

Abstract
We previously demonstrated that renal tubular peptidylarginine deiminase-4 (PAD4) is induced after ischemia-reperfusion (IR) injury and this induction of PAD4 exacerbates ischemic acute kidney injury (AKI) by promoting renal tubular inflammation and neutrophil infiltration. However, the mechanisms of renal tubular PAD4 induction after IR remain unknown. Here, we tested the hypothesis that ATP, a proinflammatory danger-associated molecular pattern (DAMP) ligand released from necrotic cells after IR injury, induces renal tubular PAD4 and exacerbates ischemic AKI via P2 purinergic receptor activation. ATP as well as ATPγS (a nonmetabolizable ATP analog) induced PAD4 mRNA, protein, and activity in human and mouse renal proximal tubule cells. Supporting the hypothesis that ATP induces renal tubular PAD4 via P2X7 receptor activation, A804598 (a selective P2X7 receptor antagonist) blocked the ATP-mediated induction of renal tubular PAD4 whereas BzATP (a selective P2X7 receptor agonist) mimicked the effects of ATP by inducing renal tubular PAD4 expression and activity. Moreover, ATP-mediated calcium influx in renal proximal tubule cells was blocked by A804598 and was mimicked by BzATP. P2X7 activation by BzATP also induced PAD4 expression and activity in mouse kidney in vivo. Finally, supporting a critical role for PAD4 in P2X7-mediated exacerbation of renal injury, BzATP exacerbated ischemic AKI in PAD4 wild-type mice but not in PAD4-deficient mice. Taken together, our studies show that ATP induces renal tubular PAD4 via P2X7 receptor activation to exacerbate renal tubular inflammation and injury after IR.
Keywords: citrullination, inflammation, protein kinase C, purinergic signaling
INTRODUCTION
Acute kidney injury (AKI) is a leading cause of mortality and morbidity in hospitalized patients costing more than $10 billion per year in the United States (7). Renal ischemia and reperfusion (IR) injury is a major cause of clinical AKI, and patients subjected to major surgery including cardiac, aortovascular, or abdominal organ transplantation frequently (~50–80%) develop ischemic AKI (4, 25). Unfortunately, despite many decades of intense research, AKI remains a major clinical problem without effective therapy (20, 32).
Kidney IR injury results in rapid renal tubular necrosis coupled together with subsequent renal tubular inflammation as well as apoptosis (23, 30, 33). Indeed, renal inflammation is a major pathogenic contributor to ischemic AKI and is mediated by induction of renal tubular proinflammatory chemokines and cytokines that attracts circulating leukocytes (including neutrophils, macrophages, lymphocytes, and natural killer cells) into the renal parenchyma to exacerbate renal tubular cell death after IR (29). In particular, neutrophils have been shown to play an important role in renal tubular inflammation and cell death after IR (1, 14, 18, 23). Consistent with these findings, neutralization or blockade of neutrophils attenuates the severity and duration of ischemic AKI (9, 13, 18).
The family of peptidyl arginine deiminases (PAD) catalyzes the conversion of peptide-bound arginine residues to peptide-bound citrulline via a calcium-dependent process (46, 61). In the kidney, two PAD mRNA subtypes have been identified, PAD2 and PAD4 (17, 58). We have shown in our previous studies that ischemic AKI results in selective induction of renal tubular PAD4 expression and activity without affecting the PAD2 expression (17). Furthermore, we showed that renal PAD4 plays a critical role in ischemic AKI as wild-type mice treated with PAD4 inhibitors or mice genetically deficient in PAD4 were significantly protected against renal IR with reduced renal tubular inflammation and injury (17, 45). In contrast, recombinant human PAD4 treatment significantly exacerbated ischemic AKI in wild-type mice subjected to renal IR injury. Further implicating a critical role for neutrophil infiltration in ischemic AKI, we showed that PAD4 activation promotes kidney neutrophil infiltration as well as renal tubular inflammation and proinflammatory transcription factor NFkB after IR (17). However, the mechanisms involved in renal tubular PAD4 induction remain unknown.
Previous studies showed that ATP released by necrotic cells serves as a danger signal via P2 purinergic receptor activation to recruit proinflammatory leukocytes (47, 51). Indeed, the effects of extracellular ATP closely mimic the effects of PAD4 (renal tubular inflammation, enhanced neutrophil chemotaxis, and proinflammatory cytokine induction). Of P2 receptor subtypes, P2X receptor subtypes (P2X4 and P2X7 in particular) have been shown to exacerbate tissue injury after IR (6, 19, 40). Therefore, in this study, we hypothesized that extracellular ATP released after IR induces renal tubular PAD4 via P2X receptor activation.
MATERIALS AND METHODS
Proximal tubule cell culture and P2 receptor modulation.
Immortalized human proximal tubular cell line (HK-2; American Type Culture Collection, Manassas, VA) was kept in low glucose DMEM/F12 medium supplemented with 10% FBS at 37°C in a humidified 5% CO2 atmosphere as described (27, 52). Mouse kidney proximal tubules were isolated using Percoll density gradient separation as described previously (60). Mouse proximal tubules were maintained in high glucose DMEM/Ham’s F12 medium plus 10% FBS, 2 mM l-glutamine, and 10 mM HEPES. After 24 h of serum deprivation, confluent cultured mouse or human proximal tubule cells were treated with drugs listed in Table 1. Proximal tubule cells were treated with ATP, ATPγS, or with selective P2 receptor agonists (Table 1) for 1–24 h. Some cells were pretreated with P2 receptor antagonist before ATP treatment. Another cohort of cells was pretreated with inhibitors of ERK MAP, AKT, or P38 MAPK signaling before P2 receptor agonist treatment.
Table 1.
