

Abstract
Hyperglycemia-induced production of endothelin (ET)-1 is a hallmark of endothelial dysfunction in diabetes. Although the detrimental vascular effects of increased ET-1 are well known, the molecular mechanisms regulating endothelial synthesis of ET-1 in the setting of diabetes remain largely unidentified. Here, we show that adapter molecule TRAF3 interacting protein 2 (TRAF3IP2) mediates high glucose-induced ET-1 production in endothelial cells and ET-1-mediated endothelial cell inflammation. Specifically, we found that high glucose upregulated TRAF3IP2 in human aortic endothelial cells, which subsequently led to activation of JNK and IKKβ. shRNA-mediated silencing of TRAF3IP2, JNK1, or IKKβ abrogated high-glucose-induced ET-converting enzyme 1 expression and ET-1 production. Likewise, overexpression of TRAF3IP2, in the absence of high glucose, led to activation of JNK and IKKβ as well as increased ET-1 production. Furthermore, ET-1 transcriptionally upregulated TRAF3IP2, and this upregulation was prevented by pharmacological inhibition of ET-1 receptor B using BQ-788, or inhibition of NADPH oxidase-derived reactive oxygen species using gp91ds-tat and GKT137831. Notably, we found that knockdown of TRAF3IP2 abolished ET-1-induced proinflammatory and adhesion molecule (IL-1β, TNF-α, monocyte chemoattractant protein 1, ICAM-1, VCAM-1, and E-selectin) expression and monocyte adhesion to endothelial cells. Finally, we report that TRAF3IP2 is upregulated and colocalized with CD31, an endothelial marker, in the aorta of diabetic mice. Collectively, findings from the present study identify endothelial TRAF3IP2 as a potential new therapeutic target to suppress ET-1 production and associated vascular complications in diabetes.
NEW & NOTEWORTHY This study provides the first evidence that the adapter molecule TRAF3 interacting protein 2 mediates high glucose-induced production of endothelin-1 by endothelial cells as well as endothelin-1-mediated endothelial cell inflammation. The findings presented herein suggest that TRAF3 interacting protein 2 may be an important therapeutic target in diabetic vasculopathy characterized by excess endothelin-1 production.
Keywords: hyperglycemia, TRAF3 interacting protein 2, endothelin-1, endothelial dysfunction
INTRODUCTION
Hyperglycemia, the principal clinical manifestation of diabetes, is a major risk factor for cardiovascular disease morbidity and mortality (6, 32, 37, 38). The link between hyperglycemia and cardiovascular disease, including atherosclerosis, is partially attributed to the direct detrimental effects of high glucose on the vascular endothelium (10, 13, 18). However, the precise molecular mechanisms by which hyperglycemia promotes a dysfunctional endothelial cell phenotype are not fully deciphered.
A key molecular signature of diabetic vasculopathy is the upregulation of endothelin (ET)-1 (9, 14, 27, 42, 46), a peptide primarily expressed in endothelial cells. The induction of ET-1 in diabetes is largely mediated by hyperglycemia (15, 28, 29, 39, 52) and contributes to the pathophysiology of endothelial dysfunction and ensuing vascular complications (40). Indeed, aside from its potent vasoconstrictive effects, overexpression of ET-1 in endothelial cells results in increased generation of reactive oxygen species (ROS), reduced nitric oxide bioavailability, enhanced expression of adhesion molecules, and accelerated atherosclerosis (2, 11, 12, 22, 23, 36, 40). Given its deleterious role in diabetic vasculopathy, a better understanding of the molecular mechanisms underlying ET-1 induction and its downstream effects on endothelial cell function is of paramount clinical relevance. Despite the focus on ET-1 in the present investigation, we acknowledge that ET-1 is not the sole mediator of vascular complications in diabetes.
Recent evidence demonstrates that endothelin converting enzyme 1 (ECE1), responsible for the conversion of big ET-1 to biologically active ET-1, is transcriptionally upregulated by activation of the proinflammatory transcription factors activator protein 1 (AP-1) and NF-κB (30). Interestingly, both AP-1 and NF-κB have also been implicated in the regulation of EDN1 (ET-1) gene transcription (1, 29). Therefore, we reasoned that an upstream master regulator of AP-1 and NF-κB could be an important modulator of ET-1 production in endothelial cells. In this regard, the recently identified cytoplasmic adapter molecule TRAF3 interacting protein 2 (TRAF3IP2) (21) has been found to mediate activation of both transcription factors in endothelial cells exposed to high glucose (49). Accordingly, we sought to test the novel hypothesis that increased expression of TRAF3IP2 mediates high glucose-induced ET-1 production in endothelial cells. Moreover, we examined whether the harmful vascular effects of increased ET-1 (2, 11, 12, 22, 23, 36, 40), such as excess generation of ROS, induction of inflammatory mediators, and endothelial-monocyte adhesion, are mediated in part via TRAF3IP2.
MATERIALS AND METHODS
Reagents.
d-Glucose, mannitol, the IKKβ inhibitor [5-(p-fluorophenyl)-2-ureido]thiophene-3-carboxamide (TPCA-1; T1452), the JNK inhibitor SP-600125 (S5567), the ET-1 receptor A (ETA) inhibitor BQ-123, the ET-1 receptor B (ETB) inhibitor BQ-788, and DMSO (D2650) were purchased from Sigma-Aldrich (St. Louis, MO). The proteasome inhibitor MG-132 (no. 1748) was obtained from Tocris (Minneapolis, MN). The NADPH oxidase (Nox)1/Nox4 inhibitor 2–2(chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-1H-pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione (GKT137831; no. 17164) was purchased from Cayman Chemical (Ann Arbor, MI). gp91ds-tat, a peptide inhibitor of NOX2 (YGRKKRRQRRRCSTRIRRQL- NH2; AS-63818) and its scrambled peptide (RKKRRQRRRCLRITRQSR-NH2; AS-63855) were purchased from AnaSpec (Fremont, CA). QCL-1000End point chromogenic Limulus amoebocyte lysate assay (no. 50-647U) was purchased from Lonza (Walkersville, MD). At the indicated concentrations and for the duration of treatment, the pharmacological inhibitors failed to modulate endothelial morphology, viability, or adherence to culture dishes (data not shown). SuperSignal West Pico Chemiluminescent Substrate (no. 34080), Restore Western blot Stripping Buffer (no. 21059), and Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (A22188) were purchased from ThermoFisher Scientific (Houston, TX).
Cell culture.
Clonetics human aortic endothelial cells (HAECs; CC-2535, Lonza) were cultured at 37°C in endothelial basal medium-2 (EBM-2; CC-3156) supplemented with EGM-2 SingleQuots (CC-4176, Lonza). HAECs were used instead of other conventional endothelial cell culture models, such as human umbilical vein endothelial cells, for consistency with our previous work in this topic (49). HAECs were used between passages 4 and 8. Absence of endotoxin was confirmed using the QCL-1000End point chromogenic Limulus amoebocyte lysate assay. At ~70% confluency, the medium was changed to EBM-2 (without supplements) containing 25 mM d-glucose (high glucose) for the indicated time periods. Five mM d-glucose + 20 mM d-mannitol served as an osmotic control. Treatments with various inhibitors, their concentrations, and duration are detailed in the figures. THP-1 cells (a human acute monocytic leukemia cell line) were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and maintained in RPMI 1640 containing 10% heat-inactivated FBS and 0.05 mM 2-mercaptoethanol.
Lenti and adenoviral transduction.
