.1469-7793.2001.00519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gromada J, Høy M, Renström E, Bokvist K, Eliasson L, Göpel S, Rorsman P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J Physiol 518: 745–759, 1999. doi: 10.1111/j.1469-7793.1999.0745p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Groop LC, Ratheiser K, Luzi L, Melander A, Simonson DC, Petrides A, Bonadonna RC, Widén E, DeFronzo RA. Effect of sulphonylurea on glucose-stimulated insulin secretion in healthy and non-insulin dependent diabetic subjects: a dose-response study. Acta Diabetol 28: 162–168, 1991. doi: 10.1007/BF00579720. [DOI] [PubMed] [Google Scholar]
- 243.Guiot Y, Stevens M, Marhfour I, Stiernet P, Mikhailov M, Ashcroft SJ, Rahier J, Henquin JC, Sempoux C. Morphological localisation of sulfonylurea receptor 1 in endocrine cells of human, mouse and rat pancreas. Diabetologia 50: 1889–1899, 2007. doi: 10.1007/s00125-007-0731-z. [DOI] [PubMed] [Google Scholar]
- 244.Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, Tsang LL, Yu MK, Huang WQ, Sun X, Chung YW, Jiang X, Sohma Y, Chan HC. Glucose-induced electrical activities and insulin secretion in pancreatic islet β-cells are modulated by CFTR. Nat Commun 5: 4420, 2014. doi: 10.1038/ncomms5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, Li C, Radda GK, Südhof TC, Han W. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci USA 105: 3992–3997, 2008. doi: 10.1073/pnas.0711700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Gustavsson N, Wang X, Wang Y, Seah T, Xu J, Radda GK, Südhof TC, Han W. Neuronal calcium sensor synaptotagmin-9 is not involved in the regulation of glucose homeostasis or insulin secretion. PLoS One 5: e15414, 2010. doi: 10.1371/journal.pone.0015414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 57: 473–508, 2005. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 248.Gylfe E, Tengholm A. Neurotransmitter control of islet hormone pulsatility. Diabetes Obes Metab 16, Suppl 1: 102–110, 2014. doi: 10.1111/dom.12345. [DOI] [PubMed] [Google Scholar]
- 249.Gyulkhandanyan AV, Lee SC, Bikopoulos G, Dai F, Wheeler MB. The Zn2+-transporting pathways in pancreatic beta-cells: a role for the L-type voltage-gated Ca2+ channel. J Biol Chem 281: 9361–9372, 2006. doi: 10.1074/jbc.M508542200. [DOI] [PubMed] [Google Scholar]
- 250.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35: 595–601, 1992. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 251.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391: 85–100, 1981. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 252.Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69: 304–316, 2011. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hanna ST, Pigeau GM, Galvanovskis J, Clark A, Rorsman P, MacDonald PE. Kiss-and-run exocytosis and fusion pores of secretory vesicles in human beta-cells. Pflugers Arch 457: 1343–1350, 2009. doi: 10.1007/s00424-008-0588-0. [DOI] [PubMed] [Google Scholar]
- 254.Hardy AB, Fox JE, Giglou PR, Wijesekara N, Bhattacharjee A, Sultan S, Gyulkhandanyan AV, Gaisano HY, MacDonald PE, Wheeler MB. Characterization of Erg K+ channels in alpha- and beta-cells of mouse and human islets. J Biol Chem 284: 30441–30452, 2009. doi: 10.1074/jbc.M109.040659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 54: 2503–2513, 2005. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 256.He LP, Mears D, Atwater I, Kitasato H. Glucagon induces suppression of ATP-sensitive K+ channel activity through a Ca2+/calmodulin-dependent pathway in mouse pancreatic beta-cells. J Membr Biol 166: 237–244, 1998. doi: 10.1007/s002329900465. [DOI] [PubMed] [Google Scholar]
- 257.Hedeskov CJ. Mechanism of glucose-induced insulin secretion. Physiol Rev 60: 442–509, 1980. [DOI] [PubMed] [Google Scholar]
- 258.Heimberg H, De Vos A, Pipeleers D, Thorens B, Schuit F. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem 270: 8971–8975, 1995. doi: 10.1074/jbc.270.15.8971. [DOI] [PubMed] [Google Scholar]
- 259.Hellman B. The frequency distribution of the number and volume of the islets Langerhans in man. I. Studies on non-diabetic adults. Acta Soc Med Ups 64: 432–460, 1959. [PubMed] [Google Scholar]
- 260.Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, Fixler Y, Shreibman D, Zamir A, Tornovsky-Babeay S, Dai C, Glaser B, Powers AC, Shapiro AM, Magnuson MA, Dor Y, Ben-Porath I. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22: 412–420, 2016. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Henquin JC. ATP-sensitive K+ channels may control glucose-induced electrical activity in pancreatic B-cells. Biochem Biophys Res Commun 156: 769–775, 1988. doi: 10.1016/S0006-291X(88)80910-0. [DOI] [PubMed] [Google Scholar]
- 262.Henquin JC. The fiftieth anniversary of hypoglycaemic sulphonamides. How did the mother compound work? Diabetologia 35: 907–912, 1992. doi: 10.1007/BF00401417. [DOI] [PubMed] [Google Scholar]
- 263.Henquin JC. Role of voltage- and Ca2+-dependent K+ channels in the control of glucose-induced electrical activity in pancreatic B-cells. Pflugers Arch 416: 568–572, 1990. doi: 10.1007/BF00382691. [DOI] [PubMed] [Google Scholar]
- 264.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49: 1751–1760, 2000. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 265.Henquin JC, Dufrane D, Kerr-Conte J, Nenquin M. Dynamics of glucose-induced insulin secretion in normal human islets. Am J Physiol Endocrinol Metab 309: E640–E650, 2015. doi: 10.1152/ajpendo.00251.2015. [DOI] [PubMed] [Google Scholar]
- 266.Henquin JC, Dufrane D, Nenquin M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes 55: 3470–3477, 2006. doi: 10.2337/db06-0868. [DOI] [PubMed] [Google Scholar]
- 267.Henquin JC, Garcia MC, Bozem M, Hermans MP, Nenquin M. Muscarinic control of pancreatic B cell function involves sodium-dependent depolarization and calcium influx. Endocrinology 122: 2134–2142, 1988. doi: 10.1210/endo-122-5-2134. [DOI] [PubMed] [Google Scholar]
- 268.Henquin JC, Ishiyama N, Nenquin M, Ravier MA, Jonas JC. Signals and pools underlying biphasic insulin secretion. Diabetes 51, Suppl 1: S60–S67, 2002. doi: 10.2337/diabetes.51.2007.S60. [DOI] [PubMed] [Google Scholar]
- 269.Henquin JC, Meissner HP. Cyclic adenosine monophosphate differently affects the response of mouse pancreatic beta-cells to various amino acids. J Physiol 381: 77–93, 1986. doi: 10.1113/jphysiol.1986.sp016314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Henquin JC, Meissner HP. Effects of amino acids on membrane potential and 86Rb+ fluxes in pancreatic beta-cells. Am J Physiol Endocrinol Metab 240: E245–E252, 1981. [DOI] [PubMed] [Google Scholar]
- 271.Henquin JC, Meissner HP. The electrogenic sodium-potassium pump of mouse pancreatic B-cells. J Physiol 332: 529–552, 1982. doi: 10.1113/jphysiol.1982.sp014429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Henquin JC, Meissner HP. Opposite effects of tolbutamide and diazoxide on 86Rb+ fluxes and membrane potential in pancreatic B cells. Biochem Pharmacol 31: 1407–1415, 1982. doi: 10.1016/0006-2952(82)90036-3. [DOI] [PubMed] [Google Scholar]
- 273.Henquin JC, Meissner HP. Significance of ionic fluxes and changes in membrane potential for stimulus-secretion coupling in pancreatic B-cells. Experientia 40: 1043–1052, 1984. doi: 10.1007/BF01971450. [DOI] [PubMed] [Google Scholar]
- 274.Henquin JC, Nenquin M. Activators of PKA and Epac distinctly influence insulin secretion and cytosolic Ca2+ in female mouse islets stimulated by glucose and tolbutamide. Endocrinology 155: 3274–3287, 2014. doi: 10.1210/en.2014-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Henquin JC, Nenquin M, Stiernet P, Ahren B. In vivo and in vitro glucose-induced biphasic insulin secretion in the mouse: pattern and role of cytoplasmic Ca2+ and amplification signals in beta-cells. Diabetes 55: 441–451, 2006. doi: 10.2337/diabetes.55.02.06.db05-1051. [DOI] [PubMed] [Google Scholar]
- 276.Henquin JC, Ravier MA, Nenquin M, Jonas JC, Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. Eur J Clin Invest 33: 742–750, 2003. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 277.Hermans MP, Schmeer W, Henquin JC. Modulation of the effect of acetylcholine on insulin release by the membrane potential of B cells. Endocrinology 120: 1765–1773, 1987. doi: 10.1210/endo-120-5-1765. [DOI] [PubMed] [Google Scholar]
- 278.Hermans MP, Schmeer W, Henquin JC. The permissive effect of glucose, tolbutamide and high K+ on arginine stimulation of insulin release in isolated mouse islets. Diabetologia 30: 659–665, 1987. [DOI] [PubMed] [Google Scholar]
- 279.Herrington J. Gating modifier peptides as probes of pancreatic beta-cell physiology. Toxicon 49: 231–238, 2007. doi: 10.1016/j.toxicon.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 280.Heusser K, Yuan H, Neagoe I, Tarasov AI, Ashcroft FM, Schwappach B. Scavenging of 14-3-3 proteins reveals their involvement in the cell-surface transport of ATP-sensitive K+ channels. J Cell Sci 119: 4353–4363, 2006. doi: 10.1242/jcs.03196. [DOI] [PubMed] [Google Scholar]
- 281.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 282.Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer, 2001, p. 814. [Google Scholar]
- 283.Hiriart M, Matteson DR. Na channels and two types of Ca channels in rat pancreatic B cells identified with the reverse hemolytic plaque assay. J Gen Physiol 91: 617–639, 1988. doi: 10.1085/jgp.91.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, Li D, Li WH, Bugliani M, Marchetti P, Bosco D, Piemonti L, Johnson P, Hughes SJ, Rutter GA. Lipotoxicity disrupts incretin-regulated human β cell connectivity. J Clin Invest 123: 4182–4194, 2013. doi: 10.1172/JCI68459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Hollingdal M, Juhl CB, Pincus SM, Sturis J, Veldhuis JD, Polonsky KS, Pørksen N, Schmitz O. Failure of physiological plasma glucose excursions to entrain high-frequency pulsatile insulin secretion in type 2 diabetes. Diabetes 49: 1334–1340, 2000. doi: 10.2337/diabetes.49.8.1334. [DOI] [PubMed] [Google Scholar]
- 286.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 87: 1409–1439, 2007. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 287.Holz GG IV, Kühtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37). Nature 361: 362–365, 1993. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Hoppa MB, Collins S, Ramracheya R, Hodson L, Amisten S, Zhang Q, Johnson P, Ashcroft FM, Rorsman P. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab 10: 455–465, 2009. doi: 10.1016/j.cmet.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Hoppa MB, Jones E, Karanauskaite J, Ramracheya R, Braun M, Collins SC, Zhang Q, Clark A, Eliasson L, Genoud C, Macdonald PE, Monteith AG, Barg S, Galvanovskis J, Rorsman P. Multivesicular exocytosis in rat pancreatic beta cells. Diabetologia 55: 1001–1012, 2012. doi: 10.1007/s00125-011-2400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486: 122–125, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol 92: 145–159, 1988. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hosker JP, Rudenski AS, Burnett MA, Matthews DR, Turner RC. Similar reduction of first- and second-phase B-cell responses at three different glucose levels in type II diabetes and the effect of gliclazide therapy. Metabolism 38: 767–772, 1989. doi: 10.1016/0026-0495(89)90064-4. [DOI] [PubMed] [Google Scholar]
- 293.Houamed KM, Sweet IR, Satin LS. BK channels mediate a novel ionic mechanism that regulates glucose-dependent electrical activity and insulin secretion in mouse pancreatic β-cells. J Physiol 588: 3511–3523, 2010. doi: 10.1113/jphysiol.2009.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Huang L, Shen H, Atkinson MA, Kennedy RT. Detection of exocytosis at individual pancreatic beta cells by amperometry at a chemically modified microelectrode. Proc Natl Acad Sci USA 92: 9608–9612, 1995. doi: 10.1073/pnas.92.21.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Hugill A, Shimomura K, Ashcroft FM, Cox RD. A mutation in KCNJ11 causing human hyperinsulinism (Y12X) results in a glucose-intolerant phenotype in the mouse. Diabetologia 53: 2352–2356, 2010. doi: 10.1007/s00125-010-1866-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Hvidt S. Insulin association in neutral solutions studied by light scattering. Biophys Chem 39: 205–213, 1991. doi: 10.1016/0301-4622(91)85023-J. [DOI] [PubMed] [Google Scholar]
- 297.Iezzi M, Eliasson L, Fukuda M, Wollheim CB. Adenovirus-mediated silencing of synaptotagmin 9 inhibits Ca2+-dependent insulin secretion in islets. FEBS Lett 579: 5241–5246, 2005. doi: 10.1016/j.febslet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 298.Iezzi M, Escher G, Meda P, Charollais A, Baldini G, Darchen F, Wollheim CB, Regazzi R. Subcellular distribution and function of Rab3A, B, C, and D isoforms in insulin-secreting cells. Mol Endocrinol 13: 202–212, 1999. doi: 10.1210/mend.13.2.0228. [DOI] [PubMed] [Google Scholar]
- 299.Iezzi M, Kouri G, Fukuda M, Wollheim CB. Synaptotagmin V and IX isoforms control Ca2+ -dependent insulin exocytosis. J Cell Sci 117: 3119–3127, 2004. doi: 10.1242/jcs.01179. [DOI] [PubMed] [Google Scholar]
- 300.Ihm SH, Matsumoto I, Sawada T, Nakano M, Zhang HJ, Ansite JD, Sutherland DE, Hering BJ. Effect of donor age on function of isolated human islets. Diabetes 55: 1361–1368, 2006. doi: 10.2337/db05-1333. [DOI] [PubMed] [Google Scholar]
- 301.Ikeuchi M, Cook DL. Glucagon and forskolin have dual effects upon islet cell electrical activity. Life Sci 35: 685–691, 1984. doi: 10.1016/0024-3205(84)90264-9. [DOI] [PubMed] [Google Scholar]
- 302.Inagaki N, Gonoi T, Clement JP IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270: 1166–1170, 1995. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 303.Irwin N, Flatt PR. Enteroendocrine hormone mimetics for the treatment of obesity and diabetes. Curr Opin Pharmacol 13: 989–995, 2013. doi: 10.1016/j.coph.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 304.Ishida H, Makino T, Kobayashi M, Tsuneoka K. Laparoscopic measurement of pancreatic blood flow. Endoscopy 15: 107–110, 1983. doi: 10.1055/s-2007-1021480. [DOI] [PubMed] [Google Scholar]
- 305.Ito K, Dezaki K, Yoshida M, Yamada H, Miura R, Rita RS, Ookawara S, Tabei K, Kawakami M, Hara K, Morishita Y, Yada T, Kakei M. Endogenous α2A-Adrenoceptor-Operated Sympathoadrenergic Tones Attenuate Insulin Secretion via cAMP/TRPM2 Signaling. Diabetes 66: 699–709, 2017. doi: 10.2337/db16-1166. [DOI] [PubMed] [Google Scholar]
- 306.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422: 173–176, 2003. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 307.Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, in ’t Veld P, Renström E, Schuit FC. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54: 2132–2142, 2005. doi: 10.2337/diabetes.54.7.2132. [DOI] [PubMed] [Google Scholar]
- 308.Iwanir S, Reuveny E. Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels. Pflugers Arch 456: 1097–1108, 2008. doi: 10.1007/s00424-008-0479-4. [DOI] [PubMed] [Google Scholar]
