

Abstract
Individuals with alcohol use disorders (AUDs) are at an increased risk of pneumonia and acute respiratory distress syndrome. Data of the lung microbiome in the setting of AUDs are lacking. The objective of this study was to determine the microbial biogeography of the upper and lower respiratory tract in individuals with AUDs compared with non-AUD subjects. Gargle, protected bronchial brush, and bronchoalveolar lavage specimens were collected during research bronchoscopies. Bacterial 16S gene sequencing and phylogenetic analysis was performed, and the alterations to the respiratory tract microbiota and changes in microbial biogeography were determined. The microbial structure of the upper and lower respiratory tract was significantly altered in subjects with AUDs compared with controls. Subjects with AUD have greater microbial diversity [P < 0.0001, effect size = 16 ± 1.7 observed taxa] and changes in microbial species relative abundances. Furthermore, microbial communities in the upper and lower respiratory tract displayed greater similarity in subjects with AUDs. Alcohol use is associated with an altered composition of the respiratory tract microbiota. Subjects with AUDs demonstrate convergence of the microbial phylogeny and taxonomic communities between distinct biogeographical sites within the respiratory tract. These results support a mechanistic pathway potentially explaining the increased incidence of pneumonia and lung diseases in patients with AUDs.
Keywords: microbiome, respiratory, alcohol, biogeography, lung
INTRODUCTION
Recent studies of the lung microbial communities reveal that a unique lower airway microbiota exists in both healthy individuals and subjects with pulmonary diseases (2, 3, 16, 22, 25, 26, 31, 41), highlighting the potential influence of microbiota on lung disease pathogenesis. Alcohol consumption has been shown to alter the gastrointestinal and oral microbial communities (24, 40); however, the impact of alcohol use on the lung microbial communities is not known. These data may be relevant as microbial alterations in the setting of alcohol may impact pulmonary immune function or enhance susceptibility to pneumonia and associated conditions, such as acute respiratory distress syndrome (ARDS).
Approximately 17 million adults in the United States meet criteria for alcohol use disorders (AUDs) (33a). AUDs are associated with numerous physiological consequences, including liver damage, cancer, immune suppression, and increased risk of infection (23, 38). Bacterial pneumonia is the most common cause of lower respiratory tract infection in patients with AUDs (23), and AUDs are an accepted risk factor for the development of bacterial pneumonia. Furthermore, patients with AUDs who develop pneumonia are frequently infected with highly virulent respiratory pathogens, and consequently experience increased morbidity and mortality compared with the general population (23). Reasons for this association remain unclear; however, host defense mechanisms are reported to be adversely affected by excessive alcohol intake. For example, excessive alcohol use suppresses acute inflammatory responses in the lung along with the ability of resident lung immune cells to kill bacteria. Additionally, alcohol consumption disturbs the bronchial mucociliary escalator, further hindering the host’s ability to efficiently eliminate invading pathogens. People with AUDs also display a significantly increased risk of oropharyngeal aspiration, suggesting that chronic ethanol exposure may render an individual more susceptible to infection due to enhanced exposure to oropharyngeal microbial communities (7, 15, 23, 27, 39).
The incidence of ARDS in patients with AUDs has been reported to be 70% compared with 31% in patients without AUDs; AUD patients are almost fourfold more likely than nonalcoholic patients to develop ARDS (28, 32). In addition, alcohol-related ARDS is associated with poorer outcomes at 90 days, including combined outcomes of death or persistent hospitalization (12). The mechanisms by which alcohol use increases susceptibility to ARDS are still being characterized. Recently, it was found that gut-associated bacteria were enriched in the lung microbiome of both an experimental sepsis model as well as in humans with ARDS, suggesting that gut-lung translocation and dysbiosis of the lung microbial communities may contribute to the development of ARDS (14).
The objective of the present study was to compare the microbiota biogeography of the upper and lower respiratory tract in otherwise healthy subjects with AUDs and matched control subjects. We hypothesized that chronic alcohol consumption would be associated with respiratory tract dysbiosis. We further hypothesized that the upper and lower respiratory tract microbial communities would be more similar in subjects with an AUD than in controls.
MATERIALS AND METHODS
Study design.
Subjects with AUDs were recruited between May 2012 and January 2014 at the Denver Comprehensive Addictions Rehabilitation and Evaluation Services center, an inpatient alcohol detoxification facility affiliated with Denver Health and Hospital System in Denver, Colorado. Control subjects without AUDs were recruited via approved print and electronic flyer advertisements in the Denver, Colorado, community. This study was approved by the Colorado Multiple Institutional Review Board. All subjects provided written, informed consent before their participation in the study.
Participants.
The Alcohol Use Disorders Identification Test (AUDIT) questionnaire was used to assess harmful alcohol use in AUD and control subjects. The AUDIT questionnaire is a well-validated and widely used 10-question screening tool used to identify hazardous and harmful patterns of alcohol consumption in the clinical setting. A score of 8 and above in men, or 5 and above in women, identifies both heavy drinkers and those with AUDs, with a sensitivity of 50–90% and a specificity of ~80% (35, 38). Subjects with AUDs were eligible to participate if they met the following criteria for study entry: 1) an AUDIT score greater than or equal to 8 for men or greater than or equal to 5 for women; 2) alcohol use within 7 days of enrollment; and 3) age greater than or equal to 21 yr. AUD subjects were not approached for enrollment until sobriety was confirmed by breathalyzer. To be eligible for the control arm of the study, subjects were required to have an AUDIT score of less than these values based on sex, corresponding to abstinence or a low risk of alcohol-related problems. Control subjects were group matched to AUD subjects on the basis of age, sex, and smoking status. Cigarette smoking history was assessed by self-report.
