

Abstract
Increases in pulmonary arterial smooth muscle cell (PASMC) intracellular Ca2+ levels and enhanced RhoA/Rho kinase-dependent Ca2+ sensitization are key determinants of PASMC contraction, migration, and proliferation accompanying the development of hypoxic pulmonary hypertension. We previously showed that acid-sensing ion channel 1a (ASIC1a)-mediated Ca2+ entry in PASMC is an important constituent of the active vasoconstriction, vascular remodeling, and right ventricular hypertrophy associated with hypoxic pulmonary hypertension. However, the enhanced ASIC1a-mediated store-operated Ca2+ entry in PASMC from pulmonary hypertensive animals is not dependent on an increase in ASIC1a protein expression, suggesting that chronic hypoxia (CH) stimulates ASIC1a function through other regulatory mechanism(s). RhoA is involved in ion channel trafficking, and levels of activated RhoA are increased following CH. Therefore, we hypothesize that activation of RhoA following CH increases ASIC1a-mediated Ca2+ entry by promoting ASIC1a plasma membrane localization. Consistent with our hypothesis, we found greater plasma membrane localization of ASIC1a following CH. Inhibition of RhoA decreased ASIC1a plasma membrane expression and largely diminished ASIC1a-mediated Ca2+ influx, whereas activation of RhoA had the opposite effect. A proximity ligation assay revealed that ASIC1a and RhoA colocalize in PASMC and that the activation state of RhoA modulates this interaction. Together, our findings show a novel interaction between RhoA and ASIC1a, such that activation of RhoA in PASMC, both pharmacologically and via CH, promotes ASIC1a plasma membrane localization and Ca2+ entry. In addition to enhanced RhoA-mediated Ca2+ sensitization following CH, RhoA can also activate a Ca2+ signal by facilitating ASIC1a plasma membrane localization and Ca2+ influx in pulmonary hypertension.
Keywords: ASIC1 trafficking, calcium sensitization, pulmonary hypertension, store-operated calcium entry
INTRODUCTION
Acid-sensing ion channel (ASIC) 1a (ASIC1a) is a proton-gated cation channel that belongs to the amiloride-sensitive degenerin/epithelial Na+ channel (ENaC) superfamily. The influx of Na+/Ca2+ contributes to membrane depolarization, activation of Ca2+/calmodulin-dependent mechanisms, and other second-messenger pathways, signifying the unique role of these channels in intracellular signaling and excitability. Our laboratory recently showed that ASIC1a, via store-operated Ca2+ entry (SOCE), is an important facilitator of pulmonary vasoconstriction in response to alveolar hypoxia and G protein-coupled receptor signaling (22, 23, 35). Furthermore, we have described a prominent role of ASIC1a in the pathophysiology of hypoxic pulmonary hypertension (22, 35). Exposure to chronic hypoxia (CH) markedly alters intracellular Ca2+-handling pathways in pulmonary arterial smooth muscle cells (PASMC) and results in augmented basal Ca2+ levels, increased SOCE and receptor-operated Ca2+ entry, and enhanced RhoA/Rho kinase-mediated Ca2+ sensitivity, all of which contribute to increased vasoconstrictor reactivity in pulmonary hypertension (5, 7, 24, 29, 55). In addition to enhanced vascular reactivity, CH causes Ca2+-dependent morphological changes, including neomuscularization, thickening of the intimal, medial, and adventitial layers of pulmonary arteries, and angiogenesis (18, 48). ASIC1a-mediated Ca2+ entry in PASMC appears to be an important constituent of the active vasoconstriction, vascular remodeling, and right ventricular hypertrophy associated with hypoxic pulmonary hypertension (35).
The enhanced SOCE observed in hypoxic pulmonary hypertension correlates with an upregulation of several proposed SOCE components, including transient receptor potential cation channel (TRPC) 1 (TRPC1), TRPC4, TRPC6, Orai1, Orai2, and stromal interaction molecule 1 (29, 37, 46, 55). Interestingly, we have not observed the same correlation between increased ASIC1a-dependent Ca2+ influx and ASIC1a gene expression, which is unaltered by exposure to CH (22, 35). These data indicate that hypoxia does not regulate ASIC1a expression at the transcriptional level and suggest that the enhanced ASIC1a-dependent SOCE in distal pulmonary arteries from pulmonary hypertensive animals is not dependent on an increase in ASIC1a protein expression. Rather, CH may stimulate ASIC1a function through other regulatory mechanism(s), such as kinase modulation of channel activity or intracellular trafficking of ASIC1a.
When the role of ASIC1a in augmenting SOCE is considered, the number of functional ASIC1a channels at the plasma membrane is critical. Moreover, altered subcellular trafficking of ASIC1a plays an essential role in several pathological conditions (reviewed in Ref. 58). The small G protein Ras homolog gene family member A (RhoA) is known to regulate diverse cellular processes. These include, but are not limited to, cytoskeletal dynamics, actomyosin contractility, transcription, cell cycle progression, and trafficking of integral plasma membrane proteins such as ion channels. Notably, RhoA increases ENaC activity by increasing the plasma membrane levels (28, 31, 39, 40, 47). Although ENaC and ASIC are members of the same ion channel family, whether ASIC1a is regulated by RhoA/Rho kinase is unknown. Furthermore, our previous data and those of other investigators demonstrate that CH is associated with greater pulmonary vascular active GTP-bound RhoA levels and RhoA/Rho kinase activity (2, 6, 12, 13, 20, 24, 33, 36). Therefore, in the present study we tested the hypothesis that activation of RhoA following CH increases ASIC1a-mediated Ca2+ entry by promoting plasma membrane localization.
METHODS
Animals and CH Exposure Protocol
Male Wistar rats (∼12 wk old; Harlan Industries) were divided into two groups: rats designated for exposure to CH were housed in a hypobaric chamber with barometric pressure maintained at ∼380 mmHg for 4 wk, and age-matched control rats were housed at ambient barometric pressure (∼630 mmHg in Albuquerque, NM). ASIC1a wild-type (ASIC1a+/+) and knockout (ASIC1a−/−) mice (Jackson Laboratory) were bred on a C57BL/6 background. Male and female mice (∼12 wk old) were used equally for each protocol. Disruption of ASIC1a was confirmed by PCR and agarose gel electrophoresis using a three-primer system to detect both wild-type and disrupted alleles (35). All protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine and conform to the National Institutes of Health guidelines for animal use.
Generation of Primary PASMC Culture
Control and CH rats and ASIC1a+/+ and ASIC1a−/− mice were anesthetized with pentobarbital sodium (200 mg/kg ip), and the heart and lungs were removed by midline thoracotomy. Intrapulmonary arteries (∼2nd–5th order) were dissected from surrounding lung parenchyma and enzymatically digested in reduced-Ca2+ Hanks’ balanced salt solution (HBSS) containing papain (26 U/ml), type I collagenase (1,750 U/ml), dithiothreitol (1 mg/ml), and BSA (2 mg/ml) at 37°C for 30 min. Single smooth muscle cells were dispersed by gentle trituration with a fire-polished pipette in Ca2+-free HBSS. The cell suspension was plated in Ham's F-12 medium supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified atmosphere of 5% CO2-95% air at 37°C. For most experiments, PASMC were cultured for only 3–4 days, because longer culture duration or passage of cells resulted in loss of phenotype in PASMC from CH rats. For some control experiments in rats and mice, PASMC were passaged once to obtain enough cells for the experiment. Cellular purity was >95%, as assessed by morphological appearance under phase-contrast microscopy and immunofluorescence staining for smooth muscle 22α.