- 309.Jackson MB, Chapman ER. The fusion pores of Ca2+-triggered exocytosis. Nat Struct Mol Biol 15: 684–689, 2008. doi: 10.1038/nsmb.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammälä CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab 6: 229–235, 2007. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. J Physiol 588: 3525–3537, 2010. doi: 10.1113/jphysiol.2010.190207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, Diez J, Berman DM, Kenyon NS, Ricordi C, Pileggi A, Molano RD, Berggren PO, Caicedo A. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc Natl Acad Sci USA 107: 6465–6470, 2010. doi: 10.1073/pnas.0908935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Jansson L, Hellerström C. Glucose-induced changes in pancreatic islet blood flow mediated by central nervous system. Am J Physiol Endocrinol Metab 251: E644–E647, 1986. [DOI] [PubMed] [Google Scholar]
- 314.Jeans AF, Oliver PL, Johnson R, Capogna M, Vikman J, Molnár Z, Babbs A, Partridge CJ, Salehi A, Bengtsson M, Eliasson L, Rorsman P, Davies KE. A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc Natl Acad Sci USA 104: 2431–2436, 2007. doi: 10.1073/pnas.0610222104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB. Metabolic cycling in control of glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 295: E1287–E1297, 2008. doi: 10.1152/ajpendo.90604.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Jewell JL, Luo W, Oh E, Wang Z, Thurmond DC. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J Biol Chem 283: 10716–10726, 2008. doi: 10.1074/jbc.M709876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, Ivarsson R, Lundquist I, Pereverzev A, Schneider T, Rorsman P, Renström E. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest 115: 146–154, 2005. doi: 10.1172/JCI200522518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Joberty G, Stabila PF, Coppola T, Macara IG, Regazzi R. High affinity Rab3 binding is dispensable for Rabphilin-dependent potentiation of stimulated secretion. J Cell Sci 112: 3579–3587, 1999. [DOI] [PubMed] [Google Scholar]
- 319.Johnson D, Shepherd RM, Gill D, Gorman T, Smith DM, Dunne MJ. Glucose-dependent modulation of insulin secretion and intracellular calcium ions by GKA50, a glucokinase activator. Diabetes 56: 1694–1702, 2007. doi: 10.2337/db07-0026. [DOI] [PubMed] [Google Scholar]
- 320.Johnson JD, Kuang S, Misler S, Polonsky KS. Ryanodine receptors in human pancreatic beta cells: localization and effects on insulin secretion. FASEB J 18: 878–880, 2004. [DOI] [PubMed] [Google Scholar]
- 321.Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, Berishvili E, Duncanson P, Watkinson M, Broichhagen J, Trauner D, Rutter GA, Hodson DJ. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab 24: 389–401, 2016. doi: 10.1016/j.cmet.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patanè G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem 274: 14112–14121, 1999. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- 323.Jow F, Zhang ZH, Kopsco DC, Carroll KC, Wang K. Functional coupling of intracellular calcium and inactivation of voltage-gated Kv1.1/Kvbeta1.1 A-type K+ channels. Proc Natl Acad Sci USA 101: 15535–15540, 2004. doi: 10.1073/pnas.0402081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Juntti-Berggren L, Larsson O, Rorsman P, Ammälä C, Bokvist K, Wåhlander K, Nicotera P, Dypbukt J, Orrenius S, Hallberg A. Increased activity of L-type Ca2+ channels exposed to serum from patients with type I diabetes. Science 261: 86–90, 1993. doi: 10.1126/science.7686306. [DOI] [PubMed] [Google Scholar]
- 325.Juntti-Berggren L, Refai E, Appelskog I, Andersson M, Imreh G, Dekki N, Uhles S, Yu L, Griffiths WJ, Zaitsev S, Leibiger I, Yang SN, Olivecrona G, Jörnvall H, Berggren PO. Apolipoprotein CIII promotes Ca2+-dependent beta cell death in type 1 diabetes. Proc Natl Acad Sci USA 101: 10090–10094, 2004. doi: 10.1073/pnas.0403551101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–846, 2006. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 327.Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, Rorsman P, Braun M. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am J Physiol Endocrinol Metab 303: E1107–E1116, 2012. doi: 10.1152/ajpendo.00207.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Kakei M, Kelly RP, Ashcroft SJ, Ashcroft FM. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett 208: 63–66, 1986. doi: 10.1016/0014-5793(86)81533-2. [DOI] [PubMed] [Google Scholar]
- 329.Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch 447: 469–479, 2004. doi: 10.1007/s00424-003-1146-4. [DOI] [PubMed] [Google Scholar]
- 330.Kanatsuka A, Makino H, Ohsawa H, Tokuyama Y, Yamaguchi T, Yoshida S, Adachi M. Secretion of islet amyloid polypeptide in response to glucose. FEBS Lett 259: 199–201, 1989. doi: 10.1016/0014-5793(89)81527-3. [DOI] [PubMed] [Google Scholar]
- 331.Kang G, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, Schwede F, Coetzee WA, Holz GG. cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J Physiol 573: 595–609, 2006. doi: 10.1113/jphysiol.2006.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Kang G, Holz GG. Amplification of exocytosis by Ca2+-induced Ca2+ release in INS-1 pancreatic beta cells. J Physiol 546: 175–189, 2003. doi: 10.1113/jphysiol.2002.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Kang L, He Z, Xu P, Fan J, Betz A, Brose N, Xu T. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab 3: 463–468, 2006. doi: 10.1016/j.cmet.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 334.Kanno T, Ma X, Barg S, Eliasson L, Galvanovskis J, Göpel S, Larsson M, Renström E, Rorsman P. Large dense-core vesicle exocytosis in pancreatic beta-cells monitored by capacitance measurements. Methods 33: 302–311, 2004. doi: 10.1016/j.ymeth.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 335.Kanno T, Rorsman P, Göpel SO. Glucose-dependent regulation of rhythmic action potential firing in pancreatic beta-cells by K(ATP)-channel modulation. J Physiol 545: 501–507, 2002. doi: 10.1113/jphysiol.2002.031344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Kasai H, Takahashi N, Tokumaru H. Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol Rev 92: 1915–1964, 2012. doi: 10.1152/physrev.00007.2012. [DOI] [PubMed] [Google Scholar]
- 337.Kasai K, Fujita T, Gomi H, Izumi T. Docking is not a prerequisite but a temporal constraint for fusion of secretory granules. Traffic 9: 1191–1203, 2008. doi: 10.1111/j.1600-0854.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 338.Kasai K, Ohara-Imaizumi M, Takahashi N, Mizutani S, Zhao S, Kikuta T, Kasai H, Nagamatsu S, Gomi H, Izumi T. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 115: 388–396, 2005. doi: 10.1172/JCI200522955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII–Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem 276: 46046–46053, 2001. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- 340.Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 52: 2461–2474, 2003. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- 341.Kato S, Ishida H, Tsuura Y, Tsuji K, Nishimura M, Horie M, Taminato T, Ikehara S, Odaka H, Ikeda I, Okada Y, Seino Y. Alterations in basal and glucose-stimulated voltage-dependent Ca2+ channel activities in pancreatic beta cells of non-insulin-dependent diabetes mellitus GK rats. J Clin Invest 97: 2417–2425, 1996. doi: 10.1172/JCI118688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kato T, Shimano H, Yamamoto T, Yokoo T, Endo Y, Ishikawa M, Matsuzaka T, Nakagawa Y, Kumadaki S, Yahagi N, Takahashi A, Sone H, Suzuki H, Toyoshima H, Hasty AH, Takahashi S, Gomi H, Izumi T, Yamada N. Granuphilin is activated by SREBP-1c and involved in impaired insulin secretion in diabetic mice. Cell Metab 4: 143–154, 2006. doi: 10.1016/j.cmet.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 343.Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest 89: 283–292, 1992. doi: 10.1172/JCI115573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Kefaloyianni E, Lyssand JS, Moreno C, Delaroche D, Hong M, Fenyö D, Mobbs CV, Neubert TA, Coetzee WA. Comparative proteomic analysis of the ATP-sensitive K+ channel complex in different tissue types. Proteomics 13: 368–378, 2013. doi: 10.1002/pmic.201200324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89: 2548–2556, 2004. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 346.Khan FA, Goforth PB, Zhang M, Satin LS. Insulin activates ATP-sensitive K(+) channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 50: 2192–2198, 2001. doi: 10.2337/diabetes.50.10.2192. [DOI] [PubMed] [Google Scholar]
- 347.Khan S, Yan-Do R, Duong E, Wu X, Bautista A, Cheley S, MacDonald PE, Braun M. Autocrine activation of P2Y1 receptors couples Ca (2+) influx to Ca (2+) release in human pancreatic beta cells. Diabetologia 57: 2535–2545, 2014. doi: 10.1007/s00125-014-3368-8. [DOI] [PubMed] [Google Scholar]
- 348.Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, Warnock G, Baekkeskov S. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes 42: 1799–1808, 1993. doi: 10.2337/diab.42.12.1799. [DOI] [PubMed] [Google Scholar]
- 349.Kinard TA, Satin LS. An ATP-sensitive Cl- channel current that is activated by cell swelling, cAMP, and glyburide in insulin-secreting cells. Diabetes 44: 1461–1466, 1995. doi: 10.2337/diab.44.12.1461. [DOI] [PubMed] [Google Scholar]
- 350.Kinard TA, Satin LS. Temperature modulates the Ca2+ current of HIT-T15 and mouse pancreatic beta-cells. Cell Calcium 20: 475–482, 1996. doi: 10.1016/S0143-4160(96)90089-5. [DOI] [PubMed] [Google Scholar]
- 351.King AJ. The use of animal models in diabetes research. Br J Pharmacol 166: 877–894, 2012. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Klein D, Misawa R, Bravo-Egana V, Vargas N, Rosero S, Piroso J, Ichii H, Umland O, Zhijie J, Tsinoremas N, Ricordi C, Inverardi L, Domínguez-Bendala J, Pastori RL. MicroRNA expression in alpha and beta cells of human pancreatic islets. PLoS One 8: e55064, 2013. doi: 10.1371/journal.pone.0055064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Koeck T, Olsson AH, Nitert MD, Sharoyko VV, Ladenvall C, Kotova O, Reiling E, Rönn T, Parikh H, Taneera J, Eriksson JG, Metodiev MD, Larsson NG, Balhuizen A, Luthman H, Stančáková A, Kuusisto J, Laakso M, Poulsen P, Vaag A, Groop L, Lyssenko V, Mulder H, Ling C. A common variant in TFB1M is associated with reduced insulin secretion and increased future risk of type 2 diabetes. Cell Metab 13: 80–91, 2011. doi: 10.1016/j.cmet.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 354.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun 301: 406–410, 2003. doi: 10.1016/S0006-291X(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 355.Kreykenbohm V, Wenzel D, Antonin W, Atlachkine V, von Mollard GF. The SNAREs vti1a and vti1b have distinct localization and SNARE complex partners. Eur J Cell Biol 81: 273–280, 2002. doi: 10.1078/0171-9335-00247. [DOI] [PubMed] [Google Scholar]
- 356.Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive K(Ca) channel is not involved in glucose-induced electrical activity in pancreatic beta-cells. J Membr Biol 119: 187–195, 1991. doi: 10.1007/BF01871418. [DOI] [PubMed] [Google Scholar]
- 357.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96: 329–339, 1999. doi: 10.1016/S0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 358.Kunii M, Ohara-Imaizumi M, Takahashi N, Kobayashi M, Kawakami R, Kondoh Y, Shimizu T, Simizu S, Lin B, Nunomura K, Aoyagi K, Ohno M, Ohmuraya M, Sato T, Yoshimura SI, Sato K, Harada R, Kim YJ, Osada H, Nemoto T, Kasai H, Kitamura T, Nagamatsu S, Harada A. Opposing roles for SNAP23 in secretion in exocrine and endocrine pancreatic cells. J Cell Biol 215: 121–138, 2016. doi: 10.1083/jcb.201604030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Kurashina T, Dezaki K, Yoshida M, Sukma Rita R, Ito K, Taguchi M, Miura R, Tominaga M, Ishibashi S, Kakei M, Yada T. The β-cell GHSR and downstream cAMP/TRPM2 signaling account for insulinostatic and glycemic effects of ghrelin. Sci Rep 5: 14041, 2015. doi: 10.1038/srep14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Kwan EP, Gaisano HY. Glucagon-like peptide 1 regulates sequential and compound exocytosis in pancreatic islet beta-cells. Diabetes 54: 2734–2743, 2005. doi: 10.2337/diabetes.54.9.2734. [DOI] [PubMed] [Google Scholar]
- 361.Lacey RJ, Berrow NS, London NJ, Lake SP, James RF, Scarpello JH, Morgan NG. Differential effects of beta-adrenergic agonists on insulin secretion from pancreatic islets isolated from rat and man. J Mol Endocrinol 5: 49–54, 1990. doi: 10.1677/jme.0.0050049. [DOI] [PubMed] [Google Scholar]
- 362.Lam AD, Ismail S, Wu R, Yizhar O, Passmore DR, Ernst SA, Stuenkel EL. Mapping dynamic protein interactions to insulin secretory granule behavior with TIRF-FRET. Biophys J 99: 1311–1320, 2010. doi: 10.1016/j.bpj.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Lang DA, Matthews DR, Burnett M, Turner RC. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 30: 435–439, 1981. doi: 10.2337/diab.30.5.435. [DOI] [PubMed] [Google Scholar]
- 364.Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem 259: 3–17, 1999. doi: 10.1046/j.1432-1327.1999.00043.x. [DOI] [PubMed] [Google Scholar]
- 365.Lang J, Fukuda M, Zhang H, Mikoshiba K, Wollheim CB. The first C2 domain of synaptotagmin is required for exocytosis of insulin from pancreatic beta-cells: action of synaptotagmin at low micromolar calcium. EMBO J 16: 5837–5846, 1997. doi: 10.1093/emboj/16.19.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Larsson O, Ammälä C, Bokvist K, Fredholm B, Rorsman P. Stimulation of the KATP channel by ADP and diazoxide requires nucleotide hydrolysis in mouse pancreatic beta-cells. J Physiol 463: 349–365, 1993. doi: 10.1113/jphysiol.1993.sp019598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Larsson O, Barker CJ, Sjöholm A, Carlqvist H, Michell RH, Bertorello A, Nilsson T, Honkanen RE, Mayr GW, Zwiller J, Berggren PO. Inhibition of phosphatases and increased Ca2+ channel activity by inositol hexakisphosphate. Science 278: 471–474, 1997. doi: 10.1126/science.278.5337.471. [DOI] [PubMed] [Google Scholar]
- 368.Larsson O, Kindmark H, Brandstrom R, Fredholm B, Berggren PO. Oscillations in KATP channel activity promote oscillations in cytoplasmic free Ca2+ concentration in the pancreatic beta cell. Proc Natl Acad Sci USA 93: 5161–5165, 1996. doi: 10.1073/pnas.93.10.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Larsson-Nyrén G, Sehlin J, Rorsman P, Renström E. Perchlorate stimulates insulin secretion by shifting the gating of L-type Ca2+ currents in mouse pancreatic B-cells towards negative potentials. Pflugers Arch 441: 587–595, 2001. doi: 10.1007/s004240000426. [DOI] [PubMed] [Google Scholar]
- 370.Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, Alvarez O. Molecular Determinants of BK Channel Functional Diversity and Functioning. Physiol Rev 97: 39–87, 2017. doi: 10.1152/physrev.00001.2016. [DOI] [PubMed] [Google Scholar]
