

Abstract
Carbon monoxide (CO) is an endogenously produced gas that has gained recognition as a biological signal transduction effector with properties similar, but not identical, to that of nitric oxide (NO). CO, which binds primarily to heme iron, may activate the hemoprotein guanylate cyclase, although with lower potency than NO. Furthermore, CO can modulate the activities of several cellular signaling molecules such as p38 MAPK, ERK1/2, JNK, Akt, NF-κB, and others. Emerging studies suggest that mitochondria, the energy-generating organelle of cells, represent a key target of CO action in eukaryotes. Dose-dependent modulation of mitochondrial function by CO can result in alteration of mitochondrial membrane potential, mitochondrial reactive oxygen species production, release of proapoptotic and proinflammatory mediators, as well as the inhibition of respiration at high concentration. CO, through modulation of signaling pathways, can impact key biological processes including autophagy, mitochondrial biogenesis, programmed cell death (apoptosis), cellular proliferation, inflammation, and innate immune responses. Inhaled CO is widely known as an inhalation hazard due to its rapid complexation with hemoglobin, resulting in impaired oxygen delivery to tissues and hypoxemia. Despite systemic and cellular toxicity at high concentrations, CO has demonstrated cyto- and tissue-protective effects at low concentration in animal models of organ injury and disease. These include models of acute lung injury (e.g., hyperoxia, hypoxia, ischemia-reperfusion, mechanical ventilation, bleomycin) and sepsis. The success of CO as a candidate therapeutic in preclinical models suggests potential clinical application in inflammatory and proliferative disorders, which is currently under evaluation in clinical trials.
Keywords: carbon monoxide, cell death, cell signaling, inflammation, lung disease, mitochondria, reactive oxygen species, sepsis
INTRODUCTION
Carbon monoxide (CO) is a gaseous molecule that occurs ubiquitously in nature as the product of organic combustion processes (171). Since the middle of the twentieth century, the medical community has recognized that CO can evolve endogenously in living organisms as the by-product of metabolic activity (30, 148, 149). In both prokaryotes and eukaryotes, biological CO arises as the result of enzymatic heme degradation, catalyzed by the heme oxygenase enzymes (Fig. 1) (163, 164, 187). The characterization of the endogenous occurrence and enzymatic production of CO, and subsequent discoveries that CO at low concentration can influence cellular signal transduction pathways, leading to altered cellular functions, including adaptive responses, has led to the recent recognition of this gas as a potential physiological mediator (88, 134–138).1
Fig. 1.

Interaction of carbon monoxide (CO) with biological systems. CO is endogenously produced in the body, primarily as the product of the erythrocyte-dependent turnover of hemoglobin-derived heme, and the cellular turnover of hemoproteins. Heme, a catalytic cofactor for hemoproteins, is degraded by the heme oxygenase (HO, EC: 1:14:99:3) enzymes. HO catalyzes the rate-limiting step in oxidative heme catabolism, to yield biliverdin-IXα, CO, and ferrous iron (Fe II), and requires 3 mol molecular oxygen (O2) and reducing equivalents from NADPH. Biliverdin-IXα produced in the HO reaction is reduced to bilirubin-IXα by NAD(P)H biliverdin reductase (BVR). Non-heme sources may make a minor contribution to exogenous CO production. In the blood, CO binds hemoglobin to form carboxyhemoglobin (CO-Hb). Therapeutic CO may be applied at low concentration through inhalation or mechanical ventilation, or by administering CO-releasing molecules (CORMs).
The term “gasotransmitter” was introduced to describe a group of biologically relevant and related gases, including CO and other small gases such as nitric oxide (NO) and hydrogen sulfide (H2S) (160, 176). NO was identified in 1986 as identical to the endothelial-derived relaxing factor responsible for regulation of blood vessel tone, laying the foundation for current acceptance of gaseous second messengers in biology (50). H2S, a third gasotransmitter, has recently emerged as a modulator of vascular function, inflammation, and other diverse cellular processes, which have been reviewed extensively elsewhere (124, 160, 176). Like NO or H2S, endogenously produced CO can exert a variety of biologic and physiologic functions (104, 134–136), which range from the regulation of vascular tone (156), mitochondrial homeostasis and biogenesis (82, 83, 157, 158), neurotransmission (169); to the modulation of inflammation (114), programmed cell death (16, 17), and cellular proliferation programs (99). Taken together, the evidence for physiological signaling roles of CO has displaced previous conceptions that endogenously derived CO represents solely a waste product of ordinary metabolism (138).
In addition to metabolic production, CO can also enter the body via the pulmonary inhalation portal (171). Environmental CO, which occurs as a common component of air pollution, wood smoke, and cigarette smoke (~5.9–17.4 mg CO/cigarette), is renowned for its systemic toxicity (21, 123, 171, 195). CO when presented at elevated concentrations can cause asphyxiation and death during accidental or occupational inhalation exposure (171). This property is directly attributed to the ability of CO to bind the heme-iron centers of hemoglobin (with an affinity ~250-fold that of oxygen), and generate carboxyhemoglobin (CO-Hb). The partial occupation of hemoglobin heme sites by CO impairs the release of O2 from the remaining heme groups, resulting in reduced O2 delivery to tissues (123, 171). The clinical toxicology of CO poisoning involves severe neurocognitive effects, cardiac dysfunction, and potential death, and has been reviewed extensively elsewhere (14, 53, 122, 123, 132).
One of the basic tenets of pharmacology is that the dosage of a compound will determine whether it potentially acts as a therapeutic or a poison (15). In this regard, research over the past two decades using animal models has uncovered a therapeutic window for this gas when applied at low concentrations, despite the well-publicized toxicity of CO at elevated concentrations. To date, an extensive collection of preclinical data in rodents and higher mammals has demonstrated salutary effects of CO in experimental models of organ injury and disease, in conditions involving aberrant inflammatory or proliferative states, and thereby has advanced the concept of CO as a gaseous therapy (104, 134, 135). These studies have collectively established a foundation for potential future clinical applications of this gas. As a result of these studies, a wealth of data has also emerged describing the physiological and cellular consequences of CO exposure in vitro and in vivo, and the underlying signaling mechanisms responsible for these effects.
Although the concept of gaseous molecule therapies, including CO (or its precedent, NO), was originally based on the proposition of direct application of the therapeutic gas by inhalation, recent years have also seen the development of chemical pro-drugs capable of delivering the gas in a controlled fashion. For example, like the native gas, CO-releasing molecules (CORMs) have also shown therapeutic potential in preclinical models of disease (103, 104). This review will focus on the cellular and molecular targets of CO in lung and vascular models, with an emphasis on cell signaling and functional outcomes. The therapeutic implications of CO will also be discussed in the context of lung and vascular diseases, as well as inflammatory disorders such as sepsis.
PHYSICOCHEMICAL PROPERTIES OF CO
Carbon monoxide (CO) is a diatomic molecule (molecular weight = 28.01), which exists as an invisible and odorless gas at ambient temperature and pressure (CO has a melting point of −205°C, and a boiling point of −191.5°C) (171). CO is water soluble (2.3 ml·100 ml−1 at 20°C) and dissolves in some organic solvents such as chloroform (171). Unlike the free radical NO, which is highly reactive in biological systems and can engage in complex redox chemistry (81), CO is an inherently stable nonradical with little biological reactivity with respect to non-iron compounds. CO can ligate with cellular heme groups at the iron center, and has been demonstrated to form complexes with cellular hemoproteins, such as cytochrome-c oxidase (18, 23, 170), cytochrome p-450 (41, 55), soluble guanylate cyclase (sGC) (49, 154, 155), inducible nitric oxide synthase (153), and other cellular cytochromes (e.g., cytochrome b558) (31).
ENDOGENOUS PRODUCTION OF CO
The endogenous production of CO is largely attributed to the degradation of heme, with the majority (~85%) originating from erythrocyte-dependent hemoglobin turnover (30, 148, 149), as well as from the turnover of cellular hemoproteins such as mitochondrial cytochromes (80, 92). A minor component of endogenous CO production is believed to originate from non-heme sources that may include lipid peroxidation and cytochrome p-450-dependent drug metabolism (172). In discoveries beginning in 1968, heme-dependent CO production was shown to directly occur as the by-product of enzyme-dependent heme degradation catalyzed by heme oxygenases (HO, EC: 1:14:99:3) (163, 164). HO activity catalyzes the oxidative cleavage of heme to biliverdin-IXα, in a reaction that liberates ferrous iron and CO. The HO reaction requires molecular oxygen and an electron donor (i.e., NADPH cytochrome p-450 reductase) (163). Bilirubin-IXα is further enzymatically reduced to biliverdin-IXα by biliverdin reductase (BVR) (163, 165). Two genetically distinct isozymes of HO have been identified and characterized, an inducible isozyme (HO-1) that is highly transcriptionally responsive to cellular stress stimuli, and a constitutively expressed isozyme (HO-2) (71, 91). The molecular regulation and function of HO-1 and related enzymes have been extensively reviewed elsewhere (2, 92, 134, 135, 137).
THERAPEUTIC CO (INHALATION AND PHARMACOLOGICAL APPLICATION)
The potential for medical application of CO originally assumed inhalation as the primary route of administration. In this case, dosimetry is controlled by adjusting the concentration of the inspired gas mixture, and can be monitored by measurement of CO-Hb in the blood. As an alternative to inhalation, metal carbonyl-based chemical CO donor compounds (CORMs) have been developed as experimental therapeutics over the past two decades (46, 65, 103, 104). CORMs can be used to deliver therapeutic CO to biological systems by injection to the systemic circulation or application to the target site. An advantage of CORMs is that they deliver CO to tissues while limiting CO-Hb buildup associated with inhaled CO (104). A key consideration of this approach is that the safety and metabolic fate of the donor molecule or backbone, in addition to that of CO, must also be evaluated (65). Further considerations for the development of new CORMs include improved water solubility, tissue-specific targeting, and the ability to induce, control, or time the release of CO (65). Similar to gaseous CO, CORMs have demonstrated vasoactive effects (47), as well as anti-inflammatory effects in various injury and disease models (22, 82, 83, 140, 159).
The first CORM to be described was Mn2CO10 (CORM-1). Subsequently, ruthenium-based compounds were introduced: tricarbonyldichlororuthenium-(II)-dimer (CORM-2) and tricarbonylchoro(glycinato)-ruthenium (II) (CORM-3) (65, 103, 104). CORM-1 and CORM-2 are hydrophobic (103), while CORM-3 is water soluble and rapidly releases CO in physiological fluids (47). CORM-2 has been utilized primarily for in vitro studies, while CORM-3 is preferred for in vivo studies. A number of new generation water-soluble CORMs have been introduced in recent years, including CORM-A1 (sodium boranocarbonate), which releases CO in a pH and temperature-dependent fashion and has demonstrated vasodilatory and hypotensive effects (105); and CORM-S1 [dicarbonyl bis(aminoethylthiolato)iron(II)], which releases CO in response to visible light activation (79). Furthermore, a novel series of hydrophobic and hydrophilic CORMs (iron-allyl carbonyls) were shown to release CO at different rates (106). More recently, a novel manganese-based CORM (CORM-401), which releases CO in response to oxidant exposure, has been shown to exert potent proangiogenic and vasodilatory properties (44). The current frontier of CORMs development has introduced compounds that facilitate controlled release of CO, and these include micellar preparations (57), enzyme-activated CORMs (ET-CORMs) (152), and new generation radiation-sensitive (photo-activated) CORMs (PHOTO-CORMs) (95, 107, 118). Additional advances have yielded bifunctional hybrid molecules (189), or CORMs conjugated to nanoparticles and other biological scaffolds (carrier conjugates) for potential improvement of drug delivery (52, 70, 142).
EFFECTS OF CO ON CELL SIGNALING
CO has been shown to impact the regulation of multiple signaling pathways leading to altered cellular functions. While the proximal target of CO action has long been hypothesized to involve a heme-binding event, such as the heme of sGC, a heme-dependent mechanism has not been unequivocally identified across all studies. Mitogen-activated protein kinases (MAPKs) and other signal transduction molecules have been identified as secondary targets, though their integration with sGC and other proximal hemoprotein targets has also not been universally established across all studies. Emerging research also identifies mitochondrial cytochromes as proximal targets of CO action, though the hierarchy of mitochondrial regulation with other cytosolic or cellular signaling events remains incompletely elucidated. Furthermore, the effects of CO on cellular signaling events can vary in cell-type and/or model-specific fashion, adding further ambiguity. Thus, no universal pathway or unifying mechanism has emerged. Nevertheless, the following sections describe the key targets of CO that have been identified in recent years in a context-specific fashion.