Determination of ASIC1 Plasma Membrane Expression
For total ASIC1 expression, control and CH primary PASMC (3–4 days of culture) were homogenized in 10 mM Tris·HCl homogenization buffer (255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 1 μM pepstatin A, and 0.3 μM aprotinin) and centrifuged at 10,000 g for 10 min at 4°C to remove insoluble debris. The PASMC lysate (10 μg) was separated by SDS-PAGE (7.5% Tris-glycine) and transferred to a polyvinylidene difluoride (PVDF) membrane. The blot was blocked for 1 h with 5% milk and incubated for 48 h at 4°C with rabbit anti-ASIC1 (1:500 dilution; catalog no. ab5674P, Millipore). We previously demonstrated the specificity of this antibody (23). For immunochemical labeling, blots were incubated for 1 h with goat anti-rabbit IgG-horseradish peroxidase (1:3,000 dilution; catalog no. 172-1019, Bio-Rad). After chemiluminescence labeling (ECL, Pierce), ASIC1 was detected by exposure of the blot to chemiluminescence-sensitive film (GeneMate). For analysis of ASIC1 bands, films were scanned using a transparency scanner (model 3200, Epson), and densitometric analysis of the scanned images was performed using ImageJ gel analysis (National Institutes of Health, Bethesda, MD). We normalized total ASIC1 expression to total protein per lane, because we have consistently observed that CH increases both mRNA and protein expression of typical “housekeeping” genes/proteins in pulmonary arterial homogenates and isolated PASMC from rats and mice. To determine total protein levels, PVDF membranes were stained with Coomassie Brilliant Blue R-250 (catalog no. 1610400, Bio-Rad). Densitometric analysis of total lane protein from scanned images was performed using ImageJ gel analysis.
To determine plasma membrane localization of ASIC1, we used a cell surface protein isolation kit (Pierce, Thermo Fisher Scientific). Control and CH primary PASMC were grown for 3–4 days until ~90% confluent. PASMC were incubated with sulfo-NHS-SS-biotin for 30 min at 4°C. The reaction was quenched, and cells were harvested and lysed with 10 mM Tris·HCl homogenization buffer and centrifuged at 10,000 g for 2 min. Clarified supernatant was added to NeutrAvidin agarose resin columns for 1 h at room temperature. The flow-through was collected as the cytosolic protein fraction, and surface protein was collected by elution with 5× sample buffer. We previously demonstrated the specificity of cell surface assay to fractionate cell surface vs. intracellular proteins (17). Surface protein (25 µl) or cytosolic protein (20 µg) lysates were separated by SDS-PAGE (7.5% Tris-glycine) and transferred to PVDF membranes. ASIC1 was detected in cell surface and cytosolic fractions by exposure of the blot to chemiluminescence-sensitive film (GeneMate). Quantification of ASIC1 bands was accomplished by densitometric analysis of scanned images (ImageJ gel analysis) and expressed as the ratio of plasma membrane to cytosolic densitometric units.
In some experiments, we determined the effect of RhoA and Rho kinase on ASIC1 plasma membrane expression by pretreating PASMC with Rho inhibitor I (1 μg/ml), Rho activator II (1 μg/ml), or the Rho kinase inhibitor Y-27632 (10 μM; Cayman Chemical). Rho inhibitor I (catalog no. CT04, Cytoskeleton), the exoenzyme C3 transferase from Clostridium botulinum (active site), covalently attached to a proprietary cell-penetrating moiety, specifically inhibits RhoA, RhoB, and RhoC proteins. Rho activator II (catalog no. CN03, Cytoskeleton) activates Rho GTPase activity and increases the level of GTP-bound RhoA by use of the catalytic domain of the bacterial cytotoxic necrotizing factor toxins and is covalently attached to a proprietary cell-penetrating moiety. Rho inhibitor I and Rho activator II were added directly to basal (no FBS) medium, and PASMC were incubated for 3 h, after which equal amounts of 10% FBS-containing medium were added to the treatments overnight (~16 h) to obtain 5% FBS and 0.5 µg/ml inhibitor/activator. Cells were incubated with Y-27632 overnight (~16 h) in medium.
Assessment of ASIC1-RhoA Colocalization
Protein-protein (ASIC1-RhoA) interactions in PASMC were determined by proximity ligation assay (PLA; Duolink in situ PLA kit, Olink Biosciences, Sigma Aldrich), as previously described (16, 17). Briefly, PASMC were plated on 18-well slides (Ibidi) and grown until ~75% confluent. In some experiments, PASMC were pretreated with Rho inhibitor I (1 μg/ml) or Rho activator II (1 μg/ml) for 16 h, as described above, before the cells were fixed with 2% paraformaldehyde to assess the effect of RhoA inhibition and activation on its interaction with ASIC1. PASMC were incubated for 30 min at 37°C with blocking buffer and then overnight with goat anti-ASIC1 (1:50 dilution; catalog no. sc-13903, Santa Cruz Biotechnology) and rabbit anti-RhoA (1:50 dilution; catalog no. 21009, New East Biosciences). Using wild-type and knockout mice, we previously determined the specificity of goat anti-ASIC1 (35). PASMC were incubated with anti-rabbit (+) and anti-goat (−) PLA probes (1:5 dilution) for 1 h at 37°C. Negative controls were completed by 1) omission of primary antibody and 2) incubation of each primary antibody individually. In addition, to show specificity of the Duolink signal, we performed this assay in PASMC from ASIC1a+/+ and ASIC1a−/− mice. Samples were amplified with Duolink orange in situ detection reagent (554/579-nm excitation/emission; Sigma Aldrich) for 100 min at 37°C. SYTOX green (1:5,000 dilution; Invitrogen) was used as a nuclear stain, and actin was stained with Alexa Fluor 647-phalloidin (1:100 dilution; Invitrogen). Samples were mounted with Duolink mounting medium, and z-stack images of the PLA interaction were acquired using a confocal microscope (model TCS SP5, Leica). Each dot was considered a positive protein-protein interaction. Using ImageJ (National Institutes of Health), we determined the number and size (pixel2) of dots per cell from five images per well.
RhoA and Rho Kinase Regulation of ASIC1a-Mediated Ca2+ Influx
PASMC were incubated with fura 2-AM (2 μM) and 0.05% pluronic acid in physiological saline solution (PSS; Molecular Probes) for 30 min at 32°C. Fura 2-loaded PASMC were alternately excited at 340 and 380 nm at a frequency of 1 Hz with a dual-excitation light source (HyperSwitch, IonOptix), and the respective 510-nm emissions were detected with a photomultiplier tube. PASMC were superfused (5 ml/min at 37°C) with HEPES-based PSS (in mM: 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 1.8 CaCl2, 6 glucose, and 3 EGTA; pH adjusted to 7.4 with NaOH).
Basal intracellular Ca2+ concentration.
To determine basal intracellular Ca2+ concentration ([Ca2+]i), we used the mathematical relationship between the measured fura 2 ratio (R) and Ca2+ concentration ([Ca]) as follows: [Ca] = Kd × (R − Rmin)/(Rmax − R) × Sf2/Sb2. A value of 225 nM was used for the dissociation constant (Kd) for the fura 2-Ca binding. Rmax and Rmin represent the ratio values, and Sb2 (bound) and Sf2 (free) are proportional to the fluorescence excited by the 380-nm wavelength measured under conditions of saturating Ca2+ levels and in the absence of Ca2+, respectively. These calibration constants were determined following each experiment with use of ionomycin (3 µM; Calbiochem) to obtain saturated and zero Ca2+ levels.