- 371.Latreille M, Hausser J, Stützer I, Zhang Q, Hastoy B, Gargani S, Kerr-Conte J, Pattou F, Zavolan M, Esguerra JL, Eliasson L, Rülicke T, Rorsman P, Stoffel M. MicroRNA-7a regulates pancreatic β cell function. J Clin Invest 124: 2722–2735, 2014. doi: 10.1172/JCI73066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, Bonner-Weir S, Weir GC. Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J Biol Chem 278: 2997–3005, 2003. doi: 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- 373.Lebreton F, Pirog A, Belouah I, Bosco D, Berney T, Meda P, Bornat Y, Catargi B, Renaud S, Raoux M, Lang J. Slow potentials encode intercellular coupling and insulin demand in pancreatic beta cells. Diabetologia 58: 1291–1299, 2015. doi: 10.1007/s00125-015-3558-z. [DOI] [PubMed] [Google Scholar]
- 374.Lebrun P, Atwater I, Claret M, Malaisse WJ, Herchuelz A. Resistance to apamin of the Ca2+-activated K+ permeability in pancreatic B-cells. FEBS Lett 161: 41–44, 1983. doi: 10.1016/0014-5793(83)80726-1. [DOI] [PubMed] [Google Scholar]
- 375.Leclercq-Meyer V, Garcia-Martinez JA, Villanueva-Peñacarrillo ML, Valverde I, Malaisse WJ. In vitro and in vivo insulinotropic action of methyl pyruvate. Horm Metab Res 27: 477–481, 1995. doi: 10.1055/s-2007-980006. [DOI] [PubMed] [Google Scholar]
- 376.Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, Azuma Y, Wang HF, Tsitsilianos N, Shafiq U, Kwon JY, Lee HJ, Lee KW, Kim JK. Transient receptor potential vanilloid type-1 channel regulates diet-induced obesity, insulin resistance, and leptin resistance. FASEB J 29: 3182–3192, 2015. doi: 10.1096/fj.14-268300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Lee HS, Moon S, Yun JH, Lee M, Hwang MY, Kim YJ, Han BG, Kim JM, Kim BJ. Genome-wide copy number variation study reveals KCNIP1 as a modulator of insulin secretion. Genomics 104: 113–120, 2014. doi: 10.1016/j.ygeno.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 378.Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science 330: 790–796, 2010. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- 379.Lee YH, Magkos F, Mantzoros CS, Kang ES. Effects of leptin and adiponectin on pancreatic β-cell function. Metabolism 60: 1664–1672, 2011. doi: 10.1016/j.metabol.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 380.Lee YH, Wang MY, Yu XX, Unger RH. Glucagon is the key factor in the development of diabetes. Diabetologia 59: 1372–1375, 2016. doi: 10.1007/s00125-016-3965-9. [DOI] [PubMed] [Google Scholar]
- 381.Leech CA, Dzhura I, Chepurny OG, Kang G, Schwede F, Genieser HG, Holz GG. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic β cells. Prog Biophys Mol Biol 107: 236–247, 2011. doi: 10.1016/j.pbiomolbio.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN, Kahn CR, de Vargas LM, Berggren PO. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol Cell 7: 559–570, 2001. doi: 10.1016/S1097-2765(01)00203-9. [DOI] [PubMed] [Google Scholar]
- 383.Leibiger IB, Leibiger B, Berggren PO. Insulin signaling in the pancreatic beta-cell. Annu Rev Nutr 28: 233–251, 2008. doi: 10.1146/annurev.nutr.28.061807.155530. [DOI] [PubMed] [Google Scholar]
- 384.Lemaire K, Ravier MA, Schraenen A, Creemers JW, Van de Plas R, Granvik M, Van Lommel L, Waelkens E, Chimienti F, Rutter GA, Gilon P, in’t Veld PA, Schuit FC. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc Natl Acad Sci USA 106: 14872–14877, 2009. doi: 10.1073/pnas.0906587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Lenzen S. A fresh view of glycolysis and glucokinase regulation: history and current status. J Biol Chem 289: 12189–12194, 2014. doi: 10.1074/jbc.R114.557314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Leung YM, Kang Y, Gao X, Xia F, Xie H, Sheu L, Tsuk S, Lotan I, Tsushima RG, Gaisano HY. Syntaxin 1A binds to the cytoplasmic C terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem 278: 17532–17538, 2003. doi: 10.1074/jbc.M213088200. [DOI] [PubMed] [Google Scholar]
- 387.Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY. SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins. Endocr Rev 28: 653–663, 2007. doi: 10.1210/er.2007-0010. [DOI] [PubMed] [Google Scholar]
- 388.Li D, Chen S, Bellomo EA, Tarasov AI, Kaut C, Rutter GA, Li WH. Imaging dynamic insulin release using a fluorescent zinc indicator for monitoring induced exocytotic release (ZIMIR). Proc Natl Acad Sci USA 108: 21063–21068, 2011. doi: 10.1073/pnas.1109773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Li DQ, Jing X, Salehi A, Collins SC, Hoppa MB, Rosengren AH, Zhang E, Lundquist I, Olofsson CS, Mörgelin M, Eliasson L, Rorsman P, Renström E. Suppression of sulfonylurea- and glucose-induced insulin secretion in vitro and in vivo in mice lacking the chloride transport protein ClC-3. Cell Metab 10: 309–315, 2009. doi: 10.1016/j.cmet.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 390.Li HZ, Li RY, Li M. A review of maternally inherited diabetes and deafness. Front Biosci (Landmark Ed) 19: 777–782, 2014. doi: 10.2741/4244. [DOI] [PubMed] [Google Scholar]
- 391.Li J, Cantley J, Burchfield JG, Meoli CC, Stöckli J, Whitworth PT, Pant H, Chaudhuri R, Groffen AJ, Verhage M, James DE. DOC2 isoforms play dual roles in insulin secretion and insulin-stimulated glucose uptake. Diabetologia 57: 2173–2182, 2014. doi: 10.1007/s00125-014-3312-y. [DOI] [PubMed] [Google Scholar]
- 392.Li L, Trifunovic A, Köhler M, Wang Y, Petrovic Berglund J, Illies C, Juntti-Berggren L, Larsson NG, Berggren PO. Defects in β-cell Ca2+ dynamics in age-induced diabetes. Diabetes 63: 4100–4114, 2014. doi: 10.2337/db13-1855. [DOI] [PubMed] [Google Scholar]
- 393.Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell 168: 101–110.e10, 2017. doi: 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 394.Li YV. Zinc and insulin in pancreatic beta-cells. Endocrine 45: 178–189, 2014. doi: 10.1007/s12020-013-0032-x. [DOI] [PubMed] [Google Scholar]
- 395.Liang Y, Jetton TL, Zimmerman EC, Najafi H, Berner DK, Matschinsky FM, Magnuson MA. Effects of glucose on insulin secretion, glucokinase activity, and transgene expression in transgenic mouse islets containing an upstream glucokinase promoter-human growth hormone fusion gene. Diabetes 43: 1138–1145, 1994. doi: 10.2337/diab.43.9.1138. [DOI] [PubMed] [Google Scholar]
- 396.Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 54: 2506–2514, 2011. doi: 10.1007/s00125-011-2204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Lindau M. High resolution electrophysiological techniques for the study of calcium-activated exocytosis. Biochim Biophys Acta 1820: 1234–1242, 2012. doi: 10.1016/j.bbagen.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B. SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc Natl Acad Sci USA 98: 12233–12238, 2001. doi: 10.1073/pnas.221450198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Liu YJ, Gylfe E. Store-operated Ca2+ entry in insulin-releasing pancreatic beta-cells. Cell Calcium 22: 277–286, 1997. doi: 10.1016/S0143-4160(97)90066-X. [DOI] [PubMed] [Google Scholar]
- 400.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372: 366–369, 1994. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 401.Lovis P, Gattesco S, Regazzi R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biol Chem 389: 305–312, 2008. doi: 10.1515/BC.2008.026. [DOI] [PubMed] [Google Scholar]
- 402.Low JT, Mitchell JM, Do OH, Bax J, Rawlings A, Zavortink M, Morgan G, Parton RG, Gaisano HY, Thorn P. Glucose principally regulates insulin secretion in mouse islets by controlling the numbers of granule fusion events per cell. Diabetologia 56: 2629–2637, 2013. doi: 10.1007/s00125-013-3019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Low JT, Zavortink M, Mitchell JM, Gan WJ, Do OH, Schwiening CJ, Gaisano HY, Thorn P. Insulin secretion from beta cells in intact mouse islets is targeted towards the vasculature. Diabetologia 57: 1655–1663, 2014. doi: 10.1007/s00125-014-3252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Luciani DS, Johnson JD. Acute effects of insulin on beta-cells from transplantable human islets. Mol Cell Endocrinol 241: 88–98, 2005. doi: 10.1016/j.mce.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 405.Lundquist I, Ericson LE. beta-Adrenergic insulin release and adrenergic innervation of mouse pancreatic islets. Cell Tissue Res 193: 73–85, 1978. doi: 10.1007/BF00221602. [DOI] [PubMed] [Google Scholar]
- 406.Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84: 1051–1095, 2004. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- 407.Ma C, Li W, Xu Y, Rizo J. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18: 542–549, 2011. doi: 10.1038/nsmb.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Maassen JA, Janssen GM, Hart LM. Molecular mechanisms of mitochondrial diabetes (MIDD). Ann Med 37: 213–221, 2005. doi: 10.1080/07853890510007188. [DOI] [PubMed] [Google Scholar]
- 409.MacDonald MJ, Longacre MJ, Langberg EC, Tibell A, Kendrick MA, Fukao T, Ostenson CG. Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52: 1087–1091, 2009. doi: 10.1007/s00125-009-1319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.MacDonald PE, Braun M, Galvanovskis J, Rorsman P. Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab 4: 283–290, 2006. doi: 10.1016/j.cmet.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 411.MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PR, Cox R, Eliasson L, Rorsman P. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 5: e143, 2007. doi: 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.MacDonald PE, Eliasson L, Rorsman P. Calcium increases endocytotic vesicle size and accelerates membrane fission in insulin-secreting INS-1 cells. J Cell Sci 118: 5911–5920, 2005. doi: 10.1242/jcs.02685. [DOI] [PubMed] [Google Scholar]
- 413.MacDonald PE, Salapatek AM, Wheeler MB. Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K(+) currents in beta-cells: a possible glucose-dependent insulinotropic mechanism. Diabetes 51, Suppl 3: S443–S447, 2002. doi: 10.2337/diabetes.51.2007.S443. [DOI] [PubMed] [Google Scholar]
- 414.MacDonald PE, Wang G, Tsuk S, Dodo C, Kang Y, Tang L, Wheeler MB, Cattral MS, Lakey JR, Salapatek AM, Lotan I, Gaisano HY. Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K(+) channels in neuroendocrine islet beta-cells through an interaction with the channel N terminus. Mol Endocrinol 16: 2452–2461, 2002. doi: 10.1210/me.2002-0058. [DOI] [PubMed] [Google Scholar]
- 415.Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch 447: 784–795, 2004. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- 416.Maechler P. Mitochondrial function and insulin secretion. Mol Cell Endocrinol 379: 12–18, 2013. doi: 10.1016/j.mce.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 417.Maechler P, Kennedy ED, Wang H, Wollheim CB. Desensitization of mitochondrial Ca2+ and insulin secretion responses in the beta cell. J Biol Chem 273: 20770–20778, 1998. doi: 10.1074/jbc.273.33.20770. [DOI] [PubMed] [Google Scholar]
- 418.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature 414: 807–812, 2001. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 419.Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402: 685–689, 1999. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- 420.Mahdi T, Hänzelmann S, Salehi A, Muhammed SJ, Reinbothe TM, Tang Y, Axelsson AS, Zhou Y, Jing X, Almgren P, Krus U, Taneera J, Blom AM, Lyssenko V, Esguerra JL, Hansson O, Eliasson L, Derry J, Zhang E, Wollheim CB, Groop L, Renström E, Rosengren AH. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab 16: 625–633, 2012. doi: 10.1016/j.cmet.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 421.Malaisse WJ, Boschero AC, Kawazu S, Hutton JC. The stimulus secretion coupling of glucose-induced insulin release. XXVII. Effect of glucose on K+ fluxes in isolated islets. Pflugers Arch 373: 237–242, 1978. doi: 10.1007/BF00580830. [DOI] [PubMed] [Google Scholar]
- 422.Malaisse WJ, Sener A. Glucose-induced changes in cytosolic ATP content in pancreatic islets. Biochim Biophys Acta 927: 190–195, 1987. doi: 10.1016/0167-4889(87)90134-0. [DOI] [PubMed] [Google Scholar]
- 423.Mandic SA, Skelin M, Johansson JU, Rupnik MS, Berggren PO, Bark C. Munc18-1 and Munc18-2 proteins modulate beta-cell Ca2+ sensitivity and kinetics of insulin exocytosis differently. J Biol Chem 286: 28026–28040, 2011. doi: 10.1074/jbc.M111.235366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet beta-cells: a comparison of islet-cell electrical activity in mouse and rat. Endocrinology 147: 4655–4663, 2006. doi: 10.1210/en.2006-0424. [DOI] [PubMed] [Google Scholar]
- 425.Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V, Piro S, Marselli L, Boggi U, Filipponi F, Tinti L, Salvini L, Wollheim CB, Purrello F, Dotta F. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 55: 3262–3272, 2012. doi: 10.1007/s00125-012-2716-9. [DOI] [PubMed] [Google Scholar]
- 426.Mariot P, Gilon P, Nenquin M, Henquin JC. Tolbutamide and diazoxide influence insulin secretion by changing the concentration but not the action of cytoplasmic Ca2+ in beta-cells. Diabetes 47: 365–373, 1998. doi: 10.2337/diabetes.47.3.365. [DOI] [PubMed] [Google Scholar]
- 427.Maritzen T, Keating DJ, Neagoe I, Zdebik AA, Jentsch TJ. Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J Neurosci 28: 10587–10598, 2008. doi: 10.1523/JNEUROSCI.3750-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Marquard J, Otter S, Welters A, Stirban A, Fischer A, Eglinger J, Herebian D, Kletke O, Klemen MS, Stožer A, Wnendt S, Piemonti L, Köhler M, Ferrer J, Thorens B, Schliess F, Rupnik MS, Heise T, Berggren PO, Klöcker N, Meissner T, Mayatepek E, Eberhard D, Kragl M, Lammert E. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat Med 21: 363–372, 2015. doi: 10.1038/nm.3822. [DOI] [PubMed] [Google Scholar]
- 429.Marselli L, Suleiman M, Masini M, Campani D, Bugliani M, Syed F, Martino L, Focosi D, Scatena F, Olimpico F, Filipponi F, Masiello P, Boggi U, Marchetti P. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 57: 362–365, 2014. doi: 10.1007/s00125-013-3098-3. [DOI] [PubMed] [Google Scholar]
- 430.Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, Marchetti P, Weir GC. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One 5: e11499, 2010. doi: 10.1371/journal.pone.0011499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 150: 687–698, 2009. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- 432.Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, Chen JZ, Shyng SL. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. eLife 6: e24149, 2017. doi: 10.7554/eLife.24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432a.Martin GM, Kandasamy B, DiMaio F, Yoshika C, Shyng S-L. Anti-diabetic drug binding site in a mammalian KATP channel revealed by crgoEM. Curr Biol 6: e31054, 20173. doi: 10.7554/eLife.31054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Masgrau R, Churchill GC, Morgan AJ, Ashcroft SJ, Galione A. NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Curr Biol 13: 247–251, 2003. doi: 10.1016/S0960-9822(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 434.Mastrolia V, Flucher SM, Obermair GJ, Drach M, Hofer H, Renström E, Schwartz A, Striessnig J, Flucher BE, Tuluc P. Loss of α2δ-1 Calcium Channel Subunit Function Increases the Susceptibility for Diabetes. Diabetes 66: 897–907, 2017. doi: 10.2337/db16-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes 51, Suppl 3: S394–S404, 2002. doi: 10.2337/diabetes.51.2007.S394. [DOI] [PubMed] [Google Scholar]