In an effort to minimize potential confounding by comorbidities, subjects were ineligible to participate in the study if they met any of the following criteria: 1) prior medical history of liver disease (documented history of cirrhosis, total bilirubin ≥2 mg/dl, or albumin <3.0 g/dl), 2) prior medical history of gastrointestinal bleeding (because of the concern of varices), 3) prior medical history of heart disease (documentation of ejection fraction <50%, myocardial infarction, or severe valvular dysfunction), 4) prior medical history of renal disease (end-stage renal disease requiring dialysis, or a serum creatinine ≥2.0 mg/dl), 5) prior medical history of lung disease defined as an abnormal chest radiograph or spirometry, 6) prior history of diabetes mellitus, 7) prior history of HIV infection, 8) pregnancy, 9) abnormal nutritional risk index (13), or 10) age >55 yr because of the potentially high prevalence of asymptomatic comorbidities in these individuals. A chest radiograph was taken of each subject before enrollment; subjects were excluded if there was any evidence of an acute process or a lesion that might necessitate clinical bronchoscopy. Subjects who failed to provide informed consent were also excluded. Whole blood samples were also obtained from all subjects on enrollment for complete blood count, complete metabolic panel, and prothrombin time with international normalized ratio. Urine samples were also obtained on enrollment to screen for cocaine, opioids, and methamphetamines. Pulmonary function tests were completed before performance of the bronchoalveolar lavage (BAL).
Sample collection and processing.
Subjects fasted and refrained from smoking and drinking for ~12 h before research sampling. Oral washes were performed by having participants gargle with 10 ml of sterile 0.9% saline for 1 min immediately before bronchoscopy. Bronchoscopy procedures were then performed in the inpatient Clinical and Translational Research Center at the University of Colorado Hospital by using standardized protocols as previously described (9). Protected specimen bronchial brush samples were collected from the right mainstem bronchus. Briefly, the brushing process occurred in a standardized fashion. The sterile brush (inside sheath) was first inserted through the bronchoscope and positioned within the takeoff of the right mainstem; the sterile brush was then extruded through its sheath through the gel barrier. After extruding the sterile brush through its sheath into the right mainstem, the brush was placed at five different positions circumferentially in the airway. In each position, 10 passes of the brush were made. On completion of brushing the five positions, the brush was pulled inside its sheath, then the entire device removed from the bronchoscope. BAL was performed in a subsegment of either the right middle lobe or the lingua. BAL was always initially attempted in the right middle lobe. If a yield of at least 15 ml was not obtainable from right middle lobe lavage, BAL could be performed in the lingula. The majority of BAL specimens were derived from the right middle lobe. Two bronchoscopists performed these procedures using an identical protocol. Two assistants aided in all procedures who had been similarly trained in the procedure.
All sample collection and processing was performed aseptically and under conditions to minimize DNA contamination. Specifically, the processing for the oral wash, bronchial brushings, and BAL microbiome samples were all performed in a laminar flow hood under DNA-free conditions. All working surfaces and tubes were decontaminated with 10% bleach before sample processing. Saline controls were collected, processed, and analyzed in parallel to all of the clinical samples.
Gargles were kept on ice after collection. All sample processing was performed as soon as possible and was always performed within 20 min of collection. The oral wash and saline control samples were transferred to sterile 15-ml conical tubes and centrifuged at 4°C, 13,000 g for 30 min. The supernatant was then discarded, and the pellet was resuspended in 0.5 ml sterile PBS and transferred to a labeled 2-ml clear microcentrifuge tube. Samples were then stored at −80°C until processed for microbiome analysis. Bronchial brushings and saline control samples were immediately placed in a labeled clear sterile 2-ml microcentrifuge tubes and vortexed for 1 min. Samples were then stored at −80°C until processed for microbiome analysis. Two milliliters of collected BAL sample was removed from whole bronchoalveolar lavage and placed into a clear 2-ml microcentrifuge tube. The samples were then centrifuged at 25 g for 1 min to settle the cells. After centrifugation, 1.5 ml of the cell-free lavage was gently removed without disturbing the cell pellet and transfer to a labeled clear 2-ml microcentrifuge tube. The sample was then centrifuged at 14,000 g for 20 min and the supernatant was removed. The BAL microbial pellet was then stored at −80°C until processed for microbiome analysis.
DNA sequencing of the 16s rRNA gene.
Sequencing and bioinformatic analysis were performed by the Louisiana State University School of Medicine Microbial Genomics Resource Group (http://metagenomics.lsuhsc.edu/mgrg), as previously described (8, 37). The respiratory tract samples were shipped to Louisiana State University Health Sciences Center, and genomic DNA extraction was performed using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA) modified to include bead-beating. Gene amplicon library sequencing (16S) was performed as previously described (8); 16S ribosomal DNA hypervariable regions V3 and V4 were amplified using gene-specific sequences, Illumina adaptors, and molecular barcodes primers. Precisely, the V3 Forward: 5′-CCTACGGGAGGCAGCAG-3′ with a forward adapter (5′-AATGATACGGCGACCACCGAGATCTACAC-3′), a forward pad: (TATGGTAATT), and a forward linker (GG), and the V4 Reverse: 5′-GGACTACHVGGGTWTCTAAT-3′ with a reverse adapter (5′- CAAGCAGAAGACGGCATACGAGAT- 3′), a reverse pad (AGTCAGTCAG), and a reverse linker (CC) were used for sequencing of microbial DNA samples. Samples were then sequenced on an Illumina MiSeq (Illumina, San Diego, CA) using the 2 × 300 bp V3 sequencing kit. In addition, internal sequencing controls (negative water control and a positive mock community control) were included to test for contamination of the sequencing runs. The read files were processed through the UPARSE pipeline (drive5, Tiburon, California), merging forward and reverse reads and then truncating merged reads that covered the V3/V4 region to a uniform length of 400 bp. Reads with quality score less than 15 in the first 400 bp were discarded. Unique reads were clustered into operational taxonomic units (OTUs) at 97% similarity. Chimeric OTUs were removed as identified by UCHIME (drive5). Finally, the original filtered reads were mapped to the OTUs by using USEARCH (drive5) at 97% identity. Quantitative Insights Into Microbial Ecology (QIIME 1.9.1, www.qiime.org) was used to pick and align a representative set (10). UCLUST (drive5) was used to assign a taxonomic classification to each read in the representative set (42). Relative abundance of each OTU was examined at phylum, class, order, family, genus, and species levels. Beta (between communities) and alpha (within a community) diversity metrics as well as taxonomic community assessments were produced using QQIIME 1.9.1 and the R package Phyloseq, as described previously (8, 37, 43).
We also evaluated the sampled microbial communities by minimum entropy decomposition oligotype clustering (available from http://oligotyping.org) as described in previous publications (17, 18). Oligotyping allows for the identification of sequence amplicon variants that differ by as little as a single nucleotide (17, 18). Node clusters with a minimum substantive abundance below 1,992 were removed per default parameters to control for nodes arising from spurious sequencing errors. After minimum substantive abundance filtering 9,958,537 reads (83% of total) remained and were subjected to downstream analysis.