Acid-induced Ca2+ entry.
ASIC1a is known to be activated by protons or a drop in extracellular pH. We previously showed the presence of acid-induced currents in PASMC (23). To determine the effect of RhoA on ASIC1a-mediated acid-induced Ca2+ entry, changes in [Ca2+]i were measured when pH of the superfusion buffer was switched from 7.4 to 6.0 in the absence or presence of Rho inhibitor I (1 µg/ml) or Rho activator II (1 µg/ml), as described above. Area under the curve was calculated as previously described (38). Some experiments were additionally conducted in the presence or absence of the ASIC1a inhibitor psalmotoxin (PcTX1, 20 nM; Phoenix Peptides) by incubation of fura 2-loaded PASMC with PcTX1 for 30 min before the acid-induced response.
Store-operated Ca2+ entry.
To determine the effect of RhoA and Rho kinase on ASIC1a-mediated SOCE, experiments were conducted in PASMC, as described previously (17, 38), in the absence or presence of Rho inhibitor I (1 μg/ml), Rho activator II (1 μg/ml), or the Rho kinase inhibitor Y-27632 (10 μM), as described above. After basal fura 2 ratios were determined, PASMC were superfused (5 ml/min at 37°C) with Ca2+-free HEPES-based PSS (in mM: 130 NaCl, 4 KC1, 1.2 MgSO4, 4 NaHCO3, 10 HEPES, 1.18 KH2PO4, 6 glucose, and 3 EGTA; pH adjusted to 7.4 with NaOH) containing 50 μM diltiazem to prevent Ca2+ entry through L-type voltage-gated Ca2+ channels. We previously demonstrated that this concentration of diltiazem blocks the increase in [Ca2+]i and vasoconstrictor response to a depolarizing concentration of KCl (50 mM) (21). In addition, arteries/cells were incubated with the sarco/endoplasmic reticulum Ca2+-ATPase inhibitor cyclopiazonic acid (10 μM) to deplete intracellular Ca2+ stores and prevent Ca2+ reuptake. Changes in [Ca2+]i were determined upon repletion of HEPES-based PSS containing 1.8 mM CaCl2 in the continued presence of diltiazem and cyclopiazonic acid. Area under the curve was calculated as previously described (38). Some experiments were additionally conducted in the presence or absence of the ASIC1a inhibitor PcTX1 (20 nM) by incubation of fura 2-loaded PASMC with PcTX1 for 30 min before the SOCE response.
Determination of GTP-Bound Activated RhoA Levels
The effect of CH and ASIC1a on active GTP-bound RhoA levels was assessed using a configuration-specific monoclonal antibody-based RhoA activation assay kit according to the manufacturer’s instructions (New East Biosciences). Intrapulmonary arteries (∼2nd–5th order) from control and CH rats were dissected from surrounding lung parenchyma and treated with vehicle or the ASIC1a inhibitor PcTX1 (20 nM) for 30 min at 37°C. Arteries were then homogenized in 10 mM Tris·HCl homogenization buffer (255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 1 μM pepstatin A, and 0.3 μM aprotinin) and centrifuged at 10,000 g for 2 min. Clarified supernatant (10 μg) was incubated with anti-active RhoA monoclonal antibody (1 µl; catalog no. 26904, New East Biosciences) and A/G agarose bead slurry (20 μl) for 1 h at 4°C. Samples were eluted with 2× sample buffer, and immunoprecipitated samples (15 µl) were separated by SDS-PAGE (12% Tris-glycine), transferred to PVDF membranes, and incubated for 4 h at 4°C with rabbit anti-RhoA (1:500 dilution; catalog no. 21009, New East Biosciences). Total protein lysates (25 µg) were separated by SDS-PAGE (12% Tris-glycine) and incubated for 24 h at 4°C with rabbit anti-RhoA (1:5,000 dilution; catalog no. ab187027, Abcam). For immunochemical labeling, blots were incubated with goat anti-rabbit IgG-horseradish peroxidase (1:3,000 or 1:1,000 dilution; Bio-Rad) for 1 h. After chemiluminescence labeling (ECL, Pierce), RhoA was detected by exposure of the blot to chemiluminescence-sensitive film (GeneMate).
Calculations and Statistics
Each individual data point is plotted within the bar graphs, which represent means ± SE; n is the number of animals in each group unless otherwise stated. Statistical significance was tested at the 95% (P < 0.05) confidence level using an unpaired t-test, one-way analysis of variance (ANOVA), or two-way ANOVA. If differences were detected by ANOVA, individual groups were compared with the Student-Newman-Keuls test.
RESULTS
ASIC1 Plasma Membrane Localization Is Greater in PASMC From CH Rats
ASIC1 expression in primary PASMC (3–4 days of culture) homogenates is similar in control and CH rats (Fig. 1, A and B). However, when examining the plasma membrane vs. cytosolic fractions, we found significantly greater plasma membrane localization of ASIC1 in PASMC from CH than control animals cultured under the same conditions (Fig. 1, C and D).
Fig. 1.

Chronic hypoxia (CH) increases acid-sensing ion channel 1 (ASIC1) plasma membrane localization in pulmonary arterial smooth muscle cells (PASMC). A and B: representative Western blot (WB) for total protein homogenate and corresponding Coomassie-stained blot and summary data detecting total ASIC1 expression (normalized to the entire lane on the Coomassie-stained blot) in PASMC from control and CH rats. C and D: representative Western blots for biotinylated (plasma membrane) and cytosolic protein and summary data showing the ratio of biotinylated to cytosolic ASIC1 expression in PASMC from control and CH rats. MW, molecular mass (kDa). ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P ≤ 0.05 vs. control (by unpaired t-test).
RhoA Regulates ASIC1 Plasma Membrane Localization in PASMC From Control and CH Rats
Although RhoA has been shown to regulate ENaC surface expression (39, 47), whether it affects ASIC1 trafficking is unknown. To determine the effect of RhoA and Rho kinase on ASIC1 plasma membrane expression, cultured PASMC (passage 1) from control animals were pretreated with Rho inhibitor I, Rho activator II, or the Rho kinase inhibitor Y-27632. Rho inhibitor I significantly diminished ASIC1 expression at the plasma membrane, with a reciprocal increase in cytosolic ASIC1 expression (Fig. 2A). Compared with vehicle, the ratio of plasma membrane to cytosolic ASIC1 expression was largely diminished in the presence of Rho inhibitor I and augmented in the presence of Rho activator II (Fig. 2B). In contrast, inhibition of Rho kinase with Y-27632 did not significantly alter ASIC1 localization in PASMC (Fig. 2B). The increase in the ratio of plasma membrane to cytosolic ASIC1 expression in primary PASMC (3–4 days of culture) from CH rats was also prevented by Rho inhibitor I (Fig. 2, C and D), suggesting that RhoA is essential for ASIC1 membrane localization.
Fig. 2.

RhoA stimulates ASIC1 plasma membrane localization in PASMC from control and CH rats. Representative Western blots for biotinylated (plasma membrane) and cytosolic (A and C) protein and summary data (B and D) showing the ratio of biotinylated to cytosolic ASIC1 expression in PASMC from control (Con, B) and CH (D) rats following treatment with Rho inhibitor I (1 µg/ml), Rho activator II (1 µg/ml), and the Rho kinase inhibitor Y-27632 (10 µM). Con vehicle and CH vehicle (D) are the same samples shown in Fig. 1D. ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P < 0.05 vs. control; #P < 0.05 vs. respective vehicle [by 1-way ANOVA and Student-Newman-Keuls test (for individual group comparisons)].