Guanylate Cyclase
The soluble form of guanylate cyclase was among the first molecules proposed as a direct molecular target of CO, activating downstream signaling pathways (49). Binding of sGC by CO, akin to that of NO, results in the activation of the enzyme and subsequent production of the second messenger molecule guanosine 3′,5′-monophosphate (cGMP), which activates protein kinase G. CO binds the heme group of sGC in a hexa-coordinate fashion, whereas the binding of NO occurs in a penta-coordinate configuration. The activation potential of CO is much lower than that of NO (approximately 80-fold less potent) (154), and may not occur in the presence of NO. Nevertheless, the activation of sGC by CO has been implicated in neurotransmission (169), regulation of vascular tone (156), and the inhibition of vascular smooth muscle (SMC) proliferation (99, 100), and platelet aggregation (19). However, sGC as the primary proximal target of CO action has not been unequivocally demonstrated in all studies, and this can also vary in a cell-type and model-specific fashion.
NADPH Oxidase
The family of NADPH oxidases (NOX1-4, DUOX1-2), which include the phagocytic NOX (NOX2, gp91phox) and a mitochondria-specific isozyme (NOX4), are cellular cytochrome-containing enzyme complexes responsible for the univalent reduction of molecular oxygen to superoxide anion (O2−) (12). The formation of O2− by NOXs is utilized for diverse processes including host defense, vascular regulation, and cellular signaling (12). NOXs have been implicated as targets of CO action (109, 130, 161, 178). Inhibition of NOX-dependent reactive oxygen species (ROS) generation has been attributed to anti-inflammatory (109), antiproliferative (161), and antiapoptotic (178) effects of CO. For example, CO was shown to inhibit LPS induced ROS-dependent trafficking of Toll-like receptor-4 (TLR4) to lipid rafts, a proximal event to activation of the NF-κB-dependent inflammatory pathway, in cultured macrophages (109). TLR4 trafficking and activation, as well as the capacity for CO to inhibit the response, were sensitive to chemical inhibition of NOX, and abolished in gp91phox-deficient macrophages (109). Experiments using CORM-2 demonstrated inhibition of cytochrome b558 (p21phox) activity in vitro, the flavocytochrome component of NOX2, thereby implicating NOX2 as a regulatory target of CO action (161). Interestingly, NOX1 has also been implicated as a target in the inhibition of vascular SMC migration by endogenous CO (130). Recent studies have also proposed that endogenous HO-derived CO can downregulate mitochondrial NOX4, which in turn inhibits matrix metalloproteinase-1 expression and apoptosis in IL-1β-activated chondrocytes (133).
Mitochondria as Targets for CO Action
Mitochondria have been strongly implicated as a primary target of CO action. Many of the mitochondrial cytochromes responsible for respiration have heme functional groups that potentially serve as binding targets for CO. The cellular toxicity of CO has long been known, such that CO at elevated concentrations can impair mitochondrial function leading to inhibition of mitochondrial respiration (23). In this regard, CO at high concentration can inhibit the terminal enzymatic step in the electron transport chain (cytochrome-c oxidase) by binding to the cytochrome a3-subunit (23). Several recent studies have demonstrated the modulation of mitochondrial (mt)ROS production and modulation of mitochondrial membrane potential by CO (13, 25, 85, 197). In macrophages, upregulation of mtROS by low concentration CO was attributed to inhibition of cytochrome-c oxidase (197), which promoted anti-inflammatory effects via p38 MAPK. These studies reported maintenance of cellular ATP and increase of mitochondrial membrane potential in the presence of CO at the concentrations used (197). Furthermore, upregulation of mtROS in macrophages was attributed to adaptive cellular signaling through upregulation of peroxisome proliferator-activated receptor-γ (PPARγ), downregulation of the proinflammatory mediator Egr-1 (13), and stabilization of the hypoxia-inducible factor-1-α (HIF-1α) (25), leading to adaptive gene expression and anti-inflammatory effects. Gaseous CO was shown to upregulate mtROS in epithelial cells, in the context of upregulating the cytoprotective autophagy program (85).
In isolated cardiac mitochondria, CO released from CORM-3 was demonstrated to exert a mitochondrial uncoupling effect. At low concentrations, CORM-3 increased mitochondrial oxygen consumption, but it limited respiration at higher concentrations via inhibition of cytochrome-c oxidase. CORM-3 also caused time-dependent decline of mitochondrial membrane potential and increase of complex I-dependent mtROS production. In contrast, CORM-3 inhibited succinate-inducible (complex II)-dependent mtROS production in isolated mitochondria (90). CO released from CORM-401 was also shown to cause mitochondrial uncoupling, accompanied by increased oxygen consumption, reduction of ATP production, and impairment of cellular glycolysis. The mitochondrial uncoupling effect of CORM-401, but not the effect on glycolysis, was further shown to be dependent on mitochondrial large-conductance calcium-regulated potassium ion channels (mitoBKCa) (68).
The administration of CO was shown to downregulate mtROS production in lung epithelial cells under conditions of hyperoxia, where ROS production was elevated due to high oxygen tension (83). CO was also observed to downregulate mtROS in macrophages in the presence of proinflammatory stimuli such as LPS and ATP (67). Treatment with LPS and ATP caused mitochondrial depolarization and cytosolic translocation of mtDNA in macrophages, whereas CO exposure abolished these effects (67).
In conclusion, the reported roles of CO effects on mitochondrial respiration, mitochondrial membrane potential, and mtROS generation are clearly dose-dependent. The detection of mtROS can be complicated by differential effects of CO on other enzymatic sources of ROS, such as NOX1-4, and can also vary in the absence or presence of inducing stimuli. The contribution of mitochondria-specific NOX (NOX4) in CO-dependent ROS flux currently remains unclear. Nevertheless, the regulation of mtROS may represent a common mechanism by which CO exerts effects on downstream signaling pathways, yet this has not been unequivocally demonstrated across all studies. Further research will be required to clarify the relative roles of mitochondrial versus cytosolic ROS production as transducing mechanisms of CO effects, both in response to CO alone or in the presence of proinflammatory stimuli.
In addition to modulation of respiratory chain activity and basic mitochondrial functions, CO has been shown to stimulate mitochondrial biogenesis, a cellular program for the de novo generation of mitochondria (82, 157, 158). CO-dependent stimulation of mitochondrial biogenesis, as demonstrated in the heart and in skeletal mitochondria, involves increased proliferator-activated receptor-γ coactivator-1α (PGC-1α), nuclear respiratory factor 1 (NRF-1), and its downstream regulator, mitochondrial transcription factor A (TFAM), factors that initiate the biogenesis program. Furthermore, the stimulation of mitochondrial biogenesis by CO is dependent on sGC, activation of the prosurvival Ser/Thr kinase Akt, and evolution of mtROS (i.e., H2O2) but independent of NO production (158). CO stimulation of cardiac mitochondrial biogenesis was shown to be protective in the context of doxorubicin-induced cardiomyopathy (157). Similarly, application of CORM-3 prevented mitochondrial function decline and stimulated mtROS-dependent activation of mitochondrial biogenesis in septic mice (82). These studies describing stimulation of mitochondrial biogenesis by CO suggest a potential novel mechanism by which CO treatment can lead to adaptive responses to systemic stress.
Ion Channels as Targets of CO Action
CO has been implicated as a regulator of ion channel activity, which may contribute to the observed signaling effects of CO towards various biological functions, including vasoregulation and neurotransmission (120, 185). One the most widely studied are high-conductance, Ca2+-dependent K+ channels (BKCa) (120, 185), which are implicated in smooth muscle relaxation and vasodilation (175). Several explanations have been proposed for the stimulatory action of CO on the BKCa channel but the precise nature of the activating mechanism remains unclear (120). There is controversial evidence for direct interactions of CO with hydrophobic protein domains, heme-iron (38, 60, 63, 120, 128, 175, 185, 188), and more recently, with a cyanide-sensitive non-heme metal binding site (162). Additional reported ion channels activated by CO include the ATP-gated purinogenic P2X2 receptor (184), and the tandem P domain channel TREK1 (K2P2.1) (33), which have been implicated in sensory neurotransmission and neuronal excitability, respectively. Variable effects of CO were reported towards the activity of voltage gated L-type Ca2+ channels, which include demonstrated activation in intestinal cells (through a mechanism involving mobilization of NO and cGMP) (89), and channel inhibition in cardiomyocytes (through a mechanism dependent on redox modification subsequent to enhanced mtROS production) (144, 166). CO has also been shown to exert variable effects on epithelial Na+ channels (ENaC) (5, 177). For example, one study has shown that CO inhibits epithelial Na+ uptake, which was associated with impaired alveolar fluid clearance in the rabbit lung (5). In contrast, in vitro studies describe activation of ENaC in normoxic mouse epithelial cells by CO (177). The authors proposed a regulatory role for endogenous HO activity as an oxygen sensor for the regulation of channel activity, based on oxygen tension-dependent variable effects of heme on channel activity (177). CO has been shown to inhibit the activities of the voltage-gated delayed rectifier K+ channel, Kv2.1 (32) and the cardiac Na+ channel Nav1.5 (40) through redox-dependent mechanisms. Curiously, CORM-2 also inhibited the purinergic P2X4 receptor, but in a mechanism independent of CO release (186). Taken together, these intriguing observations suggest a regulatory role for CO, as well as pharmaceutical effects of CO-donor compounds by modulating ion channel activity in diverse model systems. To date, few studies have related the modulation of ion channel activity by CO with its cytoprotective effects. For example, inhibition of Kv2.1 channels by CO was related to antiapoptotic effects in neurons (4, 32). Downregulation of L-type channels by CO was associated with protection from ischemic necrosis in cardiomyocytes (166). In contrast, potentiation of L-type channels by CO in astrocytes was reported to stimulate VEGF-dependent angiogenesis through a mechanism dependent on AMP-activated protein kinase (AMPK)-α, NAD-dependent deacetylase sirtuin-1, and stabilization of PGC-1α (27). Further research is needed to determine how ion-channel dependent responses are integrated with other signaling targets of CO to coordinate cytoprotective responses. The potential relevance of these systems to lung physiology and pathophysiology remains incompletely understood.
Nrf2-Dependent Responses
CO can activate a feedback mechanism to further stimulate its endogenous production through the upregulation of HO-1 gene expression via activation of the transcription factor NF-E2-related factor-2 (Nrf2). Nrf2, a master regulator of the antioxidant response, is anchored in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1). Cellular exposure to electrophiles and other inducing stimuli promote the dissociation of Keap1, thus permitting activation and translocation of Nrf2 to the nucleus (194). By binding to antioxidant-responsive elements/stress-responsive elements (ARE/StRE) in the 5′-regulatory region of HO-1 (HMOX1, Hmox1) genes, Nrf2 acts as a major transcriptional regulator of HO-1 gene expression (2, 3). Furthermore, Nrf2 orchestrates the cellular antioxidant response by regulating the expression of other genes involved in detoxification and antioxidant responses: including those encoding NAD(P)H quinone oxidoreductase, glutathione reductase, glutathione peroxidase-2, glutamate–cysteine ligase complex, glutathione-S-transferases, ferritin heavy and light chains, and others (54, 102). Nrf2 has been implicated as an important mediator of antioxidant defenses in the lung (194). For example, mice genetically deficient in Nrf2 (Nrf2−/−) were sensitized to inflammatory responses and emphysema induced by chronic cigarette smoke exposure (62, 126), and were also sensitized to asthma and airway inflammation (127).
Endogenous CO, generated from HO-1, can activate Nrf2 nuclear translocation through upregulation of phosphoinositide 3-kinase (PI3K)/Akt and consequent inhibition of glycogen synthase kinase-3β. This pathway results in further stimulation of HO-1, and activation of the mitochondrial biogenesis program through the stimulation of NRF-1 (121). A pathway dependent on Src, epidermal growth factor receptor, and downstream PI3K/Akt-signaling was shown to mediate the activation of Nrf2 and subsequent HO-1 gene expression in human tracheal smooth muscle cells in response to CORM-2 treatment (190). The neuroprotective effects conferred by inhaled CO (250 ppm) in a murine ischemic shock model were associated with HO-1 induction and were abolished in Nrf2−/− mice (173). Furthermore, the anti-inflammatory effects of CORM-2 in mice, with respect to reduction of proinflammatory cytokine production and protection from LPS-induced mortality, were abrogated in Nrf2−/− mice (125). Taken together, these studies indicate that the therapeutic potential of CO is coupled to Nrf2-dependent antioxidant responses, including the stimulation of endogenous CO production via HO-1, and these mechanisms may be significant in adaptive responses of the lung and other organs.
EFFECTS OF CO ON CELLULAR INFLAMMATION RESPONSES
Cytokine Regulation
CO has been demonstrated to exert anti-inflammatory effects when applied to cultured macrophages at low concentration, including downregulation of proinflammatory cytokines (e.g., IL-1β, TNF-α) and upregulation of the anti-inflammatory cytokine IL-10 (114). CO confers these effects through MAPKs, which are critical mediators of inflammatory and stress responses (114) (Fig. 2). The anti-inflammatory effect of CO, as demonstrated in LPS-stimulated macrophages, required activation of the MKK3/p38β MAPK pathway (114). Additional studies have implicated downregulation of JNK or ERK1/2 in the downregulation of IL-6 by CO in macrophages and pulmonary epithelial cells, respectively (101, 112). In addition to modulation of cytokines, CO was shown to inhibit TLR4 trafficking and activation through mechanisms involving inhibition of NOX-dependent ROS generation (109) and the modulation of caveolin-1-TLR4 interactions at the plasma membrane (180). CO was also proposed to confer anti-inflammatory effects in macrophages via the mtROS-dependent induction of PPARγ, which led to downregulation of the proinflammatory mediator Egr-1 (13). The anti-inflammatory effects of CO have potential therapeutic value in diseases where aberrant inflammation is implicated in the pathogenic response, including acute lung injury, transplant rejection, and sepsis.