Lentiviral particles expressing shRNA against human TRAF3IP2 (sc-29634-V), IKKβ (sc-35645-V), JNK1 (sc-29380-V), enhanced green fluorescence protein (eGFP; sc-45924-V), and Polybrene (sc-134220) were from Santa Cruz Biotechnology (Santa Cruz, CA) and were as previously described (49). Lentiviral shRNA was used at the multiplicity of infection (MOI) of 0.5 for 48 h in complete medium. To increase efficiency of lentiviral particles expressing shRNA, cells were cotreated with the cationic polymer Polybrene (5 µg/ml in water). Knockdown of respective target proteins was confirmed by immunoblot analysis. shRNA or Polybrene had no off-target effects and, at the indicated MOI and for the duration of infection, did not affect HAEC adherence, shape, or viability (Trypan blue dye exclusion). Adenovirus expressing full-length human TRAF3IP2 (GenBank Accession No. NM_147200.2) was custom generated at Vector Biolabs (Malvern, PA). Ad.eGFP (no. 1060) was purchased from Vector Biolabs. At ~70 confluency, HAECs were infected at ambient temperature with adenoviruses in PBS at the indicated MOI. After 1 h, the medium containing adenovirus was replaced with fresh culture medium. Assays were carried out after 24 h.
Superoxide and H2O2 production.
Superoxide (O2•−) generation was quantified using the lucigenin-enhanced chemiluminescence assay as previously described (47). After the subtraction of background luminescence, results were expressed as picomoles of superoxide per minute per milligram of protein. Experiments were also performed after treatment of cells with the NOX2 inhibitor gp91ds-tat (1 µM for 1 h). A corresponding scrambled peptide served as the control. H2O2 production was measured according to the manufacturer’s instructions using a commercially available kit in the presence of horseradish peroxidase (0.1 U/ml, Amplex red, 50 µM). Fluorescence was recorded at 530-nm excitation and 590-nm emission wavelengths (CytoFluor II, Applied Biosystems, Foster City, CA). Standard curves were generated using known concentrations of H2O2. Experiments were also performed after treatment with the NOX1/4 dual inhibitor GKT137831. Results are expressed as micromolars of H2O2 produced per 106 cells.
Gene expression.
Total RNA was isolated from HAECs using TRIzol reagent (Sigma), and 1 μg of RNA was reverse transcribed into cDNA using a reverse transcription kit (Agilent Technologies, Salt Lake City, UT). mRNA expression was analyzed by quantitative RT-PCR (33, 47, 49) using the following best coverage TaqMan probes from ThermoFisher Scientific-Applied Biosystems: TRAF3IP2 (Hs00974570_m1), ECE1 (Hs01043735_m1), ETA (EDNRA; Hs03988672_m1), ETB (EDNRB; Hs00240747_m1), IL-1β (Hs01555410_m1), TNF-α (Hs00174128_m1), monocyte chemoattractant protein 1 [MCP-1; chemokine (C-C motif) ligand 2; Hs00234140_m1], ICAM-1 (Hs00164932_m1), VCAM-1 (Hs01003372_m1), selectin E (Hs00174057_m1), and GAPDH (Hs02786624_g1). All data were normalized to the corresponding GAPDH mRNA level and analyzed using the method, where Ct is threshold cycle (33, 47, 49).
Promoter-reporter activity.
The 200-bp fragment of the 5′-flanking region of the human TRAF3IP2 gene (−183 to +17) in pGL3-basic vector with and without mutations in interferon regulatory factor 1 (IRF1), CCAAT/enhancer binding protein-β (c/EBPb), and AP-1 has been previously described (49). A 682-bp human ECE1 promoter in pGL3-basic vector and a deletion construct lacking AP-1 and NF-κB-binding sites were as previously described (26, 31). HAECs were transfected with the indicated promoter reporter vector (3 μg) and 100 ng of the control Renilla luciferase vector pRL-TK using the Neon transfection system (MPK-5000, Invitrogen, Carlsbad, CA) under the following conditions: pulse voltage, 1,400 V; pulse width, 20 ms; pulse number, 2; and tip type, 10 μl. Cell viability was 92%, and the transfection efficiency was 71%. Cells were harvested at the indicated time period for the dual luciferase reporter assay. pGL3-Basic served as a vector control. Data were normalized by dividing firefly luciferase activity with that of the corresponding Renilla luciferase. All plasmid preparations were endotoxin free.
Immunoblot analysis and ELISA.
Preparation of whole cell homogenates, immunoblot analysis, detection of the immunoreactive bands by enhanced chemiluminescence, and quantification by densitometry have all been previously described (33, 47, 49). Immunoblot analysis was performed on at least three separate occasions (biological and not intra-assay variables), and a representative immunoblot is shown in the figures. The source and concentration of antibodies used in the immunoblot analysis were as follows: TRAF3IP2 (1 μg/ml, IMG-563, Novus Biologicals, Littleton, CO), tubulin (1:1,000, no. 2144, Cell Signaling Technology, Beverly, MA), phospho-JNK (Thr183/Tyr185, 1:1,000, no. 9251, Cell Signaling Technology), JNK (1:1,000, no. 9252, Cell Signaling Technology), phospho-c-Jun (Ser63, 1:1,000, no. 9261, Cell Signaling Technology), c-Jun (1:1,000, no. 9165, Cell Signaling Technology), apoptosis signal-regulating kinase 1 (ASK1) (1:1,000, no. 8622, Cell Signaling Technology), Akt (1 μg/ml, no. 9272, Cell Signaling Technology), phospho-IKKβ (1:1,000, no. 2697, Cell Signaling Technology), IKKβ (1:1,000, no. 2370, Cell Signaling Technology), phospho-p65 (1:1,000, no. 3033, Cell Signaling Technology), p65 (1:1,000, no. 3034, Cell Signaling Technology), ECE1 (1 μg/ml, ab71829, Abcam, Cambridge, MA), ICAM-1 (1:400, sc-8439, Santa Cruz Biotechnology), VCAM-1 (1:200, sc-8304, Santa Cruz Biotechnology), and cleaved caspase-3 (1 μg/ml, no. 9664, Cell Signaling Technology).
ET-1, IL-1β, and TNF-α levels in equal amounts of culture supernatants were quantified by ELISA using the following kits: ET-1 (Human Endothelin-1 QuantiGlo ELISA kit, QEST00B, sensitivity: 0.102 pg/ml, R&D Systems, Minneapolis, MN), IL-1β (Human IL-1 beta/IL-1F2 Quantikine HS ELISA Kit, HSLB00D, sensitivity: 0.063 pg/ml, R&D Systems), and TNF-α (Human TNF-alpha Quantikine ELISA Kit, DTA00C, sensitivity: 5.5 pg/ml, R&D Systems).
Endothelial-monocyte adhesion.
Monocyte adhesion to endothelial cells was analyzed as previously described using the Vybrant Cell Adhesion assay kit (49). In brief, HAECs were plated in a 24-well flat-bottomed plate. At ~70% confluency, the complete medium was replaced with EBM-2 containing 25 mM d-glucose for 2 h. THP-1 cells were labeled with 5 μM calcein AM in serum-free RPMI 1640 for 30 min at 37°C, washed twice with prewarmed RPMI 1640, and resuspended in the same medium. The labeled cells (5 × 104 cells) were then added to HAECs and incubated for 1 h at 37°C. The nonadherent cells were removed carefully, and the cell layer was washed three times with ice-cold PBS. Finally, 200 μl PBS was added to each well. Monocyte adhesion to endothelial cells was quantified by measuring fluorescence at excitation and emission wavelengths of 485 and 535 nm, respectively, using BioTek Fluorimeter. Wells containing HAECs without THP-1 cells served as blanks.
Cell death.
To investigate whether the pharmacological inhibition and transduction with lentiviral shRNA particles or adenoviral expression vectors modulate cell viability, we analyzed active caspase-3 levels as markers of cell death in whole cell homogenates by immunoblot analysis. Importantly, no evidence of cell death was noted for any of the experimental conditions.
Animals.
The investigations conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the Institutional Animal Care and Use Committees of Tulane University, New Orleans, and University of Missouri-Columbia. As models of type 1 diabetes, we used male Akita (10 wk old), female nonobese diabetic (18 wk old), and male streptozotocin (STZ; STZ in sodium citrate buffer, pH 4.5, intraperitoneal, 60 mg/kg body wt, daily for 4 days, 10 wk old)-administered mouse models. The blood glucose levels were ~308, ~430, and ~490 mg/dl at 3, 5, and 10 days post-STZ administration. All these models were as previously described (49). As a model of type 2 diabetes, we used 20-wk-old female db/db mice (BKS.Cg-Dock7m+/+Leprdb/J) that were hyperglycemic (fasting glucose ~280 mg/dl and HbA1c >10%). Both diabetic db/db mice and age-matched nondiabetic control female mice (C57BL6/J) were purchased from the Jackson Laboratory (Bar Harbor, ME). All animals were housed in a temperature- (22°C) and light-controlled (12:12-h light-dark) atmosphere with ad libitum access to water and food. Aortas were collected, snap frozen, and stored at −80°C for subsequent analysis of TRAF3IP2 protein expression via immunoblot analysis or immunofluorescence.