Activation of RhoA Promotes Colocalization With ASIC1
To demonstrate the specificity of the PLA, we examined colocalization of ASIC1 and RhoA in PASMC from ASIC1+/+ and ASIC1−/− mice. Whereas we observed distinct puncta in PASMC from ASIC1+/+ mice, PASMC from ASIC1−/− mice lack a PLA signal (Fig. 3A). In rat PASMC, Rho activator II significantly increased the average number of puncta per cell but did not change the size of the puncta (Fig. 3, B and C) as assessed by Duolink PLA. Rho inhibitor I decreased the average number of puncta per cell and the size of the puncta (Fig. 3, D and E), suggesting diminished colocalization and clustering, respectively, of ASIC1 and RhoA when RhoA is inhibited.
Fig. 3.
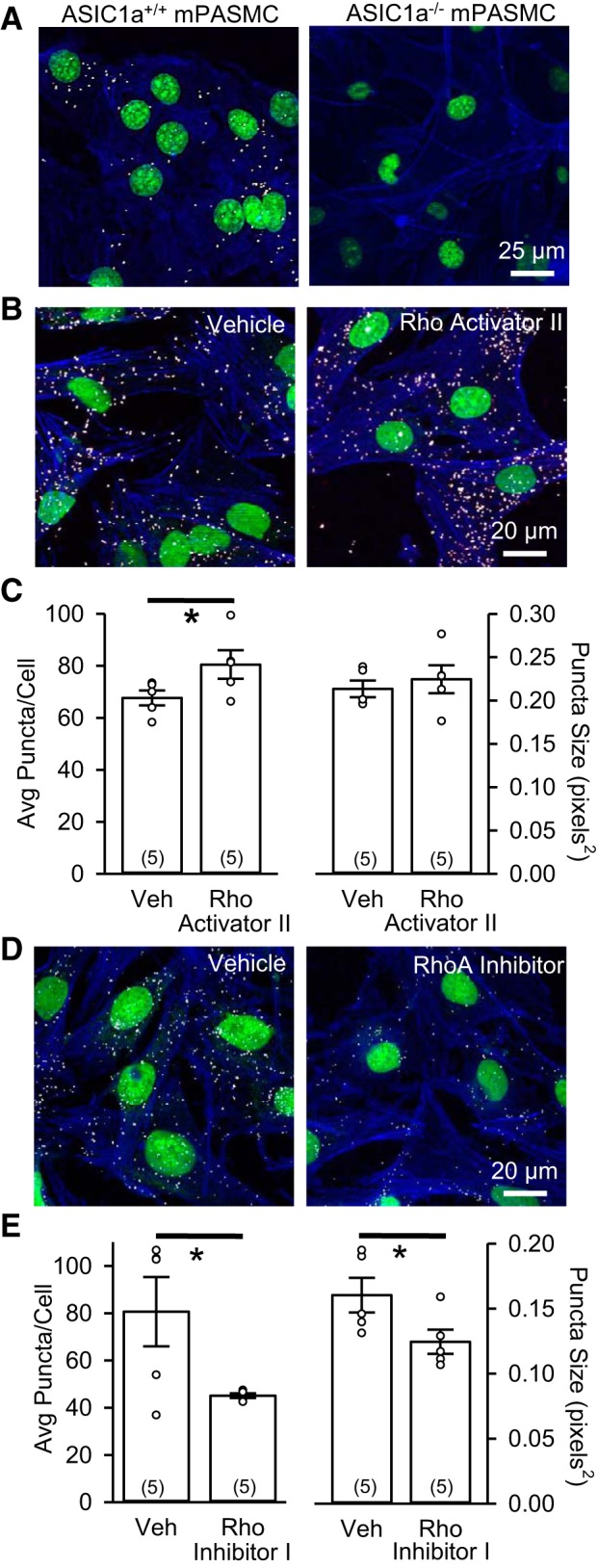
ASIC1 and RhoA colocalize in PASMC. A: specificity of proximity ligation assay (PLA) as demonstrated by ASIC1-RhoA interaction (white puncta) in PASMC from ASIC1 wild-type (ASIC1+/+) and ASIC1 knockout (ASIC1−/−) mice (mPASMC). B−E: representative confocal images and summary data showing Duolink PLA of the interaction between goat anti-ASIC1 and rabbit anti-RhoA (white puncta) in rat PASMC upon incubation with vehicle (Veh), Rho activator II, or Rho inhibitor I. Summary data are shown for average number of puncta per cell and puncta size. Actin is labeled with Alexa Fluor 647-phalloidin (blue), and nuclei are labeled with SYTOX (green). ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P < 0.05 vs. Veh (by unpaired t-test).
RhoA Facilitates ASIC1-Mediated Ca2+ Influx in PASMC
To determine if RhoA alters the function of ASIC1a in PASMC, we examined two different stimuli for ASIC1a: acid and depletion of intracellular Ca2+ stores. Decreasing pH to 6.0 resulted in a transient increase in [Ca2+]i in rat and mouse PASMC (Fig. 4). Treatment with Rho inhibitor I attenuated (Fig. 4, A and B, middle) and Rho activator II augmented (Fig. 4, A and B, right), acid-induced increases in [Ca2+]i in rat PASMC. Inhibition of ASIC1a with the pharmacological inhibitor PcTX1 (Fig. 4, A and B) largely inhibited acid-induced increases in [Ca2+]i in all groups. Similarly, Rho inhibitor I reduced (Fig. 4, C and D, middle) and Rho activator II augmented (Fig. 4, C and D, right) acid-induced increases in [Ca2+]i in PASMC from ASIC1+/+ mice, but neither had an effect on blunted acid-induced responses in PASMC from ASIC1−/− mice (Fig. 4, C and D).
Fig. 4.

RhoA facilitates ASIC1a-dependent acid-induced responses in PASMC. A and B: representative traces and summary data showing acid-induced (pH 6.0) increases in intracellular Ca2+ concentration ([Ca2+]i) in rat PASMC following treatment with vehicle, Rho inhibitor I, or Rho activator II in the absence (−) or presence (+) of psalmotoxin (PcTX1). C and D: representative traces and summary data showing acid-induced (pH 6.0) increases in [Ca2+]i in PASMC from ASIC1a+/+ and ASIC1a−/− mice following treatment with vehicle, Rho inhibitor I, or Rho activator II. F, fluorescence units; AUC, area under the curve. ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P < 0.05 vs. (−)PcTX1 or ASIC1a+/+; #P < 0.05 vs. respective vehicle [by 2-way ANOVA and Student-Newman-Keuls test (for individual group comparisons)].
We previously demonstrated that ASIC1a contributes to SOCE in PASMC (22, 23, 35, 38). Consistent with these previous findings, inhibition of ASIC1a with the pharmacological inhibitor PcTX1 (Fig. 5, A and B, left) or genetic knockout (Fig. 5, C and D, left) largely inhibited SOCE. After treatment with Rho inhibitor I, SOCE was reduced in rat PASMC, and PcTX1 did not reduce SOCE beyond the level achieved by treatment with Rho inhibitor I alone (Fig. 5, A and B, middle). PcTX1 had a similar effect to reduce the augmented SOCE in rat PASMC treated with Rho activator II (Fig. 5, A and B, right). Furthermore, Rho inhibitor I decreased (Fig. 5, C and D, middle) and Rho activator II increased (Fig. 5, C and D, right) SOCE in PASMC from ASIC1+/+ mice, but neither had an effect on SOCE responses in PASMC from ASIC1−/− mice (Fig. 5, C and D). These data suggest that modulation of acid-induced responses and SOCE by RhoA is ASIC1a-dependent and consistent with the effect of RhoA on ASIC1 plasma membrane expression.