Fig. 2.

Effect of CO on cellular inflammation pathways. CO has been implicated in the downregulation of inflammatory pathways. CO can inhibit the production of the proinflammatory cytokines (IL-1β, IL-6, TNF-α) and upregulate anti-inflammatory cytokine IL-10, through a mechanism dependent on activation of the MKK3/p38 MAPK pathway. Additionally, CO has been shown to modulate the trafficking and activation of Toll-like receptor-4 (TLR4) at the plasma membrane. CO has been proposed as an inhibitor of NOD-, leucine rich region- and pyrin domain-containing-3 (NLRP3) inflammasome activation, through the stabilization of mitochondria, leading to modulation of mitochondrial reactive oxygen species (mtROS) and inhibition of mtDNA release, key triggers for inflammasome activation under proinflammatory conditions.
Inflammasome Regulation
Inflammasomes are cytosolic protein complexes responsible for the regulation of caspase-1. Activation of caspase-1 by proteolytic cleavage governs the maturation and secretion of proinflammatory cytokines, including IL-1β (IL-1β) and IL-18. Among the known inflammasomes, the NOD-, leucine rich region- and pyrin domain-containing-3 (NLRP3) inflammasome has been recently implicated in the pathogenesis of several acute or chronic inflammatory diseases (35, 84, 143). Current research suggests that CO can regulate the activation of the NLRP3 inflammasome. Our recent studies demonstrate that application of CO inhibited caspase-1 activation and secretion of IL-1β and IL-18 in bone marrow-derived macrophages (BMDMs) in response to LPS and ATP treatment, a NLRP3 inflammasome activation model (67). CO also inhibited IL-18 secretion in BMDMs in response to LPS and nigericin, an alternate NLRP3 inflammasome activation model. LPS and ATP stimulation induced the formation of molecular complexes between NLRP3 and the adaptor ASC, or NLRP3 and caspase-1, whereas CO inhibited the formation of inflammasome complexes. In this model, the presence of CO inhibited mitochondrial depolarization induced by LPS and ATP in macrophages (67). These results suggest that low concentration CO negatively regulated NLRP3 inflammasome activation by preserving mitochondrial function (67). Application of CORM-2 downregulated caspase-1 activation and IL-1β maturation and secretion during endoplasmic reticulum stress-induced inflammation (76). In contrast to these observations, another report demonstrates that CO can promote ATP secretion from bacteria, which in turn can upregulate NLRP3 inflammasome activation by stimulating the purinergic receptor (P2X7R) in bacteria-exposed macrophages (183). The reasons for these contrasting findings remain unclear at present, but they may be related to differences between bacterial sepsis and inflammation induced by cell-free LPS. Although further studies will be needed to clarify the precise mechanisms, CO shows potential as a regulator of the NLRP3 inflammasome and secretion of IL-1β and IL-18, which may contribute to its modulatory effects on inflammation and innate immune responses.
Adaptive Immune Responses
In addition to modulation of inflammasome-dependent and independent cytokine responses, CO has been implicated in the modulation of adaptive immune functions. CO has been implicated in tolerogenic responses to endotoxin, decreasing the surface expression of TLR4/myeloid differentiation factor-2 (MD2) in dendritic cells (DC) and neutrophils. CO preconditioning conferred protection in endotoxin induced mice and was associated with reduced recruitment of DCs and neutrophils to peripheral blood (129). Recently, CO has been shown to induce immunoresponsive gene-1 in macrophages, which promotes LPS tolerance by increasing A20 expression via modulation of ROS production (64). Treatment with exogenous CO inhibited LPS-induced maturation of DCs and protected against in vivo and in vitro antigen-specific inflammation (24). Application of CO also blocked antigen presentation by DCs at the step of endosome-lysosome fusion (147). The ex vivo treatment of DCs with CO was shown to augment dendritic-cell based therapy in a type 1 diabetes model. Application of CO-conditioned DCs inhibited the accumulation and pathogenic activity of autoreactive CD8+ T cells in the pancreas (147). Taken together, these examples illustrate the complex and incompletely understood effects of CO in adaptive immune response, and warrant further investigation.
Resolution of Inflammation
Recent findings suggest that therapeutic CO can also modulate the resolution of inflammation, which represents a relatively new area of study. Lipid mediators (LMs), which include eicosanoids and recently described omega-3-derived “specialized pro-resolution mediators” (SPMs), represent the key signaling molecules in resolution of inflammation (20). The therapeutic potential of CO was tested against Streptococcus pneumoniae pneumonia in baboons (34). Plasma obtained from S. pneumoniae-infected baboons displayed significantly reduced levels of LMs/SPMs, including eicosapentaenoic acid-derived E-series resolvins (RvE) and lipoxins, suggesting that pneumonia can deregulate pro-resolution programs in baboons. CO inhalation increased the levels of plasma RvE and lipoxins relative to control levels in infected baboons. These results suggest that low-dose CO can restore altered SPM profiles during pneumonia (34). CO inhalation therapy was also shown to reduce neutrophil infiltration in the lung in a mouse model of ischemia-reperfusion (I/R)-induced lung injury and conferred additive protection when combined with intravenous application of the pro-resolving mediator resolvin D1 (RvD1). Both CO and RvD1 application were shown to reduce neutrophil-platelet aggregates in mouse blood, and reduce leukotrienes and thromboxane B2 production in I/R lungs. CO also reduced neutrophil-platelet aggregation in human whole blood (146). These intriguing studies suggest that the activation of pro-resolving programs may add an additional underlying mechanism to the anti-inflammatory protection afforded by CO.
EFFECTS OF CO ON CELLULAR PROLIFERATION
Carbon monoxide (CO) is endogenously produced by vascular smooth muscle cells (SMCs) under conditions of hypoxia and can modulate cGMP levels in both endothelial cells and SMCs (98–100). Further studies by this group demonstrated that endogenous or exogenous CO can inhibit the proliferation of SMCs via upregulation of cGMP and downregulation of the cell cycle regulatory factor E2F-1 (99). In addition, SMC-derived CO was shown to have paracrine antiproliferative effects on endothelial cells by downregulating endothelial cell expression of endothelin-1 and platelet-derived growth factor-B (98). Further studies have identified cGMP-dependent activation of p38 MAPK as a signaling pathway leading to the upregulation of p21Waf1/Cip1, a regulator of cell cycle progression, in the antiproliferative effects of CO in vascular SMCs (73, 117). The p38 MAPK-dependent upregulation of the lipid raft-associated tumor suppressor protein caveolin-1 was also implicated in the antiproliferative response (73). Experiments in human airway SMCs demonstrated a role for downregulation of ERK1/2 and cyclin D and for upregulation of p21Waf1/Cip1 and mtROS in the antiproliferative effects of CO (151, 161). In experiments using CORM-2, both NOX2 and mitochondria were implicated as regulatory targets of CO action, resulting in downregulation of NOX-derived ROS and simultaneous upregulation of mtROS. In support of this hypothesis, the NOX inhibitors diphenylene iodonium or apocynin and the respiration inhibitor rotenone (which increases mtROS) were shown to exert antiproliferative effects in this model (161). CO exposure also exerted antiproliferative and antifibrotic effects in pulmonary fibroblasts through the downregulation of cyclins A/D expression, upregulation of p21Waf1/Cip1, and inhibition of extracellular matrix formation (196). Studies in cancer models demonstrate that CO can also inhibit tumor growth by inducing growth arrest in cancer cells (181). The observed antiproliferative effects of CO have potential therapeutic value in diseases where aberrant cell proliferation and/or tissue remodeling has been implicated, including vascular injury (73, 117), pulmonary fibrosis (196), pulmonary hypertension (198), and cancer (181).
EFFECTS OF CO ON ENDOTHELIAL CELL GROWTH, VASCULAR REPAIR, AND ANGIOGENESIS
In contrast to the antiproliferative effects observed in fibroblasts and SMCs, both exogenous CO and HO-1-derived CO can act as potent regulators of endothelial cell growth and angiogenesis. Exposure to CO gas or CORM treatment promoted endothelial cell growth and motility in vitro (61, 182). CO induced endothelial cell growth through NO-dependent phosphorylation of retinoblastoma protein. In vivo, CO application as gas or CORM-2 promoted endothelial repair after vascular injury in mice and rats (61, 182). Furthermore, CO was shown to drive reendothelization via endothelial nitric oxide synthase (eNOS)-dependent NO production and subsequent NO-dependent mobilization of endothelial progenitor cells from bone marrow (182). Both CORM-2 and chemical activators of HO-1 (i.e., hemin, Δ-12 prostaglandin-J2) stimulated VEGF production in endothelial cells (66). Furthermore, proangiogenic activity in endothelial cells was upregulated by CORM-2 treatment and HO-1 expression and downregulated by inhibition of HO activity (66). HO-1 was demonstrated as an essential intermediate in the activity of the angiogenic factor stromal cell-derived factor 1 (SDF-1). Exogenous CO was shown to compensate for HO-1 deficiency, in maintaining the proangiogenic effect of SDF-1 in isolated aorta from Hmox1−/− mice (36). In contrast, antiangiogenic properties of CO have also been reported. For example, CO was shown to inhibit VEGF-induced proliferation and proangiogenic activity in human umbilical vein endothelial cells. In this model, CO suppressed VEGF-induced phosphorylation of Akt and VEGF receptor-2. CO inhibited basic fibroblast growth factor-2-dependent angiogenesis in vivo (1). Taken together, these studies suggest that CO can exert complex effects on endothelial cell growth and angiogenesis, which require additional resolution.
EFFECTS OF CO ON PROGRAMMED CELL DEATH
CO has been shown to impact cell death and survival in a cell- and stimulant-dependent manner in experimental cell death models including TNF-α stimulation, I/R and/or anoxia-reoxygenation (A/R)-induced injury, and hyperoxia exposure. CO has been shown to protect against inducers of apoptosis, including TNF-α exposure and I/R injury through the activation of the MKK3/p38 MAPK and NF-κB pathways (16, 17, 75, 191–193) (Fig. 3). For example, the antiapoptotic effect of CO in endothelial cells against TNF-α required p38 MAPK- and NF-κB-dependent expression of antiapoptotic genes (16, 17). CO exerts its antiapoptotic effects through MKK3/p38 MAPK-mediated downregulation of Fas/Fas ligand interaction and by maintaining levels of antiapoptotic proteins Bcl-2 and Bcl-XL (17, 191). CO exposure also protected rat hepatocytes from TNF-α-induced apoptosis modulation of the NF-κB pathway (75). CO exposure stimulated mtROS production, which activated PI3K and subsequent Akt phosphorylation, leading to degradation of I-κB and activation of NF-κB (75). CO-induced activation of the PI3K/Akt pathway also protected against TNF-α-induced apoptosis in cerebral microvascular endothelial cells (10). The PI3K/Akt pathway attenuated endothelial cell apoptosis during A/R injury through activation of signal transducer and activator of transcription-3 (STAT3) and attenuation of Fas expression and of caspase-3 activity (192). CO also protected pancreatic β-cells against TNF-α-induced apoptosis in a cGMP-dependent manner (56).
Fig. 3.
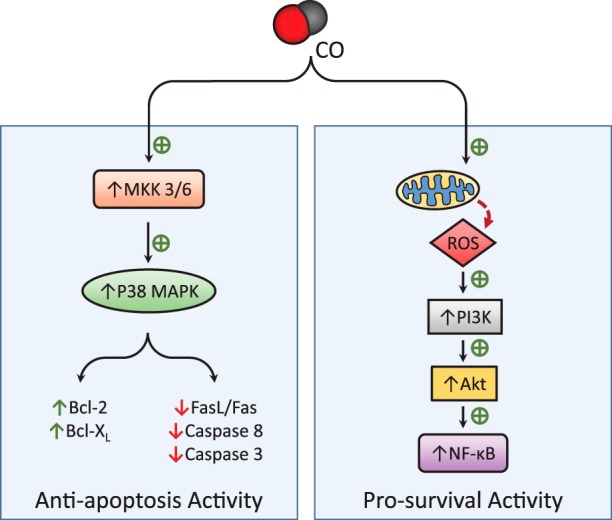
Effect of CO on cell death pathways. CO can modulate apoptotic signaling and prosurvival pathways leading to cytoprotection. CO-dependent antiapoptotic effects have been related to the upregulation of the MKK3/p38 MAPK pathway, resulting in modulation of the expression and activation of apoptotic effector molecules. CO may target mitochondria, leading to modulation of mtROS flux and downstream adaptive signaling. Furthermore, the stimulation of phosphoinositide 3-kinase (PI3K)/Akt and NF-κB-dependent prosurvival pathways has been associated with the protective effects of CO.