Immunofluorescence.
To analyze expression and localization of TRAF3IP2 in endothelial cells, we performed immunofluorescence using anti-TRAF3IP2 and antibodies against CD31 (platelet and endothelial cell adhesion molecule 1), an endothelial cell-specific marker, as previously described (3, 53). In brief, db/db mice were perfused with physiological saline, and aortas were dissected, embedded in optimal cutting temperature compound, frozen, and sectioned at 5 μm thickness using a cryostat. Sections were fixed in methanol-acetone (1:1) at −20°C for 5 min, washed in PBS, blocked in a blocking buffer (protein block, no. ab64226, Abcam) for 1 h at room temperature, and washed in PBS. Sections were then incubated with mouse TRAF3IP2 antibody (1:50, no. sc-100647, Santa Cruz Biotechnology)/goat CD31 antibody (1:25, no. AF3628, R&D Systems) mixture in an antibody dilution buffer (no. 559148, BD PharMingen, Franklin Lakes, NJ) for 1 h at room temperature. After being washed in Tris-buffered saline with Tween 20 (TBST), sections were incubated with biotinylated anti-mouse IgG 1:200 (anti-mouse-biotin, BA-2000, Vector Laboratories) for 45 min at room temperature. After being washed with TBST on a rocker, sections were then incubated with streptavidin Alexa Fluor 594 conjugate (S32356, 1:400, Life Technologies, Carlsbad, CA)/donkey anti-goat Alexa Fluor 488 conjugate (A11055, 1:400, Life Technologies) and Hoechst 33342 (1:400, H3570, Life Technologies) in antibody dilution buffer for 45 min at room temperature. After being washed in TBST, sections were mounted with antifadant (P36961, ThermoFisher Scientific) and stored in the dark until imaging. Images were captured using Nikon Eclipse E600 microscope (Tokyo, Japan) with an Olympus DP72 camera. The fluorescence intensity was measured using ImageJ 1.49v (National Institutes of Health) and presented as arbitrary units.
Statistical analysis.
Comparisons between controls and various treatments were performed by ANOVA with post hoc Dunnett’s t-tests. Significance was accepted at P ≤ 0.05. All assays were performed at least three times, and error bars in the figures indicate SEs. Densitometric results are shown as ratios and fold changes from untreated control conditions. The numbers at the bottom of each panel in all figures denote lane numbers.
RESULTS
TRAF3IP2 mediates high glucose-induced activation of JNK and AP-1.
Exposure of endothelial cells to high glucose increased TRAF3IP2 protein expression (Fig. 1, A and B) as well as activation of JNK (Fig. 1C). High glucose-induced activation of JNK and c-Jun was abrogated with shRNA-mediated silencing of TRAF3IP2 (Fig. 1, D and E). Furthermore, high glucose-induced activation of c-Jun was reduced by knockdown of JNK1 as well as by SP-600125, a pharmacological JNK inhibitor (Fig. 1E). Together, these findings indicate that high glucose-induced upregulation in TRAF3IP2 expression mediates JNK activation and consequent downstream activation of the transcription factor AP-1.
Fig. 1.

High glucose (HG) induces activation of activator protein 1 (AP-1) in part via TRAF3 interacting protein 2 (TRAF3IP2) and JNK in human aortic endothelial cells (HAECs). A: HG (25 mM d-glucose) induces time-dependent TRAF3IP2 expression. At 70% confluency, the complete medium on HAECs was replaced with endothelial basal medium-2 (without supplements) for 2 h, and cells were then incubated with HG for the indicated time periods and analyzed for TRAF3IP2 protein expression by immunoblot analysis. B: HG (15 and 25 mM), but not mannitol, induces TRAF3IP2 expression. HAECs treated as in A with HG or mannitol for 30 min were analyzed for TRAF3IP2 by immunoblot analysis. C: HG (25 mM) induces time-dependent JNK activation. HAECs treated as in A with HG were analyzed for JNK activation using activation-specific antibodies. Total JNK served as a control. D: HG (25 mM) induces JNK activation via TRAF3IP2. At 50% confluency, HAECs were infected with lentiviral TRAF3IP2 or control enhanced green fluorescent protein (eGFP) shRNA (multiplicity of infection 0.5 for 48 h). Cells were then treated with HG for 60 min. Total and activated JNK were analyzed as in C. Knockdown of TRAF3IP2 was confirmed by immunoblot analysis and is shown on the right. ASK1 served as an off-target control. E: HG (25 mM) induced AP-1 activation via TRAF3IP2 and JNK. HAECs infected with TRAF3IP2 or JNK1 shRNA or pretreated with the JNK inhibitor SP600125 (20 µM for 30 min) were incubated with HG for 60 min and analyzed for AP-1 activation by immunoblot analysis using antibodies that specifically detect phosphorylated c-Jun at Ser73. Knockdown of JNK1 is shown in the top right. Bar graphs in A–E represent densitometric analyses from 3 independent experiments. *P ≤ 0.05 vs. control (i.e., open bar); †P ≤ 0.05 vs. HG (n = 3).
TRAF3IP2 mediates high glucose-induced activation of IKKβ and NF-κB.
IKKβ, a catalytic subunit of the IKK signalosome, is an upstream regulator of NF-κB. We and others have previously demonstrated that induction of TRAF3IP2 induces IKKβ activation in multiple cell types, including endothelial cells (49). In fact, using coimmunoprecipitation and immunoblot analysis, Siebenlist and colleagues (20) demonstrated that TRAF3IP2 binds IKKβ and that their physical interaction induces activation of NF-κB. Similarly, Li and colleagues (24) demonstrated that TRAF3IP2 associates with and activates IKK, leading ultimately to the liberation of NF-κB from its binding with IκB. Later, it was demonstrated that the helix-loop-helix (HLH) region in the SEFIR domain of TRAF3IP2 binds the HLH region of IKKβ (25). Together, these reports demonstrated that TRAF3IP2 induces NF-κB activation via its physical interaction with IKKβ. Therefore, we next examined whether high glucose-induced IKKβ activation is TRAF3IP2 dependent. Exposure of endothelial cells to high glucose caused activation of IKKβ, which was abrogated by the knockdown of TRAF3IP2 (Fig. 2, A and B). Furthermore, high glucose-induced activation of p65 was reduced by knockdown of IKKβ or TRAF3IP2 (Fig. 2C). These findings indicate that high glucose-induced expression of TRAF3IP2 causes activation of IKKβ and consequent downstream activation of the transcription factor NF-κB.
Fig. 2.

High glucose (HG) induces NF-κB activation in part via TRAF3 interacting protein 2 (TRAF3IP2) and IKKβ in aortic endothelial cells (HAECs). A: HG (25 mM) induces time-dependent IKKβ activation. At 70% confluency, the complete medium on HAECs was replaced with endothelial basal medium-2 (without supplements) for 2 h, and cells were then incubated with HG for the indicated time periods and analyzed for IKKβ activation using activation-specific antibodies. Total IKKβ served as a control. B: HG (25 mM) induces IKKβ activation via TRAF3IP2. At 50% confluency, HAECs were infected with lentiviral TRAF3IP2 or control enhanced green fluorescent protein (eGFP) shRNA [multiplicity of infection (MOI) 0.5 for 48 h]. Cells were then incubated with HG for 60 min. Total and activated IKKβ were analyzed by immunoblot analysis. C: HG (25 mM) induces NF-κB activation via TRAF3IP2 and IKKβ. HAECs infected with TRAF3IP2 or IKKβ shRNA (MOI 0.5 for 48 h) were incubated with HG for 60 min and then analyzed for NF-κB activation by immunoblot analysis using antibodies that specifically detect phosphorylated p65 at Ser536. Knockdown of IKKβ was confirmed by immunoblot analysis, as shown on the right. Akt served as an off-target control. Bar graphs in A–C represent densitometric analyses from 3 independent experiments. *P ≤ 0.05 vs. control (i.e., open bar); †P ≤ 0.05 vs. HG (n = 3).