- 436.Matsumoto M, Miki T, Shibasaki T, Kawaguchi M, Shinozaki H, Nio J, Saraya A, Koseki H, Miyazaki M, Iwanaga T, Seino S. Noc2 is essential in normal regulation of exocytosis in endocrine and exocrine cells. Proc Natl Acad Sci USA 101: 8313–8318, 2004. doi: 10.1073/pnas.0306709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab 104: 648–653, 2011. doi: 10.1016/j.ymgme.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 438.McDermott AM, Sharp GW. Gi2 and Gi3 proteins mediate the inhibition of adenylyl cyclase by galanin in the RINm5F cell. Diabetes 44: 453–459, 1995. [DOI] [PubMed] [Google Scholar]
- 439.Mears D, Rojas E. Properties of voltage-gated Ca2+ currents measured from mouse pancreatic beta-cells in situ. Biol Res 39: 505–520, 2006. doi: 10.4067/S0716-97602006000300012. [DOI] [PubMed] [Google Scholar]
- 440.Meda P, Atwater I, Gonçalves A, Bangham A, Orci L, Rojas E. The topography of electrical synchrony among beta-cells in the mouse islet of Langerhans. Q J Exp Physiol 69: 719–735, 1984. doi: 10.1113/expphysiol.1984.sp002864. [DOI] [PubMed] [Google Scholar]
- 441.Meissner HP. Electrophysiological evidence for coupling between beta cells of pancreatic islets. Nature 262: 502–504, 1976. doi: 10.1038/262502a0. [DOI] [PubMed] [Google Scholar]
- 442.Meissner HP, Atwater IJ. The kinetics of electrical activity of beta cells in response to a “square wave” stimulation with glucose or glibenclamide. Horm Metab Res 8: 11–16, 1976. doi: 10.1055/s-0028-1093685. [DOI] [PubMed] [Google Scholar]
- 443.Meissner HP, Schmidt H. The electrical activity of pancreatic beta-cells of diabetic mice. FEBS Lett 67: 371–374, 1976. doi: 10.1016/0014-5793(76)80567-4. [DOI] [PubMed] [Google Scholar]
- 444.Merrins MJ, Stuenkel EL. Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J Physiol 586: 5367–5381, 2008. doi: 10.1113/jphysiol.2008.158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Michael DJ, Geng X, Cawley NX, Loh YP, Rhodes CJ, Drain P, Chow RH. Fluorescent cargo proteins in pancreatic beta-cells: design determines secretion kinetics at exocytosis. Biophys J 87: L03–L05, 2004. doi: 10.1529/biophysj.104.052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Michael DJ, Ritzel RA, Haataja L, Chow RH. Pancreatic beta-cells secrete insulin in fast- and slow-release forms. Diabetes 55: 600–607, 2006. doi: 10.2337/diabetes.55.03.06.db05-1054. [DOI] [PubMed] [Google Scholar]
- 447.Michael DJ, Xiong W, Geng X, Drain P, Chow RH. Human insulin vesicle dynamics during pulsatile secretion. Diabetes 56: 1277–1288, 2007. doi: 10.2337/db06-0367. [DOI] [PubMed] [Google Scholar]
- 448.Michaelevski I, Chikvashvili D, Tsuk S, Singer-Lahat D, Kang Y, Linial M, Gaisano HY, Fili O, Lotan I. Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. J Biol Chem 278: 34320–34330, 2003. doi: 10.1074/jbc.M304943200. [DOI] [PubMed] [Google Scholar]
- 449.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394: 192–195, 1998. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 450.Mikhailov MV, Campbell JD, de Wet H, Shimomura K, Zadek B, Collins RF, Sansom MS, Ford RC, Ashcroft FM. 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J 24: 4166–4175, 2005. doi: 10.1038/sj.emboj.7600877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95: 10402–10406, 1998. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Misler S, Barnett DW, Gillis KD, Pressel DM. Electrophysiology of stimulus-secretion coupling in human beta-cells. Diabetes 41: 1221–1228, 1992. doi: 10.2337/diab.41.10.1221. [DOI] [PubMed] [Google Scholar]
- 453.Misler S, Gee WM, Gillis KD, Scharp DW, Falke LC. Metabolite-regulated ATP-sensitive K+ channel in human pancreatic islet cells. Diabetes 38: 422–427, 1989. doi: 10.2337/diab.38.4.422. [DOI] [PubMed] [Google Scholar]
- 454.Misler S, Silva AM, Barnett D, Dickey AS. Phasic and tonic modes of depolarization-exocytosis coupling in beta-cells of porcine islets of Langerhans. Channels (Austin) 3: 101–109, 2009. doi: 10.4161/chan.3.2.7866. [DOI] [PubMed] [Google Scholar]
- 455.Misler S, Zhou Z, Dickey AS, Silva AM, Pressel DM, Barnett DW. Electrical activity and exocytotic correlates of biphasic insulin secretion from beta-cells of canine islets of Langerhans: contribution of tuning two modes of Ca2+ entry-dependent exocytosis to two modes of glucose-induced electrical activity. Channels (Austin) 3: 181–193, 2009. doi: 10.4161/chan.3.3.8972. [DOI] [PubMed] [Google Scholar]
- 456.Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y, Kubota A, Fujimoto S, Kajikawa M, Kuroe A, Tsuda K, Hashimoto H, Yamashita T, Jomori T, Tashiro F, Miyazaki J, Seino Y. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA 96: 14843–14847, 1999. doi: 10.1073/pnas.96.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Mizuno K, Fujita T, Gomi H, Izumi T. Granuphilin exclusively mediates functional granule docking to the plasma membrane. Sci Rep 6: 23909, 2016. doi: 10.1038/srep23909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Moghadam PK, Jackson MB. The functional significance of synaptotagmin diversity in neuroendocrine secretion. Front Endocrinol (Lausanne) 4: 124, 2013. doi: 10.3389/fendo.2013.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A. Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes 63: 2714–2726, 2014. doi: 10.2337/db13-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Monaghan AS, Benton DC, Bahia PK, Hosseini R, Shah YA, Haylett DG, Moss GW. The SK3 subunit of small conductance Ca2+-activated K+ channels interacts with both SK1 and SK2 subunits in a heterologous expression system. J Biol Chem 279: 1003–1009, 2004. doi: 10.1074/jbc.M308070200. [DOI] [PubMed] [Google Scholar]
- 461.Monterrat C, Grise F, Benassy MN, Hémar A, Lang J. The calcium-sensing protein synaptotagmin 7 is expressed on different endosomal compartments in endocrine, neuroendocrine cells or neurons but not on large dense core vesicles. Histochem Cell Biol 127: 625–632, 2007. doi: 10.1007/s00418-007-0271-0. [DOI] [PubMed] [Google Scholar]
- 462.Moriyama Y, Hayashi M. Glutamate-mediated signaling in the islets of Langerhans: a thread entangled. Trends Pharmacol Sci 24: 511–517, 2003. doi: 10.1016/j.tips.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 463.Moss CE, Glass LL, Diakogiannaki E, Pais R, Lenaghan C, Smith DM, Wedin M, Bohlooly-Y M, Gribble FM, Reimann F. Lipid derivatives activate GPR119 and trigger GLP-1 secretion in primary murine L-cells. Peptides 77: 16–20, 2016. doi: 10.1016/j.peptides.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Müller A, Kukley M, Uebachs M, Beck H, Dietrich D. Nanodomains of single Ca2+ channels contribute to action potential repolarization in cortical neurons. J Neurosci 27: 483–495, 2007. doi: 10.1523/JNEUROSCI.3816-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Mziaut H, Trajkovski M, Kersting S, Ehninger A, Altkrüger A, Lemaitre RP, Schmidt D, Saeger HD, Lee MS, Drechsel DN, Müller S, Solimena M. Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nat Cell Biol 8: 435–445, 2006. doi: 10.1038/ncb1395. [DOI] [PubMed] [Google Scholar]
- 466.Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc Natl Acad Sci USA 97: 11603–11608, 2000. doi: 10.1073/pnas.97.21.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Nadal A, Rovira JM, Laribi O, Leon-quinto T, Andreu E, Ripoll C, Soria B. Rapid insulinotropic effect of 17beta-estradiol via a plasma membrane receptor. FASEB J 12: 1341–1348, 1998. [DOI] [PubMed] [Google Scholar]
- 468.Nagamatsu S, Nakamichi Y, Watanabe T, Matsushima S, Yamaguchi S, Ni J, Itagaki E, Ishida H. Localization of cellubrevin-related peptide, endobrevin, in the early endosome in pancreatic beta cells and its physiological function in exo-endocytosis of secretory granules. J Cell Sci 114: 219–227, 2001. [DOI] [PubMed] [Google Scholar]
- 469.Nakazaki M, Crane A, Hu M, Seghers V, Ullrich S, Aguilar-Bryan L, Bryan J. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes 51: 3440–3449, 2002. doi: 10.2337/diabetes.51.12.3440. [DOI] [PubMed] [Google Scholar]
- 470.Namkung Y, Skrypnyk N, Jeong MJ, Lee T, Lee MS, Kim HL, Chin H, Suh PG, Kim SS, Shin HS. Requirement for the L-type Ca(2+) channel alpha(1D) subunit in postnatal pancreatic beta cell generation. J Clin Invest 108: 1015–1022, 2001. doi: 10.1172/JCI200113310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91: 301–307, 1993. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389–399, 1998. doi: 10.1016/S0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 473.Nesher R, Cerasi E. Modeling phasic insulin release: immediate and time-dependent effects of glucose. Diabetes 51, Suppl 1: S53–S59, 2002. doi: 10.2337/diabetes.51.2007.S53. [DOI] [PubMed] [Google Scholar]
- 474.Ng B, Kang Y, Elias CL, He Y, Xie H, Hansen JB, Wahl P, Gaisano HY. The actions of a novel potent islet beta-cell specific ATP-sensitive K+ channel opener can be modulated by syntaxin-1A acting on sulfonylurea receptor 1. Diabetes 56: 2124–2134, 2007. doi: 10.2337/db07-0030. [DOI] [PubMed] [Google Scholar]
- 475.Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res 23: 1554–1562, 2013. doi: 10.1101/gr.150706.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Nicholls DG. The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol Rev 96: 1385–1447, 2016. doi: 10.1152/physrev.00009.2016. [DOI] [PubMed] [Google Scholar]
- 477.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 478.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol 59: 171–191, 1997. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 479.Nichols CG, Remedi MS. The diabetic β-cell: hyperstimulated vs. hyperexcited. Diabetes Obes Metab 14, Suppl 3: 129–135, 2012. doi: 10.1111/j.1463-1326.2012.01655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Niki I, Ashcroft FM, Ashcroft SJ. The dependence on intracellular ATP concentration of ATP-sensitive K-channels and of Na,K-ATPase in intact HIT-T15 beta-cells. FEBS Lett 257: 361–364, 1989. doi: 10.1016/0014-5793(89)81572-8. [DOI] [PubMed] [Google Scholar]
- 481.Nilsson T, Arkhammar P, Rorsman P, Berggren PO. Suppression of insulin release by galanin and somatostatin is mediated by a G-protein. An effect involving repolarization and reduction in cytoplasmic free Ca2+ concentration. J Biol Chem 264: 973–980, 1989. [PubMed] [Google Scholar]
- 482.North RA. Molecular physiology of P2X receptors. Physiol Rev 82: 1013–1067, 2002. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 483.O’Rahilly S, Turner RC, Matthews DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 318: 1225–1230, 1988. doi: 10.1056/NEJM198805123181902. [DOI] [PubMed] [Google Scholar]
- 484.Obermüller S, Lindqvist A, Karanauskaite J, Galvanovskis J, Rorsman P, Barg S. Selective nucleotide-release from dense-core granules in insulin-secreting cells. J Cell Sci 118: 4271–4282, 2005. doi: 10.1242/jcs.02549. [DOI] [PubMed] [Google Scholar]
- 485.Oçal G, Flanagan SE, Hacihamdioğlu B, Berberoğlu M, Siklar Z, Ellard S, Savas Erdeve S, Okulu E, Akin IM, Atasay B, Arsan S, Yağmurlu A. Clinical characteristics of recessive and dominant congenital hyperinsulinism due to mutation(s) in the ABCC8/KCNJ11 genes encoding the ATP-sensitive potasium channel in the pancreatic beta cell. J Pediatr Endocrinol Metab 24: 1019–1023, 2011. doi: 10.1515/JPEM.2011.347. [DOI] [PubMed] [Google Scholar]
- 486.Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes 58: 1165–1174, 2009. doi: 10.2337/db08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Ohara-Imaizumi M, Fujiwara T, Nakamichi Y, Okamura T, Akimoto Y, Kawai J, Matsushima S, Kawakami H, Watanabe T, Akagawa K, Nagamatsu S. Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis. J Cell Biol 177: 695–705, 2007. doi: 10.1083/jcb.200608132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Olofsson CS, Collins S, Bengtsson M, Eliasson L, Salehi A, Shimomura K, Tarasov A, Holm C, Ashcroft F, Rorsman P. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of granule fusion with plasma membrane. Diabetes 56: 1888–1897, 2007. doi: 10.2337/db06-1150. [DOI] [PubMed] [Google Scholar]
- 489.Olofsson CS, Göpel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch 444: 43–51, 2002. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 490.Olofsson CS, Håkansson J, Salehi A, Bengtsson M, Galvanovskis J, Partridge C, SörhedeWinzell M, Xian X, Eliasson L, Lundquist I, Semb H, Rorsman P. Impaired insulin exocytosis in neural cell adhesion molecule-/- mice due to defective reorganization of the submembrane F-actin network. Endocrinology 150: 3067–3075, 2009. doi: 10.1210/en.2008-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Olofsson CS, Salehi A, Holm C, Rorsman P. Palmitate increases L-type Ca2+ currents and the size of the readily releasable granule pool in mouse pancreatic beta-cells. J Physiol 557: 935–948, 2004. doi: 10.1113/jphysiol.2004.066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Olsen HL, Hoy M, Zhang W, Bertorello AM, Bokvist K, Capito K, Efanov AM, Meister B, Thams P, Yang SN, Rorsman P, Berggren PO, Gromada J. Phosphatidylinositol 4-kinase serves as a metabolic sensor and regulates priming of secretory granules in pancreatic beta cells. Proc Natl Acad Sci USA 100: 5187–5192, 2003. doi: 10.1073/pnas.0931282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 55: 435–440, 2006. doi: 10.2337/diabetes.55.02.06.db04-1575. [DOI] [PubMed] [Google Scholar]
- 494.Otani K, Kulkarni RN, Baldwin AC, Krutzfeldt J, Ueki K, Stoffel M, Kahn CR, Polonsky KS. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am J Physiol Endocrinol Metab 286: E41–E49, 2004. doi: 10.1152/ajpendo.00533.2001. [DOI] [PubMed] [Google Scholar]
- 495.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipilä I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 52: 199–204, 2003. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 496.Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol 2: 805–811, 2000. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- 497.Ozanne SE, Guest PC, Hutton JC, Hales CN. Intracellular localization and molecular heterogeneity of the sulphonylurea receptor in insulin-secreting cells. Diabetologia 38: 277–282, 1995. doi: 10.1007/BF00400631. [DOI] [PubMed] [Google Scholar]
- 498.Ozcan S. Minireview: microRNA function in pancreatic β cells. Mol Endocrinol 28: 1922–1933, 2014. doi: 10.1210/me.2014-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Pace CS, Goldsmith KT. Effect of substitution of a permeable weak acid for the permissive role of glucose in amino acid-induced electrical activity in B-cells. Endocrinology 119: 2433–2438, 1986. doi: 10.1210/endo-119-6-2433. [DOI] [PubMed] [Google Scholar]
- 500.Panten U, Kriegstein E, Poser W, Schönborn J, Hasselblatt A. Effects of l-leucine and alpha-ketoisocaproic acid upon insulin secretion and metabolism of isolated pancreatic islets. FEBS Lett 20: 225–228, 1972. doi: 10.1016/0014-5793(72)80801-9. [DOI] [PubMed] [Google Scholar]