P2 receptor drugs and signaling inhibitors: abbreviations and doses used in this study
| Drug | Dose |
|---|---|
| Adenosine triphosphate (ATP, P2 purinergic receptor agonist) | 0.5–1 mM |
| Adenosine-5′-(γ-thio)-triphosphate (ATPγS. P2 purinergic receptor agonist, a nonhydrolyzable analog of ATP) | 100–500 µM |
| Adenosine 5′-O-2-thiodiphosphate (ADP-β-S, P2 receptor agonist) | 50–100 µM |
| 2-Methylthioadenosine triphosphate (2-MT ATP, P2Y receptor agonist) | 10–20 µM |
| 2′(3′)-O-(4-Benzoylbenzoyl)adenosine-5′-triphosphate (BzATP, selective P2X7 receptor agonist) | 10–50 µM |
| Pyridoxalphosphate-6-azophenyl-2′,5′-disulfonic acid (Iso-PPADS, P2X-purinoceptor antagonist) | 10–50 µM |
| N-cyano-N′′-[(1S)-1-phenylethyl]-N′-5-quinolinyl-guanidine (A804598, selective P2X7 antagonist) | 10–25 µM |
| Bisindolylmaleimide (BIS, selective protein kinase C inhibitor) | 50–200 nM |
| 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98058, MEK1 inhibitor to block ERK MAPK signaling) | 20–50 µM |
| 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002, PI3 kinase inhibitor to inhibit AKT signaling) | 5–10 µM |
Renal IR injury in mice.
PAD4-deficient mice and wild-type control mice (on a C57BL/6 albino background) were provided by Dr. Kerri Mowen (Dept. of Pharmacology and Chemical Physiology, Scripps Research Institute, La Jolla, CA). PAD4-deficient mice were created by mating PAD4 floxed mice (flanked at exons 9 and 10 with LoxP sites introduced into the introns) with CMV Cre mice. PAD4-deficient mice bred normally and had no anatomical abnormalities.
After Columbia University Institutional Animal Care and Use Committee approval, adult PAD4 wild-type mice or PAD4-deficient mice weighing ~20–25 g were anesthetized intraperitoneally with pentobarbital sodium (Sigma, St Louis, MO: 50 mg/kg body weight or to effect). Mice were then subjected to right nephrectomy and 30 min left renal ischemia as described previously. For pain management, all mice received 0.5–1 mg/kg sc buprenorphine SR before surgery. Sham-operated animals underwent anesthesia followed by laparotomy, right nephrectomy, bowel manipulations, and wound closure without renal ischemia. Body temperature of all mice was sustained at ~37°C using a surgical heating pad during surgery as well as during recovery from anesthesia.
Measurement of renal function after kidney IR injury.
Twenty-four hours after renal IR injury, plasma creatinine was measured using an enzymatic creatinine reagent kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA). This creatinine measurement method eliminates the interferences from mouse plasma chromagens known to occur in the Jaffe method.
Histological detection of kidney injury.
Twenty-four hours after renal IR injury, kidneys were fixed in 10% formalin overnight. After automated dehydration through graded alcohol series, tissues were embedded in paraffin, sectioned at 5 µm, and stained with hematoxylin-eosin (H&E). Kidney H&E sections after renal IR surgery or sham surgery were assessed by a pathologist (V. D’Agati) who was unaware of the treatment that each animal had received. An established grading scale of kidney necrotic IR injury to the proximal tubules (0–4, Renal Injury Score) was used for the histopathological assessment as outlined by Jablonski et al. (22) and as described in our previous studies (27, 28).
Detection of kidney neutrophil infiltration.
Infiltrating neutrophils after renal IR injury were detected with immunohistochemistry with rat anti-mouse Ly6B monoclonal antibody (AbD Serotec, Raleigh, NC) as described (43, 44). Primary IgG2a antibody (MCA1212, AbD Serotec) was utilized as a negative isotype control. Quantification of kidney infiltrating neutrophils was performed using 5–7 randomly chosen 200× microscope image fields (corticomedullary junction for kidney neutrophils), and results are expressed as neutrophils counted per 200× field.
PAD4 activity assay.
Mouse kidney PAD4 activity was measured 24 h after renal IR injury or sham surgery. Activity was measured according to methods described by Wildeman and Pires with slight modifications as described (17, 62). This fluorescence-based assay utilizes small molecular mimics of a natural substrate of PAD4 followed by trypsin cleavage unmasking the fluorophore. Briefly, kidney homogenates were incubated with the enzyme substrate (Benzoyl-dl-arginine-7-amino-4-methylcoumarine, Santa Cruz Biotechnology, Santa Cruz, CA) plus 10 mM CaCl2 at 37°C for 10 min with or without 200 µM 2-chloroamidine (a PAD4 inhibitor) in 50 mM Tris·HCl buffer (pH 7.6). Following trypsin hydrolysis, the reaction was quenched in liquid nitrogen. The supernatant was centrifuged at 14,000 g and analyzed with HPLC to measure the product of PAD4 enzyme (7-amino-4-methylcoumarine) on a C18 reversed-phase column with a binary low-pressure gradient elution system with a fluorescence detector set to 441 nm upon excitation with light at 342 nm.
PAD RT-PCR and immunoblotting.
After treatment with P2 receptor modulators with or without signaling inhibitors, RT-PCR for PAD2/4 mRNA was performed as described previously (17, 45) with the primers listed in Table 2. Both conventional semiquantitative and quantitative real-time RT-PCR were performed. Primer design was based on published GenBank sequences. To confirm equal RNA loading, GAPDH mRNA expression was also measured. For PAD4 protein detection, human and mouse renal proximal tubule cells were homogenized in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris·HCl, 1 mM EDTA, 1% Triton X-100, pH 7.4) and centrifuged for 10 min at 1,000 g. The supernatant was collected, and anti-PAD4 antibody (Abcam, Cambridge, MA) detected PAD4 expression. For protein loading control, membranes were stripped and reprobed for mouse anti-β-actin (Sigma).
Table 2.
Primers used in quantitative reverse transcription polymerase chain reactions to amplify mouse cDNAs
| Primers | Sequence (Sense/Antisense) | AnnealingTemp (°C) |
|---|---|---|
| Mouse | 5′-GAGCAAGGATGGCCCAAGG-3′ | 65 |
| PAD4 | 5′-GACAGTTCCACCCCAGTGAT-3′ | |
| Mouse | 5′-AAAATATGCTGCGGGAACGG-3′ | 65 |
| PAD2 | 5′-TAGTTGACAGTGACCTTGTCG-3′ | |
| Human | 5′-CACCTTGACTCAGCTTGACA-3′ | 64 |
| PAD4 | 5′-GGTCCAGGTCCTCTGATCTT-3′ | |
| Mouse/ | 5′-ACCACAGTCCATGCCATCAC-3′ | 65 |
| Human | 5′-CACCACCCTGTTGCTGTAGCC-3′ | |
| GAPDH |
Primer sequences are based on published GenBank sequences for mice and humans. Annealing temperatures used for each primer are also provided.