Fig. 5.

RhoA facilitates ASIC1a-mediated store-operated Ca2+ entry (SOCE) in PASMC. A and B: representative traces and summary data showing SOCE responses in rat PASMC following treatment with vehicle, Rho inhibitor I, or Rho activator II in the absence (−) or presence (+) of PcTX1. C and D: representative traces and summary data showing SOCE responses in PASMC from ASIC1a+/+ and ASIC1a−/− mice following treatment with vehicle, Rho inhibitor I, or Rho activator II. ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P < 0.05 vs. (−)PcTX1 or ASIC1a+/+; #P < 0.05 vs. respective vehicle [by 2-way ANOVA and Student-Newman-Keuls test (for individual group comparisons)].
CH-Mediated Increases in SOCE Are RhoA-Dependent but Rho Kinase-Independent
Basal [Ca2+]i levels were significantly higher in PASMC from CH than control rats (Fig. 6A). Treatment with Rho inhibitor I, Rho activator II, or Y-27632 did not significantly alter basal [Ca2+]i levels in PASMC from control or CH rats (Fig. 6A).
Fig. 6.
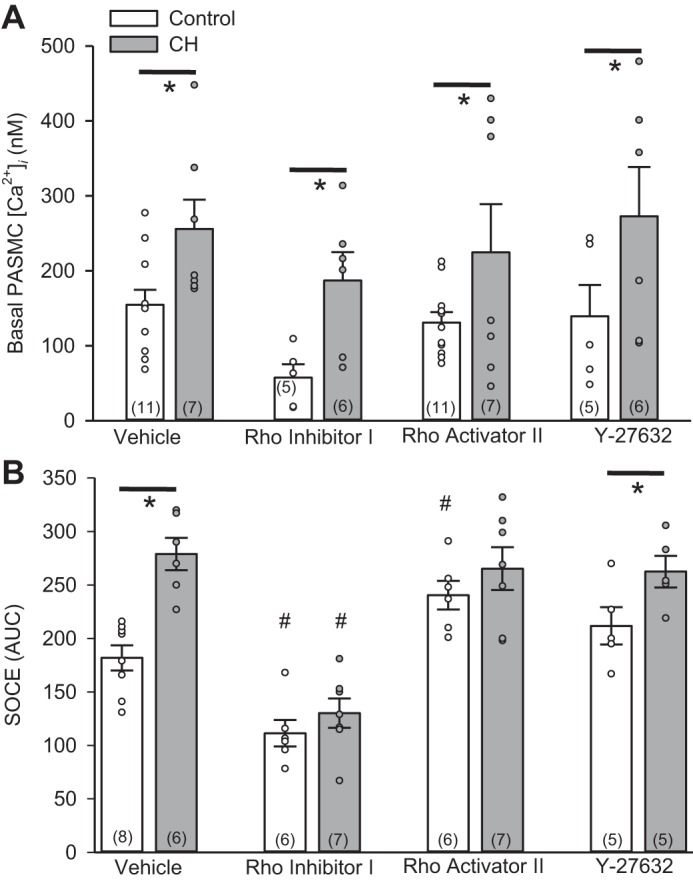
CH-mediated increase in SOCE is RhoA-dependent, but Rho kinase-independent. A and B: basal [Ca2+]i and SOCE responses in the presence of vehicle, Rho inhibitor I, Rho activator II, or the Rho kinase inhibitor Y-27632 in PASMC from control or CH rats. ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P < 0.05 vs. control; #P < 0.05 vs. respective vehicle [by 2-way ANOVA and Student-Newman-Keuls test (for individual group comparisons)].
SOCE was largely augmented in PASMC from CH rats (Fig. 6B). Rho inhibitor I reduced SOCE in PASMC from control and CH rats and normalized responses between groups. In contrast, Rho activator II increased SOCE in PASMC from control rats but did not further augment SOCE in PASMC from CH rats. The Rho kinase inhibitor Y-27632 did not significantly alter SOCE in PASMC from control or CH rats. These data suggest that RhoA, but not Rho kinase, is an important facilitator of SOCE in PASMC.
Inhibition of ASIC1 Does Not Influence CH-Induced Activation of RhoA in Isolated Pulmonary Arteries
RhoA can be activated by increases in [Ca2+]i following receptor stimulation (42, 43, 54); therefore, we determined whether ASIC1a-mediated Ca2+ entry leads to activation of RhoA by examining GTP-bound RhoA levels in the presence of PcTX1. Consistent with our previous data (6, 24), CH increases the levels of activated RhoA (Fig. 7A) without increasing total RhoA (Fig. 7B). PcTX1 had no effect on GTP-bound RhoA levels in PASMC from control or CH rats (Fig. 7C), suggesting that ASIC1a does not alter activation of RhoA.
Fig. 7.
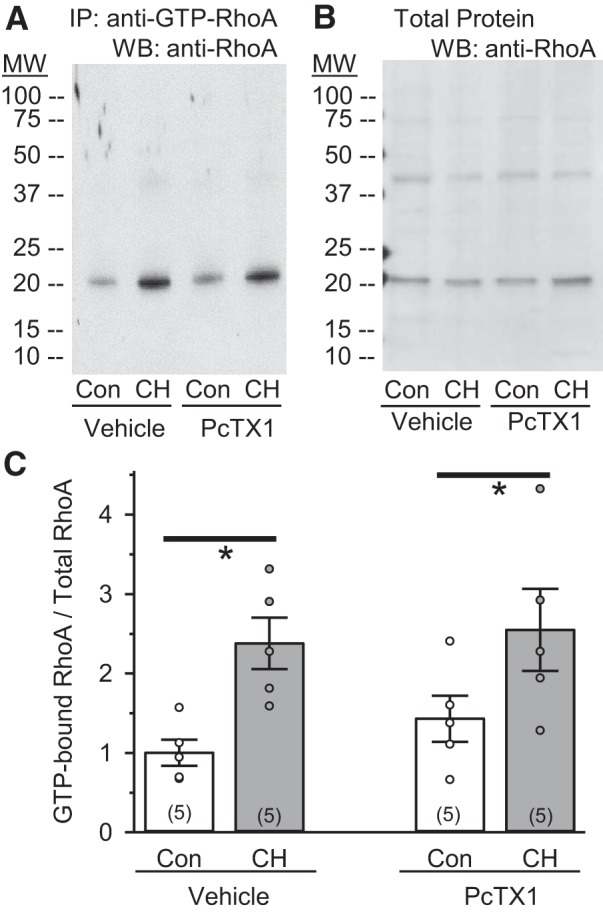
Inhibition of ASIC1a does not influence CH-induced activation of RhoA in isolated pulmonary arteries. A and B: representative images of GTP-bound activated RhoA (~22 kDa) and total RhoA (~22 kDa) in intrapulmonary arterial homogenates from control (Con) and CH rats preincubated in the presence of vehicle or PcTX1. IP, immunoprecipitation. C: summary data showing expression of GTP bound to total RhoA. ○, Individual data points; bars, means ± SE of number of animals shown in parentheses. *P ≤ 0.05 vs. corresponding Con [by 2-way ANOVA and Student-Newman-Keuls test (for individual group comparisons)].