- 501.Park SH, Ryu SY, Yu WJ, Han YE, Ji YS, Oh K, Sohn JW, Lim A, Jeon JP, Lee H, Lee KH, Lee SH, Berggren PO, Jeon JH, Ho WK. Leptin promotes K(ATP) channel trafficking by AMPK signaling in pancreatic β-cells. Proc Natl Acad Sci USA 110: 12673–12678, 2013. doi: 10.1073/pnas.1216351110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Parra LA, Baust T, El Mestikawy S, Quiroz M, Hoffman B, Haflett JM, Yao JK, Torres GE. The orphan transporter Rxt1/NTT4 (SLC6A17) functions as a synaptic vesicle amino acid transporter selective for proline, glycine, leucine, and alanine. Mol Pharmacol 74: 1521–1532, 2008. doi: 10.1124/mol.108.050005. [DOI] [PubMed] [Google Scholar]
- 503.Pasyk EA, Kang Y, Huang X, Cui N, Sheu L, Gaisano HY. Syntaxin-1A binds the nucleotide-binding folds of sulphonylurea receptor 1 to regulate the KATP channel. J Biol Chem 279: 4234–4240, 2004. doi: 10.1074/jbc.M309667200. [DOI] [PubMed] [Google Scholar]
- 504.Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Søvik O, Polak M, Hattersley AT; Neonatal Diabetes International Collaborative Group . Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 355: 467–477, 2006. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 505.Penner R, Neher E. The patch-clamp technique in the study of secretion. Trends Neurosci 12: 159–163, 1989. doi: 10.1016/0166-2236(89)90059-3. [DOI] [PubMed] [Google Scholar]
- 506.Perez-Armendariz E, Atwater I. Glucose-evoked changes in [K+] and [Ca2+] in the intercellular spaces of the mouse islet of Langerhans. Adv Exp Med Biol 211: 31–51, 1986. doi: 10.1007/978-1-4684-5314-0_3. [DOI] [PubMed] [Google Scholar]
- 507.Perez-Armendariz E, Atwater I, Rojas E. Glucose-induced oscillatory changes in extracellular ionized potassium concentration in mouse islets of Langerhans. Biophys J 48: 741–749, 1985. doi: 10.1016/S0006-3495(85)83832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Pérez-Armendariz M, Roy C, Spray DC, Bennett MV. Biophysical properties of gap junctions between freshly dispersed pairs of mouse pancreatic beta cells. Biophys J 59: 76–92, 1991. doi: 10.1016/S0006-3495(91)82200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117–161, 2003. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 510.Pertusa JA, Sanchez-Andres JV, Martín F, Soria B. Effects of calcium buffering on glucose-induced insulin release in mouse pancreatic islets: an approximation to the calcium sensor. J Physiol 520: 473–483, 1999. doi: 10.1111/j.1469-7793.1999.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22: 549–558, 1999. doi: 10.1016/S0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 512.Pfeiffer T, Kraushaar U, Düfer M, Schönecker S, Haspel D, Günther E, Drews G, Krippeit-Drews P. Rapid functional evaluation of beta-cells by extracellular recording of membrane potential oscillations with microelectrode arrays. Pflugers Arch 462: 835–840, 2011. doi: 10.1007/s00424-011-1029-z. [DOI] [PubMed] [Google Scholar]
- 513.Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J Biol Chem 281: 26932–26942, 2006. doi: 10.1074/jbc.M601225200. [DOI] [PubMed] [Google Scholar]
- 514.Plant TD. Na+ currents in cultured mouse pancreatic B-cells. Pflugers Arch 411: 429–435, 1988. doi: 10.1007/BF00587723. [DOI] [PubMed] [Google Scholar]
- 515.Plant TD. Properties and calcium-dependent inactivation of calcium currents in cultured mouse pancreatic B-cells. J Physiol 404: 731–747, 1988. doi: 10.1113/jphysiol.1988.sp017316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Poulsen CR, Bokvist K, Olsen HL, Høy M, Capito K, Gilon P, Gromada J. Multiple sites of purinergic control of insulin secretion in mouse pancreatic beta-cells. Diabetes 48: 2171–2181, 1999. doi: 10.2337/diabetes.48.11.2171. [DOI] [PubMed] [Google Scholar]
- 517.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432: 226–230, 2004. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 518.Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci USA 106: 5813–5818, 2009. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Prasad RB, Groop L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel) 6: 87–123, 2015. doi: 10.3390/genes6010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18: 162–185, 2013. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 521.Pressel DM, Misler S. Role of voltage-dependent ionic currents in coupling glucose stimulation to insulin secretion in canine pancreatic islet B-cells. J Membr Biol 124: 239–253, 1991. doi: 10.1007/BF01994357. [DOI] [PubMed] [Google Scholar]
- 522.Prokopenko I, McCarthy MI, Lindgren CM. Type 2 diabetes: new genes, new understanding. Trends Genet 24: 613–621, 2008. doi: 10.1016/j.tig.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Proks P, de Wet H, Ashcroft FM. Activation of the K(ATP) channel by Mg-nucleotide interaction with SUR1. J Gen Physiol 136: 389–405, 2010. doi: 10.1085/jgp.201010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Proks P, de Wet H, Ashcroft FM. Sulfonylureas suppress the stimulatory action of Mg-nucleotides on Kir6.2/SUR1 but not Kir6.2/SUR2A KATP channels: a mechanistic study. J Gen Physiol 144: 469–486, 2014. doi: 10.1085/jgp.201411222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525.Proks P, Eliasson L, Ammälä C, Rorsman P, Ashcroft FM. Ca(2+)- and GTP-dependent exocytosis in mouse pancreatic beta-cells involves both common and distinct steps. J Physiol 496: 255–264, 1996. doi: 10.1113/jphysiol.1996.sp021682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Proks P, Puljung MC, Vedovato N, Sachse G, Mulvaney R, Ashcroft FM. Running out of time: the decline of channel activity and nucleotide activation in adenosine triphosphate-sensitive K-channels. Philos Trans R Soc Lond B Biol Sci 371: 20150426, 2016. doi: 10.1098/rstb.2015.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157: 447–458, 2014. doi: 10.1016/j.cell.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Quesada I, Fuentes E, Andreu E, Meda P, Nadal A, Soria B. On-line analysis of gap junctions reveals more efficient electrical than dye coupling between islet cells. Am J Physiol Endocrinol Metab 284: E980–E987, 2003. doi: 10.1152/ajpendo.00473.2002. [DOI] [PubMed] [Google Scholar]
- 529.Quesada I, Todorova MG, Alonso-Magdalena P, Beltrá M, Carneiro EM, Martin F, Nadal A, Soria B. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified alpha- and beta-cells within intact human islets of Langerhans. Diabetes 55: 2463–2469, 2006. doi: 10.2337/db06-0272. [DOI] [PubMed] [Google Scholar]
- 530.Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem 284: 25749–25760, 2009. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods 37: 15–26, 1991. doi: 10.1016/0165-0270(91)90017-T. [DOI] [PubMed] [Google Scholar]
- 532.Ragvin A, Moro E, Fredman D, Navratilova P, Drivenes Ø, Engström PG, Alonso ME, de la Calle Mustienes E, Gómez Skarmeta JL, Tavares MJ, Casares F, Manzanares M, van Heyningen V, Molven A, Njølstad PR, Argenton F, Lenhard B, Becker TS. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc Natl Acad Sci USA 107: 775–780, 2010. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10, Suppl 4: 32–42, 2008. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 534.Ramalingam L, Oh E, Thurmond DC. Doc2b enrichment enhances glucose homeostasis in mice via potentiation of insulin secretion and peripheral insulin sensitivity. Diabetologia 57: 1476–1484, 2014. doi: 10.1007/s00125-014-3227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, Johnson PR, Rorsman P, Braun M. Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes 59: 2198–2208, 2010. doi: 10.2337/db09-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Randle PJ. Glucokinase and candidate genes for type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 36: 269–275, 1993. doi: 10.1007/BF00400227. [DOI] [PubMed] [Google Scholar]
- 537.Ravier MA, Güldenagel M, Charollais A, Gjinovci A, Caille D, Söhl G, Wollheim CB, Willecke K, Henquin JC, Meda P. Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54: 1798–1807, 2005. doi: 10.2337/diabetes.54.6.1798. [DOI] [PubMed] [Google Scholar]
- 538.Reimann F, Huopio H, Dabrowski M, Proks P, Gribble FM, Laakso M, Otonkoski T, Ashcroft FM. Characterisation of new KATP-channel mutations associated with congenital hyperinsulinism in the Finnish population. Diabetologia 46: 241–249, 2003. doi: 10.1007/s00125-002-1014-3. [DOI] [PubMed] [Google Scholar]
- 539.Reinbothe TM, Alkayyali S, Ahlqvist E, Tuomi T, Isomaa B, Lyssenko V, Renström E. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia 56: 340–349, 2013. doi: 10.1007/s00125-012-2758-z. [DOI] [PubMed] [Google Scholar]
- 540.Remedi MS, Agapova SE, Vyas AK, Hruz PW, Nichols CG. Acute sulfonylurea therapy at disease onset can cause permanent remission of KATP-induced diabetes. Diabetes 60: 2515–2522, 2011. doi: 10.2337/db11-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Remedi MS, Rocheleau JV, Tong A, Patton BL, McDaniel ML, Piston DW, Koster JC, Nichols CG. Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia 49: 2368–2378, 2006. doi: 10.1007/s00125-006-0367-4. [DOI] [PubMed] [Google Scholar]
- 542.Remizov O, Jakubov R, Düfer M, Krippeit Drews P, Drews G, Waring M, Brabant G, Wienbergen A, Rustenbeck I, Schöfl C. Palmitate-induced Ca2+-signaling in pancreatic beta-cells. Mol Cell Endocrinol 212: 1–9, 2003. doi: 10.1016/j.mce.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 543.Renström E, Ding WG, Bokvist K, Rorsman P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron 17: 513–522, 1996. doi: 10.1016/S0896-6273(00)80183-X. [DOI] [PubMed] [Google Scholar]
- 544.Renström E, Eliasson L, Bokvist K, Rorsman P. Cooling inhibits exocytosis in single mouse pancreatic B-cells by suppression of granule mobilization. J Physiol 494: 41–52, 1996. doi: 10.1113/jphysiol.1996.sp021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Renström E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol 502: 105–118, 1997. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YH, Johnson JD, Kieffer TJ. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32: 1121–1133, 2014. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 547.Ribalet B, Beigelman PM. Cyclic variation of K+ conductance in pancreatic beta-cells: Ca2+ and voltage dependence. Am J Physiol Cell Physiol 237: C137–C146, 1979. [DOI] [PubMed] [Google Scholar]
- 548.Ribalet B, Beigelman PM. Effects of divalent cations on beta-cell electrical activity. Am J Physiol Cell Physiol 241: C59–C67, 1981. [DOI] [PubMed] [Google Scholar]
- 549.Ribalet B, Beigelman PM. Effects of sodium on beta-cell electrical activity. Am J Physiol Cell Physiol 242: C296–C303, 1982. [DOI] [PubMed] [Google Scholar]
- 550.Richards P, Parker HE, Adriaenssens AE, Hodgson JM, Cork SC, Trapp S, Gribble FM, Reimann F. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63: 1224–1233, 2014. doi: 10.2337/db13-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Ricordi C, Zeng YJ, Alejandro R, Tzakis A, Venkataramanan R, Fung J, Bereiter D, Mintz DH, Starzl TE. In vivo effect of FK506 on human pancreatic islets. Transplantation 52: 519–522, 1991. doi: 10.1097/00007890-199109000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Riedel MJ, Steckley DC, Light PE. Current status of the E23K Kir6.2 polymorphism: implications for type-2 diabetes. Hum Genet 116: 133–145, 2005. doi: 10.1007/s00439-004-1216-5. [DOI] [PubMed] [Google Scholar]
- 553.Riz M, Braun M, Wu X, Pedersen MG. Inwardly rectifying Kir2.1 currents in human β-cells control electrical activity: characterisation and mathematical modelling. Biochem Biophys Res Commun 459: 284–287, 2015. doi: 10.1016/j.bbrc.2015.02.099. [DOI] [PubMed] [Google Scholar]
- 554.Rocheleau JV, Remedi MS, Granada B, Head WS, Koster JC, Nichols CG, Piston DW. Critical role of gap junction coupled KATP channel activity for regulated insulin secretion. PLoS Biol 4: e26, 2006. doi: 10.1371/journal.pbio.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, Caicedo A. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14: 45–54, 2011. doi: 10.1016/j.cmet.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Rodriguez-Diaz R, Caicedo A. Neural control of the endocrine pancreas. Best Pract Res Clin Endocrinol Metab 28: 745–756, 2014. doi: 10.1016/j.beem.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 557.Rodriguez-Diaz R, Dando R, Huang YA, Berggren PO, Roper SD, Caicedo A. Real-time detection of acetylcholine release from the human endocrine pancreas. Nat Protoc 7: 1015–1023, 2012. doi: 10.1038/nprot.2012.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO, Caicedo A. Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17: 888–892, 2011. doi: 10.1038/nm.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Rodriguez-Diaz R, Menegaz D, Caicedo A. Neurotransmitters act as paracrine signals to regulate insulin secretion from the human pancreatic islet. J Physiol 592: 3413–3417, 2014. doi: 10.1113/jphysiol.2013.269910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Rojas E, Stokes CL, Mears D, Atwater I. Single-microelectrode voltage clamp measurements of pancreatic beta-cell membrane ionic currents in situ. J Membr Biol 143: 65–77, 1995. doi: 10.1007/BF00232524. [DOI] [PubMed] [Google Scholar]
- 561.Rolland JF, Henquin JC, Gilon P. G protein-independent activation of an inward Na(+) current by muscarinic receptors in mouse pancreatic beta-cells. J Biol Chem 277: 38373–38380, 2002. doi: 10.1074/jbc.M203888200. [DOI] [PubMed] [Google Scholar]
- 562.Ropero AB, Soria B, Nadal A. A nonclassical estrogen membrane receptor triggers rapid differential actions in the endocrine pancreas. Mol Endocrinol 16: 497–505, 2002. doi: 10.1210/mend.16.3.0794. [DOI] [PubMed] [Google Scholar]
- 563.Rorsman P, Ammälä C, Berggren PO, Bokvist K, Larsson O. Cytoplasmic calcium transients due to single action potentials and voltage-clamp depolarizations in mouse pancreatic B-cells. EMBO J 11: 2877–2884, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Rorsman P, Arkhammar P, Bokvist K, Hellerström C, Nilsson T, Welsh M, Welsh N, Berggren PO. Failure of glucose to elicit a normal secretory response in fetal pancreatic beta cells results from glucose insensitivity of the ATP-regulated K+ channels. Proc Natl Acad Sci USA 86: 4505–4509, 1989. doi: 10.1073/pnas.86.12.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Rorsman P, Ashcroft FM, Trube G. Single Ca channel currents in mouse pancreatic B-cells. Pflugers Arch 412: 597–603, 1988. doi: 10.1007/BF00583760. [DOI] [PubMed] [Google Scholar]
- 566.Rorsman P, Bokvist K, Ammälä C, Arkhammar P, Berggren PO, Larsson O, Wåhlander K. Activation by adrenaline of a low-conductance G protein-dependent K+ channel in mouse pancreatic B cells. Nature 349: 77–79, 1991. doi: 10.1038/349077a0. [DOI] [PubMed] [Google Scholar]
- 567.Rorsman P, Bokvist K, Ammälä C, Arkhammar P, Berggren PO, Larsson O, Wåhlander K. Activation by adrenaline of a low-conductance G protein-dependent K+ channel in mouse pancreatic B cells. Nature 349: 77–79, 1991. doi: 10.1038/349077a0. [DOI] [PubMed] [Google Scholar]
- 568.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 75: 155–179, 2013. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 569.Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 51: 300–308, 2012. doi: 10.1016/j.ceca.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 107: 224–235, 2011. doi: 10.1016/j.pbiomolbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 571.Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46: 1029–1045, 2003. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 572.Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J Physiol 374: 531–550, 1986. doi: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Rorsman P, Trube G. Glucose dependent K+-channels in pancreatic beta-cells are regulated by intracellular ATP. Pflugers Arch 405: 305–309, 1985. doi: 10.1007/BF00595682. [DOI] [PubMed] [Google Scholar]