TRAF3IP2 mediates high glucose-induced expression of ECE1.
Exposure of endothelial cells to high glucose increased expression of ECE1 at the mRNA and protein levels (Fig. 3, A and B). Silencing of TRAF3IP2, JNK1, or IKKβ largely, but not completely, abrogated the induction of ECE1 mRNA and protein caused by high glucose (Fig. 3, A and B). This abrogation was also achieved by pharmacological inhibition of JNK by SP600125 and IKKβ by TPCA-1 or by proteasome inhibition by MG-132 (Fig. 3, C and D). Luciferase reporter assay revealed that, in the setting of high glucose, ECE1 is transcriptionally regulated by both AP-1 and NF-κB (Fig. 3E).
Fig. 3.

High glucose (HG) induces endothelin converting enzyme 1 (ECE1) expression in part via TRAF3 interacting protein 2 (TRAF3IP2) and its downstream signaling intermediates. A and B: HG (25 mM) induces ECE1 mRNA and protein expression in part via TRAF3IP2, IKKβ, and JNK. At 50% confluency, human aortic endothelial cells (HAECs) infected with lentiviral TRAF3IP2, IKKβ, JNK1, or control eGFP shRNA [multiplicity of infection (MOI) 0.5] for 48 h were treated with HG for 120 min. ECE1 mRNA expression (A) was analyzed by quantitative RT-PCR and protein levels (B) by immunoblot analysis. C and D: pharmacological inhibition of IKKβ, NF-κB, and JNK inhibit HG (25 mM)-induced ECE1 expression. At 70% confluency, the complete medium on HAECs was replaced with endothelial basal medium-2 (without supplements) for 2 h, pretreated with TPCA-1 (5 µM in DMSO), MG-132 (5 µM in DMSO for 1h), SP600125 (20 µM for 30 min), or DMSO vehicle, and then incubated with HG and analyzed for ECE1 mRNA (C) and protein expression (D) after 2 h as in A. E: HG (25 mM) stimulates ECE1 promoter-dependent reporter gene activation via NF-κB and AP-1. HAECs were transfected with a reporter vector containing a 682-bp fragment of the 5′-flanking region of the human ECE1 gene (3 μg for 24 h) with and without the deletions. pGL3-Basic served as a vector control. Cells were cotransfected with the Renilla luciferase vector (100 ng). After transfection, cells were treated with HG for 12 h and harvested for the dual-luciferase assay (n = 6). Firefly luciferase data were normalized to that of corresponding Renilla luciferase activity. Bar graphs in A–D represent densitometric analyses from at least 3 independent experiments. A–E: *P ≤ 0.01 vs. control (i.e., first open bar); †P ≤ 0.05 vs. HG (n = 3–6).
Overexpression of TRAF3IP2 activates JNK and IKKβ.
Ectopic expression of TRAF3IP2 (Fig. 4A), in the absence of high glucose, caused activation of IKKβ and JNK and downstream activation of p65 and c-Jun (Fig. 4B–E). Activation of p65 with overexpression of TRAF3IP2 was blunted with pharmacological inhibition of IKKβ using TPCA-1 (Fig. 4C). Inhibition of TRAF3IP2-induced activation of p65 was also achieved by proteasome inhibition by MG-132 (Fig. 4C). Similarly, pharmacological inhibition of JNK using SP600125 blunted TRAF3IP2-induced activation of JNK and c-Jun (Fig. 4, D and E).
Fig. 4.

Overexpression of TRAF3 interacting protein 2 (TRAF3IP2), by itself, activates IKKβ/NF-κB and JNK/activator protein 1 (AP-1). A: adenoviral transduction of TRAF3IP2. At 70% confluency, human aortic endothelial cells (HAECs) were infected at the indicated multiplicity of infection (MOI) with an adenoviral vector expressing TRAF3IP2. After 24 h, TRAF3IP2 protein levels were analyzed by immunoblot analysis. B: ectopic expression of TRAF3IP2 activates IKKβ. HAECs infected with Ad.TRAF3IP2 (MOI 10 for 24 h) or incubated with TPCA-1 (5 µM in DMSO) were analyzed for IKKβ activation by immunoblot analysis. C: ectopic expression of TRAF3IP2 activates NF-κB via IKKβ and IκB degradation. HAECs treated as in B but with TPCA-1 (5 µM in DMSO) or MG-132 (5 µM in DMSO) were analyzed for NF-κB activation by immunoblot analysis using activation-specific anti-p65 antibodies. D: ectopic expression of TRAF3IP2 activates JNK. HAECs infected with Ad.TRAF3IP2 (MOI 10 for 24 h) and incubated with SP600125 (20 µM in DMSO for 30 min) were analyzed for JNK activation by immunoblot analysis. E: ectopic expression of TRAF3IP2 activates AP-1 via JNK. HAECs treated as in D were analyzed for AP-1 activation by immunoblot analysis using activation-specific anti-c-Jun antibodies. Bar graphs in A–E represent densitometric analyses from 3 independent experiments. *P ≤ 0.05 vs. control (i.e., open bar); †P ≤ 0.05 vs. HG (n = 3).
TRAF3IP2 mediates high glucose-induced production of ET-1.
Exposure of endothelial cells to high glucose increased production of ET-1 that was largely abolished by knockdown of TRAF3IP2, JNK1, or IKKβ (Fig. 5A). Moreover, overexpression of TRAF3IP2 led to increased ET-1 production, thus recapitulating the effects of high glucose (Fig. 5A). These findings indicate that high glucose-induced production of ET-1 in endothelial cells is in part mediated by TRAF3IP2. Our results also demonstrated that neither pharmacological inhibitors nor silencing of TRAF3IP2, IKKβ, or JNK1 induced cell death, as evidenced by the low levels of cleaved caspase-3 (Fig. 5B).
Fig. 5.

High glucose (HG) as well as ectopic expression of TRAF3 interacting protein 2 (TRAF3IP2) by itself stimulate endothelin-1 (ET-1) production. A: human aortic endothelial cells (HAECs) infected with lentiviral TRAF3IP2, IKKβ, JNK1, or control eGFP shRNA [multiplicity of infection (MOI) 0.5 for 48 h] or infected with adenoviral vector expressing TRAF3IP2 or control eGFP (MOI 10 for 24 h) were treated with HG (25 mM) for 24 h. ET-1 levels in culture supernatants were analyzed by ELISA. B: demonstration that neither pharmacological inhibitors nor silencing of TRAF3IP2, IKKβ, or JNK1 induced cell death, as evidenced by the low levels of cleaved caspase-3. *P ≤ 0.001 vs. control (i.e., first open bar); †P ≤ 0.05 vs. HG (n = 12); #P < 0.001 vs. Ad.eGFP (n = 12).
ET-1 upregulates TRAF3IP2.
Treatment of endothelial cells with ET-1 increased TRAF3IP2 at the protein and mRNA levels (Fig. 6, A–C). The promoter-reporter assays revealed that TRAF3IP2 is transcriptionally regulated by ET-1 (Fig. 6D).
Fig. 6.