DISCUSSION
The increase in PASMC [Ca2+]i associated with hypoxic pulmonary hypertension is an important determinant of contraction, migration, and proliferation. We previously showed that the enhanced ASIC1a-dependent SOCE in distal pulmonary arteries from pulmonary hypertensive animals is not dependent on an increase in ASIC1 protein expression (35). The goal of this study was to determine whether CH increases SOCE by stimulating plasma membrane localization of ASIC1. Although total ASIC1 expression is not different between PASMC from control rats and PASMC from CH rats, our findings demonstrate greater plasma membrane localization of ASIC1 following CH. RhoA is important to integral membrane protein trafficking, and levels of activated RhoA are increased following CH. Therefore, we next determined the contribution of RhoA to ASIC1 plasma membrane localization. Inhibition of RhoA decreased ASIC1 plasma membrane expression and largely diminished ASIC1a-mediated acid-induced Ca2+ influx and SOCE, whereas activation of RhoA had the opposite effect. Furthermore, we show that ASIC1 and RhoA colocalize in PASMC and that this colocalization is dependent on the activation state of RhoA. In addition to enhanced RhoA-mediated Ca2+ sensitization of the contractile apparatus following CH, the findings from the current study demonstrate that RhoA can also activate a Ca2+ signal by facilitating ASIC1a plasma membrane localization and Ca2+ influx in pulmonary hypertensive animals.
There are several possibilities by which CH may lead to enhanced ASIC1a-dependent SOCE. First, CH could upregulate ASIC1 expression. In mice, neither ASIC1 protein expression nor mRNA levels were different in pulmonary arteries from normoxic and CH mice (35). In rats, mRNA expression is unchanged during the course of CH exposure; however, we observed an increase in ASIC1 protein expression in isolated pulmonary arterial lysates from 4-wk-old CH rats (22). Since multiple cell types are present in the isolated arterial lysate, it is unclear whether this reflects a change specifically in PASMC ASIC1 expression. Our current findings show that that total ASIC1 expression is not different in PASMC from control and CH rats; however, ASIC1a-dependent SOCE responses are largely augmented. These data strongly suggest that an increase in ASIC1 expression is not required for enhanced ASIC1a-mediated SOCE following CH and have led to the current investigation of alternative possibilities by which CH increases ASIC1a activity. Through direct association or protein modification, various effector proteins and/or signaling molecules have been shown to regulate the function of ASIC1a (58) by altering 1) the kinetics of the channel, 2) the coupling to up- or downstream effectors, and/or 3) the proportion of ASIC1a at the membrane. Consistent with the latter, our current data suggest that CH promotes ASIC1a plasma membrane localization. However, despite advances in identification of these signaling molecules, the molecular mechanism(s) that govern trafficking of ASIC1a remains largely unknown.
Our previous investigations examined the interaction between ASIC1 and the PDZ domain-containing protein interacting with C kinase-1 (PICK1) (16, 17). The COOH terminus of ASIC1 shares homology with type II PDZ-binding motifs and binds the scaffolding protein PICK1 (11, 19). In addition to the PDZ domain, PICK1 contains a larger BAR (Bin/amphiphysin/Rvs) domain that directly binds to lipids and is important in membrane localization (26). Although the interaction between PICK1 and ASIC1 has been shown to increase surface expression of ASIC1 in other cells (11, 19, 26), inhibition of PICK1 did not change plasma membrane expression of ASIC1 in PASMC under basal conditions (17). We did, however, find that PICK1 is an important scaffolding protein that facilitates modulation of ASIC1 by PKA, PKC, and calcineurin (17). Furthermore, PICK1 is essential for CH-induced, ASIC1-dependent activation of the nuclear factor of activated T cells isoform c3 transcription factor (16).
In addition to PICK1, annexin II light chain p11 (also known as S100A10) has also been associated with ASIC1 surface expression (10). Coexpression of p11 and ASIC1 in CHO-K1 cells led to a twofold increase in plasma membrane expression of ASIC1 and ASIC1-dependent currents (10). The p11 adaptor protein forms a complex with annexin II (52) and together have been shown to regulate the trafficking of several ion channels, including volume-activated Cl− channels, TASK-1 background K+ channels, voltage-gated Na+ channels, and vanilloid TRPC5 and TRPC6 (15, 34, 41, 53). Annexin II is also an actin-binding protein involved in recruitment of various members of the Rho family of small GTPases, including RhoA, to the actin cytoskeleton (1, 3, 14). RhoA localizes to the plasma membrane, as well as to other intracellular membrane compartments, where it is recognized to play a regulatory role in cytoskeletal rearrangement (44). In addition to, or possibly because of, effects on the cytoskeleton, RhoA plays a major role in membrane and vesicle trafficking of several ion channels (8, 27, 32, 39, 40, 49, 50).
Although there is little prior evidence linking RhoA with ASIC1a, several studies have shown that small GTPases, including RhoA, are involved in the regulation of both gating and localization of the related family member ENaC to the apical plasma membrane in epithelial cells (28, 31, 39, 40, 47). Moreover, an intact cytoskeleton appears to be required for RhoA-dependent translocation and activation of ENaC (28). The current study suggests that RhoA similarly regulates ASIC1a localization at the plasma membrane in PASMC. Furthermore, this appears to be an effect of RhoA, per se, and not the downstream target Rho kinase. However, important questions remain unanswered regarding the mechanism of action of RhoA on ASIC1: does RhoA promote ASIC1 trafficking to the plasma membrane or, rather, impede internalization of the channel from the membrane? Moreover, the contribution of RhoA-dependent cytoskeletal rearrangement on ASIC1 membrane localization remains undefined. Our current findings demonstrate specific colocalization between ASIC1 and RhoA that increases and decreases by activation and inhibition of RhoA, respectively. These data support a direct interaction between ASIC1 and RhoA; however, we cannot rule out the role of cytoskeletal dynamics in this response.
RhoA also interacts with and regulates TRPC1-mediated SOCE. In epithelial cells, RhoA has been shown to physically associate with TRPC1 and enhance SOCE and epithelial restoration after wounding (9). In endothelial cells, inactivation of RhoA diminishes actin polymerization and assembly and surface expression of the TRPC1/inositol 1,4,5-trisphosphate receptor complex and decreases SOCE (32). Considering that TRPC1 has also been shown to mediate SOCE in PASMC and play a role in the development of pulmonary hypertension (30, 51, 56), these studies suggest that RhoA may have other targets as well. Our current findings show that inhibition of RhoA reduces RhoA-ASIC1 colocalization, decreases ASIC1 plasma membrane expression, and diminishes SOCE; however, our findings do not preclude a potential role for RhoA to regulate other SOCE components in PASMC. How ASIC1 contributes to the SOCE response and its interactions with other known mediators of SOCE, such as stromal interaction molecule 1, TRPC1, or Orai1, is of interest and an area we are currently investigating.