Predicted functional capacity.
Inferred metagenomes were generated using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) v0.9.0 (29a). Closed reference OTUs were picked using QIIME 1.9.1 and the GreenGenes v13.5 16S reference genome database and subjected to PICRUSt analysis. Closed reference OTUs were normalized for estimated 16S copy number and annotated for inferred genetic content by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (27a). Inferred relative gene abundances were subsequently binned into pathways and functional categories defined by the KEGG BRITE hierarchy. Nonmicrobial categories (e.g., organismal systems and human diseases) were excluded from analysis.
Data repository.
All data generated from sequencing have been deposited in the National Center for Biotechnology Information of the National Institutes of Health via the Sequence Read Archive with accession number (PRJNA378267).
Statistical analysis.
AUD and control groups matched for age, sex, and smoking history were compared on outcome variables. Sequencing runs were included as a potentially confounding covariate (2 runs in total). Where AUD n = 16 and control n = 23 is indicated, plotted data represents a single sequencing run, which is representative of both sequencing runs and the full cohort n = 78. For all alpha diversity (α-diversity) data, OTU tables were rarefied to 2,000 sequences/sample to account for variations in sequencing depth before analysis. α-Diversity statistics in Table 2 were calculated using a nonparametric t-test with Monte Carlo simulations. Using DESeq2, a negative binomial mixture model to account for overdispersion in sequenced-read count data was used to detect differentially abundant OTUs between control and AUD subjects (Fig. 3) (30). All unweighted UniFrac beta diversity (β-diversity) data were generated from unrarefied OTUs in which outcome variable P values were adjusted for differences in sample sequenced-read count. All weighted β-diversity data, as well as the data in Fig. 6, A, D, and G, were normalized by variance stabilizing transformation of raw read counts by using DESeq2. Unweighted UniFrac β-diversity (considers presence/absence of species) and weighted UniFrac β-diversity (considers abundance of each species) distances were plotted using nonmetric multidimensional scaling. For inferred metagenomic data, statistically significant differences in relative KEGG gene abundances were tested using the Mann-Whitney U-test with correction for multiple comparison via false discovery rate. Significant group mean differences in UniFrac distance were assessed by Mann-Whitney U-test. Significant group clustering by UniFrac distance was determined by permutational analysis of variance (ADONIS). Fisher’s exact tests were used for statistical analysis on categorical variables. Spearman ρ correlation coefficients were calculated to estimate oligotype abundance covariation between sampled sites as a function of AUD status. Differences in AUD and control group median ρ correlation coefficient were tested using the Mann-Whitney U-test. This analysis was repeated using the more conservative Kendall τ correlation coefficient to control skew in the correlation estimates. Bootstrapped median correlation coefficient 95% confidence intervals (CI) were generated on 5,000 resampling trials. Spearman ρ, Kendall τ, and CI calculations as well as the Mann-Whitney U-test were performed using the R software package. A P value < 0.05 and Q value < 0.1 were considered statistically significant where indicated in the text.
Table 2.
Comparison of α-diversity metrics in alcohol use and no alcohol use cohorts
| Alcohol Use |
No Alcohol Use |
||||
|---|---|---|---|---|---|
| Diversity Metric | Mean | SD | Mean | SD | P Value |
| Chao1 | 183.3064 | 46.02512 | 164.5138 | 47.12525 | 0.0651 |
| PD_Whole Tree | 14.18034 | 2.289357 | 12.68001 | 2.710997 | 0.0069 |
| Shannon | 5.185907 | 0.542789 | 4.91016 | 0.501214 | 0.0179 |
| Simpson | 0.938822 | 0.03643 | 0.931001 | 0.029871 | 0.2858 |
Fig. 3.

Chronic alcohol use alters individual OTU relative abundances of upper and lower airway microbiota. Taxa with significantly different relative abundance in AUD cohorts compared with controls at each distinct biogeographical site [gargle (A), brush (B), and BAL (C)] were determined using DESeq2. Bars represent the mean log2 fold change in relative abundance ± SE for AUD subjects compared with healthy controls as determined by DESeq2 analysis. All taxa are significantly differentially abundant following correction for multiple comparisons (Q < 0.05). Repeated taxonomic classifications represent different OTUs.
Fig. 6.

Chronic alcohol use is associated with a disrupted airway microbial biogeography. Mean unweighted UniFrac (phylogenetic) distances were computed for AUD and control subjects between each sample site. Between-site correlations in taxon relative abundance were estimated using Spearman and Kendall correlation coefficients. Gargle vs. brush (A, B, and C) sample shifts in community similarity between healthy control and AUD are plotted alongside brush vs. BAL (D, E, and F) and gargle vs. BAL (G, H, and I). Boxplots denote first and third quartiles. Whiskers are plotted by Tukey’s method. *P < 0.05 for healthy controls vs. AUD cohorts via Mann-Whitney U-test. Bars represent the median correlation magnitude of coabundant taxa between different sites for AUD and healthy controls. *P < 0.05 by Mann-Whitney U-test between healthy controls and AUD cohorts (AUD, n = 41; control, n = 37).
RESULTS
Description of AUD and controls.
Seventy-eight participants were included in this study (Table 1), including 41 individuals with an AUD and 37 controls. Controls and AUD subjects were similar in age, weight, height, and smoking status. Controls and AUD subjects were also similar in ethnicity; however, there were more AUD subjects who were of Hispanic/Latino and American Indian descent. The percentage of smokers was also similar between the AUD and control cohorts, as was the pack-year smoking history. There was a higher percentage of men in the alcohol group.
Table 1.
Demographic and clinical characteristics of the control and alcohol use disorder groups
| Parameters | Alcohol Use Disorder | Healthy Control | P Value |
|---|---|---|---|
| n | 41 | 37 | |
| Sex, male, % | 87.8 (n = 36) | 64.8 (n = 24) | 0.0296 |
| Age, yr | 43.38 ± 7.02 | 40.81 ± 7.27 | 0.115 |
| Height, m | 1.73 ± 0.11 | 1.73 ± 0.10 | 0.913 |
| Weight, kg | 76.09 ± 13.80 | 85.01 ± 17.21 | 0.279 |
| Race | |||
| Hispanic/Latino, n = 16 | 12 | 4 | |
| American Indian, n = 9 | 9 | 0 | |
| Asian, n = 1 | 0 | 1 | |
| Black, n = 10 | 4 | 6 | |
| Native Hawaiian, n = 1 | 0 | 1 | |
| White, n = 58 | 28 | 30 | |
| AUDIT | 29.70 ± 6.99 | 2.78 ± 2.08 | <0.001 |
| Smokers | 61% (n = 25) | 51% (n = 19) | 0.398 |
| Pack-years, median, IQR | 13, 20.125 | 15, 9 | 0.964 |
Values are means ± SD. AUDIT, Alcohol Use Disorders Identification Test; IQR, interquartile range.