Endothelin-1 (ET-1) induces TRAF3 interacting protein 2 (TRAF3IP2) expression. A and B: ET-1 induces TRAF3IP2 expression in a dose- and time-dependent manner. At 70% confluency, the complete medium on human aortic endothelial cells (HAECs) was replaced with endothelial basal medium-2 (without supplements) for 2 h, and cells were then treated with ET-1 at the indicated concentrations for 1 h (A) or for up to 6 h with ET-1 at 10 nM (B). TRAF3IP2 expression was analyzed by immunoblot analysis. C: ET-1 induces TRAF3IP2 mRNA expression. HAECs were treated with ET-1 (10 nM) for up to 6 h and then analyzed for TRAF3IP2 mRNA expression by quantitative RT-PCR. D: ET-1 stimulates TRAF3IP2 promoter-dependent reporter gene activation in part via CCAAT/enhancer binding protein-β (c/EBPb) and activator protein 1. HAECs were transfected with a reporter vector containing a 200-bp fragment of the 5′-flanking region of the human TRAF3IP2 gene (3 µg for 24 h) with and without mutations (n = 6). pGL3-basic served as a vector control. Cells were cotransfected with the Renilla luciferase vector (100 ng). After transfection, cells were treated with ET-1 (10 nM) for 12 h and harvested for the dual-luciferase assay. Firefly luciferase data were normalized to that of corresponding Renilla luciferase activity. Bar graphs in A and B represent densitometric analyses from 3 independent experiments. A–D: *P ≤ 0.01 vs. control (i.e., open bar); †P ≤ 0.05 vs. control or untreated HAECs transfected with the intact TRAF3IP2 promoter reporter construct (n = 3–6).
ET-1-induced upregulation of TRAF3IP2 is ROS mediated.
ET-1-induced expression of TRAF3IP2 was blunted with pharmacological inhibition of ETB receptors using BQ-788, whereas inhibition of ETA receptors using BQ-123 did not significantly alter expression of TRAF3IP2 (Fig. 7A). Supporting these results, quantitative RT-PCR revealed that endothelial cells predominantly express ETB receptors (Fig. 7A). ET-1-induced superoxide generation and H2O2 production were abolished using gp91ds-tat and GKT137831, respectively (Fig. 7, B and C). Furthermore, gp91ds-tat and GKT137831 abolished ET-1-induced upregulation of TRAF3IP2 (Fig. 7D). Together, these findings indicate that TRAF3IP2 is regulated by ET-1-induced ROS.
Fig. 7.

Oxidative stress mediates endothelin-1 (ET-1)-induced TRAF3 interacting protein 2 (TRAF3IP2) expression. A: ET-1 induces TRAF3IP2 expression in part via ET-1 receptor B (ETB). At 70% confluency, the complete medium on human aortic endothelial cells (HAECs) was replaced with endothelial basal medium-2 (without supplements) for 2 h, and cells were treated with BQ-123, BQ-788 (1 µM for 1 h), or solvent control DMSO and then with ET-1 (10 nM for 1 h). TRAF3IP2 expression was analyzed by immunoblot analysis (left and middle). ETA and ETB expression at basal conditions was analyzed by quantitative RT-PCR (right). B and C: ET-1 induces oxidative stress. HAECs were treated with the NADPH oxidase (NOX)2 inhibitor gp91ds-tat (1 µM for 1 h) or NOX1/4 dual inhibitor GKT137831 (5 µM in DMSO for 15 min) followed by ET-1 (10 nM) for 15 (B) or 30 (C) min. Superoxide anion (O2−·; B) and H2O2 (C) production were analyzed by cytochrome c assay and Amplex red assay, respectively (n = 6). D: ET-1-induced TRAF3IP2 expression is oxidative stress responsive. HAECs treated as in B and C but for 1 h with ET-1 (10 nM) were analyzed for TRAF3IP2 expression by immunoblot analysis. Bar graphs in A and D represent densitometric analyses from 3 independent experiments. A–D: *P ≤ 0.01 vs. control (i.e., open bar); †P ≤ 0.05 vs. ET-1 (n = 3–6).
ET-1-induced endothelial inflammation is mediated by TRAF3IP2.
Treatment of endothelial cells with ET-1 increased mRNA expression of multiple proinflammatory and adhesion molecules, including IL-1β, TNF-α, MCP-1, ICAM-1, VCAM-1, and selectin E (Fig. 8A) as well as secretion of IL-1β and TNF-α (Fig. 8B). In addition, ET-1 also caused an increase in ICAM-1 and VCAM-1 protein content (Fig. 8C). Notably, all these effects were abrogated with knockdown of TRAF3IP2 (Fig. 8, A–C).
Fig. 8.

Endothelin-1 (ET-1) induces the expression of multiple inflammatory mediators in human aortic endothelial cells (HAECs) in part via TRAF3 interacting protein 2 (TRAF3IP2). A–C: ET-1 stimulates proinflammatory cytokine and adhesion molecule expression. At 70% confluency, the complete medium on HAECs was replaced with endothelial basal medium-2 (without supplements) for 2 h, and cells were then incubated with ET-1 (10 nM) for 2 h (A and C) or 24 h (B). mRNA expression was analyzed by quantitative RT-PCR (n = 6), protein expression by immunoblot analysis (n = 3), and secreted cytokine levels by ELISA (data are expressed as absolute values or fold changes; n = 6). Bar graphs in C represent densitometric analyses from 3 independent experiments. A–C: *P ≤ 0.01 vs. control (i.e., open bar); †P ≤ 0.05 vs. ET-1 (n = 3–6).
ET-1-induced endothelial cell-monocyte adhesion is mediated by TRAF3IP2.
Treatment of endothelial cells with ET-1 increased monocyte adhesion to endothelial cells. This effect was abrogated with knockdown of TRAF3IP2 (Fig. 9).
Fig. 9.

Endothelin-1 (ET-1) promotes monocyte-endothelial cell adhesion in part via TRAF3 interacting protein 2 (TRAF3IP2). Human aortic endothelial cells (HAECs) at 50% confluency were infected with lentiviral particles expressing TRAF3IP2 or control eGFP shRNA [multiplicity of infection (MOI) 0.5 for 48 h], treated with ET-1 (10 nM) for 12 h, and then incubated with calcein AM-loaded THP-1 monocytic cells for 1 h. Monocyte-endothelial cell adhesion was quantified by measuring fluorescence at excitation and emission wavelengths of 485 and 535 nm, and relative fluorescence intensity was quantified. Wells containing HAECs without THP-1 cells served as blanks. *P ≤ 0.01 vs. control (i.e., open bar); †P ≤ 0.01 vs. ET-1 (n = 6).
Aortic TRAF3IP2 expression is increased in mouse models of diabetes.
TRAF3IP2 expression was increased in aortas from type 1 (Fig. 10, A and B) and type 2 (Fig. 10C) diabetic mice. Figure 10C also shows that TRAF3IP2 is upregulated (~3-fold) and colocalized with CD31, an endothelial marker, in aortas of diabetic mice.
Fig. 10.

TRAF3 interacting protein 2 (TRAF3IP2) expression is increased in aortic tissues from type 1 and type 2 diabetic mouse models. A and B: TRAF3IP2 expression in aortas from type 1 diabetic Akita and nonobese diabetic (NOD) mice (A) and streptozotocin (STZ)-induced diabetes (B). TRAF3IP2 expression was analyzed by immunoblot analysis. C: colocalization of TRAF3IP2 and CD31, an endothelial cell-specific marker, in aortas from type 2 diabetic db/db mice by immunofluorescence. Positive TRAF3IP2 expression was detected not only in endothelial cells (arrows) but also in the media (arrowheads) and adventitia (block arrows). Magnification: ×10 and ×60. Colocalization of TRAF3IP2 and CD31 demonstrated an ~3-fold increase in TRAF3IP2 expression in CD31-positive endothelial cells (bottom right). A.U., arbitrary units. Scale bars = 10 and 75 µm.
DISCUSSION
Increased formation of ET-1 is a hallmark of endothelial dysfunction in diabetes (9, 14, 27, 42, 46). Although the harmful vascular effects of increased ET-1 are well recognized (2, 11, 12, 22, 23, 36, 40), the molecular mechanisms governing endothelial synthesis of ET-1 in the setting of diabetes remain largely unknown. Here, we provide the initial evidence that the adapter molecule TRAF3IP2 mediates high glucose-induced ET-1 production in endothelial cells. Furthermore, we demonstrate, for the first time, that ET-1 signaling, in turn, upregulates TRAF3IP2 in endothelial cells and that ET-1-induced endothelial inflammation and monocyte adhesion are mediated by TRAF3IP2. Of further significance, we demonstrate that TRAF3IP2 is upregulated in aortic endothelial cells of diabetic mice. Collectively, findings from the present study identify endothelial TRAF3IP2 as a potential new therapeutic target to suppress ET-1 production and associated vascular complications in diabetes.