While CO has consistently been shown to protect against cell death from multiple injuries, the precise pathways have been conflicting and have exhibited cell type-specific variations. During hypoxia, unlike TNF-α-induced apoptosis, CO exposure protected against cell death by decreasing hyperoxia-induced NOX-dependent ROS formation, leading to inhibition of Fas-dependent apoptosis and modulation of Bcl-2-related proteins (178). CO exposure was protective against apoptosis in retinal I/R injury, but variable effects on p38 phosphorylation were reported dependent on experimental conditions (141, 167).
CO suppressed Fas-activating antibody (Jo2)-induced apoptosis of murine lung endothelial cells through both inhibition of Fas-associated protein with death domain (FADD) phosphorylation via JNK and inhibition of caspase-8 activation via cellular FLICE (FADD-like IL-1β-converting enzyme)-like inhibitory protein (c-FLIP) (179). CO also conferred cytoprotection against apoptosis through activation of ERK1/2 and NF-κB prosurvival pathways in this model (179). In BMDMs, CO also protects against A/R injury through mtROS-mediated HIF-1α activation and increased expression of anti-inflammatory cytokine transforming growth factor-β (25). CO has also been shown to protect against hypoxia-induced necrosis in cardiac myocytes by inhibiting L-type Ca2+ channel-mediated influx of Ca2+ (166). To date, few studies have examined the effect of CO on cellular necroptosis, a programmed form of necrotic cell death. Treatment with CORM-A1 reduced hepatocyte necroptotic cell death after ethanol exposure (9). The inhibitory potential of CO on cell death has been recapitulated using in vivo models of I/R and organ transplantation as discussed in subsequent sections.
EFFECTS OF CO ON CELLULAR AUTOPHAGY
Autophagy is a genetically regulated cellular homeostatic program for the lysosome-dependent turnover of cellular organelles and proteins that can be regulated by cellular stress and nutrient starvation (26). The processing of autophagy substrates is dependent on their recognition and sequestration in double-membraned compartments called autophagosomes, which subsequently fuse to lysosomes where the cargoes are digested by lysosomal enzymes. Recent studies from this laboratory have demonstrated that CO can stimulate the autophagy program in cultured epithelial cells. CO exposure increased the expression and activation of the autophagy protein microtubule-associated protein-1 light chain 3B (LC3B) in mouse lung and in cultured human alveolar or bronchial epithelial cells. Moreover, CO was shown to increase autophagosome formation in pulmonary epithelial cells (85). In this context, CO was shown to stimulate the production of mtROS in the absence of other inducing stimuli typically required for activation of autophagy. The enhanced autophagy response to CO exposure was inhibited by both cellular antioxidants (e.g., N-acetyl-l-cysteine) and mitochondria-specific antioxidants (e.g., mito-TEMPO) (85). Recent studies have also explored protective effects of CO application to mesenchymal stromal cells (MSCs) with the goal of enhancing the therapeutic potential of MSC-based therapies (51). CO enhanced autophagy in MSCs and protected the cells from oxidative stress and apoptosis, whereas CO preconditioning failed to confer protection in MSCs derived from autophagy-deficient mice (51). These observations, taken together, support a role for autophagy as an underlying mechanism for the cytoprotective effects of CO.
PROTECTIVE EFFECTS OF CO IN ORGAN INJURY MODELS
CO has been shown to confer protection in preclinical animal models of lung injury and disease. Although the mode of protection and the underlying mechanisms have been reported to vary in a model-specific faction, the observed protection phenotypes have been primarily associated with anti-inflammatory effects, and with the modulation of apoptotic and proliferation programs. Additional mechanisms have also been implicated including systemic effects on vasoregulation, autophagy, and innate immune responses. The following sections highlight major findings in this area.
Oxidative Lung Injury
Inhalation of supplemental oxygen (hyperoxia) is commonly used as supportive therapy for patients with respiratory failure. However, hyperoxia has also been shown to be a consistent experimental model of lung injury. Hyperoxia (>95% fraction of inspired O2) in rodents causes acute lung injury associated with an excessive inflammatory response. Inhalation of CO at low concentrations (e.g., 250 ppm) has been shown to extend survival and protect the lung against the effects of hyperoxia in both rats and mice (115, 116). Compared with hyperoxia-only controls, the application of CO ameliorated lung injury markers including neutrophil influx in the airways, fibrin deposition, edema, protein accumulation in the bronchioalveolar lavage (BAL) fluid, and lung tissue apoptosis after hyperoxia exposure (115, 116). The protection afforded by CO treatment against the lethal effects of hyperoxia was associated with the inhibition of proinflammatory cytokine (e.g., TNFα, IL-1β, IL-6) production and was dependent on the upregulation of the MKK3/p38β MAPK pathway (116). Similarly, CO was also shown to confer anti-inflammatory and tissue-protective effects in a model of hyperoxia-induced bronchopulmonary dysplasia in neonatal mice (8, 45). CO ameliorated alveolar simplification in this model and was associated with the reduction of inflammatory cell influx including monocytes (F4/80+, CD11c+, and CD11b+) and neutrophils (Gr-1+), and inhibition of proinflammatory cytokines, including C-C motif chemokine ligand 2 (8).
Ischemia-Reperfusion Injury
Tissue-protective effects of CO have been demonstrated in a model of pulmonary I/R injury in mice which can cause lung injury associated with activation of Fas-dependent apoptosis in situ (191, 193). In this model, CO conferred antiapoptotic effects through upregulation of the MKK3/p38α MAPK pathway, resulting in the inhibition of apoptotic tissue markers, including the expression of Fas/FasL and Bcl-2 family proteins, activation of caspases-3, -8, -9, cleavage of PARP, and release of cytochrome c (191, 193). Consistently, the therapeutic effect of CO was compromised in Mkk3−/− mice or by chemical inhibition of p38 MAPK (191). Furthermore, inhaled CO was shown to rescue HO-1-deficient mice (hmox-1−/−) from mortality induced by pulmonary ischemia (48). CO conferred protection in this model by promoting fibrinolysis (48). These effects of CO were dependent on the downregulation of cGMP and ERK1/2-dependent expression of the proinflammatory factor Egr-1, resulting in the reduced expression of the inhibition of plasminogen activator inhibitor-1 (PAI-1), an inhibitor of fibrinolysis (96). Consistently, PAI-1-deficient mice (Serpine1−/−) were protected from ischemic insult and CO afforded no further protection to these mice (96). In hepatic I/R injury, CO has also been shown to attenuate apoptosis through activation of the miR-34a/SIRT1 pathway (72).
Transplant Rejection
Organ transplantation has been a frequently used model of I/R injury in preclinical studies. I/R injury may also represent an underlying pathogenic mechanism of graft failure during organ transplantation. CO, when applied at low concentration (e.g., 250 ppm), has been demonstrated to reduce apoptosis, inflammation, and transplant-associated I/R injury, thus reducing the incidence of graft rejection and preserving graft function. In a rat model of orthotopic left lung transplantation, exogenous application of CO significantly protected the graft and reduced hemorrhage, fibrosis, and thrombosis (150). Furthermore, CO inhibited lung cell apoptosis after transplantation and attenuated lung and systemic proinflammatory cytokine production (e.g., macrophage inflammatory protein-1α, macrophage migration inhibitory factor, and IL-6) (150). Furthermore, protection against I/R injury was conferred in syngeneic rat orthotopic lung transplantation by inhaled CO administered to either the donor or the recipient (77). Delivery of CO to lung grafts by saturation of the preservation media also reduced I/R injury and inflammation in syngeneic rat orthotopic lung transplantation (78). In a vascular transplantation model, aortic graft transplant recipients exposed to CO displayed reduced vascular remodeling and reduced leukocyte, macrophage, and T cell infiltration in the graft (117). Preexposure of murine pancreatic islet cells to CO improved islet cell survival and function after transplant (56). The mechanisms underlying the protective effects of CO in organ transplantation are currently believed to involve primarily anti-inflammatory, immunomodulatory, and antiapoptotic effects, though other mechanisms such as inhibition of cell proliferation and improved perfusion may contribute to the observed protection (6, 7, 110, 117, 119). In addition to lung and vascular transplantation, the protective effects of CO are now substantiated in transplantation models of the pancreas, heart, intestine, kidney, and liver (7, 42, 56, 69, 87, 110, 111, 139, 174).
Mechanical Ventilation-Induced Lung Injury
Mechanical ventilation (MV) in rodent lungs causes cyclical stretch and activation of sterile inflammatory pathways, resulting in ventilator-induced lung injury (VILI). This experimental model is used as an animal model of acute respiratory distress syndrome (ARDS) in humans. The therapeutic potential of CO has been demonstrated in rodent models of MV-induced VILI (37, 58, 59). In mice subjected to MV at moderate tidal volume, CO conferred tissue protection when administered through the ventilator circuit (37, 58, 59). CO reduced MV-induced proinflammatory cytokine and chemokine production and reduced markers of lung injury such as alveolar protein and cellular influx, lung neutrophil recruitment, and pulmonary edema (58, 59). The protective effects of CO were shown independently to require caveolin-1 (59) and the activation of PPARγ, leading to the inhibition of Egr-1 (58). These studies, taken together, suggest that MV in the presence of CO may provide protection in VILI. However, more studies are required to elucidate the precise mechanisms underlying the therapeutic potential of CO in VILI models.
Bleomycin-Induced Lung Injury (Fibrosis)
Idiopathic pulmonary fibrosis (IPF) is a fatal disease with no known etiology characterized by scarring or thickening of lung tissues associated with fibroblast hyperproliferation and extracellular matrix remodeling (145). Bleomycin, a redox cycling compound that generates O2− and H2O2, is a frequently used chemotherapeutic agent that is a known inducer of pulmonary fibrosis in humans. Intratracheal administration of bleomycin in mice is used to model IPF in animals. Exogenous CO treatment can provide protection against bleomycin-induced fibrotic lung injury in mice (196). In mice treated with bleomycin intratracheally and then exposed to CO or ambient air, the lungs from CO-treated animals displayed reduced lung hydroxyproline, collagen, and fibronectin levels relative to air-treated bleomycin-injured controls. The protective effect of CO in this model was associated with an antiproliferative effect of CO on fibroblast proliferation associated with the increased expression of p21Waf1/Cip1 and inhibition of cyclins A/D expression (196).
Pulmonary Hypertension
Pulmonary arterial hypertension (PAH) is characterized by progressive pulmonary arteriole smooth muscle remodeling, resulting in increases in pulmonary vascular resistance and, ultimately, right ventricular failure (43). Administration of CO was shown to provide protection in rodent models of monocrotaline-induced and hypoxia-induced PAH. Compared with controls, intermittent exposure to CO reversed established PAH and right ventricular hypertrophy, restored right ventricular and pulmonary arterial pressures, as well as pulmonary vascular morphology. The ability of CO to reverse PAH was dependent on endothelial nitric oxide synthase (eNOS)-dependent NO generation. The protective effect of CO was endothelial cell-dependent and was associated with increased apoptosis and decreased cellular proliferation of vascular SMCs (198). Further studies have shown that CO decreased pulmonary artery vascular resistance and inhibited hypoxic vasoconstriction, through activation of the sGC/cGMP, and the hyperpolarization of potassium channels (39).
Endotoxemia and Sepsis
CO has demonstrated anti-inflammatory effects in animal models of endotoxin shock and sepsis (114). CO preconditioning reduced the production of serum TNF-α, IL-1β, IL-6, and increased the production of the anti-inflammatory cytokine IL-10. Furthermore, CO preconditioning also reduced organ injury and prolonged survival following LPS challenge (101, 114). Anti-inflammatory effects of CO through modulation of pro- or anti-inflammatory cytokine production were diminished in heat shock factor-1 knockout (hsf1−/−) mice, suggesting a role for the heat shock response in vivo (74). Anti-inflammatory effects of CO have been recently documented in higher mammals including swine and non-human primates (NHPs) (94, 97). CO pretreatment improved organ function and reduced lung inflammation in LPS-challenged swine. CO inhibited serum levels of the proinflammatory IL-1β in response to LPS, while inducing the anti-inflammatory cytokine IL-10 after LPS challenge in this model (94). Anti-inflammatory effects were also reported in NHPs subjected to LPS inhalation (97). CO exposure following LPS inhalation decreased TNF-α release in BAL fluid and reduced pulmonary neutrophilia without affecting IL-6 and IL-8. The anti-inflammatory effects of CO in this model required elevated doses (500 ppm) that were associated with excessive CO-Hb buildup (97). These studies highlight interspecies variation in the dosimetry and efficacy of CO therapy with respect to modulation of inflammation.