Microbial composition comparisons between control and AUD cohorts.
The number of observed OTUs was used to evaluate the α-diversity of the oropharyngeal (gargle), bronchial mucosa (brush), and distal lung alveolar (BAL) microbial communities between AUD and controls (Fig. 1). Across sites, an increase in OTU α-diversity was observed in AUD populations over control populations [P < 0.0001, median control = 114 ± 3.3 95% CI, median alcohol = 134 ± 3.35 95% CI, effect size (ES) = 20 ± 2.38 observed OTUs; Fig. 1A]. Specifically, there were significant increases in α-diversity in the gargle (P < 0.0001, median of control = 135 ± 3.8 95% CI, median of alcohol = 149 ± 2.2 95% CI, ES = 14 ± 2.22 observed OTUs; Fig. 1B), brush (P < 0.0001, median of control = 116 ± 3.4 95% CI, median of alcohol = 133 ± 3.85 95% CI, ES = 17 ± 2.60 observed OTUs; Fig. 1C), and BAL (P < 0.0001, median of control = 72 ± 4.17 95% CI, median of alcohol = 90 ± 6.75 95% CI, ES = 18 ± 4.01 observed OTUs; Fig. 1D) communities between AUD cohorts and controls. Additional α-diversity metrics were significantly increased in AUD compared with control populations (Table 2). β-Diversity of the gargle, brush, and BAL communities in AUD and controls was also assessed. Significant differences in the β-diversity (unweighted UniFrac) across gargle (P = 0.006; Fig. 2A), brush (P = 0.07; Fig. 2B), and BAL (P = 0.004; Fig. 2C) communities of AUD and control cohorts were observed. Significant differences were also observed in the weighted UniFrac β-diversity between AUD and control cohorts (data not shown).
Fig. 1.

Chronic alcohol use is associated with increased α-diversity of the respiratory tract microbiota. The observed operational taxonomic unit (OUT) α-diversity of the respiratory tract microbial communities was compared between healthy controls and alcohol use disorder (AUD) cohorts at each distinct biogeographical site. We evaluated the effects of alcohol use on the respiratory tract microbiota at all three distinct sites combined (A), the upper airway (gargle) (B), the middle airway (brush) (C), and the lower airway (BAL) (D). Boxplots denote top quartile and bottom quartile. Whiskers are plotted by Tukey’s method, *P < 0.05 for healthy controls vs. AUD cohorts (AUD subjects, n = 16; control subjects, n = 23) via Mann-Whitney U-test. Data are representative of two sequencing runs, which make up the full cohort; n = 78.
Fig. 2.
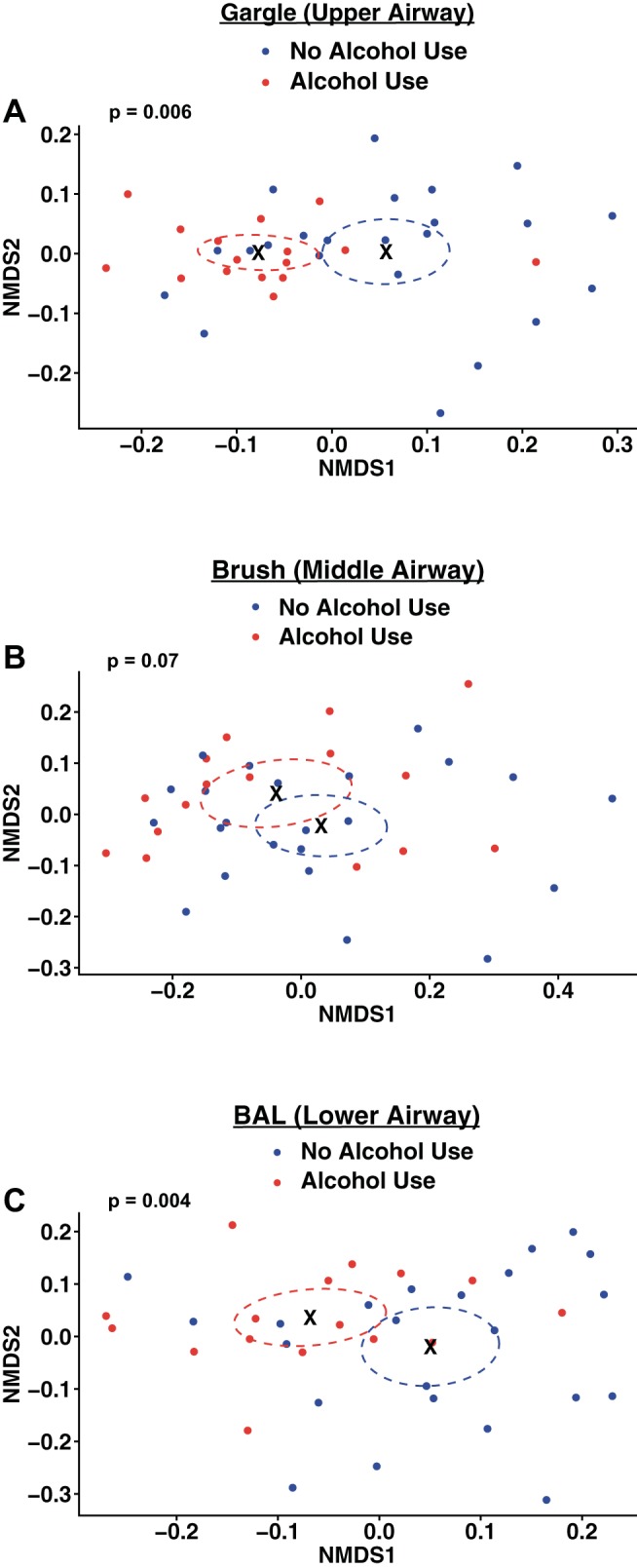
Chronic alcohol use is associated with changes in the community composition of the respiratory tract microbiota. Unweighted β-diversity of the respiratory tract microbial communities was compared between healthy controls and AUD cohorts at each distinct biogeographical site. Unweighted UniFrac distances were plotted using nonmetric multidimensional scaling (NMDS) ordination. We evaluated the effects of alcohol use on the respiratory tract microbiota at the upper airway (gargle) (A), the middle airway (brush) (B), and the lower airway (BAL) (C). Points represent individual subject samples. Circles outline the 95% confidence intervals around the sample group centroid indicated by X. P values indicate significant clustering of healthy control and AUD cohorts (AUD, n = 16; control, n = 23) as calculated by ADONIS multivariate permutational analysis of variance. Data are representative of two sequencing runs, which makeup the full cohort; n = 78.