Confocal PAD4 Immunohistochemistry.
Confocal PAD4 Immunohistochemistry was performed on mouse kidneys treated with vehicle (5% DMSO) or with BzATP (5 mg/kg ip) 24 h before being subjected to sham surgery or to 30 min renal ischemia and 24 h reperfusion. Kidneys were fixed with 4% paraformaldehyde, dehydrated with 30% sucrose, frozen in O.C.T (Tissue-Tek, Torrance, CA) and cryosectioned (5 µm). After washes, sections were blocked with 2% BSA for 1 h at room temperature and stained with rabbit anti-PAD4 antibody (Abcam) overnight followed by Alexa Fluor 594-conjugated goat anti-rabbit (Invitrogen, Carlsbad, CA) secondary antibody. The cryosections were mounted in ProLong Gold anti-fade reagent-containing 4′,6-diamidino-2-phenylindole from Molecular Probes (Invitrogen). Kidney sections were imaged under confocal microscopy (Nikon Eclipse), and images were acquired with NIS software version 4.10. Two negative-control experiments were performed on serial sections by omitting primary antibody or by using nonspecific isotype control primary antibody.
Measurement of calcium influx in HK-2 cells with P2 receptor activation.
Confluent HK-2 cells were incubated in modified 100 μl/well HBSS (in mM: 138 NaCl, 5.3 KCl, 2 CaCl2, 1 MgSO4, 0.34 Na2HPO4, 4.2 NaHCO3, 0.44 KH2PO4, 5.5 dextrose, and 20 HEPES, pH 7.4) containing 4.6 μM fluo-4 AM (DMSO vehicle final concentration: 0.5%) and 0.05% Pluronic F-127 (DMSO vehicle final concentration: 0.25%) for 60 min at 37°C. After the cells were loaded, they were washed 3 times with modified HBSS and left for an additional 30 min at room temperature to allow complete deesterification of the intracellular acetoxy methyl esters. After a stable baseline was acquired for 120 s, the cells were treated with 500–1,000 μM ATP, with 500 μM ATPγS, or with μM BzATP. Some cells were pretreated with P2 receptor antagonist iso-PPADS or with A804598 before ATP treatment (Table 1), and the fluorescence was recorded for 120 s at an excitation wavelength of 490 nm and an emission wavelength of 520 nm using a microplate reader (Synergy HT; BioTek Instruments, Winooski, VT). Triplicate wells were simultaneously measured, and values were averaged for each data point. In all studies, the fluorescence after treatments is presented as the percent change from baseline fluorescence.
Statistical analysis.
The data were analyzed with Student's t-test when comparing means between two groups or one-way ANOVA plus Tukey’s post hoc multiple comparison test when comparing multiple groups. The Mann-Whitney nonparametric test was used to analyze ordinal values of renal injury scores. In all cases, a probability statistic P < 0.05 was taken to indicate significance. All data are expressed throughout the text as means ± SE.
RESULTS
ATP induces renal tubular PAD4 expression and activity in human and mouse renal proximal tubule cells.
We first determined whether ATP and its nonmetabolizable analog (ATPγS) induce PAD4 protein expression and activity in human (HK-2) and mouse proximal tubule cells. Initially, HK-2 cells were treated with vehicle or with ATPγS (100 µM or 500 µM) for 6 h and RT-PCR for PAD4 and GAPDH performed. Figure 1A shows that ATPγS induced PAD4 mRNA expression in HK-2 cells (representative of 6 experiments). Consistent with these mRNA changes, ATP (1 mM for 16 h, Fig. 1B) or ATPγS (100 μM for 16 h, Fig. 1C) induced PAD4 protein in HK-2 cells (representative of 4 experiments). In addition, a nonselective P2 receptor agonist 2-methylthio ATP (2-MT ATP, 10 μM for 16 h) induced PAD4 protein expression in HK-2 cells (Fig. 1D, representative of 4 experiments). Moreover, ATP (1 mM for 16 h) induced PAD4 expression in primary culture of mouse proximal tubules (Fig. 1F, representative of 4 experiments). Finally, ATPγS significantly increased PAD4 activity in human (N = 4, Fig. 1G) and mouse proximal tubule cells (Fig. 1H).
Fig. 1.

ATP induces PAD4 in human and mouse proximal tubules. A: ATPγS (a nonmetabolizable analog of ATP) induced PAD4 mRNA expression in human proximal tubule (HK-2) cells. HK-2 cells were treated with vehicle or with ATPγS (100 µM or 500 µM) for 6 h and RT-PCR performed (representative of 6 experiments per group). GAPDH was used as a normalizing control. Top panel shows representative RT-PCR images, and bottom panel shows band intensity quantification for PAD4 mRNA normalized to GAPDH. B–E: ATP (1 mM for 16 h, B) or ATPγS (100 μM for 16 h, C) induced PAD4 protein in HK-2 cells (representative of 4–6 experiments per group). Moreover, a nonselective P2 receptor agonist 2-methylthio ATP (2-MT ATP, 10 μM for 16 h) induced PAD4 protein expression in HK-2 cells (D). Band intensity quantifications for PAD4 protein normalized to β-actin are shown in E. F: ATP (1 mM for 16 h) also induced PAD4 expression in primary culture of mouse proximal tubules (representative of 4 experiments per group). Top panel represents representative PAD4 immunoblot images, and bottom panel shows band intensity quantification for PAD4 protein normalized to β-actin. G and H: ATPγS significantly increased PAD4 activity in human (N = 4–6, G) and mouse proximal tubule cells (N = 4, H). *P < 0.05 vs. vehicle.
ATP induces PAD4 expression via P2X receptor activation in human proximal tubule cells.