In addition to mechanisms related to downregulation of cytokine and inflammatory mediator production, CO has been shown to improve bacterial clearance in sepsis models, though the mechanisms remain incompletely understood (29, 113, 183). The pharmacological application of CORM-2 enhanced bacterial phagocytosis in vivo and rescued HO-1-deficient mice from sepsis-induced mortality after cecal-ligation and puncture (CLP) surgery (29). Furthermore, CORM treatment using the compound ALF-186 improved intestinal bacterial clearance in a model of chronic colitis in Il10−/− mice, and enhanced bactericidal activity in wild-type and HO-1-deficient macrophages (113). In an Escherichia coli infection model, endogenous HO-derived CO was associated with enhanced macrophage phagocytosis and this was shown to require NLRP3 and caspase-1-dependent immune responses (183). Our laboratory has recently demonstrated that CO inhalation (250 ppm) either as pretreatment or as posttreatment improved mouse survival in the CLP model (86). The protective effects of CO in CLP were dependent on the induction of Beclin 1-dependent autophagy and phagocytosis, the reduction of inflammation, and enhanced bacterial clearance from lung, liver, and blood. The prosurvival effects of CO in CLP were dependent on the autophagy program, as they were reduced in autophagy-compromised (Becn1+/−) mice (86).
POTENTIAL FOR INHALATION CO AS A THERAPY FOR HUMAN DISEASE
CO has been demonstrated as an effective anti-inflammatory modulator in preclinical models of organ injury and disease, both in rodents and in higher animals (e.g., swine and primates). On the basis of these observations, continuing efforts are underway to develop inhaled CO (iCO) as a therapy for clinical application. Previous clinical trials have demonstrated the safety of iCO application in humans. In a randomized, double-blinded, placebo-controlled, two-way crossover trial, experimental endotoxemia was induced in healthy volunteers by injection of 2 ng/kg LPS, and inhalation CO (iCO) was applied at 500 ppm CO for 1 hour (which increased CO-Hb to 7%), with air inhalation as the placebo control. Under these conditions, iCO did not modulate the inflammatory response as measured by systemic cytokine production, with no adverse effects of iCO reported (93). In a different human clinical trial, iCO was applied to patients with chronic obstructive pulmonary disease (COPD) (11). In this study, ex-smoking patients with stable COPD were subjected to iCO (100–125 ppm, 2 hours/day for 4 days), which increased CO-Hb levels to 4.5%. Application of iCO to patients with stable COPD did not cause adverse effects, and patients exhibited a trend in reduction of sputum eosinophils and improvement of methacholine responsiveness (11). We have recently completed a multicenter phase IIa, double-blinded, sham-controlled, clinical trial, in which IPF patients were randomized to treatment with inhaled CO at 100–200 ppm or to inhaled 21% oxygen for 2 hours/day, twice/week, for 12 weeks (131). Application of iCO did not affect serum concentration of matrix metalloproteinase-7 (MMP7), the primary study end point, following 12 weeks of treatment, relative to controls. No differences were observed in physiologic measures, incidence of acute exacerbations, hospitalization, death or patient-reported outcomes, with no differences in the distribution of adverse events between the groups. We concluded that inhaled CO is well tolerated and can be safely administered to IPF patients, albeit with no significant changes in study end points (131). Our findings warrant further testing the efficacy of iCO in future IPF clinical trials.
CONCLUSIONS
Much has been learned about the cellular signaling networks that are modulated by exposure to CO, including major signal transduction pathways associated with the regulation of inflammation, cell death, and cellular proliferation programs. An emerging concept is that mitochondria may represent a common target of CO action, such that the resulting flux in mitochondria-derived mediators and mtROS generation may be associated with the regulation of downstream signaling effects of CO. Although the heme moieties of cellular and mitochondrial cytochromes have been identified as candidate proximal targets of CO, not all studies have unequivocally identified a heme-dependent mechanism. Furthermore, model and tissue-specific variation in molecular pathways have left some ambiguity and unanswered questions such that no unifying mechanism has emerged. CO has now been shown to confer cyto- and tissue protection in a number of animal models of organ injury and diseases. Further research is needed to identify how discrete signaling targets of CO are integrated in a context-specific fashion to coordinate cytoprotection. In addition to the lung and vascular models covered in this review, salutary effects of CO therapy have been upheld in other diverse models of disease, including metabolic syndrome (83), traumatic brain injury (28), preeclampsia (168), and others. The successful demonstration of cyto- and tissue-protective effects of inhaled CO in preclinical animal models using rodents and higher mammals has led to continued efforts toward the clinical application of CO, both as iCO, or through pharmacological delivery of CORMs. Human clinical trials now in progress attempt to harness the therapeutic effects of CO observed in animal models and aim to characterize the safety and efficacy of iCO as a therapeutic drug. Barriers to progress include differences in pulmonary physiology between humans and experimental animals, as well as regulatory issues. Nevertheless, several studies have now been completed that lend support for the safety and feasibility of iCO therapy (11, 93, 131).
Further progress in therapeutic use of iCO is dependent on the completion and analysis of several ongoing clinical trials in organ transplantation, pulmonary fibrosis, and critical illness (sepsis/ARDS) (104, 108). At present the therapeutic potential of CO remains to be realized in the clinical setting.
Glossary
- AMPK
AMP-activated protein kinase
- ARDS
Acute respiratory distress syndrome
- BKCa
Ca2+-dependent K+ channels
- BMDM
Bone-marrow-derived macrophages
- BVR
Biliverdin reductase
- CLP
Cecal ligation and puncture
- CO
Carbon monoxide
- CO-Hb
Carboxy-hemoglobin
- CORM
Carbon monoxide releasing molecule
- cGMP
Guanosine 3′,5′-monophosphate
- Egr-1
Early growth response-1
- EnaC
Epithelial Na+ channels
- HO-1
Heme oxygenase-1
- HIF-1α
Hypoxia-inducible factor-1-α
- H2S
Hydrogen sulfide
- iCO
Inhaled CO
- IL
Interleukin
- I/R
Ischemia-reperfusion
- JNK
c-Jun NH2-terminal kinase
- MAPK
Mitogen-activated protein kinase
- MSCs
Mesenchymal stromal cells
- mtROS
Mitochondrial reactive oxygen species
- MV
Mechanical ventilation
- NHP
Non-human primate
- NO
Nitric oxide
- NOX
NADPH oxidase
- NF-κB
Nuclear factor-κB
- NLRP3
NOD, leucine rich region- and pyrin domain-containing-3
- NRF-1
Nuclear respiratory factor-1
- Nrf2
NF-E2-related factor-2
- sGC
Soluble guanylate cyclase
- p21Waf1/Cip1
Cyclin-dependent kinase inhibitor-1
- PGC-1α
Proliferator-activated receptor-γ coactivator-1α
- PI3K/Akt
Phosphoinositide 3-kinase/Akt
- PPARγ
Peroxisome proliferator-activated receptor-γ
- ROS
Reactive oxygen species
- RvD1
Resolvin-D1
- SMC
Smooth muscle cell
- TLR4
Toll-like receptor-4
- VILI
Ventilator-induced lung injury
GRANTS
This work was supported by National Institutes of Health-National Heart, Lung, and Blood Institute Grants P01 HL-108801, R01 HL-079904, and R01 HL-060234 (A. M. K. Choi).
DISCLOSURES
A.M.K. Choi is a co-founder of Proterris, Inc., and is on the Scientific Advisory Board at Proterris. No other conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.C.M. prepared figures; S.W.R. and K.C.M. drafted manuscript; S.W.R., K.C.M., and A.M.K.C. edited and revised manuscript; S.W.R. and A.M.K.C. approved final version of manuscript.
Footnotes
Glossary of abbreviations appears at the end of the article.
REFERENCES
- 1.Ahmad S, Hewett PW, Fujisawa T, Sissaoui S, Cai M, Gueron G, Al-Ani B, Cudmore M, Ahmed SF, Wong MK, Wegiel B, Otterbein LE, Vítek L, Ramma W, Wang K, Ahmed A. Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation. Thromb Haemost 113: 329–337, 2015. doi: 10.1160/TH14-01-0002. [DOI] [PubMed] [Google Scholar]
- 2.Alam J, Igarashi K, Immenschuh S, Shibahara S, Tyrrell RM. Regulation of heme oxygenase-1 gene transcription: recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid Redox Signal 6: 924–933, 2004. doi: 10.1089/ars.2004.6.924. [DOI] [PubMed] [Google Scholar]
- 3.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071–26078, 1999. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 4.Al-Owais MM, Dallas ML, Boyle JP, Scragg JL, Peers C. Heme oxygenase-1 influences apoptosis via CO-mediated inhibition of K+ channels. Adv Exp Med Biol 860: 343–351, 2015. doi: 10.1007/978-3-319-18440-1_39. [DOI] [PubMed] [Google Scholar]
- 5.Althaus M, Fronius M, Buchäckert Y, Vadász I, Clauss WG, Seeger W, Motterlini R, Morty RE. Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am J Respir Cell Mol Biol 41: 639–650, 2009. doi: 10.1165/rcmb.2008-0458OC. [DOI] [PubMed] [Google Scholar]
- 6.Amano MT, Camara NO. The immunomodulatory role of carbon monoxide during transplantation. Med Gas Res 3: 1, 2013. doi: 10.1186/2045-9912-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amersi F, Shen XD, Anselmo D, Melinek J, Iyer S, Southard DJ, Katori M, Volk HD, Busuttil RW, Buelow R, Kupiec-Weglinski JW. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology 35: 815–823, 2002. doi: 10.1053/jhep.2002.32467. [DOI] [PubMed] [Google Scholar]
- 8.Anyanwu AC, Bentley JK, Popova AP, Malas O, Alghanem H, Goldsmith AM, Hershenson MB, Pinsky DJ. Suppression of inflammatory cell trafficking and alveolar simplification by the heme oxygenase-1 product carbon monoxide. Am J Physiol Lung Cell Mol Physiol 306: L749–L763, 2014. doi: 10.1152/ajplung.00236.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bakhautdin B, Das D, Mandal P, Roychowdhury S, Danner J, Bush K, Pollard K, Kaspar JW, Li W, Salomon RG, McMullen MR, Nagy LE. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J Hepatol 61: 1029–1037, 2014. doi: 10.1016/j.jhep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basuroy S, Tcheranova D, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-α-induced apoptosis. Am J Physiol Cell Physiol 300: C256–C265, 2011. doi: 10.1152/ajpcell.00272.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, van der Toorn M, Boezen HM, Kerstjens HA. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J 30: 1131–1137, 2007. doi: 10.1183/09031936.00163206. [DOI] [PubMed] [Google Scholar]
- 12.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 13.Bilban M, Bach FH, Otterbein SL, Ifedigbo E, de Costa d’Avila JC, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O, Otterbein LE. Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 24: 601–610, 2006. doi: 10.1016/j.immuni.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Bleecker ML. Carbon monoxide intoxication. Handb Clin Neurol 131: 191–203, 2015. doi: 10.1016/B978-0-444-62627-1.00024-X. [DOI] [PubMed] [Google Scholar]
- 15.Borzelleca JF. Paracelsus: herald of modern toxicology. Toxicol Sci 53: 2–4, 2000. doi: 10.1093/toxsci/53.1.2. [DOI] [PubMed] [Google Scholar]
- 16.Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-κ B to protect endothelial cells from tumor necrosis factor-α-mediated apoptosis. J Biol Chem 277: 17950–17961, 2002. doi: 10.1074/jbc.M108317200. [DOI] [PubMed] [Google Scholar]
- 17.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med 192: 1015–1026, 2000. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown SD, Piantadosi CA. Reversal of carbon monoxide-cytochrome c oxidase binding by hyperbaric oxygen in vivo. Adv Exp Med Biol 248: 747–754, 1989. doi: 10.1007/978-1-4684-5643-1_84. [DOI] [PubMed] [Google Scholar]
- 19.Brüne B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol 32: 497–504, 1987. [PubMed] [Google Scholar]
- 20.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40: 315–327, 2014. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calafat AM, Polzin GM, Saylor J, Richter P, Ashley DL, Watson CH. Determination of tar, nicotine, and carbon monoxide yields in the mainstream smoke of selected international cigarettes. Tob Control 13: 45–51, 2004. doi: 10.1136/tc.2003.003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cepinskas G, Katada K, Bihari A, Potter RF. Carbon monoxide liberated from carbon monoxide-releasing molecule CORM-2 attenuates inflammation in the liver of septic mice. Am J Physiol Gastrointest Liver Physiol 294: G184–G191, 2008. doi: 10.1152/ajpgi.00348.2007. [DOI] [PubMed] [Google Scholar]