Over the last two decades, the number of new cases of diabetes has almost tripled in the United States, and current projections estimate that one in three Americans will have diabetes by 2050 (8). This epidemic of diabetes also contributes to the staggering rates of cardiovascular disease and mortality (35). Indeed, 8 of 10 patients with diabetes die from cardiovascular diseases (20), and this is largely attributed to the proatherogenic effects of hyperglycemia. In this regard, data accrued over the last decade indicate that the risk of macrovascular complications rises with the severity of hyperglycemia, suggesting that the relation between metabolic disturbances and vascular damage is roughly linear (6, 32, 37, 38). Glycemic dysregulation and consequent generation of ET-1 in endothelial cells is considered central to the pathogenesis of atherosclerosis in diabetes (5, 16, 34, 43, 44, 48, 51). ET-1 contributes to vascular oxidative stress, inflammation, reduced nitric oxide bioavailability, impaired endothelium-dependent dilation, and development of atherosclerosis (2, 11, 12, 22, 23, 36, 40). Therefore, an understanding of the molecular mechanisms regulating production of ET-1 in endothelial cells is crucial for identification of new targets and strategies to prevent and treat vascular derangements related to diabetes.
In the present investigation, we identify TRAF3IP2 as an important transducer of ET-1 production. Indeed, results from the present study demonstrate that high glucose transcriptionally upregulates TRAF3IP2 and that this induction largely mediates ET-1 production. More specifically, as shown in Fig. 11, we provide evidence that high glucose-induced upregulation of TRAF3IP2 causes activation of JNK and IKKβ, which consequently leads to AP-1 and NF-κB-mediated transcription of ECE1, the enzyme responsible for the conversion of big ET-1 to biologically active ET-1.
Fig. 11.
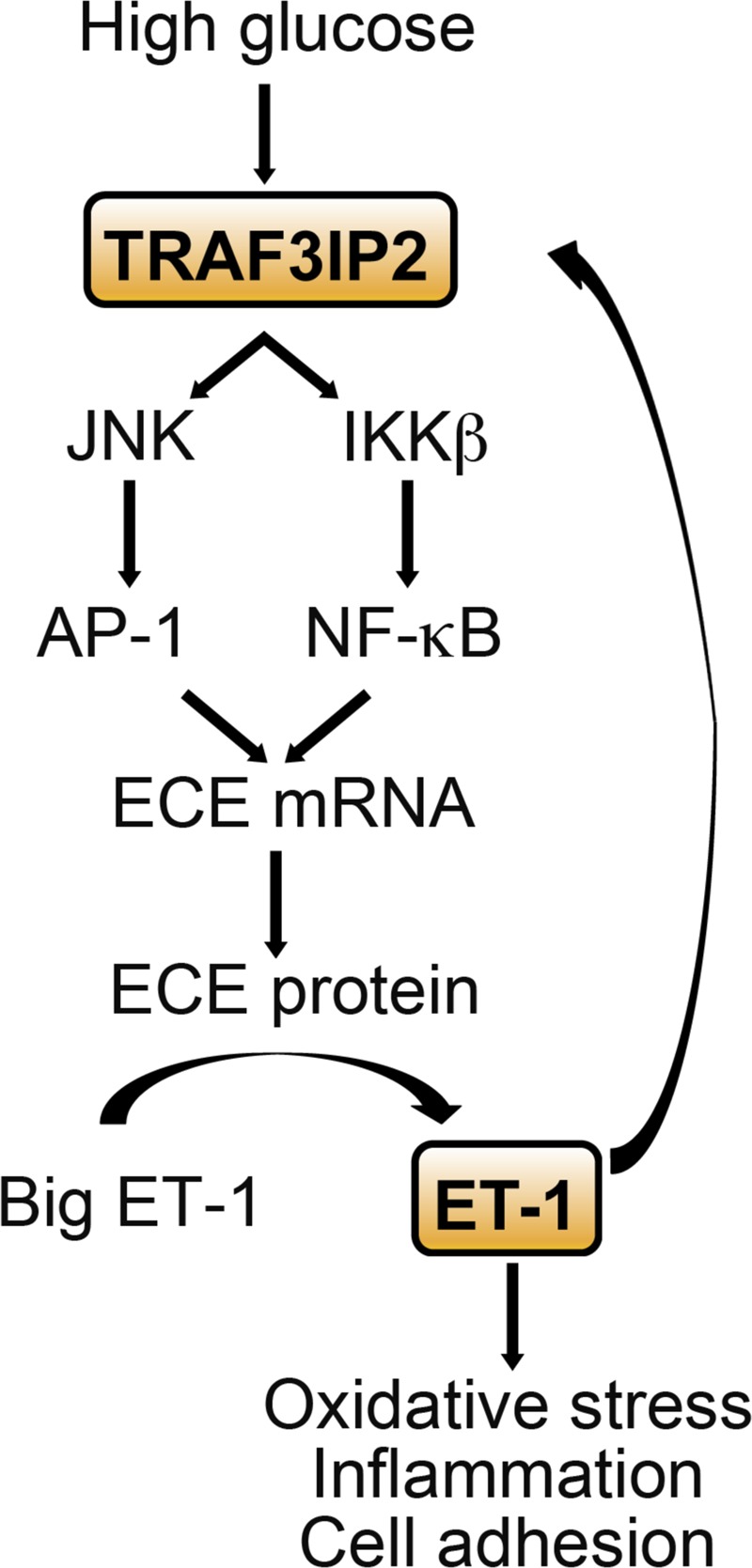
Summary of findings. Schematic illustrating a vicious cycle resulting from the cross talk between high glucose- and endothelin (ET)-1-mediated TRAF3 interacting protein 2 (TRAF3IP2) induction and TRAF3IP2-dependent ET-converting enzyme 1 (ECE1) transcription, ET-1 production, and ET-1-induced upregulation of proinflammatory and adhesion molecule expression as well as increased monocyte-endothelial cell adhesion. Together, these findings suggest that TRAF3IP2 may be an important therapeutic target in diabetic vasculopathy characterized by excess ET-1 formation. AP-1, activator protein 1.
Although silencing of TRAF3IP2, IKKβ, or JNK1 largely reduced ET-1 production in response to high glucose (Fig. 5), it should be noted that the abrogation was incomplete. This finding suggests that other mechanisms, independent of TRAF3IP2 downstream signaling events, may also be implicated in the production of ET-1 from endothelial cells. In this regard, data are available demonstrating that high glucose activates PKC in endothelial cells and that its pharmacological blockade inhibits high glucose-induced ECE1 and ET-1 expression (19, 39), indicating that activation of PKC also plays an important role in high glucose-induced ET-1 production. Whether PKC and TRAF3IP2 interact in the production of ET-1 and the mechanisms by which this occurs need further investigation.
Another salient and novel finding of the present investigation is that ET-1 transcriptionally upregulates TRAF3IP2, and this upregulation is predominantly mediated via endothelial ETB receptors and ensuing generation of ROS. Indeed, for the first time, we demonstrate that NOX2 and NOX1/4 inhibitors markedly inhibit ET-1-induced upregulation of TRAF3IP2. However, although most of ET-1-induced ROS appears to be NOX dependent, we cannot rule out the contribution of other sources of ROS. Notably, we provide evidence in cell culture that silencing TRAF3IP2 abolishes ET-1-induced endothelial inflammation and monocyte adhesion, although future studies should confirm these findings in vivo. Nevertheless, results from the present study support the existence of a self-perpetuating vicious cycle whereby high glucose promotes expression of TRAF3IP2, leading to ET-1 production, which, in turn, further promotes the upregulation of TRAF3IP2 (Fig. 11). Although this feedforward mechanism between ET-1 and TRAF3IP2 signaling under high glucose conditions is interesting and novel, it should be acknowledged that activation of downstream signaling molecules (e.g., JNK and IKKβ) only increased transiently in response to high glucose, suggesting that counterregulatory mechanisms may be in place. Importantly, silencing of TRAF2PI2 not only abrogated ET-1 production but also prevented its downstream proinflammatory effects. Taken together, these data support TRAF3IP2 as a potential new therapeutic target to inhibit both ET-1 production and ET-1-induced vascular inflammation. In fact, rodent and human studies have demonstrated that pharmacological ET-1 receptor antagonists exert vascular protective effects (4, 7, 17, 41, 45, 50). However, future research is needed to determine the role of TRAF3PI2 inhibition in reducing ET-1 production and vascular complications in preclinical models of diabetes and diabetic patients.