- 574.Rosario LM, Atwater I, Rojas E. Membrane potential measurements in islets of Langerhans from ob/ob obese mice suggest an alteration in [Ca2+]i-activated K+ permeability. Q J Exp Physiol 70: 137–150, 1985. doi: 10.1113/expphysiol.1985.sp002885. [DOI] [PubMed] [Google Scholar]
- 575.Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, Olivotto M, Wanke E. Glucose- and arginine-induced insulin secretion by human pancreatic beta-cells: the role of HERG K(+) channels in firing and release. FASEB J 14: 2601–2610, 2000. doi: 10.1096/fj.00-0077com. [DOI] [PubMed] [Google Scholar]
- 576.Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, Zhang E, Almgren P, Ladenvall C, Axelsson AS, Edlund A, Pedersen MG, Jonsson A, Ramracheya R, Tang Y, Walker JN, Barrett A, Johnson PR, Lyssenko V, McCarthy MI, Groop L, Salehi A, Gloyn AL, Renström E, Rorsman P, Eliasson L. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 61: 1726–1733, 2012. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, Nagaraj V, Reinbothe TM, Tuncel J, Eliasson L, Groop L, Rorsman P, Salehi A, Lyssenko V, Luthman H, Renström E. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 327: 217–220, 2010. doi: 10.1126/science.1176827. [DOI] [PubMed] [Google Scholar]
- 578.Sabourin J, Le Gal L, Saurwein L, Haefliger JA, Raddatz E, Allagnat F. Store-operated Ca2+ Entry Mediated by Orai1 and TRPC1 Participates to Insulin Secretion in Rat β-Cells. J Biol Chem 290: 30530–30539, 2015. doi: 10.1074/jbc.M115.682583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Sadoul K, Berger A, Niemann H, Weller U, Roche PA, Klip A, Trimble WS, Regazzi R, Catsicas S, Halban PA. SNAP-23 is not cleaved by botulinum neurotoxin E and can replace SNAP-25 in the process of insulin secretion. J Biol Chem 272: 33023–33027, 1997. doi: 10.1074/jbc.272.52.33023. [DOI] [PubMed] [Google Scholar]
- 580.Saisho Y. β-Cell dysfunction: its critical role in prevention and management of type 2 diabetes. World J Diabetes 6: 109–124, 2015. doi: 10.4239/wjd.v6.i1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Saito T, Okada S, Yamada E, Ohshima K, Shimizu H, Shimomura K, Sato M, Pessin JE, Mori M. Syntaxin 4 and Synip (syntaxin 4 interacting protein) regulate insulin secretion in the pancreatic beta HC-9 cell. J Biol Chem 278: 36718–36725, 2003. doi: 10.1074/jbc.M305114200. [DOI] [PubMed] [Google Scholar]
- 582.Sakamaki J, Fu A, Reeks C, Baird S, Depatie C, Al Azzabi M, Bardeesy N, Gingras AC, Yee SP, Screaton RA. Role of the SIK2-p35-PJA2 complex in pancreatic β-cell functional compensation. Nat Cell Biol 16: 234–244, 2014. doi: 10.1038/ncb2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Sakura H, Ammälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett 377: 338–344, 1995. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- 584.Sakura H, Ashcroft SJ, Terauchi Y, Kadowaki T, Ashcroft FM. Glucose modulation of ATP-sensitive K-currents in wild-type, homozygous and heterozygous glucokinase knock-out mice. Diabetologia 41: 654–659, 1998. doi: 10.1007/s001250050964. [DOI] [PubMed] [Google Scholar]
- 585.Sakura H, Bond C, Warren-Perry M, Horsley S, Kearney L, Tucker S, Adelman J, Turner R, Ashcroft FM. Characterization and variation of a human inwardly-rectifying-K-channel gene (KCNJ6): a putative ATP-sensitive K-channel subunit. FEBS Lett 367: 193–197, 1995. doi: 10.1016/0014-5793(95)00498-X. [DOI] [PubMed] [Google Scholar]
- 586.Sakura H, Wat N, Horton V, Millns H, Turner RC, Ashcroft FM. Sequence variations in the human Kir6.2 gene, a subunit of the beta-cell ATP-sensitive K-channel: no association with NIDDM in while Caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia 39: 1233–1236, 1996. doi: 10.1007/BF02658512. [DOI] [PubMed] [Google Scholar]
- 587.Sánchez-Andrés JV, Gomis A, Valdeolmillos M. The electrical activity of mouse pancreatic beta-cells recorded in vivo shows glucose-dependent oscillations. J Physiol 486: 223–228, 1995. doi: 10.1113/jphysiol.1995.sp020804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Sandström PE, Sehlin J. Furosemide reduces insulin release by inhibition of Cl− and Ca2+ fluxes in beta-cells. Am J Physiol Endocrinol Metab 255: E591–E596, 1988. [DOI] [PubMed] [Google Scholar]
- 589.Sansbury FH, Flanagan SE, Houghton JA, Shuixian Shen FL, Al-Senani AM, Habeb AM, Abdullah M, Kariminejad A, Ellard S, Hattersley AT. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 55: 2381–2385, 2012. doi: 10.1007/s00125-012-2595-0. [DOI] [PubMed] [Google Scholar]
- 590.Santos RM, Rojas E. Muscarinic receptor modulation of glucose-induced electrical activity in mouse pancreatic B-cells. FEBS Lett 249: 411–417, 1989. doi: 10.1016/0014-5793(89)80669-6. [DOI] [PubMed] [Google Scholar]
- 591.Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Soria B, Valdeolmillos M. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflugers Arch 418: 417–422, 1991. doi: 10.1007/BF00550880. [DOI] [PubMed] [Google Scholar]
- 592.Sassmann A, Gier B, Gröne HJ, Drews G, Offermanns S, Wettschureck N. The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice. J Clin Invest 120: 2184–2193, 2010. doi: 10.1172/JCI41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Satin LS, Cook DL. Calcium current inactivation in insulin-secreting cells is mediated by calcium influx and membrane depolarization. Pflugers Arch 414: 1–10, 1989. doi: 10.1007/BF00585619. [DOI] [PubMed] [Google Scholar]
- 594.Satin LS, Tavalin SJ, Smolen PD. Inactivation of HIT cell Ca2+ current by a simulated burst of Ca2+ action potentials. Biophys J 66: 141–148, 1994. doi: 10.1016/S0006-3495(94)80759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Sato Y, Aizawa T, Komatsu M, Okada N, Yamada T. Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes 41: 438–443, 1992. doi: 10.2337/diab.41.4.438. [DOI] [PubMed] [Google Scholar]
- 596.Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci USA 105: 5683–5686, 2008. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Schaefer JH. The normal weight of the pancreas in the adult human being: a biometric study. Anat Rec 32: 119–132, 1926. doi: 10.1002/ar.1090320204. [DOI] [Google Scholar]
- 598.Scheen AJ, Castillo MJ, Lefèbvre PJ. Assessment of residual insulin secretion in diabetic patients using the intravenous glucagon stimulatory test: methodological aspects and clinical applications. Diabetes Metab 22: 397–406, 1996. [PubMed] [Google Scholar]
- 599.Schmidli RS, Colman PG, Bonifacio E, Bottazzo GF, Harrison LC. High level of concordance between assays for glutamic acid decarboxylase antibodies. The First International Glutamic Acid Decarboxylase Antibody Workshop. Diabetes 43: 1005–1009, 1994. doi: 10.2337/diab.43.8.1005. [DOI] [PubMed] [Google Scholar]
- 600.Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415: 321–326, 2002. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- 601.Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122, 2001. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 602.Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, Prentki M. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem 272: 18572–18579, 1997. doi: 10.1074/jbc.272.30.18572. [DOI] [PubMed] [Google Scholar]
- 603.Schulla V, Renström E, Feil R, Feil S, Franklin I, Gjinovci A, Jing XJ, Laux D, Lundquist I, Magnuson MA, Obermüller S, Olofsson CS, Salehi A, Wendt A, Klugbauer N, Wollheim CB, Rorsman P, Hofmann F. Impaired insulin secretion and glucose tolerance in beta cell-selective Ca(v)1.2 Ca2+ channel null mice. EMBO J 22: 3844–3854, 2003. doi: 10.1093/emboj/cdg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Schwanstecher C, Neugebauer B, Schulz M, Schwanstecher M. The common single nucleotide polymorphism E23K in K(IR)6.2 sensitizes pancreatic beta-cell ATP-sensitive potassium channels toward activation through nucleoside diphosphates. Diabetes 51, Suppl 3: S363–S367, 2002. doi: 10.2337/diabetes.51.2007.S363. [DOI] [PubMed] [Google Scholar]
- 605.Schwanstecher C, Schwanstecher M. Nucleotide sensitivity of pancreatic ATP-sensitive potassium channels and type 2 diabetes. Diabetes 51, Suppl 3: S358–S362, 2002. doi: 10.2337/diabetes.51.2007.S358. [DOI] [PubMed] [Google Scholar]
- 606.Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, Smith DM, Kasper M, Ämmälä C, Sandberg R. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab 24: 593–607, 2016. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem 275: 9270–9277, 2000. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- 608.Sehlin J, Meissner HP. Effects of Cl− deficiency on the membrane potential in mouse pancreatic beta-cells. Biochim Biophys Acta 937: 309–318, 1988. doi: 10.1016/0005-2736(88)90253-2. [DOI] [PubMed] [Google Scholar]
- 609.Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342, 2005. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 610.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest 121: 2118–2125, 2011. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB, . et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J Biol Chem 269: 4895–4902, 1994. [PubMed] [Google Scholar]
- 612.Seufert J, Kieffer TJ, Habener JF. Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc Natl Acad Sci USA 96: 674–679, 1999. doi: 10.1073/pnas.96.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Sha L, Westerlund J, Szurszewski JH, Bergsten P. Amplitude modulation of pulsatile insulin secretion by intrapancreatic ganglion neurons. Diabetes 50: 51–55, 2001. doi: 10.2337/diabetes.50.1.51. [DOI] [PubMed] [Google Scholar]
- 614.Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J Neurosci 20: 1710–1721, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Shibasaki T, Sunaga Y, Fujimoto K, Kashima Y, Seino S. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J Biol Chem 279: 7956–7961, 2004. doi: 10.1074/jbc.M309068200. [DOI] [PubMed] [Google Scholar]
- 616.Shibasaki T, Sunaga Y, Seino S. Integration of ATP, cAMP, and Ca2+ signals in insulin granule exocytosis. Diabetes 53, Suppl 3: S59–S62, 2004. doi: 10.2337/diabetes.53.suppl_3.S59. [DOI] [PubMed] [Google Scholar]
- 617.Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 104: 19333–19338, 2007. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina MV, Hastoy B, Philippaert K, Reinbothe T, Rorsman N, Salehi A, Sones WR, Vergari E, Weston C, Gorelik J, Katsura M, Nikolaev VO, Vennekens R, Zaccolo M, Galione A, Johnson PR, Kaku K, Ladds G, Rorsman P. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J Clin Invest 125: 4714–4728, 2015. doi: 10.1172/JCI81975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Shimomura K, Tusa M, Iberl M, Brereton MF, Kaizik S, Proks P, Lahmann C, Yaluri N, Modi S, Huopio H, Ustinov J, Otonkoski T, Laakso M, Ashcroft FM. A mouse model of human hyperinsulinism produced by the E1506K mutation in the sulphonylurea receptor SUR1. Diabetes 62: 3797–3806, 2013. doi: 10.2337/db12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, Berggren PO, Magnuson MA. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem 277: 37176–37183, 2002. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- 621.Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol 110: 643–654, 1997. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282: 1138–1141, 1998. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- 623.Silva AM, Rodrigues RJ, Tomé AR, Cunha RA, Misler S, Rosário LM, Santos RM. Electrophysiological and immunocytochemical evidence for P2X purinergic receptors in pancreatic beta cells. Pancreas 36: 279–283, 2008. doi: 10.1097/MPA.0b013e31815a8473. [DOI] [PubMed] [Google Scholar]
- 624.Silva JP, Köhler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 26: 336–340, 2000. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 625.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renström E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschütz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig J. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430–1439, 2004. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626.Skovso S, Dionne DA, Elghazi L, Li H, Hutchinson D, Hu X, Taghizadeh F, Bernal-Mizrachi E, Johnson JD. Long-term study of mice with beta cell specific insulin resistance. Diabetologia 59: 1–581, 2016. 27539147 [Google Scholar]
- 627.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885, 2007. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 628.Slepchenko KG, Daniels NA, Guo A, Li YV. Autocrine effect of Zn2+ on the glucose-stimulated insulin secretion. Endocrine 50: 110–122, 2015. doi: 10.1007/s12020-015-0568-z. [DOI] [PubMed] [Google Scholar]
- 629.Smismans A, Schuit F, Pipeleers D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia 40: 1411–1415, 1997. doi: 10.1007/s001250050843. [DOI] [PubMed] [Google Scholar]
- 630.Smith PA, Aschroft FM, Fewtrell CM. Permeation and gating properties of the L-type calcium channel in mouse pancreatic beta cells. J Gen Physiol 101: 767–797, 1993. doi: 10.1085/jgp.101.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Smith PA, Ashcroft FM, Rorsman P. Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K(+)-currents in isolated mouse pancreatic beta-cells. FEBS Lett 261: 187–190, 1990. doi: 10.1016/0014-5793(90)80667-8. [DOI] [PubMed] [Google Scholar]
- 632.Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rorsman P. Delayed rectifying and calcium-activated K+ channels and their significance for action potential repolarization in mouse pancreatic beta-cells. J Gen Physiol 95: 1041–1059, 1990. doi: 10.1085/jgp.95.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Smith PA, Bokvist K, Rorsman P. Demonstration of A-currents in pancreatic islet cells. Pflugers Arch 413: 441–443, 1989. doi: 10.1007/BF00584497. [DOI] [PubMed] [Google Scholar]
- 634.Smith PA, Duchen MR, Ashcroft FM. A fluorimetric and amperometric study of calcium and secretion in isolated mouse pancreatic beta-cells. Pflugers Arch 430: 808–818, 1995. doi: 10.1007/BF00386180. [DOI] [PubMed] [Google Scholar]
- 635.Smith PA, Rorsman P, Ashcroft FM. Modulation of dihydropyridine-sensitive Ca2+ channels by glucose metabolism in mouse pancreatic beta-cells. Nature 342: 550–553, 1989. doi: 10.1038/342550a0. [DOI] [PubMed] [Google Scholar]
- 636.Smith PA, Sakura H, Coles B, Gummerson N, Proks P, Ashcroft FM. Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic beta-cells. J Physiol 499: 625–635, 1997. doi: 10.1113/jphysiol.1997.sp021955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75–86, 2003. doi: 10.1016/S0092-8674(03)00477-X. [DOI] [PubMed] [Google Scholar]
- 638.Speidel D, Salehi A, Obermueller S, Lundquist I, Brose N, Renström E, Rorsman P. CAPS1 and CAPS2 regulate stability and recruitment of insulin granules in mouse pancreatic beta cells. Cell Metab 7: 57–67, 2008. doi: 10.1016/j.cmet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 639.Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M. Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56: 1078–1086, 2007. doi: 10.2337/db06-0232. [DOI] [PubMed] [Google Scholar]