To further investigate the effect of RhoA on ASIC1a, we examined the conventional activation of ASIC1a by assessing acid-induced changes in [Ca2+]i in PASMC. We observed a similar effect of Rho inhibitor I to reduce and Rho activator II to augment acid-induced increases in [Ca2+]i, supporting the findings that RhoA is indeed modulating ASIC1a function as opposed to other components of the SOCE complex. It is evident from previous data examining whole cell current in PASMC (23) and the present data assessing [Ca2+]i that activation of ASIC1a by acid is markedly distinct from ASIC1a activation by store depletion and/or a G protein-coupled agonist. Whereas acid induces a transient response, store depletion results in a more sustained activation of ASIC1a. Although the conventional activation of ASICs is widely known to be extracellular acidosis, the current studies support reports that ASICs can be regulated by various nonproton ligands, effector proteins, and signaling molecules independent of acidosis (57, 58).
In addition to augmented PASMC [Ca2+]i levels, CH is also associated with a RhoA/Rho kinase-mediated increase in Ca2+ sensitivity of the contractile apparatus (2, 6, 12, 13, 20, 24, 33, 36). Rho kinase plays prominent roles in mediating Ca2+ sensitivity and pulmonary vasoconstriction not only by inhibiting myosin light chain phosphatase but also by promoting actin polymerization and dynamic reorganization of the cytoskeleton, which leads to increased force generation for a given level of Ca2+. Whereas this enhanced Ca2+ sensitization following CH is dependent on Rho kinase, our data would suggest that subcellular trafficking of ASIC1a occurs specifically through RhoA, and not Rho kinase. It is also possible that the Ca2+ signal mediated by ASIC1a activates the RhoA/Rho kinase pathway (42, 43, 54). However, our findings that inhibition of ASIC1a did not alter the activation state of RhoA are consistent with RhoA acting upstream, rather than downstream, of ASIC1a.
RhoA and Rho kinase mediate a broad range of cellular responses, such as stress fiber formation, cell proliferation, migration, contraction, and apoptosis (4, 45). In smooth muscle, most attention has focused on the RhoA and Rho kinase inhibitory effect on myosin light chain phosphatase, leading to increased Ca2+ sensitization and contraction. However, emerging evidence suggests that RhoA and Rho kinase can additionally regulate smooth muscle contraction by controlling the activities of ion channels (25). Our findings show a novel interaction between RhoA and ASIC1a in which activation of RhoA in PASMC promotes ASIC1a plasma membrane localization and increased SOCE. These studies demonstrate the crucial importance of ASIC1a subcellular localization, rather than altered expression, in determining cellular signaling and function. Furthermore, these findings expand our mechanistic understanding of the vital role of RhoA in the development of hypoxic pulmonary hypertension.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL-111084 (to N. L. Jernigan) and R01 HL-132883 (to T. C. Resta).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.M.H. and N.L.J. performed experiments; L.M.H. and N.L.J. analyzed data; L.M.H., T.C.R., and N.L.J. interpreted results of experiments; L.M.H. and N.L.J. prepared figures; L.M.H. and N.L.J. drafted manuscript; L.M.H., T.C.R., and N.L.J. edited and revised manuscript; L.M.H., T.C.R., and N.L.J. approved final version of manuscript; T.C.R. and N.L.J. conceived and designed research.
REFERENCES
- 1.Babbin BA, Parkos CA, Mandell KJ, Winfree LM, Laur O, Ivanov AI, Nusrat A. Annexin 2 regulates intestinal epithelial cell spreading and wound closure through Rho-related signaling. Am J Pathol 170: 951–966, 2007. doi: 10.2353/ajpath.2007.060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barman SA. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves Rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol 293: L472–L479, 2007. doi: 10.1152/ajplung.00101.2006. [DOI] [PubMed] [Google Scholar]
- 3.Benaud C, Le Dez G, Mironov S, Galli F, Reboutier D, Prigent C. Annexin A2 is required for the early steps of cytokinesis. EMBO Rep 16: 481–489, 2015. doi: 10.15252/embr.201440015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 348: 241–255, 2000. doi: 10.1042/bj3480241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonnet S, Belus A, Hyvelin J-M, Roux E, Marthan R, Savineau J-P. Effect of chronic hypoxia on agonist-induced tone and calcium signaling in rat pulmonary artery. Am J Physiol Lung Cell Mol Physiol 281: L193–L201, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010. doi: 10.1152/ajplung.00276.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broughton BRS, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 8.Cheng J, Wang H, Guggino WB. Regulation of cystic fibrosis transmembrane regulator trafficking and protein expression by a Rho family small GTPase TC10. J Biol Chem 280: 3731–3739, 2005. doi: 10.1074/jbc.M410026200. [DOI] [PubMed] [Google Scholar]
- 9.Chung HK, Rathor N, Wang SR, Wang J-Y, Rao JN. RhoA enhances store-operated Ca2+ entry and intestinal epithelial restitution by interacting with TRPC1 after wounding. Am J Physiol Gastrointest Liver Physiol 309: G759–G767, 2015. doi: 10.1152/ajpgi.00185.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donier E, Rugiero F, Okuse K, Wood JN. Annexin II light chain p11 promotes functional expression of acid-sensing ion channel ASIC1a. J Biol Chem 280: 38666–38672, 2005. doi: 10.1074/jbc.M505981200. [DOI] [PubMed] [Google Scholar]
- 11.Duggan A, Garcia-Anoveros J, Corey DP. The PDZ domain protein PICK1 and the sodium channel BNaC1 interact and localize at mechanosensory terminals of dorsal root ganglion neurons and dendrites of central neurons. J Biol Chem 277: 5203–5208, 2002. doi: 10.1074/jbc.M104748200. [DOI] [PubMed] [Google Scholar]
- 12.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 13.Gao Y, Portugal AD, Negash S, Zhou W, Longo LD, Usha Raj J. Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high altitude hypoxia. Am J Physiol Lung Cell Mol Physiol 292: L678–L684, 2007. doi: 10.1152/ajplung.00178.2006. [DOI] [PubMed] [Google Scholar]
- 14.Garrido-Gómez T, Dominguez F, Quiñonero A, Estella C, Vilella F, Pellicer A, Simon C. Annexin A2 is critical for embryo adhesiveness to the human endometrium by RhoA activation through F-actin regulation. FASEB J 26: 3715–3727, 2012. doi: 10.1096/fj.12-204008. [DOI] [PubMed] [Google Scholar]
- 15.Girard C, Tinel N, Terrenoire C, Romey G, Lazdunski M, Borsotto M. p11, an annexin II subunit, an auxiliary protein associated with the background K+ channel, TASK-1. EMBO J 21: 4439–4448, 2002. doi: 10.1093/emboj/cdf469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez Bosc LV, Plomaritas DR, Herbert LM, Giermakowska W, Browning C, Jernigan NL. ASIC1-mediated calcium entry stimulates NFATc3 nuclear translocation via PICK1 coupling in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 311: L48–L58, 2016. doi: 10.1152/ajplung.00040.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbert LM, Nitta CH, Yellowhair TR, Browning C, Gonzalez Bosc LV, Resta TC, Jernigan NL. PICK1/calcineurin suppress ASIC1-mediated Ca2+ entry in rat pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 310: C390–C400, 2016. doi: 10.1152/ajpcell.00091.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133–145, 2003. doi: 10.1113/jphysiol.2002.030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hruska-Hageman AM, Wemmie JA, Price MP, Welsh MJ. Interaction of the synaptic protein PICK1 (protein interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain Na+ channel 1) and ASIC (acid-sensing ion channel). Biochem J 361: 443–450, 2002. doi: 10.1042/bj3610443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyvelin J-M, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97: 185–191, 2005. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 21.Jernigan NL, Broughton BR, Walker BR, Resta TC. Impaired NO-dependent inhibition of store- and receptor-operated calcium entry in pulmonary vascular smooth muscle after chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 290: L517–L525, 2006. doi: 10.1152/ajplung.00308.2004. [DOI] [PubMed] [Google Scholar]