Comparisons between the gargle, brush, and BAL communities of AUD and controls demonstrated significant increases in the relative abundance of specific OTUs in the gargle (e.g., Neisseria, Fig. 3A), brush (e.g., Porphyromonas, Fig. 3B), and BAL (e.g., Actinobacillus, Fig. 3C) samples in AUD subjects over controls, as determined by DESeq2.
We also assessed the change in the abundance of OTUs mapping to Gram-negative taxa between AUD and controls in each respiratory tract biogeographical site. We found that the relative abundance of Gram-negative counts was significantly increased in the AUD cohort compared with controls in the gargle (Fig. 4A), brush (Fig. 4B), and BAL (Fig. 4C) samples. Additionally, increased abundance of specific Gram-negative taxa such as Hemophilus in BAL samples and Prevotella in brush and BAL samples (Fig. 4D) was observed in AUD subjects over controls. Furthermore, the inferred functional capacity of the respiratory microbial communities of AUD and controls was also assessed. Differences in inferred biochemical pathway gene content of all three biogeographical sites was not significantly associated with AUD status (data not shown).
Fig. 4.

Chronic alcohol use is associated with increases in the relative read counts of genus-agglomerated OTUs classified to Gram-negative organisms. The number of sequencing reads of OTUs that map to known Gram-negative genera was significantly increased in AUD populations compared with controls in the gargle (A), brush (B), and BAL samples (C). Boxplot whiskers 95% confidence interval of the mean ± SE number of Gram-negative OTUs in the different sites for AUD and healthy controls; *P < 0.05 for healthy controls vs. AUD cohorts (AUD, n = 41; control, n = 37). D: differentially abundant, genus-agglomerated Gram-negative OTUs in each biogeographical site in AUD subjects over healthy controls.
Finally, we assessed the core taxa (taxa found in 100% of the samples within an experimental group) present in the gargle, brush, and BAL communities between the AUD and controls. We found that 37 taxa were shared in the gargle (Fig. 5A), 13 shared in the brush (Fig. 5B), and 49 shared in the BAL (Fig. 5C), between AUD and controls. In addition, 15 taxa in the gargle, 11 in the brush, and 5 in the BAL samples were unique to subjects with an AUD. Conversely, 3 taxa in the gargle, 3 in the brush, and 2 in the BAL samples were unique to controls. The unique and shared taxa observed between AUD and controls in each biogeographical site is shown in Supplemental Table S1.
Fig. 5.

Core microbiome of controls and AUD cohorts at each distinct sampling site. Core taxa present in 100% of subjects within a cohort were compared. Taxa in light gray were found in the full cohort of samples, taxa in dark gray were only found in AUD subject samples, and taxa in gray were only found in healthy control samples. Taxa found in each section are shown in Supplemental Table S1.
AUD and the microbial biogeography of the respiratory tract.
To examine the association of AUD with the phylogenetic compositional similarity of upper and lower airway microbial communities, we compared the unweighted β-diversity in AUD and control subjects as a function of biogeographical sampling site. β-diversity analysis demonstrated that all three sampling sites represent unique microbial communities in both control and AUD subjects (data not shown). Interestingly, there was a convergence in the microbial communities between biogeographical sites in the AUD cohort. To quantify this we employed two methods. First, we calculated the unweighted UniFrac phylogenetic distance between each sample site (gargle to brush, gargle to BAL, and brush to BAL) within each individual subject. We then compared the mean distances between the control and AUD cohorts. This analysis demonstrated that AUDs were associated with a significantly decreased phylogenetic distance between all sampling sites (P = 0.001, control median = 0.624 ± 0.042 95% CI, AUD median = 0.514 ± 0.037 95% CI; Fig. 6A; P = 0.018, median of control = 0.531 ± 0.034 95% CI, AUD median = 0.488 ± 0.029 95% CI; Fig. 6D; and P = 0.024, control median = 0.466 ± 0.041 95% CI, AUD median = 0.422 ± 0.042 95% CI; Fig. 6G) compared with control subjects. In contradistinction, no significant difference was observed between the UniFrac distance of smokers and nonsmokers (data not shown). Weighted UniFrac distance between sampling sites was also significantly decreased in the AUD cohorts compared with the control subjects (data not shown). Second, to complement our data on differences in community composition, we assessed whether individual taxa that are shared between sample sites correlate in relative abundance across sample sites more so in AUD subjects or controls. We generated strain-level sequence nodes by using the minimum entropy decomposition oligotyping method (19) and calculated same-node, between-site correlation coefficients in the AUD and control cohorts. We found that median correlation coefficients were positive in both AUD and control groups, suggesting that changes in the relative abundance of a node in one site for both groups tended to reflect changes in the relative abundance of that node in other sites. In addition, AUD cohort correlation coefficients were significantly right shifted compared with control cohort coefficients, suggesting that changes in specific node relative abundances are more closely shared by the sample sites of AUD subjects over control. Figure 6, B and C, shows the median correlation coefficients in both AUD and control groups for gargle to brush samples (Spearman ρ: P < 2 × 10−13, control median = 0.394 ± 0.032 95% CI and AUD median = 0.478 ± 0.041 95% CI; Kendall τ: P < 3 × 10−6, control median = 0.279 ± 0.025 95% CI and AUD median = 0.333 ± 0.025 95% CI). Figure 6, E and F, shows the median correlation coefficients in both AUD and control groups for brush to BAL (P < 3 × 10−6, control median = 0.248 ± 0.029 95% CI, AUD median = 0.521 ± 0.036 95% CI; Kendall τ: P < 3 × 10−16, control median = 0.169 ± 0.027 95% CI, AUD median = 0.392 ± 0.025 95% CI). Figure 6, H and I, shows the median correlation coefficients in both AUD and control groups for gargle to BAL (Spearman ρ, P < 3 × 10−16, control median = 0.321 ± 0.023 95% CI, AUD median = 0.441 ± 0.032 95% CI; Kendall τ: P < 2 × 10−13, control median = 0.230 ± 0.018 95% CI, AUD median = 0.317 ± 0.017 95% CI). We repeated this analysis comparing smokers to nonsmokers. In contrast to subjects with AUD, smokers exhibited a decreased median correlation coefficient between oligotype abundance in gargle and BAL (ρ: P = 0.003; τ: P = 0.001) as well as BAL and brush (ρ: P < 3 × 10−16; τ: P = < 3 × 10−16) sampled communities compared with control (data not shown). Median coefficients between gargle and brush were slightly greater in smokers over nonsmokers (ρ: P = 0.015; τ: P = 0.002; data not shown).