In conclusion, we demonstrate the novel findings that TRAF3IP2 mediates ECE1 transcription, ET-1 production, and ET-1-induced endothelial cell inflammation (i.e., upregulation of proinflammatory and adhesion molecules as well as increased monocyte adhesion). In addition, we show that diabetic mice exhibit increased TRAF3IP2 expression in the aortic wall, including the endothelium. Together, the elucidation of these findings suggests that TRAF3IP2 may be an important therapeutic target in diabetic vasculopathy characterized by excess ET-1 formation.
GRANTS
B. Chandrasekar is a recipient of a Department of Veterans Affairs Research Career Scientist award. This work was supported by United States Department of Veterans Affairs, Office of Research and Development-Biomedical Laboratory Research and Development Service Award I01-BX002255 (to B. Chandrasekar) and National Institutes of Health (NIH) Grants K01-HL-125503 and R01-HL-137769 (to J. Padilla). V. G. DeMarco is supported by unrestricted funding from Boehringer Ingelheim. U. Siebenlist is supported by the Intramural Research Program of the NIH.
DISCLAIMERS
The contents of this report do not represent the views of the Department of Veterans Affairs or the United States government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.P., A.C., N.A.D., S.L.-O., L.A.M.-L., U.S., V.G.D., and B.C. conceived and designed research; J.P., A.C., N.A.D., H.K.K., S.L.-O., L.A.M.-L., U.S., V.G.D., and B.C. interpreted results of experiments; J.P., H.K.K., and B.C. prepared figures; J.P. drafted manuscript; J.P., A.C., N.D.D., S.L.-O., L.A.M.-L., U.S., V.G.D., and B.C. edited and revised manuscript; J.P., A.C., N.A.D., H.K.K., S.L.-O., L.A.M.-L., U.S., V.G.D., and B.C. approved final version of manuscript; N.A.D., H.K.K. and B.C. performed experiments; N.A.D., H.K.K. and B.C. analyzed data.
REFERENCES
- 1.Adamopoulos C, Piperi C, Gargalionis AN, Dalagiorgou G, Spilioti E, Korkolopoulou P, Diamanti-Kandarakis E, Papavassiliou AG. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-kappaB and JNK-AP-1 signaling pathways. Cell Mol Life Sci 73: 1685–1698, 2016. doi: 10.1007/s00018-015-2091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amiri F, Virdis A, Neves MF, Iglarz M, Seidah NG, Touyz RM, Reudelhuber TL, Schiffrin EL. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation 110: 2233–2240, 2004. doi: 10.1161/01.CIR.0000144462.08345.B9. [DOI] [PubMed] [Google Scholar]
- 3.Aroor AR, Habibi J, Kandikattu HK, Garro-Kacher M, Barron B, Chen D, Hayden MR, Whaley-Connell A, Bender SB, Klein T, Padilla J, Sowers JR, Chandrasekar B, DeMarco VG. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc Diabetol 16: 61, 2017. doi: 10.1186/s12933-017-0544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babaei S, Picard P, Ravandi A, Monge JC, Lee TC, Cernacek P, Stewart DJ. Blockade of endothelin receptors markedly reduces atherosclerosis in LDL receptor deficient mice: role of endothelin in macrophage foam cell formation. Cardiovasc Res 48: 158–167, 2000. doi: 10.1016/S0008-6363(00)00169-3. [DOI] [PubMed] [Google Scholar]
- 5.Bansilal S, Farkouh ME, Fuster V. Role of insulin resistance and hyperglycemia in the development of atherosclerosis. Am J Cardiol 99: 6B–14B, 2007. doi: 10.1016/j.amjcard.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JA, Paneni F, Cosentino F, Creager MA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J 34: 2444–2452, 2013. doi: 10.1093/eurheartj/eht142. [DOI] [PubMed] [Google Scholar]
- 7.Bohm F, Ahlborg G, Pernow J. Endothelin-1 inhibits endothelium-dependent vasodilatation in the human forearm: reversal by ETA receptor blockade in patients with atherosclerosis. Clin Sci 102: 321–327, 2002. [PubMed] [Google Scholar]
- 8.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 8: 29, 2010. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardillo C, Campia U, Bryant MB, Panza JA. Increased activity of endogenous endothelin in patients with type II diabetes mellitus. Circulation 106: 1783–1787, 2002. doi: 10.1161/01.CIR.0000032260.01569.64. [DOI] [PubMed] [Google Scholar]
- 10.Ceriello A. Hyperglycaemia and the vessel wall: the pathophysiological aspects on the atherosclerotic burden in patients with diabetes. Eur J Cardiovasc Prev Rehab 17, Suppl 1: S15–S19, 2010. doi: 10.1097/01.hjr.0000368193.24732.66. [DOI] [PubMed] [Google Scholar]
- 11.Dong F, Zhang X, Wold LE, Ren Q, Zhang Z, Ren J. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br J Pharmacol 145: 323–333, 2005. doi: 10.1038/sj.bjp.0706193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 269: 713–717, 2000. doi: 10.1006/bbrc.2000.2354. [DOI] [PubMed] [Google Scholar]
- 13.Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des 19: 5695–5703, 2013. doi: 10.2174/1381612811319320005. [DOI] [PubMed] [Google Scholar]
- 14.Gogg S, Smith U, Jansson PA. Increased MAPK activation and impaired insulin signaling in subcutaneous microvascular endothelial cells in type 2 diabetes: the role of endothelin-1. Diabetes 58: 2238–2245, 2009. doi: 10.2337/db08-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan Q, Liu W, Liu Y, Fan Y, Wang X, Yu C, Zhang Y, Wang S, Liu J, Zhao J, Gao L. High glucose induces the release of endothelin-1 through the inhibition of hydrogen sulfide production in HUVECs. Int J Mol Med 35: 810–814, 2015. doi: 10.3892/ijmm.2014.2059. [DOI] [PubMed] [Google Scholar]
- 16.Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 3: 853–876, 2007. [PMC free article] [PubMed] [Google Scholar]
- 17.Halcox JP, Nour KR, Zalos G, Quyyumi AA. Coronary vasodilation and improvement in endothelial dysfunction with endothelin ETA receptor blockade. Circ Res 89: 969–976, 2001. doi: 10.1161/hh2301.100980. [DOI] [PubMed] [Google Scholar]
- 18.Hansen NW, Hansen AJ, Sams A. The endothelial border to health: Mechanistic evidence of the hyperglycemic culprit of inflammatory disease acceleration. IUBMB Life 69: 148–161, 2017. doi: 10.1002/iub.1610. [DOI] [PubMed] [Google Scholar]
- 19.Khamaisi M, Dahan R, Hamed S, Abassi Z, Heyman SN, Raz I. Role of protein kinase C in the expression of endothelin converting enzyme-1. Endocrinology 150: 1440–1449, 2009. doi: 10.1210/en.2008-0524. [DOI] [PubMed] [Google Scholar]
- 20.Laakso M. Cardiovascular disease in type 2 diabetes: challenge for treatment and prevention. J Intern Med 249: 225–235, 2001. doi: 10.1046/j.1365-2796.2001.00789.x. [DOI] [PubMed] [Google Scholar]
- 21.Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci USA 97: 10494–10499, 2000. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung JW, Wong WT, Koon HW, Mo FM, Tam S, Huang Y, Vanhoutte PM, Chung SS, Chung SK. Transgenic mice over-expressing ET-1 in the endothelial cells develop systemic hypertension with altered vascular reactivity. PLoS One 6: e26994, 2011. doi: 10.1371/journal.pone.0026994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li MW, Mian MO, Barhoumi T, Rehman A, Mann K, Paradis P, Schiffrin EL. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 33: 2306–2315, 2013. doi: 10.1161/ATVBAHA.113.302028. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, Haag M, Stark GR. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci USA 97: 10489–10493, 2000. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal 2: ra63, 2009. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Ongil S, Saura M, Zaragoza C, Gónzalez-Santiago L, Rodríguez-Puyol M, Lowenstein CJ, Rodríguez-Puyol D. Hydrogen peroxide regulation of bovine endothelin-converting enzyme-1. Free Radic Biol Med 32: 406–413, 2002. doi: 10.1016/S0891-5849(01)00822-X. [DOI] [PubMed] [Google Scholar]