- 23.Chance B, Erecinska M, Wagner M. Mitochondrial responses to carbon monoxide toxicity. Ann NY Acad Sci 174: 193–204, 1970. doi: 10.1111/j.1749-6632.1970.tb49786.x. [DOI] [PubMed] [Google Scholar]
- 24.Chauveau C, Rémy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, Tesson L, Brion R, Beriou G, Gregoire M, Josien R, Cuturi MC, Anegon I. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 106: 1694–1702, 2005. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- 25.Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, Gallo D, Cszimadia E, Bach FH, Lee PJ, Otterbein LE. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci USA 104: 5109–5114, 2007. doi: 10.1073/pnas.0609611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 368: 651–662, 2013. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 27.Choi YK, Kim JH, Lee DK, Lee KS, Won MH, Jeoung D, Lee H, Ha KS, Kwon YG, Kim YM. Carbon monoxide potentiation of L-type Ca2+ channel activity increases HIF-1α-independent VEGF expression via an AMPKα/SIRT1-mediated PGC-1α/ERRα axis. Antioxid Redox Signal 27: 21–36, 2017. doi: 10.1089/ars.2016.6684. [DOI] [PubMed] [Google Scholar]
- 28.Choi YK, Maki T, Mandeville ET, Koh SH, Hayakawa K, Arai K, Kim YM, Whalen MJ, Xing C, Wang X, Kim KW, Lo EH. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat Med 22: 1335–1341, 2016. doi: 10.1038/nm.4188. [DOI] [PubMed] [Google Scholar]
- 29.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest 118: 239–247, 2008. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coburn RF, Blakemore WS, Forster RE. Endogenous carbon monoxide production in man. J Clin Invest 42: 1172–1178, 1963. doi: 10.1172/JCI104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross AR, Higson FK, Jones OT, Harper AM, Segal AW. The enzymic reduction and kinetics of oxidation of cytochrome b-245 of neutrophils. Biochem J 204: 479–485, 1982. doi: 10.1042/bj2040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dallas ML, Boyle JP, Milligan CJ, Sayer R, Kerrigan TL, McKinstry C, Lu P, Mankouri J, Harris M, Scragg JL, Pearson HA, Peers C. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J 25: 1519–1530, 2011. doi: 10.1096/fj.10-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dallas ML, Scragg JL, Peers C. Modulation of hTREK-1 by carbon monoxide. Neuroreport 19: 345–348, 2008. doi: 10.1097/WNR.0b013e3282f51045. [DOI] [PubMed] [Google Scholar]
- 34.Dalli J, Kraft BD, Colas RA, Shinohara M, Fredenburgh LE, Hess DR, Chiang N, Welty-Wolf KE, Choi AM, Piantadosi CA, Serhan CN. The regulation of proresolving lipid mediator profiles in baboon pneumonia by inhaled carbon monoxide. Am J Respir Cell Mol Biol 53: 314–325, 2015. doi: 10.1165/rcmb.2014-0299OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735, 2011. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med 204: 605–618, 2007. doi: 10.1084/jem.20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolinay T, Szilasi M, Liu M, Choi AM. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med 170: 613–620, 2004. doi: 10.1164/rccm.200401-023OC. [DOI] [PubMed] [Google Scholar]
- 38.Dong DL, Zhang Y, Lin DH, Chen J, Patschan S, Goligorsky MS, Nasjletti A, Yang BF, Wang WH. Carbon monoxide stimulates the Ca2+-activated big conductance K channels in cultured human endothelial cells. Hypertension 50: 643–651, 2007. doi: 10.1161/HYPERTENSIONAHA.107.096057. [DOI] [PubMed] [Google Scholar]
- 39.Dubuis E, Potier M, Wang R, Vandier C. Continuous inhalation of carbon monoxide attenuates hypoxic pulmonary hypertension development presumably through activation of BKCa channels. Cardiovasc Res 65: 751–761, 2005. doi: 10.1016/j.cardiores.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Elies J, Dallas ML, Boyle JP, Scragg JL, Duke A, Steele DS, Peers C. Inhibition of the cardiac Na+ channel Nav1.5 by carbon monoxide. J Biol Chem 289: 16421–16429, 2014. doi: 10.1074/jbc.M114.569996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Estabrook RW, Franklin MR, Hildebrandt AG. Factors influencing the inhibitory effect of carbon monoxide on cytochrome P-450-catalyzed mixed function oxidation reactions. Ann NY Acad Sci 174: 218–232, 1970. doi: 10.1111/j.1749-6632.1970.tb49788.x. [DOI] [PubMed] [Google Scholar]
- 42.Faleo G, Neto JS, Kohmoto J, Tomiyama K, Shimizu H, Takahashi T, Wang Y, Sugimoto R, Choi AM, Stolz DB, Carrieri G, McCurry KR, Murase N, Nakao A. Carbon monoxide ameliorates renal cold ischemia-reperfusion injury with an upregulation of vascular endothelial growth factor by activation of hypoxia-inducible factor. Transplantation 85: 1833–1840, 2008. doi: 10.1097/TP.0b013e31817c6f63. [DOI] [PubMed] [Google Scholar]
- 43.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 351: 1655–1665, 2004. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 44.Fayad-Kobeissi S, Ratovonantenaina J, Dabiré H, Wilson JL, Rodriguez AM, Berdeaux A, Dubois-Randé JL, Mann BE, Motterlini R, Foresti R. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem Pharmacol 102: 64–77, 2016. doi: 10.1016/j.bcp.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Gonzalez A, Alex Mitsialis S, Liu X, Kourembanas S. Vasculoprotective effects of heme oxygenase-1 in a murine model of hyperoxia-induced bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 302: L775–L784, 2012. doi: 10.1152/ajplung.00196.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Foresti R, Bani-Hani MG, Motterlini R. Use of carbon monoxide as a therapeutic agent: promises and challenges. Intensive Care Med 34: 649–658, 2008. doi: 10.1007/s00134-008-1011-1. [DOI] [PubMed] [Google Scholar]
- 47.Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe A, Green CJ, Motterlini R. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol 142: 453–460, 2004. doi: 10.1038/sj.bjp.0705825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7: 598–604, 2001. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- 49.Furchgott RF, Jothianandan D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels 28: 52–61, 1991. [DOI] [PubMed] [Google Scholar]
- 50.Furchgott RF. Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Biosci Rep 19: 235–251, 1999. doi: 10.1023/A:1020537506008. [DOI] [PubMed] [Google Scholar]
- 51.Ghanta S, Tsoyi K, Liu X, Nakahira K, Ith B, Coronata AA, Fredenburgh LE, Englert JA, Piantadosi CA, Choi AM, Perrella MA. Mesenchymal stromal cells deficient in autophagy proteins are susceptible to oxidative injury and mitochondrial dysfunction. Am J Respir Cell Mol Biol 56: 300–309, 2017. doi: 10.1165/rcmb.2016-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gläser S, Mede R, Görls H, Seupel S, Bohlender C, Wyrwa R, Schirmer S, Dochow S, Reddy GU, Popp J, Westerhausen M, Schiller A. Remote-controlled delivery of CO via photoactive CO-releasing materials on a fiber optical device. Dalton Trans 45: 13222–13233, 2016. doi: 10.1039/C6DT02011A. [DOI] [PubMed] [Google Scholar]
- 53.Gorman D, Drewry A, Huang YL, Sames C. The clinical toxicology of carbon monoxide. Toxicology 187: 25–38, 2003. doi: 10.1016/S0300-483X(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 54.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947, 2013. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 55.Guengerich FP, Ballou DP, Coon MJ. Purified liver microsomal cytochrome P-450. Electron-accepting properties and oxidation-reduction potential. J Biol Chem 250: 7405–7414, 1975. [PubMed] [Google Scholar]
- 56.Günther L, Berberat PO, Haga M, Brouard S, Smith RN, Soares MP, Bach FH, Tobiasch E. Carbon monoxide protects pancreatic beta-cells from apoptosis and improves islet function/survival after transplantation. Diabetes 51: 994–999, 2002. doi: 10.2337/diabetes.51.4.994. [DOI] [PubMed] [Google Scholar]
- 57.Hasegawa U, van der Vlies AJ, Simeoni E, Wandrey C, Hubbell JA. Carbon monoxide-releasing micelles for immunotherapy. J Am Chem Soc 132: 18273–18280, 2010. doi: 10.1021/ja1075025. [DOI] [PubMed] [Google Scholar]
- 58.Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, Alber S, Kaynar AM, Schmidt R, Ryter SW, Choi AM. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med 177: 1223–1232, 2008. doi: 10.1164/rccm.200708-1265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoetzel A, Schmidt R, Vallbracht S, Goebel U, Dolinay T, Kim HP, Ifedigbo E, Ryter SW, Choi AM. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med 37: 1708–1715, 2009. doi: 10.1097/CCM.0b013e31819efa31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou S, Xu R, Heinemann SH, Hoshi T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc Natl Acad Sci USA 105: 4039–4043, 2008. doi: 10.1073/pnas.0800304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu QS, Chen YX, Huang QS, Deng BQ, Xie SL, Wang JF, Nie RQ. Carbon monoxide releasing molecule accelerates reendothelialization after carotid artery balloon injury in rat. Biomed Environ Sci 28: 253–262, 2015. doi: 10.3967/bes2015.036. [DOI] [PubMed] [Google Scholar]
- 62.Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Hegab AE, Homma S, Nomura A, Sakamoto T, Shimura M, Yoshida A, Yamamoto M, Sekizawa K. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10: 1113–1125, 2005. doi: 10.1111/j.1365-2443.2005.00905.x. [DOI] [PubMed] [Google Scholar]
- 63.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res 97: 805–812, 2005. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jamal Uddin M, Joe Y, Kim SK, Oh Jeong S, Ryter SW, Pae HO, Chung HT. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell Mol Immunol 13: 170–179, 2016. doi: 10.1038/cmi.2015.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ji X, Damera K, Zheng Y, Yu B, Otterbein LE, Wang B. Toward carbon monoxide-based therapeutics: critical drug delivery and developability issues. J Pharm Sci 105: 406–416, 2016. doi: 10.1016/j.xphs.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Józkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid Redox Signal 5: 155–162, 2003. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- 67.Jung SS, Moon JS, Xu JF, Ifedigbo E, Ryter SW, Choi AM, Nakahira K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am J Physiol Lung Cell Mol Physiol 308: L1058–L1067, 2015. doi: 10.1152/ajplung.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaczara P, Motterlini R, Rosen GM, Augustynek B, Bednarczyk P, Szewczyk A, Foresti R, Chlopicki S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: a role for mitoBKCa channels. Biochim Biophys Acta 1847: 1297–1309, 2015. doi: 10.1016/j.bbabio.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 69.Kaizu T, Nakao A, Tsung A, Toyokawa H, Sahai R, Geller DA, Murase N. Carbon monoxide inhalation ameliorates cold ischemia/reperfusion injury after rat liver transplantation. Surgery 138: 229–235, 2005. doi: 10.1016/j.surg.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 70.Kautz AC, Kunz PC, Janiak C. CO-releasing molecule (CORM) conjugate systems. Dalton Trans 45: 18045–18063, 2016. doi: 10.1039/C6DT03515A. [DOI] [PubMed] [Google Scholar]
- 71.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA 86: 99–103, 1989. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim HJ, Joe Y, Yu JK, Chen Y, Jeong SO, Mani N, Cho GJ, Pae HO, Ryter SW, Chung HT. Carbon monoxide protects against hepatic ischemia/reperfusion injury by modulating the miR-34a/SIRT1 pathway. Biochim Biophys Acta 1852: 1550–1559, 2015. doi: 10.1016/j.bbadis.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 73.Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, Ryter SW, Choi AMK. Caveolin-1 expression by means of p38β mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci USA 102: 11319–11324, 2005. doi: 10.1073/pnas.0501345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim HP, Wang X, Zhang J, Suh GY, Benjamin I, Ryter SW, Choi AM. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38 beta MAPK and heat shock factor-1. J Immunol 175: 2622–2629, 2005. doi: 10.4049/jimmunol.175.4.2622. [DOI] [PubMed] [Google Scholar]
- 75.Kim HS, Loughran PA, Rao J, Billiar TR, Zuckerbraun BS. Carbon monoxide activates NF-κB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am J Physiol Gastrointest Liver Physiol 295: G146–G152, 2008. doi: 10.1152/ajpgi.00105.2007. [DOI] [PubMed] [Google Scholar]
- 76.Kim S, Joe Y, Jeong SO, Zheng M, Back SH, Park SW, Ryter SW, Chung HT. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun 20: 799–815, 2014. doi: 10.1177/1753425913508593. [DOI] [PubMed] [Google Scholar]