We also assessed the core taxa between biogeographical sites in AUD and control subjects. Thirty-one taxa in the gargle, 40 in the brush, and 0 in the BAL samples were unique to the specific sampling site (Fig. 7A). Analysis of the core microbiota from the AUD cohort revealed 7 taxa that were common between all three sites, 12 taxa that were common between the gargle and the brush, 5 taxa common to the brush and BAL, and 2 common between the BAL and the gargle (Fig. 7A). In the control subjects, 27 taxa in the gargle, 39 in the brush, and 0 in the BAL samples were unique to the specific sampling site (Fig. 7B). The core microbiota from the controls revealed 5 taxa that were common between all three sites, 6 taxa that were common between the gargle and the brush, 5 taxa common to the brush and BAL, and 2 common between the BAL and the gargle (Fig. 7B). The taxa unique and shared between each biogeographical site are shown in Supplemental Table S2.
Fig. 7.

Core microbiome between biogeographical sites of controls and AUD cohorts. Core taxa present in 100% of subjects for each treatment condition were compared. Taxa in gray mesh were found in all of the samples, whereas taxa found in gray hatches were found in both the gargle and the brush, taxa in gray dashes were found in the brush and the BAL, and taxa in gray dots were found in the BAL and gargle. Similarly, taxa in dark gray were only found in gargle, taxa in gray were only found in BAL, and taxa in light gray were only found in brush samples. Taxa found in each section are shown in Supplemental Table S2.
DISCUSSION
This study examines the microbial composition of the oropharyngeal cavity, the bronchial mucosa, and the distal lung in a cohort of controls and subjects with AUDs. We found that there were significant differences in the microbial communities of the oropharyngeal cavity, the bronchial mucosa, and the distal lung in AUD subjects compared with controls. In contrast, lung microbial communities did not differ significantly in overall analyses between smokers and nonsmokers, similar to previous reports (31). Overall, our findings suggest that AUDs influence the microbial composition across the oropharyngeal cavity, the bronchial mucosa, and the distal lung alveoli. It remains possible that alterations in the microbial composition we report have the potential to impact infectious susceptibility commonly observed among people with AUDs.
Previous investigations have demonstrated that the oropharyngeal microbial communities in AUD subjects differ from those in controls (33, 40). In addition, these groups have reported enrichment of certain organisms in AUDs as well as a reduction in other normal oropharyngeal microbes (33, 40). We found significant differences in the diversity of populations in the oropharynx of AUD subjects compared with controls. Several taxa, including Neisseria, Prevotella, Porphyromonas, and Veillonella, were more common in AUD subjects. We also found an increased relative abundance of taxa such as Actinobacillus in the AUD population. These taxa are members of the normal oral microbiota, and other studies have reported increased relative abundance of Neisseria in AUD subjects (33). Similarly, Porphyromonas, a bacteria linked to periodontal disease (11), was significantly increased in AUD oropharyngeal samples. Periodontal disease has been linked to several systemic illnesses such as cardiac disease and rheumatoid arthritis, suggesting that the microbial communities associated with periodontal disease may influence broader aspects of health (20, 29). Overall, these data support previous work suggesting that alcohol use is associated with a disrupted community structure in the oropharynx.
Our results provide important insights into the lung microbial communities in AUD subjects and control populations. Although similar studies have been performed to evaluate the lung microbial populations in other pulmonary conditions, including smoking, cystic fibrosis, COPD, and asthma (3, 11, 16, 31, 34), to our knowledge this is the first study that evaluates the lung microbiota in the context of AUD. AUDs have been implicated in infectious lung conditions, such as community-acquired pneumonia, and ARDS. Establishing alterations in lung microbial communities related to AUDs while accounting for other potential confounders, including smoking, suggests pulmonary dysbiosis as a novel mechanism underlying alcohol-lung disease associations. Importantly, we found significant differences in the microbial diversity of populations in the bronchial mucosa and the distal lung alveoli of AUD subjects compared with controls that are similar to those seen in other pulmonary diseases, such as cystic fibrosis, COPD, and asthma, all of which showed changes in relative abundance and microbial diversity associated with the disease state (16, 25, 34).
In these investigations, AUDs were associated with a disrupted biogeography of lung microbial communities. We observed that subjects with AUDs had more similar microbial communities across the three biogeographical sampling sites. Specifically, changes in relative abundance of oligotypes in any one sample site more closely reflected changes in relative abundance of the same oligotypes in all other sites in subjects with AUDs over control. These data suggest enhanced communication between biogeographical sites of the respiratory tract that may be influenced by alcohol consumption. Reasons for this similarity in oligotypes across biogeographical sites among subjects with AUDs are unknown, but several potential explanations exist. First, patients with AUDs have significantly higher rates of aspiration (21, 28) that could contribute to deposition of oligotypes across the three compartments. Furthermore, alcohol use impairs ciliary function and may therefore impede clearance of aspirated organisms, further fostering the propensity for oropharyngeal organisms in the upper airway to migrate distally. Clinically, the propensity for aspiration coupled with impaired mucociliary clearance may manifest as decreased resistance to colonization by known pathogens in people with AUDs. Alternatively, alcohol use suppresses host immunity and the acute inflammatory response, leading to an impaired ability to clear pathogens in the distal airspaces. Clinical implications of persistent pathogens in the AUD setting might include increased pulmonary inflammation from retained bacterial products, particularly Gram-negative organisms (i.e., LPS), and immune cells which are hypo-responsive because of chronic antigen stimulation. These two factors may in turn contribute to a more permissive environment for the alcohol-associated bacterial pathogens (Streptococcus pneumoniae, Klebsiella pneumoniae, and Legionella pneumophilia). To our knowledge, our work represents the first attempt to compared microbial communities across three biogeographical sites. Collectively, our findings suggest that alcohol use has a substantial impact on the microbial composition of the upper respiratory tract and lower respiratory tract microbial communities. However, additional investigation will be necessary to determine if these changes to respiratory tract microbiota related to AUDs contribute to susceptibility to bacterial pneumonia among these individuals.