Next, we determined which P2 receptor subtype (P2X or P2Y) is responsible for ATP-mediated renal proximal tubular PAD4 induction. We show in Fig. 2A that a P2Y receptor agonist adenosine 5′-(β-thio)diphosphate (ADP-β-S, 50–100 μM for 16 h) failed to induce PAD4 in HK-2 cells (representative of 4 experiments). In contrast, pretreating HK-2 cells with 50 μM iso-PPADS (a P2X antagonist) prevented ATP (1 mM for 16 h)-induced increase in PAD4 expression (Fig. 2B, representative of 4 experiments).
Fig. 2.
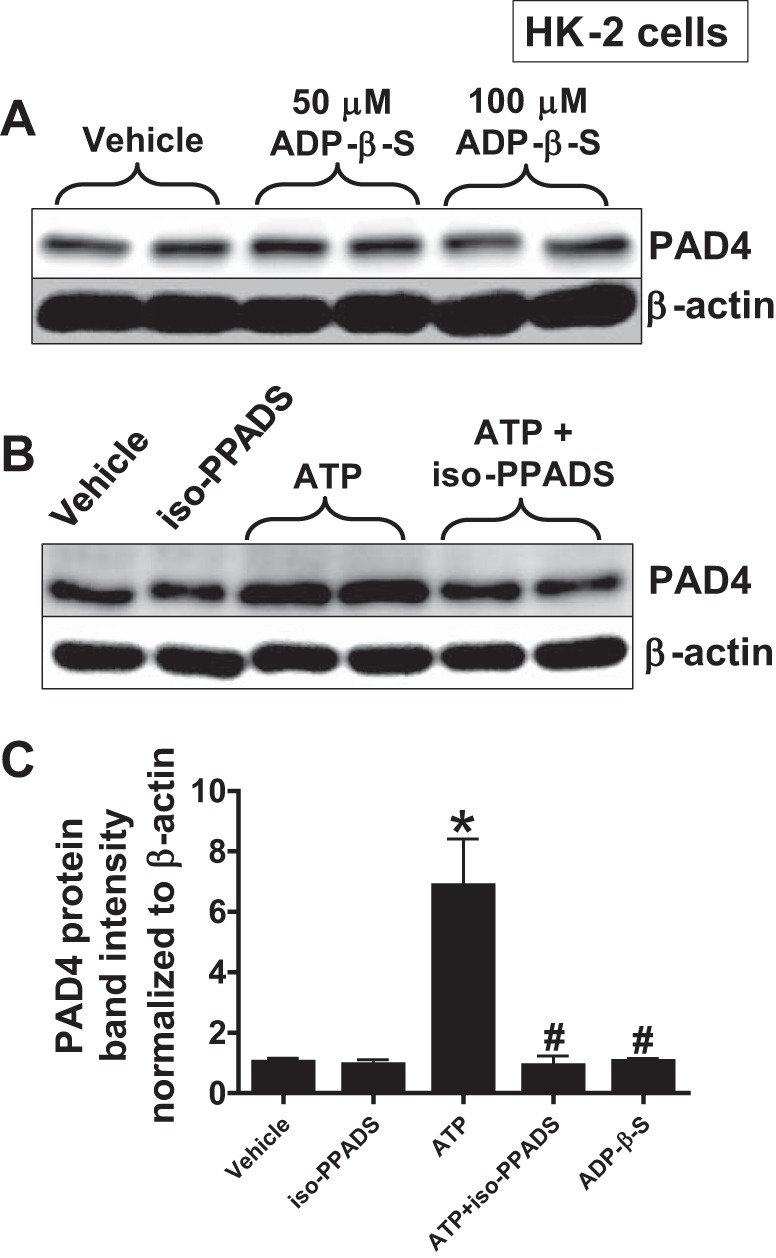
ATP induces PAD4 expression via P2X receptor activation in HK-2 cells. A: a P2Y receptor agonist, adenosine 5′-(β-thio)diphosphate (ADP-β-S, 50–100 μM for 16 h), failed to induce PAD4 in HK-2 cells. B: in contrast, a P2X antagonist, iso-PPADS (50 μM), prevented ATP (1 mM for 16 h)-mediated increases in PAD4 expression. Iso-PPADS was added 30 min before ATP or vehicle treatment. β-Actin was probed as a protein loading control (representative of 4 experiments per group). C: band intensity quantifications for PAD4 protein expression in HK-2 cells demonstrating that ATP-mediated induction of PAD4 in HK-2 cells was attenuated by a P2X receptor antagonist, iso-PPADS, whereas a P2Y agonist, ADP-β-S (100 μM), had no effect on PAD4 expression. *P < 0.05 vs. vehicle. #P < 0.05 vs. ATP.
ATP-mediated renal proximal tubular PAD4 induction is mediated by P2X7 receptor activation.
As we previously showed that renal tubular PAD4 induction exacerbates renal inflammation and injury after ischemic AKI and P2X7 receptor has been shown to play a role in tissue injury and inflammation including in the kidney (6, 40, 63), we next tested whether ATP induces renal proximal tubular PAD4 via P2X7 receptor activation. Our results support this hypothesis as ATP-mediated induction of PAD4 in HK-2 cells was attenuated by pretreatment with a selective P2X7 receptor antagonist, A804598 (20 μM, Fig. 3A). In addition, a selective P2X7 agonist, BzATP (10 μM), induced PAD4 mRNA (Fig. 3B) and protein expression (Fig. 3C) as well as PAD4 activity (Fig. 3D) in HK-2 cells (N = 4–6).
Fig. 3.