- 77.Kohmoto J, Nakao A, Kaizu T, Tsung A, Ikeda A, Tomiyama K, Billiar TR, Choi AM, Murase N, McCurry KR. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 140: 179–185, 2006. doi: 10.1016/j.surg.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 78.Kohmoto J, Nakao A, Sugimoto R, Wang Y, Zhan J, Ueda H, McCurry KR. Carbon monoxide-saturated preservation solution protects lung grafts from ischemia-reperfusion injury. J Thorac Cardiovasc Surg 136: 1067–1075, 2008. doi: 10.1016/j.jtcvs.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kretschmer R, Gessner G, Görls H, Heinemann SH, Westerhausen M. Dicarbonyl-bis(cysteamine)iron(II): a light induced carbon monoxide releasing molecule based on iron (CORM-S1). J Inorg Biochem 105: 6–9, 2011. doi: 10.1016/j.jinorgbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 80.Kutty RK, Daniel RF, Ryan DE, Levin W, Maines MD. Rat liver cytochrome P-450b, P-420b, and P-420c are degraded to biliverdin by heme oxygenase. Arch Biochem Biophys 260: 638–644, 1988. doi: 10.1016/0003-9861(88)90492-4. [DOI] [PubMed] [Google Scholar]
- 81.Lancaster JR., Jr Nitric oxide: a brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci OA 1: FSO59, 2015. doi: 10.4155/fso.15.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R, Neviere R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J Pharmacol Exp Ther 329: 641–648, 2009. doi: 10.1124/jpet.108.148049. [DOI] [PubMed] [Google Scholar]
- 83.Lancel S, Montaigne D, Marechal X, Marciniak C, Hassoun SM, Decoster B, Ballot C, Blazejewski C, Corseaux D, Lescure B, Motterlini R, Neviere R. Carbon monoxide improves cardiac function and mitochondrial population quality in a mouse model of metabolic syndrome. PLoS One 7: e41836, 2012. doi: 10.1371/journal.pone.0041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol 54: 151–160, 2016. doi: 10.1165/rcmb.2015-0231TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol 45: 867–873, 2011. doi: 10.1165/rcmb.2010-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal 20: 432–442, 2014. doi: 10.1089/ars.2013.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, Bach FH, Wang H. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J 21: 3450–3457, 2007. doi: 10.1096/fj.07-8472com. [DOI] [PubMed] [Google Scholar]
- 88.Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol 301: H1–H11, 2011. doi: 10.1152/ajpheart.00230.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lim I, Gibbons SJ, Lyford GL, Miller SM, Strege PR, Sarr MG, Chatterjee S, Szurszewski JH, Shah VH, Farrugia G. Carbon monoxide activates human intestinal smooth muscle L-type Ca2+ channels through a nitric oxide-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 288: G7–G14, 2005. doi: 10.1152/ajpgi.00205.2004. [DOI] [PubMed] [Google Scholar]
- 90.Lo Iacono L, Boczkowski J, Zini R, Salouage I, Berdeaux A, Motterlini R, Morin D. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic Biol Med 50: 1556–1564, 2011. doi: 10.1016/j.freeradbiomed.2011.02.033. [DOI] [PubMed] [Google Scholar]
- 91.Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 261: 411–419, 1986. [PubMed] [Google Scholar]
- 92.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37: 517–554, 1997. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 93.Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med 171: 354–360, 2005. doi: 10.1164/rccm.200404-446OC. [DOI] [PubMed] [Google Scholar]
- 94.Mazzola S, Forni M, Albertini M, Bacci ML, Zannoni A, Gentilini F, Lavitrano M, Bach FH, Otterbein LE, Clement MG. Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J 19: 2045–2047, 2005. doi: 10.1096/fj.05-3782fje. [DOI] [PubMed] [Google Scholar]
- 95.Mede R, Klein M, Claus RA, Krieck S, Quickert S, Görls H, Neugebauer U, Schmitt M, Gessner G, Heinemann SH, Popp J, Bauer M, Westerhausen M. CORM-EDE1: a highly water-soluble and nontoxic manganese-based photoCORM with a biogenic ligand sphere. Inorg Chem 55: 104–113, 2016. doi: 10.1021/acs.inorgchem.5b01904. [DOI] [PubMed] [Google Scholar]
- 96.Mishra S, Fujita T, Lama VN, Nam D, Liao H, Okada M, Minamoto K, Yoshikawa Y, Harada H, Pinsky DJ. Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci USA 103: 5191–5196, 2006. doi: 10.1073/pnas.0600241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mitchell LA, Channell MM, Royer CM, Ryter SW, Choi AM, McDonald JD. Evaluation of inhaled carbon monoxide as an anti-inflammatory therapy in a nonhuman primate model of lung inflammation. Am J Physiol Lung Cell Mol Physiol 299: L891–L897, 2010. doi: 10.1152/ajplung.00366.2009. [DOI] [PubMed] [Google Scholar]
- 98.Morita T, Kourembanas S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest 96: 2676–2682, 1995. doi: 10.1172/JCI118334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem 272: 32804–32809, 1997. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 100.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci USA 92: 1475–1479, 1995. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, Otterbein SL, Otterbein LE, Choi AM. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem 278: 36993–36998, 2003. doi: 10.1074/jbc.M302942200. [DOI] [PubMed] [Google Scholar]
- 102.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10: 549–557, 2004. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 103.Motterlini R, Mann BE, Foresti R. Therapeutic applications of carbon monoxide-releasing molecules. Expert Opin Investig Drugs 14: 1305–1318, 2005. doi: 10.1517/13543784.14.11.1305. [DOI] [PubMed] [Google Scholar]
- 104.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov 9: 728–743, 2010. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 105.Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J 19: 284–286, 2005. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- 106.Motterlini R, Sawle P, Hammad J, Mann BE, Johnson TR, Green CJ, Foresti R. Vasorelaxing effects and inhibition of nitric oxide in macrophages by new iron-containing carbon monoxide-releasing molecules (CO-RMs). Pharmacol Res 68: 108–117, 2013. doi: 10.1016/j.phrs.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 107.Nagel C, McLean S, Poole RK, Braunschweig H, Kramer T, Schatzschneider U. Introducing [Mn(CO)3(tpa-κ(3)N)](+) as a novel photoactivatable CO-releasing molecule with well-defined iCORM intermediates—synthesis, spectroscopy, and antibacterial activity. Dalton Trans 43: 9986–9997, 2014. doi: 10.1039/c3dt51848e. [DOI] [PubMed] [Google Scholar]
- 108.Nakahira K, Choi AM. Carbon monoxide in the treatment of sepsis. Am J Physiol Lung Cell Mol Physiol 309: L1387–L1393, 2015. doi: 10.1152/ajplung.00311.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakao A, Choi AM, Murase N. Protective effect of carbon monoxide in transplantation. J Cell Mol Med 10: 650–671, 2006. doi: 10.1111/j.1582-4934.2006.tb00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakao A, Kimizuka K, Stolz DB, Seda Neto J, Kaizu T, Choi AM, Uchiyama T, Zuckerbraun BS, Bauer AJ, Nalesnik MA, Otterbein LE, Geller DA, Murase N. Protective effect of carbon monoxide inhalation for cold-preserved small intestinal grafts. Surgery 134: 285–292, 2003. doi: 10.1067/msy.2003.238. [DOI] [PubMed] [Google Scholar]
- 112.Ning W, Choi AM, Li C. Carbon monoxide inhibits IL-17-induced IL-6 production through the MAPK pathway in human pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 289: L268–L273, 2005. doi: 10.1152/ajplung.00168.2004. [DOI] [PubMed] [Google Scholar]
- 113.Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, Mackey LC, Hansen JJ, Moeser AJ, Rawls JF, Borst LB, Otterbein LE, Plevy SE. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 144: 789–798, 2013. doi: 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6: 422–428, 2000. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 115.Otterbein LE, Mantell LL, Choi AM. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 276: L688–L694, 1999. [DOI] [PubMed] [Google Scholar]
- 116.Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA, Choi AM. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol 163: 2555–2563, 2003. doi: 10.1016/S0002-9440(10)63610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, Stachulak C, Bodyak N, Smith RN, Csizmadia E, Tyagi S, Akamatsu Y, Flavell RJ, Billiar TR, Tzeng E, Bach FH, Choi AM, Soares MP. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 9: 183–190, 2003. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 118.Pai S, Hafftlang M, Atongo G, Nagel C, Niesel J, Botov S, Schmalz HG, Yard B, Schatzschneider U. New modular manganese(I) tricarbonyl complexes as PhotoCORMs: in vitro detection of photoinduced carbon monoxide release using COP-1 as a fluorogenic switch-on probe. Dalton Trans 43: 8664–8678, 2014. doi: 10.1039/c4dt00254g. [DOI] [PubMed] [Google Scholar]
- 119.Pannen BH, Köhler N, Hole B, Bauer M, Clemens MG, Geiger KK. Protective role of endogenous carbon monoxide in hepatic microcirculatory dysfunction after hemorrhagic shock in rats. J Clin Invest 102: 1220–1228, 1998. doi: 10.1172/JCI3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Peers C, Boyle JP, Scragg JL, Dallas ML, Al-Owais MM, Hettiarachichi NT, Elies J, Johnson E, Gamper N, Steele DS. Diverse mechanisms underlying the regulation of ion channels by carbon monoxide. Br J Pharmacol 172: 1546–1556, 2015. doi: 10.1111/bph.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 103: 1232–1240, 2008. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Piantadosi CA. Carbon monoxide poisoning. N Engl J Med 347: 1054–1055, 2002. doi: 10.1056/NEJMp020104. [DOI] [PubMed] [Google Scholar]
- 123.Piantadosi CA. Diagnosis and treatment of carbon monoxide poisoning. Respir Care Clin N Am 5: 183–202, 1999. [PubMed] [Google Scholar]
- 124.Polhemus DJ, Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 114: 730–737, 2014. doi: 10.1161/CIRCRESAHA.114.300505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qin S, Du R, Yin S, Liu X, Xu G, Cao W. Nrf2 is essential for the anti-inflammatory effect of carbon monoxide in LPS-induced inflammation. Inflamm Res 64: 537–548, 2015. doi: 10.1007/s00011-015-0834-9. [DOI] [PubMed] [Google Scholar]
- 126.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114: 1248–1259, 2004. doi: 10.1172/JCI200421146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202: 47–59, 2005. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Riesco-Fagundo AM, Pérez-García MT, González C, López-López JR Jr. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res 89: 430–436, 2001. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- 129.Riquelme SA, Bueno SM, Kalergis AM. Carbon monoxide down-modulates Toll-like receptor 4/MD2 expression on innate immune cells and reduces endotoxic shock susceptibility. Immunology 144: 321–332, 2015. doi: 10.1111/imm.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rodriguez AI, Gangopadhyay A, Kelley EE, Pagano PJ, Zuckerbraun BS, Bauer PM. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler Thromb Vasc Biol 30: 98–104, 2010. doi: 10.1161/ATVBAHA.109.197822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rosas IO, Goldberg HJ, Collard HR, El-Chemaly S, Flaherty K, Hunninghake GM, Lasky JA, Lederer DJ, Machado R, Martinez FJ, Maurer R, Teller D, Noth I, Peters E, Raghu G, Garcia JGN, Choi AM. A Phase II clinical trial of low-dose inhaled carbon monoxide in idiopathic pulmonary fibrosis. Chest 153: 94–104, 2018. doi: 10.1016/j.chest.2017.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rose JJ, Wang L, Xu Q, McTiernan CF, Shiva S, Tejero J, Gladwin MT. Carbon monoxide poisoning: pathogenesis, management and future directions of therapy. Am J Respir Crit Care Med 195: 596–606, 2017. doi: 10.1164/rccm.201606-1275CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rousset F, Nguyen MV, Grange L, Morel F, Lardy B. Heme oxygenase-1 regulates matrix metalloproteinase MMP-1 secretion and chondrocyte cell death via Nox4 NADPH oxidase activity in chondrocytes. PLoS One 8: e66478, 2013. doi: 10.1371/journal.pone.0066478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650, 2006. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 135.Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res 167: 7–34, 2016. doi: 10.1016/j.trsl.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ryter SW, Morse D, Choi AM. Carbon monoxide: to boldly go where NO has gone before. Sci STKE 2004: RE6, 2004. doi: 10.1126/stke.2302004re6. [DOI] [PubMed] [Google Scholar]
- 137.Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem 234-235: 249–263, 2002. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine. BioEssays 26: 270–280, 2004. doi: 10.1002/bies.20005. [DOI] [PubMed] [Google Scholar]
- 139.Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MP. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol 166: 4185–4194, 2001. doi: 10.4049/jimmunol.166.6.4185. [DOI] [PubMed] [Google Scholar]