Notably, we employed strict inclusion and exclusion criteria to ensure that participants had no significant comorbid conditions that might confound our analysis. Nevertheless, our work is not without limitations. Systematic oral examination was not performed; therefore we could not determine the precise impact of periodontal disease or tooth condition on the oral or lung microbiome. We took multiple steps to minimize contamination during sample collection, including protected specimen airway brushing. However, the possibility of contamination cannot be completely eliminated. Systematic contamination of biological specimens is a likely and common occurrence (36); however, we suspect that both AUD and control specimens are influenced equally by contaminants. Finally, we assayed biological samples without controlling for bacterial viability. However, dead bacteria are capable of provoking an immune response (1).
In summary, we investigated changes in the biogeography of respiratory tract microbial communities in human subjects with AUDs compared with controls. We determined that AUDs are associated with an altered composition of microbial communities throughout the respiratory tree including the oropharynx, mainstem bronchus, and lower bronchoalveolar spaces. In addition, chronic alcohol use appeared to drive a relative convergence of microbial phylogeny among otherwise distinct anatomical sites within the respiratory tract. The role of these changes and the nature of the immune response to them will be important areas of future research to illuminate mechanisms of pneumonia pathogenesis and prevention in subjects with AUDs.
GRANTS
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants R24-AA-019661, P60-AA-009803, and T32-AA-07577; National Heart Lung and Blood Institute Grant P01-HL076100; NIH National Center for Advancing Translational Sciences Grant UL1TR001082; National Center for Research Resources Grant CTSI-ULI-RR025780; and National Institute of General Medical (which funds the Louisiana Clinical and Translational Science Center) Grant U54 GM-104940. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.R.S., E.L.B., R.W.V., M.L., and C.M.T. performed experiments; D.R.S., V.J.M., E.E.B., J.E.S., M.L., and C.M.T. analyzed data; D.R.S., E.L.B., V.J.M., R.W.V., E.E.B., J.E.S., M.L., C.M.T., and D.A.W. interpreted results of experiments; D.R.S., V.J.M., E.E.B., and C.M.T. prepared figures; D.R.S. and D.A.W. drafted manuscript; D.R.S., E.L.B., V.J.M., R.W.V., E.E.B., J.E.S., M.L., C.M.T., and D.A.W. edited and revised manuscript; D.R.S., E.L.B., V.J.M., R.W.V., E.E.B., J.E.S., M.L., C.M.T., and D.A.W. approved final version of manuscript; E.L.B., R.W.V., and D.A.W. conceived and designed research.
Supplemental Data
The effects of alcohol use on the core microbiota of the upper and lower airway
The effects of alcohol use on the core microbiota of the respiratory tract microbial biogeography
ACKNOWLEDGMENTS
The authors thank clients and staff at Denver Comprehensive Addictions Rehabilitation and Evaluation Services, as well as personnel from the University of Colorado Hospital Inpatient Clinical and Translational Research Center.
REFERENCES
- 1.Adams CA. The probiotic paradox: live and dead cells are biological response modifiers. Nutr Res Rev 23: 37–46, 2010. doi: 10.1017/S0954422410000090. [DOI] [PubMed] [Google Scholar]
- 2.Beck JM, Schloss PD, Venkataraman A, Twigg H III, Jablonski KA, Bushman FD, Campbell TB, Charlson ES, Collman RG, Crothers K, Curtis JL, Drews KL, Flores SC, Fontenot AP, Foulkes MA, Frank I, Ghedin E, Huang L, Lynch SV, Morris A, Palmer BE, Schmidt TM, Sodergren E, Weinstock GM, Young VB; Lung HIV Microbiome Project . Multicenter comparison of lung and oral microbiomes of HIV-infected and HIV-uninfected individuals. Am J Respir Crit Care Med 192: 1335–1344, 2015. doi: 10.1164/rccm.201501-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res 160: 258–266, 2012. doi: 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkowitz H, Reichel J, Shim C. The effect of ethanol on the cough reflex. Clin Sci Mol Med 45: 527–531, 1973. [DOI] [PubMed] [Google Scholar]
- 8.Bruce-Keller AJ, Salbaum JM, Luo M. Blanchard E 4th, Taylor CM, Welsh DA, Berthoud HR. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol Psychiatry 77: 607–615, 2014. doi: 10.1016/j.biopsych.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burnham EL, Phang TL, House R, Vandivier RW, Moss M, Gaydos J. Alveolar macrophage gene expression is altered in the setting of alcohol use disorders. Alcohol Clin Exp Res 35: 284–294, 2011. doi: 10.1111/j.1530-0277.2010.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336, 2010. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charlson ES, Chen J, Custers-Allen R, Bittinger K, Li H, Sinha R, Hwang J, Bushman FD, Collman RG. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5: e15216, 2010. doi: 10.1371/journal.pone.0015216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark BJ, Williams A, Feemster LM, Bradley KA, Macht M, Moss M, Burnham EL; NHLBI ARDS Network Investigators . Alcohol screening scores and 90-day outcomes in patients with acute lung injury. Crit Care Med 41: 1518–1525, 2013. doi: 10.1097/CCM.0b013e318287f1bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Detsky AS, Baker JP, Mendelson RA, Wolman SL, Wesson DE, Jeejeebhoy KN. Evaluating the accuracy of nutritional assessment techniques applied to hospitalized patients: methodology and comparisons. JPEN J Parenter Enteral Nutr 8: 153–159, 1984. doi: 10.1177/0148607184008002153. [DOI] [PubMed] [Google Scholar]