ATP induces renal proximal tubule PAD4 expression via P2X7 receptor activation. A: a selective P2X7 receptor antagonist, A804598 (20 μM, 30 min pretreatment), prevented ATP-mediated induction of PAD4 in human renal proximal tubule (HK-2) cells. HK-2 cells were treated with 1 mM ATP for 16 h. B and C: a selective P2X7 agonist BzATP (10 μM) induced PAD4 mRNA (6 h treatment, B) and protein (16 h treatment, C) expression in HK-2 cells. GAPDH and β-actin were probed as a loading control for mRNA and protein, respectively. Representative gel images are shown in top panel and band intensity quantifications are shown in bottom panel. D: BzATP (10 μM for 16 h) significantly induced PAD4 activity in HK-2 cells. Representative of 4–6 experiments per group. E: ATP (1 mM for 6 h) significantly increased PAD4 activity in HK-2 cells, and this increase in activity was attenuated by a selective P2X7 receptor antagonist, A804598 (20 μM). *P < 0.05 vs. vehicle. #P < 0.05 vs. ATP. Vertical lines between the lanes in A indicate that these lanes were located on the same immunoblot but were not located in neighboring lanes on the original gel and immunoblot image.
ATP induces calcium influx in human proximal tubule cells by P2X7 receptor activation.
We next determined whether ATP induces calcium influx via P2X7 receptor activation in HK-2 cells as P2X receptors are ligand-gated transmembrane cation channels allowing extracellular cation influx including calcium (6, 53). Figure 4 shows a representative relative delta fluorescent unit (RFU) tracing of Fluo-4 over time as an indicator of calcium influx (Fig. 4A) as well as averaged maximal delta RFU (Fig. 4B) in HK-2 cells (N = 4) demonstrating ATP (1 mM) rapidly caused calcium influx in HK-2 cells. Again implicating a role for P2X receptors in ATP-mediated calcium influx in HK-2 cells, a selective P2X receptor antagonist iso-PPADS (50 μM) attenuated the ATP-mediated calcium influx. In subsequent studies, we show that ATP-mediated calcium influx in HK-2 cells is also mediated by the P2X7 receptor activation as a selective P2X7 antagonist, A804598 (a P2X7 receptor antagonist, 20 µM), blocked and a selective P2X7 agonist, BzATP (50 µM, a selective P2X7 receptor agonist), mimicked the effects of ATP on calcium influx in HK-2 cells, respectively (Fig. 4C, N = 4–8).
Fig. 4.
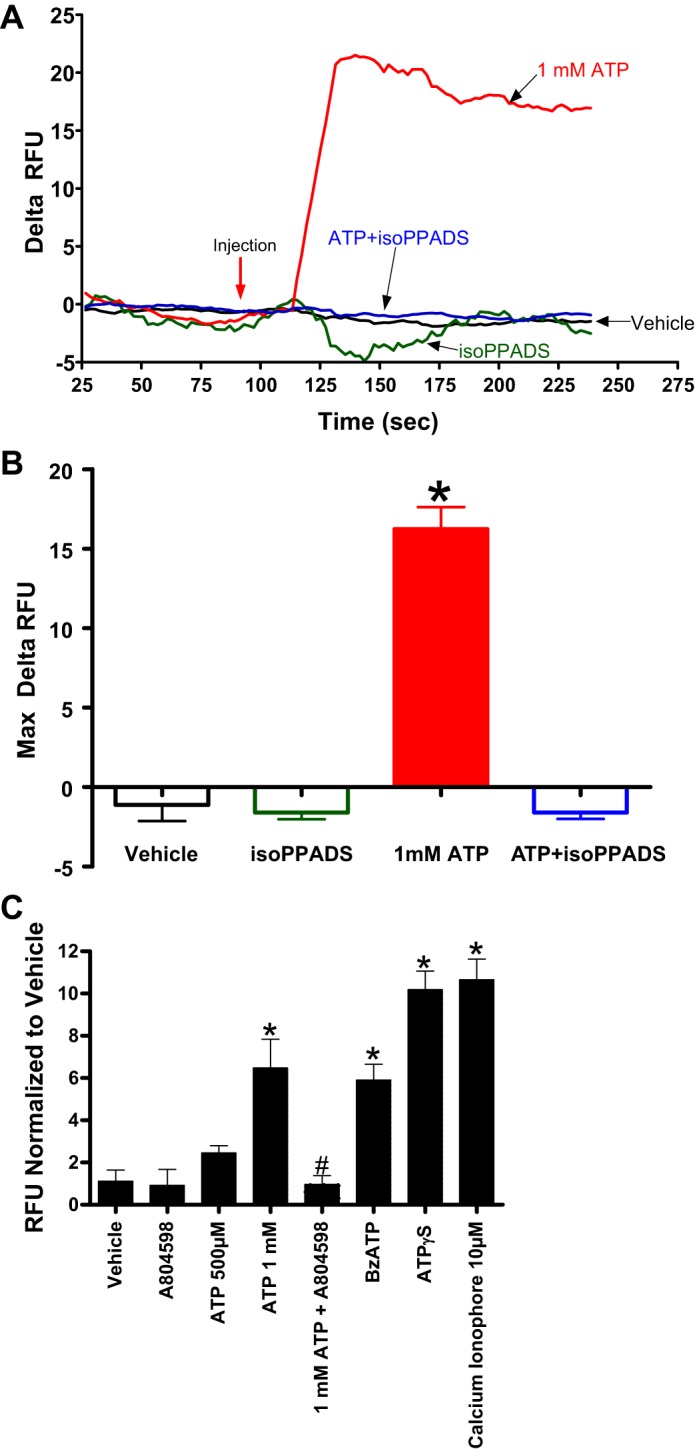
ATP induces calcium influx in human kidney proximal tubule (HK-2) cells via P2X7 receptor activation. Representative relative delta fluorescent unit (RFU) tracing of Fluo-4 (A) over time as an indicator of calcium influx as well as averaged maximal delta RFU (B) in HK-2 cells (N = 4). HK-2 cells were treated with vehicle or with 50 μM iso-PPADS (a P2X receptor antagonist) 15 min before 1 mM ATP treatment. *P < 0.05 vs. vehicle group. Red arrow indicates when ATP or vehicle was injected to the cells. C: averaged maximal delta RFU (normalized to vehicle) indicating calcium influx in HK-2 cells treated with ATP (500 µM or 1 mM), ATPγS (500 µM), and BzATP (50 µM, a selective P2X7 receptor agonist). Some cells were pretreated with A804598 (a P2X7 receptor antagonist, 20 µM) before ATP treatment. Calcium ionophore (10 µM) was used as a positive control. ATP-mediated calcium influx in HK-2 cells is mediated by P2X7 receptor activation as a selective P2X7 antagonist, A804598, blocked and a selective P2X7 agonist, BzATP, mimicked the effects of ATP on calcium influx in HK-2 cells. *P < 0.01 vs. vehicle. #P < 0.05 vs. ATP 1 mM. N = 4–8.