- 27.Mahmoud AM, Szczurek MR, Blackburn BK, Mey JT, Chen Z, Robinson AT, Bian JT, Unterman TG, Minshall RD, Brown MD, Kirwan JP, Phillips SA, Haus JM. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol Rep 4: e12895, 2016. doi: 10.14814/phy2.12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manea SA, Manea A, Heltianu C. Inhibition of JAK/STAT signaling pathway prevents high-glucose-induced increase in endothelin-1 synthesis in human endothelial cells. Cell Tissue Res 340: 71–79, 2010. doi: 10.1007/s00441-010-0936-1. [DOI] [PubMed] [Google Scholar]
- 29.Manea SA, Todirita A, Manea A. High glucose-induced increased expression of endothelin-1 in human endothelial cells is mediated by activated CCAAT/enhancer-binding proteins. PLoS One 8: e84170, 2013. doi: 10.1371/journal.pone.0084170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martínez-Miguel P, Medrano-Andrés D, Griera-Merino M, Ortiz A, Rodríguez-Puyol M, Rodríguez-Puyol D, López-Ongil S. Tweak up-regulates endothelin-1 system in mouse and human endothelial cells. Cardiovasc Res 113: 207–221, 2017. doi: 10.1093/cvr/cvw239. [DOI] [PubMed] [Google Scholar]
- 31.Martínez-Miguel P, Raoch V, Zaragoza C, Valdivielso JM, Rodríguez-Puyol M, Rodríguez-Puyol D, López-Ongil S. Endothelin-converting enzyme-1 increases in atherosclerotic mice: potential role of oxidized low density lipoproteins. J Lipid Res 50: 364–375, 2009. doi: 10.1194/jlr.M800215-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Mellbin LG, Anselmino M, Rydén L. Diabetes, prediabetes and cardiovascular risk. Eur J Cardiovasc Prev Rehabil 17, Suppl 1: S9–S14, 2010. doi: 10.1097/01.hjr.0000368192.24732.2f. [DOI] [PubMed] [Google Scholar]
- 33.Mummidi S, Das NA, Carpenter AJ, Kandikattu H, Krenz M, Siebenlist U, Valente AJ, Chandrasekar B. Metformin inhibits aldosterone-induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt-induced cardiac fibrosis in vivo. J Mol Cell Cardiol 98: 95–102, 2016. doi: 10.1016/j.yjmcc.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 34.Nakagami H, Kaneda Y, Ogihara T, Morishita R. Endothelial dysfunction in hyperglycemia as a trigger of atherosclerosis. Curr Diabetes Rev 1: 59–63, 2005. doi: 10.2174/1573399052952550. [DOI] [PubMed] [Google Scholar]
- 35.Nathan DM, Meigs J, Singer DE. The epidemiology of cardiovascular disease in type 2 diabetes mellitus: how sweet it is…or is it? Lancet 350, Suppl 1: SI4–SI9, 1997. doi: 10.1016/S0140-6736(97)90021-0. [DOI] [PubMed] [Google Scholar]
- 36.Nishiyama SK, Zhao J, Wray DW, Richardson RS. Vascular function and endothelin-1: tipping the balance between vasodilation and vasoconstriction. J Appl Physiol 122: 354–360, 2017. doi: 10.1152/japplphysiol.00772.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J 34: 2436–2443, 2013. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paneni F, Lüscher TF. Cardiovascular protection in the treatment of type 2 diabetes: a review of clinical trial results across drug classes. Am J Med 130: S18–S29, 2017. doi: 10.1016/j.amjmed.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Park JY, Takahara N, Gabriele A, Chou E, Naruse K, Suzuma K, Yamauchi T, Ha SW, Meier M, Rhodes CJ, King GL. Induction of endothelin-1 expression by glucose: an effect of protein kinase C activation. Diabetes 49: 1239–1248, 2000. doi: 10.2337/diabetes.49.7.1239. [DOI] [PubMed] [Google Scholar]
- 40.Pernow J, Shemyakin A, Böhm F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci 91: 507–516, 2012. doi: 10.1016/j.lfs.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 41.Rafnsson A, Shemyakin A, Pernow J. Selective endothelin ETA and dual ET(A)/ET(B) receptor blockade improve endothelium-dependent vasodilatation in patients with type 2 diabetes and coronary artery disease. Life Sci 118: 435–439, 2014. doi: 10.1016/j.lfs.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 42.Reynolds LJ, Credeur DP, Manrique C, Padilla J, Fadel PJ, Thyfault JP. Obesity, type 2 diabetes, and impaired insulin-stimulated blood flow: role of skeletal muscle NO synthase and endothelin-1. J Appl Physiol 122: 38–47, 2017. doi: 10.1152/japplphysiol.00286.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res 10: 472–482, 2013. doi: 10.1177/1479164113500680. [DOI] [PubMed] [Google Scholar]
- 44.Ross R. Atherosclerosis−an inflammatory disease. N Engl J Med 340: 115–126, 1999. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 45.Symons JD, Rendig SV, Fu LW, Longhurst JC. Endothelin-1 limits increases in blood flow to native and collateral-dependent myocardium. Am J Physiol 273: R41–R48, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Tang ST, Su H, Zhang Q, Tang HQ, Wang CJ, Zhou Q, Wei W, Zhu HQ, Wang Y. Sitagliptin inhibits endothelin-1 expression in the aortic endothelium of rats with streptozotocin-induced diabetes by suppressing the nuclear factor-κB/IκBα system through the activation of AMP-activated protein kinase. Int J Mol Med 37: 1558–1566, 2016. doi: 10.3892/ijmm.2016.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med 70: 117–128, 2014. doi: 10.1016/j.freeradbiomed.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf) 219: 22–96, 2017. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 49.Venkatesan B, Valente AJ, Das NA, Carpenter AJ, Yoshida T, Delafontaine JL, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction. Cell Signal 25: 359–371, 2013. doi: 10.1016/j.cellsig.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watson AM, Li J, Schumacher C, de Gasparo M, Feng B, Thomas MC, Allen TJ, Cooper ME, Jandeleit-Dahm KA. The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 53: 192–203, 2010. doi: 10.1007/s00125-009-1540-3. [DOI] [PubMed] [Google Scholar]
- 51.Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149–1160, 2003. doi: 10.1016/S0735-1097(03)00994-X. [DOI] [PubMed] [Google Scholar]
- 52.Yamauchi T, Ohnaka K, Takayanagi R, Umeda F, Nawata H. Enhanced secretion of endothelin-1 by elevated glucose levels from cultured bovine aortic endothelial cells. FEBS Lett 267: 16–18, 1990. doi: 10.1016/0014-5793(90)80276-O. [DOI] [PubMed] [Google Scholar]
- 53.Yariswamy M, Yoshida T, Valente AJ, Kandikattu HK, Sakamuri SS, Siddesha JM, Sukhanov S, Saifudeen Z, Ma L, Siebenlist U, Gardner JD, Chandrasekar B. Cardiac-restricted overexpression of TRAF3 interacting protein 2 (TRAF3IP2) results in spontaneous development of myocardial hypertrophy, fibrosis, and dysfunction. J Biol Chem 291: 19425–19436, 2016. doi: 10.1074/jbc.M116.724138. [DOI] [PMC free article] [PubMed] [Google Scholar]