- 14.Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, Huffnagle GB. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol 1: 16113, 2016. doi: 10.1038/nmicrobiol.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott MK, Sisson JH, Wyatt TA. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am J Respir Cell Mol Biol 36: 452–459, 2007. doi: 10.1165/rcmb.2005-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6: e16384, 2011. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eren AM, Borisy GG, Huse SM, Mark Welch JL. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci USA 111: E2875–E2884, 2014. doi: 10.1073/pnas.1409644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, Sogin ML. Oligotyping: Differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol 4: 1111–1119, 2013. doi: 10.1111/2041-210X.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J 9: 968–979, 2015. doi: 10.1038/ismej.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farquharson D, Butcher JP, Culshaw S. Periodontitis, Porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol 5: 112–120, 2012. doi: 10.1038/mi.2011.66. [DOI] [PubMed] [Google Scholar]
- 21.Fuxench-López Z, Ramírez-Ronda CH. Pharyngeal flora in ambulatory alcoholic patients: prevalence of gram-negative bacilli. Arch Intern Med 138: 1815–1816, 1978. doi: 10.1001/archinte.1978.03630370033017. [DOI] [PubMed] [Google Scholar]
- 22.Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, Nicod LP, Lloyd CM, Marsland BJ. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med 20: 642–647, 2014. doi: 10.1038/nm.3568. [DOI] [PubMed] [Google Scholar]
- 23.Happel KI, Nelson S. Alcohol, immunosuppression, and the lung. Proc Am Thorac Soc 2: 428–432, 2005. doi: 10.1513/pats.200507-065JS. [DOI] [PubMed] [Google Scholar]
- 24.Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res 39: 763–775, 2015. doi: 10.1111/acer.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO. Disordered microbial communities in asthmatic airways. PLoS One 5: e8578, 2010. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hosgood HD III, Sapkota AR, Rothman N, Rohan T, Hu W, Xu J, Vermeulen R, He X, White JR, Wu G, Wei F, Mongodin EF, Lan Q. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environ Mol Mutagen 55: 643–651, 2014. doi: 10.1002/em.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson WD., Jr Impaired defense mechanisms associated with acute alcoholism. Ann N Y Acad Sci 252: 343–347, 1975. doi: 10.1111/j.1749-6632.1975.tb19177.x. [DOI] [PubMed] [Google Scholar]
- 27a.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–D114, 2012. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kershaw CD, Guidot DM. Alcoholic Lung Disease (Online). National Institute on Alcohol Abuse and Alcoholism; http://pubs.niaaa.nih.gov/publications/arh311/66-75.htm [date of access, 06/06/2016]. [PMC free article] [PubMed] [Google Scholar]
- 29.Kjellström B, Rydén L, Klinge B, Norhammar A. Periodontal disease - important to consider in cardiovascular disease prevention. Expert Rev Cardiovasc Ther 14: 987–989, 2016. doi: 10.1080/14779072.2016.1202112. [DOI] [PubMed] [Google Scholar]
- 29a.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821 2013. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luthold RV, Fernandes GR, Franco-de-Moraes AC, Folchetti LG, Ferreira SR. Gut microbiota interactions with the immunomodulatory role of vitamin D in normal individuals. Metabolism 69: 76–86, 2017. doi: 10.1016/j.metabol.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L, Jablonski K, Kleerup E, Lynch SV, Sodergren E, Twigg H, Young VB, Bassis CM, Venkataraman A, Schmidt TM, Weinstock GM; Lung HIV Microbiome Project . Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187: 1067–1075, 2013. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med 31, Suppl: S207–S212, 2003. doi: 10.1097/01.CCM.0000057845.77458.25. [DOI] [PubMed] [Google Scholar]
- 33.Muto M, Hitomi Y, Ohtsu A, Shimada H, Kashiwase Y, Sasaki H, Yoshida S, Esumi H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: implications for carcinogenesis in upper aerodigestive tract. Int J Cancer 88: 342–350, 2000. doi: 10.1002/1097-0215(20001101)88:3<342::AID-IJC4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 33a.National Institute on Alcohol Abuse and Alcoholism Alcohol Use Disorder (Online). https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-use-disorders [date of access, 29/05/2016].
- 34.Rabin HR, Surette MG. The cystic fibrosis airway microbiome. Curr Opin Pulm Med 18: 622–627, 2012. doi: 10.1097/MCP.0b013e328358d49a. [DOI] [PubMed] [Google Scholar]
- 35.Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test (AUDIT): a review of recent research. Alcohol Clin Exp Res 26: 272–279, 2002. doi: 10.1111/j.1530-0277.2002.tb02534.x. [DOI] [PubMed] [Google Scholar]
- 36.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12: 87, 2014. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samuelson DR, de la Rua NM, Charles TP, Ruan S, Taylor CM, Blanchard EE, Luo M, Ramsay AJ, Shellito JE, Welsh DA. Oral immunization of mice with live Pneumocystis murina protects against Pneumocystis pneumonia. J Immunol 196: 2655–2665, 2016. doi: 10.4049/jimmunol.1502004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuckit MA. Alcohol-use disorders. Lancet 373: 492–501, 2009. doi: 10.1016/S0140-6736(09)60009-X. [DOI] [PubMed] [Google Scholar]
- 39.Sisson JH. Alcohol and airways function in health and disease. Alcohol 41: 293–307, 2007. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas AM, Gleber-Netto FO, Fernandes GR, Amorim M, Barbosa LF, Francisco AL, de Andrade AG, Setubal JC, Kowalski LP, Nunes DN, Dias-Neto E. Alcohol and tobacco consumption affects bacterial richness in oral cavity mucosa biofilms. BMC Microbiol 14: 250, 2014. doi: 10.1186/s12866-014-0250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Twigg HL III, Knox KS, Zhou J, Crothers KA, Nelson DE, Toh E, Day RB, Lin H, Gao X, Dong Q, Mi D, Katz BP, Sodergren E, Weinstock GM. Effect of advanced HIV Infection on the respiratory microbiome. Am J Respir Crit Care Med 194: 226–235, 2016. doi: 10.1164/rccm.201509-1875OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267, 2007. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wicks S, Taylor CM, Luo M, Blanchard E IV, Ribnicky DM, Cefalu WT, Mynatt RL, Welsh DA. Artemisia supplementation differentially affects the mucosal and luminal ileal microbiota of diet-induced obese mice. Nutrition 30, Suppl: S26–S30, 2014. doi: 10.1016/j.nut.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effects of alcohol use on the core microbiota of the upper and lower airway
The effects of alcohol use on the core microbiota of the respiratory tract microbial biogeography