- 640.Speier S, Rupnik M. A novel approach to in situ characterization of pancreatic beta-cells. Pflugers Arch 446: 553–558, 2003. doi: 10.1007/s00424-003-1097-9. [DOI] [PubMed] [Google Scholar]
- 641.Speier S, Yang SB, Sroka K, Rose T, Rupnik M. KATP-channels in beta-cells in tissue slices are directly modulated by millimolar ATP. Mol Cell Endocrinol 230: 51–58, 2005. doi: 10.1016/j.mce.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 642.Spurlin BA, Thurmond DC. Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol Endocrinol 20: 183–193, 2006. doi: 10.1210/me.2005-0157. [DOI] [PubMed] [Google Scholar]
- 643.Srinivas M, Rozental R, Kojima T, Dermietzel R, Mehler M, Condorelli DF, Kessler JA, Spray DC. Functional properties of channels formed by the neuronal gap junction protein connexin36. J Neurosci 19: 9848–9855, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Stanley CA. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J Clin Endocrinol Metab 101: 815–826, 2016. doi: 10.1210/jc.2015-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Stansfeld PJ, Hopkinson R, Ashcroft FM, Sansom MS. PIP(2)-binding site in Kir channels: definition by multiscale biomolecular simulations. Biochemistry 48: 10926–10933, 2009. doi: 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Stegink LD, Filer LJ Jr, Baker GL. Plasma amino acid concentrations in normal adults fed meals with added monosodium l-glutamate and aspartame. J Nutr 113: 1851–1860, 1983. [DOI] [PubMed] [Google Scholar]
- 647.Stein DT, Stevenson BE, Chester MW, Basit M, Daniels MB, Turley SD, McGarry JD. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest 100: 398–403, 1997. doi: 10.1172/JCI119546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab 23: 770–784, 2016. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Stiernet P, Guiot Y, Gilon P, Henquin JC. Glucose acutely decreases pH of secretory granules in mouse pancreatic islets. Mechanisms and influence on insulin secretion. J Biol Chem 281: 22142–22151, 2006. doi: 10.1074/jbc.M513224200. [DOI] [PubMed] [Google Scholar]
- 650.Suckale J, Solimena M. The insulin secretory granule as a signaling hub. Trends Endocrinol Metab 21: 599–609, 2010. doi: 10.1016/j.tem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 651.Südhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol 4: a011353, 2012. doi: 10.1101/cshperspect.a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80: 675–690, 2013. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Südhof TC. The presynaptic active zone. Neuron 75: 11–25, 2012. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–477, 2009. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Sugawara K, Shibasaki T, Mizoguchi A, Saito T, Seino S. Rab11 and its effector Rip11 participate in regulation of insulin granule exocytosis. Genes Cells 14: 445–456, 2009. doi: 10.1111/j.1365-2443.2009.01285.x. [DOI] [PubMed] [Google Scholar]
- 656.Sugita S, Shin OH, Han W, Lao Y, Südhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J 21: 270–280, 2002. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589: 1515–1525, 2011. doi: 10.1113/jphysiol.2010.201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Svendsen B, Pais R, Engelstoft MS, Milev NB, Richards P, Christiansen CB, Egerod KL, Jensen SM, Habib AM, Gribble FM, Schwartz TW, Reimann F, Holst JJ. GLP1- and GIP-producing cells rarely overlap and differ by bombesin receptor-2 expression and responsiveness. J Endocrinol 228: 39–48, 2016. doi: 10.1530/JOE-15-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, Bourinet E, Lory P, Miller RJ, Nargeot J, Monteil A. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep 10: 873–880, 2009. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Takahashi H, Shibasaki T, Park JH, Hidaka S, Takahashi T, Ono A, Song DK, Seino S. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes 64: 1262–1272, 2015. doi: 10.2337/db14-0576. [DOI] [PubMed] [Google Scholar]
- 661.Takahashi N, Hatakeyama H, Okado H, Noguchi J, Ohno M, Kasai H. SNARE conformational changes that prepare vesicles for exocytosis. Cell Metab 12: 19–29, 2010. doi: 10.1016/j.cmet.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 662.Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GC, Miyashita Y, Kasai H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc Natl Acad Sci USA 96: 760–765, 1999. doi: 10.1073/pnas.96.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Takahashi N, Kadowaki T, Yazaki Y, Miyashita Y, Kasai H. Multiple exocytotic pathways in pancreatic beta cells. J Cell Biol 138: 55–64, 1997. doi: 10.1083/jcb.138.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science 297: 1349–1352, 2002. doi: 10.1126/science.1073806. [DOI] [PubMed] [Google Scholar]
- 665.Takahashi N, Sawada W, Noguchi J, Watanabe S, Ucar H, Hayashi-Takagi A, Yagishita S, Ohno M, Tokumaru H, Kasai H. Two-photon fluorescence lifetime imaging of primed SNARE complexes in presynaptic terminals and β cells. Nat Commun 6: 8531, 2015. doi: 10.1038/ncomms9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Takahashi T, Shibasaki T, Takahashi H, Sugawara K, Ono A, Inoue N, Furuya T, Seino S. Antidiabetic sulfonylureas and cAMP cooperatively activate Epac2A. Sci Signal 6: ra94, 2013. doi: 10.1126/scisignal.2004581. [DOI] [PubMed] [Google Scholar]
- 667.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150: 1223–1234, 2012. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668.Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, Shimizu T, Mita T, Kanazawa A, Imaizumi MO, Abe T, Kiyonari H, Hojyo S, Fukada T, Kawauchi T, Nagamatsu S, Hirano T, Kawamori R, Watada H. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest 123: 4513–4524, 2013. doi: 10.1172/JCI68807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Tamarina NA, Wang Y, Mariotto L, Kuznetsov A, Bond C, Adelman J, Philipson LH. Small-conductance calcium-activated K+ channels are expressed in pancreatic islets and regulate glucose responses. Diabetes 52: 2000–2006, 2003. doi: 10.2337/diabetes.52.8.2000. [DOI] [PubMed] [Google Scholar]
- 670.Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, Salehi A, Korsgren O, Renström E, Groop L, Birnir B. γ-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 55: 1985–1994, 2012. doi: 10.1007/s00125-012-2548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Taneera J, Lang S, Sharma A, Fadista J, Zhou Y, Ahlqvist E, Jonsson A, Lyssenko V, Vikman P, Hansson O, Parikh H, Korsgren O, Soni A, Krus U, Zhang E, Jing XJ, Esguerra JL, Wollheim CB, Salehi A, Rosengren A, Renström E, Groop L. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab 16: 122–134, 2012. doi: 10.1016/j.cmet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 672.Tang Y, Axelsson AS, Spégel P, Andersson LE, Mulder H, Groop LC, Renström E, Rosengren AH. Genotype-based treatment of type 2 diabetes with an α2A-adrenergic receptor antagonist. Sci Transl Med 6: 257ra139, 2014. doi: 10.1126/scitranslmed.3009934. [DOI] [PubMed] [Google Scholar]
- 673.Taniguchi H, Okada Y, Shimada C, Baba S. GABA in pancreatic islets. Arch Histol Jpn 40, Suppl: 87–97, 1977. doi: 10.1679/aohc1950.40.Supplement_87. [DOI] [PubMed] [Google Scholar]
- 674.Tarasov AI, Girard CA, Ashcroft FM. ATP sensitivity of the ATP-sensitive K+ channel in intact and permeabilized pancreatic beta-cells. Diabetes 55: 2446–2454, 2006. doi: 10.2337/db06-0360. [DOI] [PubMed] [Google Scholar]
- 675.Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 52: 28–35, 2012. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Tarasov AI, Semplici F, Li D, Rizzuto R, Ravier MA, Gilon P, Rutter GA. Frequency-dependent mitochondrial Ca(2+) accumulation regulates ATP synthesis in pancreatic β cells. Pflugers Arch 465: 543–554, 2013. doi: 10.1007/s00424-012-1177-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One 7: e39722, 2012. doi: 10.1371/journal.pone.0039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Tarvin JT, Pace CS. Glucose-induced electrical activity in the pancreatic beta-cell: effect of veratridine. Am J Physiol Cell Physiol 240: C127–C134, 1981. [DOI] [PubMed] [Google Scholar]
- 679.Terauchi Y, Sakura H, Yasuda K, Iwamoto K, Takahashi N, Ito K, Kasai H, Suzuki H, Ueda O, Kamada N, Jishage K, Komeda K, Noda M, Kanazawa Y, Taniguchi S, Miwa I, Akanuma Y, Kodama T, Yazaki Y, Kadowaki T. Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem 270: 30253–30256, 1995. doi: 10.1074/jbc.270.51.30253. [DOI] [PubMed] [Google Scholar]
- 680.Teubner B, Degen J, Söhl G, Güldenagel M, Bukauskas FF, Trexler EB, Verselis VK, De Zeeuw CI, Lee CG, Kozak CA, Petrasch-Parwez E, Dermietzel R, Willecke K. Functional expression of the murine connexin 36 gene coding for a neuron-specific gap junctional protein. J Membr Biol 176: 249–262, 2000. doi: 10.1007/s00232001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268: 426–429, 1995. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- 682.Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58: 221–232, 2015. doi: 10.1007/s00125-014-3451-1. [DOI] [PubMed] [Google Scholar]
- 683.Thorens B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes Metab 16, Suppl 1: 87–95, 2014. doi: 10.1111/dom.12346. [DOI] [PubMed] [Google Scholar]
- 684.Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, Lemaire K, Schraenen A, Van Lommel L, Lehnert S, Aguayo-Mazzucato C, Cheng-Xue R, Gilon P, Van Mechelen I, Bonner-Weir S, Lemaigre F, Schuit F. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res 21: 95–105, 2011. doi: 10.1101/gr.109173.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685.Toreson WE. Glycogen infiltration (so-called hydropic degeneration) in the pancreas in human and experimental diabetes mellitus. Am J Pathol 27: 327–347, 1951. [PMC free article] [PubMed] [Google Scholar]
- 686.Torii S, Zhao S, Yi Z, Takeuchi T, Izumi T. Granuphilin modulates the exocytosis of secretory granules through interaction with syntaxin 1a. Mol Cell Biol 22: 5518–5526, 2002. doi: 10.1128/MCB.22.15.5518-5526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J, Lory P. Subunit-specific modulation of T-type calcium channels by zinc. J Physiol 578: 159–171, 2007. doi: 10.1113/jphysiol.2006.114496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Trajkovski M, Mziaut H, Altkrüger A, Ouwendijk J, Knoch KP, Müller S, Solimena M. Nuclear translocation of an ICA512 cytosolic fragment couples granule exocytosis and insulin expression in beta-cells. J Cell Biol 167: 1063–1074, 2004. doi: 10.1083/jcb.200408172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Trajkovski M, Mziaut H, Schubert S, Kalaidzidis Y, Altkrüger A, Solimena M. Regulation of insulin granule turnover in pancreatic beta-cells by cleaved ICA512. J Biol Chem 283: 33719–33729, 2008. doi: 10.1074/jbc.M804928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Trapani JG, Andalib P, Consiglio JF, Korn SJ. Control of single channel conductance in the outer vestibule of the Kv2.1 potassium channel. J Gen Physiol 128: 231–246, 2006. doi: 10.1085/jgp.200509465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J Gen Physiol 112: 333–349, 1998. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Travers ME, Mackay DJ, Dekker Nitert M, Morris AP, Lindgren CM, Berry A, Johnson PR, Hanley N, Groop LC, McCarthy MI, Gloyn AL. Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes 62: 987–992, 2013. doi: 10.2337/db12-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496, 2010. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Trube G, Rorsman P. Calcium and potassium currents recorded from pancreatic beta-cells under voltage clamp control. Adv Exp Med Biol 211: 167–175, 1986. doi: 10.1007/978-1-4684-5314-0_14. [DOI] [PubMed] [Google Scholar]
- 695.Trube G, Rorsman P, Ohno-Shosaku T. Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic beta-cells. Pflugers Arch 407: 493–499, 1986. doi: 10.1007/BF00657506. [DOI] [PubMed] [Google Scholar]
- 696.Tschritter O, Stumvoll M, Machicao F, Holzwarth M, Weisser M, Maerker E, Teigeler A, Häring H, Fritsche A. The prevalent Glu23Lys polymorphism in the potassium inward rectifier 6.2 (KIR6.2) gene is associated with impaired glucagon suppression in response to hyperglycemia. Diabetes 51: 2854–2860, 2002. doi: 10.2337/diabetes.51.9.2854. [DOI] [PubMed] [Google Scholar]
- 697.Tsuboi T, Ravier MA, Parton LE, Rutter GA. Sustained exposure to high glucose concentrations modifies glucose signaling and the mechanics of secretory vesicle fusion in primary rat pancreatic beta-cells. Diabetes 55: 1057–1065, 2006. doi: 10.2337/diabetes.55.04.06.db05-1577. [DOI] [PubMed] [Google Scholar]
- 698.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387: 179–183, 1997. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 699.Uenishi E, Shibasaki T, Takahashi H, Seki C, Hamaguchi H, Yasuda T, Tatebe M, Oiso Y, Takenawa T, Seino S. Actin dynamics regulated by the balance of neuronal Wiskott-Aldrich syndrome protein (N-WASP) and cofilin activities determines the biphasic response of glucose-induced insulin secretion. J Biol Chem 288: 25851–25864, 2013. doi: 10.1074/jbc.M113.464420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jørgensen T, Sandbaek A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40: 1098–1102, 2008. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 701.Valdeolmillos M, Santos RM, Contreras D, Soria B, Rosario LM. Glucose-induced oscillations of intracellular Ca2+ concentration resembling bursting electrical activity in single mouse islets of Langerhans. FEBS Lett 259: 19–23, 1989. doi: 10.1016/0014-5793(89)81484-X. [DOI] [PubMed] [Google Scholar]
- 702.Varadi A, Ainscow EK, Allan VJ, Rutter GA. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic beta-cells. J Cell Sci 115: 4177–4189, 2002. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- 703.Varadi A, Tsuboi T, Johnson-Cadwell LI, Allan VJ, Rutter GA. Kinesin I and cytoplasmic dynein orchestrate glucose-stimulated insulin-containing vesicle movements in clonal MIN6 beta-cells. Biochem Biophys Res Commun 311: 272–282, 2003. doi: 10.1016/j.bbrc.2003.09.208. [DOI] [PubMed] [Google Scholar]
- 704.Vasseur M, Debuyser A, Joffre M. Sensitivity of pancreatic beta cell to calcium channel blockers. An electrophysiologic study of verapamil and nifedipine. Fundam Clin Pharmacol 1: 95–113, 1987. doi: 10.1111/j.1472-8206.1987.tb00549.x. [DOI] [PubMed] [Google Scholar]
- 705.Vedovato N, Cliff E, Proks P, Poovazhagi V, Flanagan SE, Ellard S, Hattersley AT, Ashcroft FM. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk. Diabetologia 59: 1430–1436, 2016. doi: 10.1007/s00125-016-3964-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Velasco JM, Petersen JU, Petersen OH. Single-channel Ba2+ currents in insulin-secreting cells are activated by glyceraldehyde stimulation. FEBS Lett 231: 366–370, 1988. doi: 10.1016/0014-5793(88)80851-2. [DOI] [PubMed] [Google Scholar]