ATP induces renal proximal tubular PAD4 via PKC activation.
As the P2X receptor has been shown to activate several intracellular kinases including PKC, ERK MAPK, and p38 MAPK (36, 50, 57), we tested whether ATP-mediated induction of renal tubular PAD4 is mediated by these kinases. Figure 5 suggests a critical role for PKC in ATP-mediated PAD4 induction as bisindolylmaleimide II (a PKC inhibitor) prevented ATP-mediated induction of PAD4 in HK-2 cells. In contrast, p38 MAPK or ERK MAPK does not appear to play a role as neither SB203680 (a p38 MAPK inhibitor) nor PD98059 (an ERK MAPK inhibitor) attenuated ATP-mediated PAD4 induction in HK-2 cells (N = 3–4).
Fig. 5.
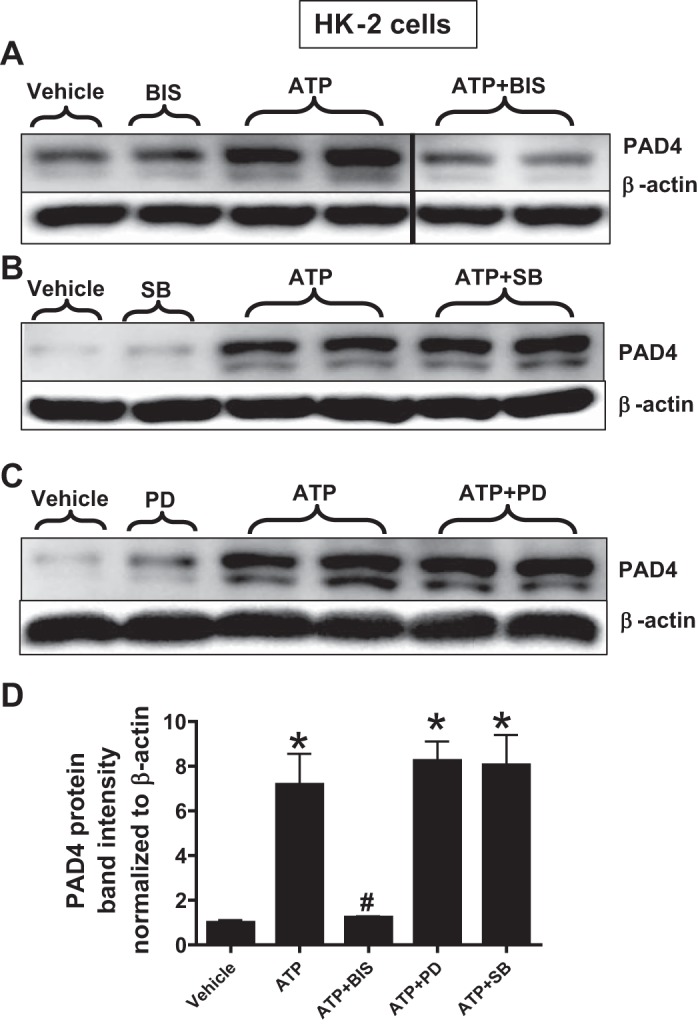
ATP induces PAD4 via PKC activation in human proximal tubule (HK-2) cells. HK-2 cells were treated with vehicle or with 1 mM ATP 16 h. Some cells were pretreated with 100 nM bisindolylmaleimide II (BIS; a PKC inhibitor, A), with 10 μM SB203680 (SB; a p38 MAPK inhibitor, B), or with 50 μM PD98059 (PD; an ERK MAPK inhibitor, C) 30 min before ATP treatment. β-Actin was probed to control for protein loading (representative of 3–4 experiments per group). D: band intensity quantifications for PAD4 protein expression in HK-2 cells demonstrating that ATP-mediated induction of PAD4 in HK-2 cells was attenuated by a PKC inhibitor (bisindolylmaleimide II) but not by a specific ERK (PD98059) or a p38 MAPK (SB203680) inhibitor. *P < 0.05 vs. vehicle. #P < 0.05 vs ATP. Vertical lines between the lanes in A indicate that these lanes were located on the same immunoblot but were not located in neighboring lanes on the original gel and immunoblot image.
P2X7 receptor activation increases mouse kidney PAD4 expression in vivo.
Next, we determined whether P2X7 receptor induces PAD4 expression in mouse kidney in vivo. Figure 6 shows that a selective P2X7 receptor agonist (BzATP) treatment (5 mg/kg ip) selectively induced PAD4 mRNA expression measured by quantitative RT-PCR without changing PAD2 mRNA expression in mouse kidney cortical tissues. As demonstrated previously, 20 min renal IR also selectively induced PAD4 mRNA expression in mouse kidney (17) and this PAD4 induction was significantly amplified with BzATP treatment. PAD2 mRNA expression did not change after renal IR with or without BzATP treatment (N = 5–7). We also show that BzATP induces PAD4 protein expression in renal tubules in vivo. Figure 7A shows representative fluorescent immunohistochemistry experiments to determine kidney PAD4 protein expression (red fluorescence, 400×, representative of 4 experiments), and Fig. 7B shows quantification of PAD4 immunofluorescence detected. Twenty-minute renal IR induced PAD4 protein expression in mouse renal tubules (vehicle 20 min RIR) and this PAD4 induction was amplified with 5 mg/kg BzATP treatment (BzATP 20 min RIR). Control isotype antibody staining demonstrates lack of staining and served as a negative control experiment.
Fig. 6.