- 22.Jernigan NL, Herbert LM, Walker BR, Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol 302: C931–C940, 2012. doi: 10.1152/ajpcell.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jernigan NL, Paffett ML, Walker BR, Resta TC. ASIC1 contributes to pulmonary vascular smooth muscle store-operated Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 297: L271–L285, 2009. doi: 10.1152/ajplung.00020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin L-M. Rock ‘n’ Rho: regulation of ion channels. Am J Physiol Heart Circ Physiol 296: H908–H909, 2009. doi: 10.1152/ajpheart.00185.2009. [DOI] [PubMed] [Google Scholar]
- 26.Jin W, Shen C, Jing L, Zha XM, Xia J. PICK1 regulates the trafficking of ASIC1a and acidotoxicity in a BAR domain lipid binding-dependent manner. Mol Brain 3: 39, 2010. doi: 10.1186/1756-6606-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones SVP. Role of the small GTPase Rho in modulation of the inwardly rectifying potassium channel Kir2.1. Mol Pharmacol 64: 987–993, 2003. doi: 10.1124/mol.64.4.987. [DOI] [PubMed] [Google Scholar]
- 28.Karpushev AV, Ilatovskaya DV, Pavlov TS, Negulyaev YA, Staruschenko A. Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na+ channel. PLoS One 5: e8827, 2010. doi: 10.1371/journal.pone.0008827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin M-J, Leung GPH, Zhang W-M, Yang X-R, Yip K-P, Tse C-M, Sham JSK. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 30.Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher-Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A, Weissmann N. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 188: 1451–1459, 2013. doi: 10.1164/rccm.201307-1252OC. [DOI] [PubMed] [Google Scholar]
- 31.Mastroberardino L, Spindler B, Forster I, Loffing J, Assandri R, May A, Verrey F. Ras pathway activates epithelial Na+ channel and decreases its surface expression in Xenopus oocytes. Mol Biol Cell 9: 3417–3427, 1998. doi: 10.1091/mbc.9.12.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem 278: 33492–33500, 2003. doi: 10.1074/jbc.M302401200. [DOI] [PubMed] [Google Scholar]
- 33.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 34.Nilius B, Gerke V, Prenen J, Szücs G, Heinke S, Weber K, Droogmans G. Annexin II modulates volume-activated chloride currents in vascular endothelial cells. J Biol Chem 271: 30631–30636, 1996. doi: 10.1074/jbc.271.48.30631. [DOI] [PubMed] [Google Scholar]
- 35.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. doi: 10.1152/ajpheart.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 37.Parrau D, Ebensperger G, Herrera EA, Moraga F, Riquelme RA, Ulloa CE, Rojas RT, Silva P, Hernandez I, Ferrada J, Diaz M, Parer JT, Cabello G, Llanos AJ, Reyes RV. Store-operated channels in the pulmonary circulation of high- and low-altitude neonatal lambs. Am J Physiol Lung Cell Mol Physiol 304: L540–L548, 2013. doi: 10.1152/ajplung.00024.2012. [DOI] [PubMed] [Google Scholar]
- 38.Plomaritas DR, Herbert LM, Yellowhair TR, Resta TC, Gonzalez Bosc LV, Walker BR, Jernigan NL. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L419–L430, 2014. doi: 10.1152/ajplung.00095.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, Staruschenko A. Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J Biol Chem 281: 26520–26527, 2006. doi: 10.1074/jbc.M603716200. [DOI] [PubMed] [Google Scholar]
- 40.Pochynyuk O, Staruschenko A, Bugaj V, Lagrange L, Stockand JD. Quantifying RhoA facilitated trafficking of the epithelial Na+ channel toward the plasma membrane with total internal reflection fluorescence-fluorescence recovery after photobleaching. J Biol Chem 282: 14576–14585, 2007. doi: 10.1074/jbc.M701348200. [DOI] [PubMed] [Google Scholar]
- 41.Poon WYL, Malik-Hall M, Wood JN, Okuse K. Identification of binding domains in the sodium channel NaV1.8 intracellular N-terminal region and annexin II light chain p11. FEBS Lett 558: 114–118, 2004. doi: 10.1016/S0014-5793(03)01512-6. [DOI] [PubMed] [Google Scholar]
- 42.Ratz PH, Berg KM, Urban NH, Miner AS. Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol 288: C769–C783, 2005. doi: 10.1152/ajpcell.00529.2004. [DOI] [PubMed] [Google Scholar]
- 43.Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res 93: 548–556, 2003. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 44.Sit S-T, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci 124: 679–683, 2011. doi: 10.1242/jcs.064964. [DOI] [PubMed] [Google Scholar]
- 45.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 46.Song M, Makino A, Yuan JXJ. STIM2 contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staruschenko A, Nichols A, Medina JL, Camacho P, Zheleznova NN, Stockand JD. Rho small GTPases activate the epithelial Na+ channel. J Biol Chem 279: 49989–49994, 2004. doi: 10.1074/jbc.M409812200. [DOI] [PubMed] [Google Scholar]
- 48.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 49.Stirling L, Williams MR, Morielli AD. Dual roles for RHOA/RHO-kinase in the regulated trafficking of a voltage-sensitive potassium channel. Mol Biol Cell 20: 2991–3002, 2009. doi: 10.1091/mbc.E08-10-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storey NM, O’Bryan JP, Armstrong DL. Rac and Rho mediate opposing hormonal regulation of the ether-a-go-go-related potassium channel. Curr Biol 12: 27–33, 2002. doi: 10.1016/S0960-9822(01)00625-X. [DOI] [PubMed] [Google Scholar]
- 51.Sun CK, Zhen YY, Lu HI, Sung PH, Chang LT, Tsai TH, Sheu JJ, Chen YL, Chua S, Chang HW, Chen YL, Lee FY, Yip HK. Reducing TRPC1 expression through liposome-mediated siRNA delivery markedly attenuates hypoxia-induced pulmonary arterial hypertension in a murine model. Stem Cells Int 2014: 316214, 2014. doi: 10.1155/2014/316214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svenningsson P, Greengard P. p11 (S100A10)—an inducible adaptor protein that modulates neuronal functions. Curr Opin Pharmacol 7: 27–32, 2007. doi: 10.1016/j.coph.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 53.van de Graaf SF, Hoenderop JG, Gkika D, Lamers D, Prenen J, Rescher U, Gerke V, Staub O, Nilius B, Bindels RJ. Functional expression of the epithelial Ca2+ channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex. EMBO J 22: 1478–1487, 2003. doi: 10.1093/emboj/cdg162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Weigand L, Foxson J, Shimoda LA, Sylvester JT. Ca2+ signaling in hypoxic pulmonary vasoconstriction: effects of myosin light chain and Rho kinase antagonists. Am J Physiol Lung Cell Mol Physiol 293: L674–L685, 2007. doi: 10.1152/ajplung.00141.2007. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 56.Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, Sham JS. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 63: 173–180, 2014. doi: 10.1161/HYPERTENSIONAHA.113.01902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu Y, Chen Z, Li WG, Cao H, Feng EG, Yu F, Liu H, Jiang H, Xu TL. A nonproton ligand sensor in the acid-sensing ion channel. Neuron 68: 61–72, 2010. doi: 10.1016/j.neuron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Mol Brain 6: 1, 2013. doi: 10.1186/1756-6606-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]