- 140.Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol 145: 800–810, 2005. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schallner N, Fuchs M, Schwer CI, Loop T, Buerkle H, Lagrèze WA, van Oterendorp C, Biermann J, Goebel U. Postconditioning with inhaled carbon monoxide counteracts apoptosis and neuroinflammation in the ischemic rat retina. PLoS One 7: e46479, 2012. doi: 10.1371/journal.pone.0046479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schatzschneider U. Novel lead structures and activation mechanisms for CO-releasing molecules (CORMs). Br J Pharmacol 172: 1638–1650, 2015. doi: 10.1111/bph.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schroder K, Tschopp J. The inflammasomes. Cell 140: 821–832, 2010. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 144.Scragg JL, Dallas ML, Wilkinson JA, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem 283: 24412–24419, 2008. doi: 10.1074/jbc.M803037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Selman M, King TE Jr, Pardo A; American Thoracic Society; European Respiratory Society; American College of Chest Physicians . Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134: 136–151, 2001. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 146.Shinohara M, Kibi M, Riley IR, Chiang N, Dalli J, Kraft BD, Piantadosi CA, Choi AM, Serhan CN. Cell-cell interactions and bronchoconstrictor eicosanoid reduction with inhaled carbon monoxide and resolvin D1. Am J Physiol Lung Cell Mol Physiol 307: L746–L757, 2014. doi: 10.1152/ajplung.00166.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Simon T, Pogu S, Tardif V, Rigaud K, Rémy S, Piaggio E, Bach JM, Anegon I, Blancou P. Carbon monoxide-treated dendritic cells decrease β1-integrin induction on CD8+ T cells and protect from type 1 diabetes. Eur J Immunol 43: 209–218, 2013. doi: 10.1002/eji.201242684. [DOI] [PubMed] [Google Scholar]
- 148.Sjöstrand T. Endogenous production of carbon monoxide in man under normal and pathophysiological conditions. Scand J Clin Lab Invest 1: 201–214, 1949. doi: 10.3109/00365514909069943. [DOI] [Google Scholar]
- 149.Sjöstrand T. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol Scand 26: 338–344, 1952. doi: 10.1111/j.1748-1716.1952.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 150.Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol 163: 231–242, 2003. doi: 10.1016/S0002-9440(10)63646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Song R, Mahidhara RS, Liu F, Ning W, Otterbein LE, Choi AM. Carbon monoxide inhibits human airway smooth muscle cell proliferation via mitogen-activated protein kinase pathway. Am J Respir Cell Mol Biol 27: 603–610, 2002. doi: 10.1165/rcmb.4851. [DOI] [PubMed] [Google Scholar]
- 152.Stamellou E, Storz D, Botov S, Ntasis E, Wedel J, Sollazzo S, Krämer BK, van Son W, Seelen M, Schmalz HG, Schmidt A, Hafner M, Yard BA. Different design of enzyme-triggered CO-releasing molecules (ET-CORMs) reveals quantitative differences in biological activities in terms of toxicity and inflammation. Redox Biol 2: 739–748, 2014. doi: 10.1016/j.redox.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stevenson TH, Gutierrez AF, Alderton WK, Lian L, Scrutton NS. Kinetics of CO binding to the haem domain of murine inducible nitric oxide synthase: differential effects of haem domain ligands. Biochem J 358: 201–208, 2001. doi: 10.1042/bj3580201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry 33: 5636–5640, 1994. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 155.Stone JR, Marletta MA. Synergistic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem Biol 5: 255–261, 1998. doi: 10.1016/S1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- 156.Suematsu M, Kashiwagi S, Sano T, Goda N, Shinoda Y, Ishimura Y. Carbon monoxide as an endogenous modulator of hepatic vascular perfusion. Biochem Biophys Res Commun 205: 1333–1337, 1994. doi: 10.1006/bbrc.1994.2811. [DOI] [PubMed] [Google Scholar]
- 157.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest 117: 3730–3741, 2007. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci 120: 299–308, 2007. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 159.Sun B, Sun H, Liu C, Shen J, Chen Z, Chen X. Role of CO-releasing molecules liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. J Surg Res 139: 128–135, 2007. doi: 10.1016/j.jss.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 160.Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov 15: 185–203, 2016. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Taillé C, El-Benna J, Lanone S, Boczkowski J, Motterlini R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J Biol Chem 280: 25350–25360, 2005. doi: 10.1074/jbc.M503512200. [DOI] [PubMed] [Google Scholar]
- 162.Telezhkin V, Brazier SP, Mears R, Müller CT, Riccardi D, Kemp PJ. Cysteine residue 911 in C-terminal tail of human BK(Ca)α channel subunit is crucial for its activation by carbon monoxide. Pflugers Arch 461: 665–675, 2011. doi: 10.1007/s00424-011-0924-7. [DOI] [PubMed] [Google Scholar]
- 163.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem 244: 6388–6394, 1969. [PubMed] [Google Scholar]
- 164.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 61: 748–755, 1968. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tenhunen R, Ross ME, Marver HS, Schmid R. Reduced nicotinamide-adenine dinucleotide phosphate dependent biliverdin reductase: partial purification and characterization. Biochemistry 9: 298–303, 1970. doi: 10.1021/bi00804a016. [DOI] [PubMed] [Google Scholar]
- 166.Uemura K, Adachi-Akahane S, Shintani-Ishida K, Yoshida K. Carbon monoxide protects cardiomyogenic cells against ischemic death through L-type Ca2+ channel inhibition. Biochem Biophys Res Commun 334: 661–668, 2005. doi: 10.1016/j.bbrc.2005.06.142. [DOI] [PubMed] [Google Scholar]
- 167.Ulbrich F, Kaufmann KB, Meske A, Lagrèze WA, Augustynik M, Buerkle H, Ramao CC, Biermann J, Goebel U. The CORM ALF-186 mediates anti-apoptotic signaling via an activation of the p38 MAPK after ischemia and reperfusion Injury in retinal ganglion cells. PLoS One 11: e0165182, 2016. doi: 10.1371/journal.pone.0165182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Venditti CC, Casselman R, Young I, Karumanchi SA, Smith GN. Carbon monoxide prevents hypertension and proteinuria in an adenovirus sFlt-1 preeclampsia-like mouse model. PLoS One 9: e106502, 2014. doi: 10.1371/journal.pone.0106502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science 259: 381–384, 1993. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 170.Volpe JA, O’Toole MC, Caughey WS. Quantitative infrared spectroscopy of CO complexes of cytochrome c oxidase, hemoglobin and myoglobin: evidence for one CO per heme. Biochem Biophys Res Commun 62: 48–53, 1975. doi: 10.1016/S0006-291X(75)80403-7. [DOI] [PubMed] [Google Scholar]
- 171.Von Burg R. Carbon monoxide. J Appl Toxicol 19: 379–386, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 172.Vreman HJ, Wong RJ, Stevenson DK. Carbon monoxide in breath, blood, and other tissues. In: Carbon Monoxide Toxicity, edited by Penney DG. Boca Raton, Fl: CRC Press, 2000, p. 19–60. doi: 10.1201/9781420039320.ch2. [DOI] [Google Scholar]
- 173.Wang B, Cao W, Biswal S, Doré S. Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke 42: 2605–2610, 2011. doi: 10.1161/STROKEAHA.110.607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang H, Lee SS, Gao W, Czismadia E, McDaid J, Ollinger R, Soares MP, Yamashita K, Bach FH. Donor treatment with carbon monoxide can yield islet allograft survival and tolerance. Diabetes 54: 1400–1406, 2005. doi: 10.2337/diabetes.54.5.1400. [DOI] [PubMed] [Google Scholar]
- 175.Wang R, Wu L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J Biol Chem 272: 8222–8226, 1997. doi: 10.1074/jbc.272.13.8222. [DOI] [PubMed] [Google Scholar]
- 176.Wang R. Gasotransmitters: growing pains and joys. Trends Biochem Sci 39: 227–232, 2014. doi: 10.1016/j.tibs.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 177.Wang S, Publicover S, Gu Y. An oxygen-sensitive mechanism in regulation of epithelial sodium channel. Proc Natl Acad Sci USA 106: 2957–2962, 2009. doi: 10.1073/pnas.0809100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem 282: 1718–1726, 2007. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 179.Wang X, Wang Y, Lee SJ, Kim HP, Choi AM, Ryter SW. Carbon monoxide inhibits Fas activating antibody-induced apoptosis in endothelial cells. Med Gas Res 1: 8, 2011. doi: 10.1186/2045-9912-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 180.Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol 182: 3809–3818, 2009. doi: 10.4049/jimmunol.0712437. [DOI] [PubMed] [Google Scholar]
- 181.Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, Penacho N, Seth P, Sukhatme V, Ahmed A, Pandolfi PP, Helczynski L, Bjartell A, Persson JL, Otterbein LE. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res 73: 7009–7021, 2013. doi: 10.1158/0008-5472.CAN-13-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wegiel B, Gallo DJ, Raman KG, Karlsson JM, Ozanich B, Chin BY, Tzeng E, Ahmad S, Ahmed A, Baty CJ, Otterbein LE. Nitric oxide-dependent bone marrow progenitor mobilization by carbon monoxide enhances endothelial repair after vascular injury. Circulation 121: 537–548, 2010. doi: 10.1161/CIRCULATIONAHA.109.887695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, Soares MP, Otterbein LE. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest 124: 4926–4940, 2014. doi: 10.1172/JCI72853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wilkinson WJ, Gadeberg HC, Harrison AW, Allen ND, Riccardi D, Kemp PJ. Carbon monoxide is a rapid modulator of recombinant and native P2X(2) ligand-gated ion channels. Br J Pharmacol 158: 862–871, 2009. doi: 10.1111/j.1476-5381.2009.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wilkinson WJ, Kemp PJ. Carbon monoxide: an emerging regulator of ion channels. J Physiol 589: 3055–3062, 2011. doi: 10.1113/jphysiol.2011.206706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wilkinson WJ, Kemp PJ. The carbon monoxide donor, CORM-2, is an antagonist of ATP-gated, human P2X4 receptors. Purinergic Signal 7: 57–64, 2011. doi: 10.1007/s11302-010-9213-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid Redox Signal 4: 603–614, 2002. doi: 10.1089/15230860260220102. [DOI] [PubMed] [Google Scholar]
- 188.Williams SE, Brazier SP, Baban N, Telezhkin V, Müller CT, Riccardi D, Kemp PJ. A structural motif in the C-terminal tail of slo1 confers carbon monoxide sensitivity to human BK Ca channels. Pflugers Arch 456: 561–572, 2008. doi: 10.1007/s00424-007-0439-4. [DOI] [PubMed] [Google Scholar]
- 189.Wilson JL, Fayad Kobeissi S, Oudir S, Haas B, Michel B, Dubois Randé JL, Ollivier A, Martens T, Rivard M, Motterlini R, Foresti R. Design and synthesis of new hybrid molecules that activate the transcription factor Nrf2 and simultaneously release carbon monoxide. Chemistry 20: 14698–14704, 2014. doi: 10.1002/chem.201403901. [DOI] [PubMed] [Google Scholar]
- 190.Yang CM, Lin CC, Lee IT, Hsu CK, Tai YC, Hsieh HL, Chi PL, Hsiao LD. c-Src-dependent transactivation of EGFR mediates CORM-2-induced HO-1 expression in human tracheal smooth muscle cells. J Cell Physiol 230: 2351–2361, 2015. doi: 10.1002/jcp.24912. [DOI] [PubMed] [Google Scholar]
- 191.Zhang X, Shan P, Alam J, Davis RJ, Flavell RA, Lee PJ. Carbon monoxide modulates Fas/Fas ligand, caspases, and Bcl-2 family proteins via the p38alpha mitogen-activated protein kinase pathway during ischemia-reperfusion lung injury. J Biol Chem 278: 22061–22070, 2003. doi: 10.1074/jbc.M301858200. [DOI] [PubMed] [Google Scholar]
- 192.Zhang X, Shan P, Alam J, Fu X-Y, Lee PJ. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J Biol Chem 280: 8714–8721, 2005. doi: 10.1074/jbc.M408092200. [DOI] [PubMed] [Google Scholar]
- 193.Zhang X, Shan P, Otterbein LE, Alam J, Flavell RA, Davis RJ, Choi AM, Lee PJ. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J Biol Chem 278: 1248–1258, 2003. doi: 10.1074/jbc.M208419200. [DOI] [PubMed] [Google Scholar]
- 194.Zhao H, Eguchi S, Alam A, Ma D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. Am J Physiol Lung Cell Mol Physiol 312: L155–L162, 2017. doi: 10.1152/ajplung.00449.2016. [DOI] [PubMed] [Google Scholar]
- 195.Zhong Q, Huang Y, Shen H, Chen Y, Chen H, Huang T, Zeng EY, Tao S. Global estimates of carbon monoxide emissions from 1960 to 2013. Environ Sci Pollut Res Int 24: 864–873, 2017. doi: 10.1007/s11356-016-7896-2. [DOI] [PubMed] [Google Scholar]
- 196.Zhou Z, Song R, Fattman CL, Greenhill S, Alber S, Oury TD, Choi AM, Morse D. Carbon monoxide suppresses bleomycin-induced lung fibrosis. Am J Pathol 166: 27–37, 2005. doi: 10.1016/S0002-9440(10)62229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zuckerbraun BS, Chin BY, Bilban M, de Costa d’Avila JC, Rao J, Billiar TR, Otterbein LE. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J 21: 1099–1106, 2007. doi: 10.1096/fj.06-6644com. [DOI] [PubMed] [Google Scholar]
- 198.Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S, Otterbein LE. Carbon monoxide reverses established pulmonary hypertension. J Exp Med 203: 2109–2119, 2006. doi: 10.1084/jem.20052267. [DOI] [PMC free article] [PubMed] [Google Scholar]