P2X7 receptor activation selectively increases mouse kidney PAD4 mRNA in vivo. PAD4 wild-type mice were treated with vehicle or with 5 mg/kg BzATP ip 15 min before sham surgery or 20 min renal ischemia and 24 h reperfusion. A: PAD4 and PAD2 mRNA expressions in kidney cortexes were measured with quantitative RT-PCR. BzATP (a selective P2X7 receptor agonist) induced PAD4 but not PAD2 mRNA expression in mouse kidney (BzATP Sham). Renal IR also selectively induced PAD4 mRNA expression in mouse kidney (Vehicle 20 min RIR) and this PAD4 induction was significantly amplified with BzATP treatment (BzATP 20 min RIR). No change in PAD2 mRNA expression was observed in mouse kidney 24 h post-RIR (N = 5–7 experiments). B: PAD4 activity in mouse kidney cortexes. BzATP as well as 20 min RIR increased kidney PAD4 activity in sham-operated mice. *P < 0.05 vs. Vehicle Sham, #P < 0.05 vs. Vehicle 20 min RIR. Error bars represent 1 SE.
Fig. 7.

P2X7 receptor activation induces mouse kidney PAD4 protein expression in vivo. Representative fluorescent immunohistochemistry experiments (A, red fluorescence, 400×) and band intensity quantifications (B) to determine kidney PAD4 expression. PAD4 wild-type mice were treated with vehicle or with 5 mg/kg BzATP ip 15 min before sham surgery or 20 min renal ischemia and 24 h reperfusion. Sham-operated mice treated with BzATP had slightly higher renal tubular PAD4 expression compared with vehicle-treated mice. Moreover, renal IR induced PAD4 mRNA expression in mouse renal tubules (Vehicle 20 min RIR) and this PAD4 induction was amplified with BzATP treatment (BzATP 20 min RIR). Representative of 4 experiments. Control isotype antibody staining served as a negative control experiment. *P < 0.05 vs. vehicle sham. #P < 0.05 vs. vehicle 20m RIR.
P2X7 receptor-mediated exacerbation of renal IR injury requires PAD4.
P2X7 receptor activation is known to exacerbate ischemic AKI (63). Moreover, we showed previously that mice treated with recombinant human PAD4 showed worse renal tubular inflammation and injury after renal IR (17). Therefore, we tested whether P2X7 receptor activation exacerbates renal IR (RIR) injury via induction of renal tubular PAD4. PAD4 wild-type (WT) mice subjected to 20 min renal IR injury again showed increased renal injury compared with PAD4-deficient (KO) mice as described previously (Fig. 8). PAD4 WT mice pretreated with BzATP (5 mg/kg ip, 24 h before ischemia) had significantly higher plasma Cr compared with vehicle-treated PAD4 WT mice (Fig. 8). Supporting a critical role for PAD4 in P2X7 receptor-mediated exacerbation of RIR injury, BzATP failed to exacerbate RIR injury in PAD4 KO mice subjected to 20 min RIR.
Fig. 8.

P2X7 receptor activation exacerbates renal IR (RIR) injury in PAD4 wild-type (WT) mice but not in PAD4-deficient (KO) mice. PAD4 WT and PAD4 KO mice were pretreated with vehicle or with 5 mg/kg BzATP ip 24 h before being subjected to sham surgery or to 20 min RIR injury. Kidney injury was assessed by analyzing plasma creatinine. Kidney function was worse in PAD4 WT mice treated with BzATP before 20 min RIR compared with WT mice treated with vehicle before 20 min RIR. As demonstrated previously, PAD4 deficiency resulted in significant protection against RIR injury compared with PAD4 WT mice. Supporting a critical role for PAD4 in P2X7 receptor-mediated exacerbation of RIR injury, BzATP failed to exacerbate RIR injury in PAD4 KO mice subjected to 20 min RIR. *P < 0.05 vs. vehicle-treated PAD4 wild-type mice subjected to sham surgery. #P < 0.05 vs. BzATP-treated PAD4 wild-type mice subjected to RIR (N = 3–8). Error bars represent 1 SE.
P2X7 receptor activation exacerbates renal tubular necrosis and kidney neutrophil infiltration after renal IR (RIR) injury in PAD4 WT mice but not in PAD4 KO mice.
Figure 9A shows representative H&E images (from 4 experiments) of PAD4 WT and PAD4 KO mice subjected to 20 min renal ischemia and 24 h reperfusion (magnification 200×) with or without 5 mg/kg BzATP treatment. The kidneys of PAD4 WT mice subjected to renal IR showed increased renal tubular injury compared with PAD4 KO mice. Moreover, BzATP-treated PAD4 WT mice had increased renal tubular necrosis and proteinaceous casts after 20 min renal IR injury compared with vehicle-treated PAD4 WT mice. Histologic grading was performed using the Jablonski scale renal injury score [scale: 0–4 (22)] to grade renal tubular necrosis 24 h after renal IR (Fig. 9B). Consistent with the plasma creatinine data, PAD4 KO mice had a decreased renal injury score compared with PAD4 WT mice subjected to 20 min renal IR injury. BzATP increased renal injury score in PAD4 WT mice after 20 min renal IR injury. In contrast, BzATP failed to exacerbate renal tubular necrosis in PAD4 KO mice subjected to 20 min RIR again suggesting that renal PAD4 is required for exacerbation of renal tubular necrosis with P2X7 receptor activation.
Fig. 9.

P2X7 receptor activation exacerbates renal tubular necrosis after renal IR (RIR) injury in PAD4 wild-type (WT) mice but not in PAD4-deficient (KO) mice. PAD4 WT and PAD4 KO mice were pretreated with vehicle or with 5 mg/kg BzATP ip 24 h before being subjected to 20 min RIR injury. A: representative H&E images (N = 4) of PAD4 mice subjected to 20 min renal ischemia and 24 h of reperfusion (magnification 200×) after vehicle or BzATP treatment. B: the renal injury score (scale: 0–4, N = 4) for histology grading was used to grade renal tubular necrosis 24 h after 20 min renal IR. Renal tubular necrosis was worse in PAD4 WT mice treated with BzATP before 20 min RIR compared with WT mice treated with vehicle before 20 min RIR. As demonstrated previously, PAD4 deficiency resulted in significant reduction in renal tubular injury score after RIR compared with PAD4 WT mice. Moreover, BzATP failed to exacerbate renal tubular necrosis in PAD4 KO mice subjected to 20 min RIR. *P < 0.05 vs. vehicle-treated PAD4 wild-type mice subjected to RIR. Error bars represent 1 SE.