- 707.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 76: 387–417, 2007. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, Jacobson DA. Type 2 Diabetes-Associated K+ Channel TALK-1 Modulates β-Cell Electrical Excitability, Second-Phase Insulin Secretion, and Glucose Homeostasis. Diabetes 64: 3818–3828, 2015. doi: 10.2337/db15-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Vignali S, Leiss V, Karl R, Hofmann F, Welling A. Characterization of voltage-dependent sodium and calcium channels in mouse pancreatic A- and B-cells. J Physiol 572: 691–706, 2006. doi: 10.1113/jphysiol.2005.102368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Vikman J, Jimenez-Feltström J, Nyman P, Thelin J, Eliasson L. Insulin secretion is highly sensitive to desorption of plasma membrane cholesterol. FASEB J 23: 58–67, 2009. doi: 10.1096/fj.08-105734. [DOI] [PubMed] [Google Scholar]
- 711.Vila-Carriles WH, Zhao G, Bryan J. Defining a binding pocket for sulfonylureas in ATP-sensitive potassium channels. FASEB J 21: 18–25, 2007. doi: 10.1096/fj.06-6730hyp. [DOI] [PubMed] [Google Scholar]
- 712.Vollmer K, Holst JJ, Baller B, Ellrichmann M, Nauck MA, Schmidt WE, Meier JJ. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 57: 678–687, 2008. doi: 10.2337/db07-1124. [DOI] [PubMed] [Google Scholar]
- 713.Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344: 9145–9149, 1993. [DOI] [PubMed] [Google Scholar]
- 714.Wagoner PK, Chen C, Worley JF, Dukes ID, Oxford GS. Amylin modulates beta-cell glucose sensing via effects on stimulus-secretion coupling. Proc Natl Acad Sci USA 90: 9145–9149, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715.Wahl MA, Plehn RJ, Landsbeck EA, Verspohl EJ, Ammon HPT. Are ionic fluxes of pancreatic beta cells a target for gastric inhibitory polypeptide? Mol Cell Endocrinol 90: 117–123, 1992. doi: 10.1016/0303-7207(92)90109-J. [DOI] [PubMed] [Google Scholar]
- 716.Walent JH, Porter BW, Martin TF. A novel 145 kd brain cytosolic protein reconstitutes Ca(2+)-regulated secretion in permeable neuroendocrine cells. Cell 70: 765–775, 1992. doi: 10.1016/0092-8674(92)90310-9. [DOI] [PubMed] [Google Scholar]
- 717.Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 13, Suppl 1: 95–105, 2011. doi: 10.1111/j.1463-1326.2011.01450.x. [DOI] [PubMed] [Google Scholar]
- 718.Wang H, Ishizaki R, Kobayashi E, Fujiwara T, Akagawa K, Izumi T. Loss of granuphilin and loss of syntaxin-1A cause differential effects on insulin granule docking and fusion. J Biol Chem 286: 32244–32250, 2011. doi: 10.1074/jbc.M111.268631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.Wang Z, Oh E, Clapp DW, Chernoff J, Thurmond DC. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J Biol Chem 286: 41359–41367, 2011. doi: 10.1074/jbc.M111.291500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Wang Z, Oh E, Thurmond DC. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem 282: 9536–9546, 2007. doi: 10.1074/jbc.M610553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis: roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 122: 893–903, 2009. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab 19: 872–882, 2014. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 74: 1318–1328, 1984. doi: 10.1172/JCI111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell 14: 4103–4113, 2003. doi: 10.1091/mbc.E03-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 725.Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53, Suppl 3: S16–S21, 2004. doi: 10.2337/diabetes.53.suppl_3.S16. [DOI] [PubMed] [Google Scholar]
- 726.Wendt A, Speidel D, Danielsson A, Esguerra JL, Bogen IL, Walaas SI, Salehi A, Eliasson L. Synapsins I and II are not required for insulin secretion from mouse pancreatic β-cells. Endocrinology 153: 2112–2119, 2012. doi: 10.1210/en.2011-1702. [DOI] [PubMed] [Google Scholar]
- 727.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91: 795–826, 2011. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 728.Wierup N, Sundler F, Heller RS. The islet ghrelin cell. J Mol Endocrinol 52: R35–R49, 2013. doi: 10.1530/JME-13-0122. [DOI] [PubMed] [Google Scholar]
- 729.Wierup N, Svensson H, Mulder H, Sundler F. The ghrelin cell: a novel developmentally regulated islet cell in the human pancreas. Regul Pept 107: 63–69, 2002. doi: 10.1016/S0167-0115(02)00067-8. [DOI] [PubMed] [Google Scholar]
- 730.Wilding JP. The importance of free fatty acids in the development of Type 2 diabetes. Diabet Med 24: 934–945, 2007. doi: 10.1111/j.1464-5491.2007.02186.x. [DOI] [PubMed] [Google Scholar]
- 731.Wiser O, Trus M, Hernández A, Renström E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA 96: 248–253, 1999. doi: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev 61: 914–973, 1981. [DOI] [PubMed] [Google Scholar]
- 733.Wollheim CB, Ullrich S, Meda P, Vallar L. Regulation of exocytosis in electrically permeabilized insulin-secreting cells. Evidence for Ca2+ dependent and independent secretion. Biosci Rep 7: 443–454, 1987. doi: 10.1007/BF01362507. [DOI] [PubMed] [Google Scholar]
- 734.Worley JF III, McIntyre MS, Spencer B, Mertz RJ, Roe MW, Dukes ID. Endoplasmic reticulum calcium store regulates membrane potential in mouse islet beta-cells. J Biol Chem 269: 14359–14362, 1994. [PubMed] [Google Scholar]
- 735.Wu B, Wei S, Petersen N, Ali Y, Wang X, Bacaj T, Rorsman P, Hong W, Südhof TC, Han W. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from β-cells. Proc Natl Acad Sci USA 112: 9996–10001, 2015. doi: 10.1073/pnas.1513004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Wu ZY, Zhu LJ, Zou N, Bombek LK, Shao CY, Wang N, Wang XX, Liang L, Xia J, Rupnik M, Shen Y. AMPA receptors regulate exocytosis and insulin release in pancreatic β cells. Traffic 13: 1124–1139, 2012. doi: 10.1111/j.1600-0854.2012.01373.x. [DOI] [PubMed] [Google Scholar]
- 737.Wuttke A, Idevall-Hagren O, Tengholm A. P2Y1 receptor-dependent diacylglycerol signaling microdomains in β cells promote insulin secretion. FASEB J 27: 1610–1620, 2013. doi: 10.1096/fj.12-221499. [DOI] [PubMed] [Google Scholar]
- 738.Xia F, Xie L, Mihic A, Gao X, Chen Y, Gaisano HY, Tsushima RG. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage-gated calcium channel function in pancreatic beta-cells. Endocrinology 149: 5136–5145, 2008. doi: 10.1210/en.2008-0161. [DOI] [PubMed] [Google Scholar]
- 739.Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, Murphy AJ, Yancopoulos GD, Lin C, Gromada J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab 24: 608–615, 2016. doi: 10.1016/j.cmet.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 740.Xu G, Kaneto H, Laybutt DR, Duvivier-Kali VF, Trivedi N, Suzuma K, King GL, Weir GC, Bonner-Weir S. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes 56: 1551–1558, 2007. doi: 10.2337/db06-1033. [DOI] [PubMed] [Google Scholar]
- 741.Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54: 567–581, 2007. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 742.Yaekura K, Julyan R, Wicksteed BL, Hays LB, Alarcon C, Sommers S, Poitout V, Baskin DG, Wang Y, Philipson LH, Rhodes CJ. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem 278: 9715–9721, 2003. doi: 10.1074/jbc.M211352200. [DOI] [PubMed] [Google Scholar]
- 743.Yamada H, Yoshida M, Ito K, Dezaki K, Yada T, Ishikawa SE, Kakei M. Potentiation of Glucose-stimulated Insulin Secretion by the GPR40-PLC-TRPC Pathway in Pancreatic β-Cells. Sci Rep 6: 25912, 2016. doi: 10.1038/srep25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Yan-Do R, Duong E, Manning Fox JE, Dai X, Suzuki K, Khan S, Bautista A, Ferdaoussi M, Lyon J, Wu X, Cheley S, MacDonald PE, Braun M. A Glycine-Insulin Autocrine Feedback Loop Enhances Insulin Secretion From Human β-Cells and Is Impaired in Type 2 Diabetes. Diabetes 65: 2311–2321, 2016. doi: 10.2337/db15-1272. [DOI] [PubMed] [Google Scholar]
- 745.Yang C, Mora S, Ryder JW, Coker KJ, Hansen P, Allen LA, Pessin JE. VAMP3 null mice display normal constitutive, insulin- and exercise-regulated vesicle trafficking. Mol Cell Biol 21: 1573–1580, 2001. doi: 10.1128/MCB.21.5.1573-1580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Yang J, Chi Y, Burkhardt BR, Guan Y, Wolf BA. Leucine metabolism in regulation of insulin secretion from pancreatic beta cells. Nutr Rev 68: 270–279, 2010. doi: 10.1111/j.1753-4887.2010.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Yang Y, Gillis KD. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J Gen Physiol 124: 641–651, 2004. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PRV, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther 340: 483–489, 2012. doi: 10.1124/jpet.111.187708. [DOI] [PubMed] [Google Scholar]
- 749.Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang HY, Tanahashi T, Nakamura N, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Takeda J, Maeda E, Shin HD, Cho YM, Park KS, Lee HK, Ng MC, Ma RC, So WY, Chan JC, Lyssenko V, Tuomi T, Nilsson P, Groop L, Kamatani N, Sekine A, Nakamura Y, Yamamoto K, Yoshida T, Tokunaga K, Itakura M, Makino H, Nanjo K, Kadowaki T, Kasuga M. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 40: 1092–1097, 2008. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 750.Yasuda T, Shibasaki T, Minami K, Takahashi H, Mizoguchi A, Uriu Y, Numata T, Mori Y, Miyazaki J, Miki T, Seino S. Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metab 12: 117–129, 2010. doi: 10.1016/j.cmet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 751.Yi Z, Yokota H, Torii S, Aoki T, Hosaka M, Zhao S, Takata K, Takeuchi T, Izumi T. The Rab27a/granuphilin complex regulates the exocytosis of insulin-containing dense-core granules. Mol Cell Biol 22: 1858–1867, 2002. doi: 10.1128/MCB.22.6.1858-1867.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Yin P, Gandasi NR, Riz M, Cortese G, Chibalina MV, Rorsman P, Sherman A, Pedersen MG, Barg S. Clustering of L-type Ca(2+)-channels promotes exocytosis of individual secretory granules. Diabetologia 58: S128, 2015. [Google Scholar]
- 753.Yoshida M, Dezaki K, Yamato S, Aoki A, Sugawara H, Toyoshima H, Ishikawa SE, Kawakami M, Nakata M, Yada T, Kakei M. Regulation of voltage-gated K+ channels by glucose metabolism in pancreatic beta-cells. FEBS Lett 583: 2225–2230, 2009. doi: 10.1016/j.febslet.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 754.Yosida M, Dezaki K, Uchida K, Kodera S, Lam NV, Ito K, Rita RS, Yamada H, Shimomura K, Ishikawa SE, Sugawara H, Kawakami M, Tominaga M, Yada T, Kakei M. Involvement of cAMP/EPAC/TRPM2 activation in glucose- and incretin-induced insulin secretion. Diabetes 63: 3394–3403, 2014. doi: 10.2337/db13-1868. [DOI] [PubMed] [Google Scholar]
- 755.Yue JTY, Burdett E, Coy DH, Giacca A, Efendic S, Vranic M. Somatostatin receptor type 2 antagonism improves glucagon and corticosterone counterregulatory responses to hypoglycemia in streptozotocin-induced diabetic rats. Diabetes 61: 197–207, 2012. doi: 10.2337/db11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Zehetner J, Danzer C, Collins S, Eckhardt K, Gerber PA, Ballschmieter P, Galvanovskis J, Shimomura K, Ashcroft FM, Thorens B, Rorsman P, Krek W. PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev 22: 3135–3146, 2008. doi: 10.1101/gad.496908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22: 537–548, 1999. doi: 10.1016/S0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 758.Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 325: 607–610, 2009. doi: 10.1126/science.1172256. [DOI] [PubMed] [Google Scholar]
- 759.Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic beta-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J Gen Physiol 126: 353–363, 2005. doi: 10.1085/jgp.200509312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Zhang Q, Bengtsson M, Partridge C, Salehi A, Braun M, Cox R, Eliasson L, Johnson PR, Renström E, Schneider T, Berggren PO, Göpel S, Ashcroft FM, Rorsman P. R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat Cell Biol 9: 453–460, 2007. doi: 10.1038/ncb1563. [DOI] [PubMed] [Google Scholar]
- 761.Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, Leiss V, Nassar MA, Welling A, Gribble FM, Reimann F, Hofmann F, Wood JN, Ashcroft FM, Rorsman P. Na+ current properties in islet α- and β-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol 592: 4677–4696, 2014. doi: 10.1113/jphysiol.2014.274209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Zhang Q, Galvanovskis J, Abdulkader F, Partridge CJ, Gopel SO, Eliasson L, Rorsman P. Cell coupling in mouse pancreatic beta-cells measured in intact islets of Langerhans. Philos Trans A Math Physiol Eng Sci 366: 3503–3523, 2008. doi: 10.1098/rsta.2008.0110. [DOI] [PubMed] [Google Scholar]
- 763.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437: 902–905, 2005. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Zhang W, Lilja L, Mandic SA, Gromada J, Smidt K, Janson J, Takai Y, Bark C, Berggren PO, Meister B. Tomosyn is expressed in beta-cells and negatively regulates insulin exocytosis. Diabetes 55: 574–581, 2006. doi: 10.2337/diabetes.55.03.06.db05-0015. [DOI] [PubMed] [Google Scholar]
- 765.Zhang Z, Bhalla A, Dean C, Chapman ER, Jackson MB. Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nat Neurosci 12: 163–171, 2009. doi: 10.1038/nn.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Zhao S, Torii S, Yokota-Hashimoto H, Takeuchi T, Izumi T. Involvement of Rab27b in the regulated secretion of pituitary hormones. Endocrinology 143: 1817–1824, 2002. doi: 10.1210/endo.143.5.8823. [DOI] [PubMed] [Google Scholar]
- 767.Zhou Y, Waanders LF, Holmseth S, Guo C, Berger UV, Li Y, Lehre AC, Lehre KP, Danbolt NC. Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J Biol Chem 289: 1329–1344, 2014. doi: 10.1074/jbc.M113.529065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768.Zhou Z, Misler S. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem 270: 3498–3505, 1995. doi: 10.1074/jbc.270.8.3498. [DOI] [PubMed] [Google Scholar]
- 769.Zhu D, Koo E, Kwan E, Kang Y, Park S, Xie H, Sugita S, Gaisano HY. Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia 56: 359–369, 2013. doi: 10.1007/s00125-012-2757-0. [DOI] [PubMed] [Google Scholar]
- 770.Zhu D, Zhang Y, Lam PP, Dolai S, Liu Y, Cai EP, Choi D, Schroer SA, Kang Y, Allister EM, Qin T, Wheeler MB, Wang CC, Hong WJ, Woo M, Gaisano HY. Dual role of VAMP8 in regulating insulin exocytosis and islet β cell growth. Cell Metab 16: 238–249, 2012. doi: 10.1016/j.cmet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 771.Zhuang H, Bhattacharjee A, Hu F, Zhang M, Goswami T, Wang L, Wu S, Berggren PO, Li M. Cloning of a T-type Ca2+ channel isoform in insulin-secreting cells. Diabetes 49: 59–64, 2000. doi: 10.2337/diabetes.49.1.59. [DOI] [PubMed] [Google Scholar]
- 772.Zou N, Wu X, Jin YY, He MZ, Wang XX, Su LD, Rupnik M, Wu ZY, Liang L, Shen Y. ATP regulates sodium channel kinetics in pancreatic islet beta cells. J Membr Biol 246: 101–107, 2013. doi: 10.1007/s00232-012-9506-7. [DOI] [PubMed] [Google Scholar]
- 773.Zünkler BJ, Lenzen S, Männer K, Panten U, Trube G. Concentration-dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP-regulated K+ currents in pancreatic B-cells. Naunyn Schmiedebergs Arch Pharmacol 337: 225–230, 1988. doi: 10.1007/BF00169252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures S1-S18 - .pdf (1.08 MB)