Figure 10 shows representative images of immunohistochemistry for neutrophils (dark brown, Fig. 10A) and counts of infiltrating kidney neutrophils (Fig. 10B, N = 4) in the kidneys of PAD4 WT and PAD4 KO mice subjected to 20 min renal ischemia and 24 h reperfusion (magnification 200×) with or without 5 mg/kg BzATP treatment. Renal tubular neutrophil infiltration was higher in PAD4 WT mice treated with BzATP compared with PAD4 WT mice treated with vehicle before 20 min RIR (Fig. 10). In addition, PAD4 deficiency resulted in significant reduction in kidney neutrophil infiltration after RIR compared with PAD4 WT mice. Moreover, BzATP failed to increase kidney neutrophil infiltration in PAD4 KO mice subjected to 20 min RIR suggesting that renal PAD4 is required for exacerbation of kidney neutrophil infiltration with P2X7 receptor activation.
Fig. 10.

P2X7 receptor activation increases kidney neutrophil infiltration after renal IR (RIR) injury in PAD4 wild-type (WT) mice but not in PAD4-deficient (KO) mice. PAD4 WT and PAD4 KO mice were pretreated with vehicle or with 5 mg/kg BzATP ip 24 h before being subjected to 20 min RIR injury. Representative images (from 4 experiments, magnification, 200×) (A) and quantifications of infiltrated neutrophils per 200× field (N = 4) (B) of immunohistochemistry of neutrophil infiltration (dark brown) in the kidneys (corticomedullary junction) of mice subjected to 20 min renal ischemia and 24 h of reperfusion with and without BzATP treatment. Renal tubular kidney infiltration was higher in PAD4 WT mice treated with BzATP before 20 min RIR compared with PAD4 WT mice treated with vehicle before 20 min RIR. In addition, PAD4 deficiency resulted in significant reduction in kidney neutrophil infiltration after RIR compared with PAD4 WT mice. Moreover, BzATP failed to increase kidney neutrophil infiltration in PAD4 KO mice subjected to 20 min RIR. *P < 0.05 vs. vehicle-treated PAD4 WT mice subjected to RIR. Error bars represent 1 SE.
DISCUSSION
The major finding of this study is that ATP, a well-recognized DAMP molecule, induced human and mouse renal proximal tubule PAD4 expression and activity via P2X7 receptor activation. ATP activation of P2X7 receptor induces calcium influx in HK-2 cells and ATP-mediated induction of PAD4 is inhibited by blocking the PKC activity. In vivo, P2X7 receptor activation selectively induces kidney PAD4 expression and further augments renal IR induced upregulation of PAD4. Finally, our studies show that a selective P2X7 receptor agonist exacerbated ischemic AKI and renal inflammation in PAD4-competent mice but not in PAD4-deficient mice, suggesting a critical role of PAD4 in P2X7 receptor-mediated exacerbation of renal injury.
PAD4 received significant clinical and scientific interest recently due to its critical role in innate immunity, infection control, and autoimmune disease (2, 5). Indeed, PAD4 activation is implicated in several autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, ulcerative colitis, and lupus. However, unlike the better characterized role for PAD4 in neutrophils responsible for the formation of neutrophil extracellular traps, the role of PAD4 in the kidney remains largely unclear. We recently showed that renal IR injury profoundly upregulates renal tubular PAD4 expression and activity, and this induction of renal PAD4 plays a major role in generating renal inflammation and injury after IR (17, 45). Renal tubular upregulation of PAD4 or exogenous recombinant PAD4 treatment induced the expression of neutrophil attracting chemokines MIP-2 and KC to exacerbate renal tubular inflammatory response after IR. Taken together, our previous studies suggest a critical role of renal tubular PAD4 in regulating renal tubular inflammation and injury after ischemic AKI. Our current study expands on our previous studies by demonstrating that ATP released by necrotic renal cells directly induces renal tubular PAD4 via P2X7 receptor activation.
Release of damage-associated molecular pattern ligands (DAMPs) after IR initiates a sterile inflammatory process via innate immune system activation (37, 47). ATP is a well-recognized DAMP molecule that rapidly initiates tissue inflammation and immune cell infiltration (39). As intracellular ATP concentration (5–10 mM) is far greater than the extracellular ATP levels (1–10 nM), IR-induced cell membrane damage rapidly releases intracellular ATP into the extracellular compartment (51, 55). ATP released by necrotic cells serves as a danger signal by recruiting inflammatory leukocytes and by promoting inflammation and cell death (47, 51). As these effects of ATP mimics the effects of renal PAD4 in exacerbating ischemic AKI, we hypothesized that extracellular ATP induces renal tubular PAD4 to enhance renal IR-induced tubular inflammation and cell death.
We used 0.5–1 mM ATP and 100–500 μM ATPγS in our studies as the human P2X7 receptor has relatively low affinity [pEC50 values ~4 (~100 μM)] for ATP and its analogs including ATPγS (10, 24, 48). Additionally, we used relatively high dose of ATP as ATP is rapidly degraded to ADP→AMP→adenosine by endogenous ectonucleotidases (21, 41). Finally, cell membrane damage can release millimolar concentrations of intracellular ATP into the extracellular compartment (51, 55). Therefore, renal proximal tubule cells can be exposed to millimolar concentrations of ATP released by dying or dead neighboring cells after renal IR injury.
Extracellular ATP subsequently binds to two distinct transmembrane anchored P2 purinergic receptor subtypes: ligand-gated ionotropic P2X receptors (P2X1-7) and metabotropic G protein-coupled P2Y receptors (P2Y1,2,4,6,11–14) (31, 49, 56). The P2X receptors (P2X7 subtype in particular) are involved in a variety of pathological responses including inflammation, tissue damage, and the graft-vs.-host disease (15, 54, 59). Our current studies suggest that ATP (most likely released by necrotic renal cells after IR injury) induces renal tubular PAD4 by activating the P2X7 receptor via PKC activation. ATP-mediated activation of P2X7 receptor is strongly supported by our findings that a selective P2X7 receptor agonist (BzATP) mimicked and a selective P2X7 receptor antagonist (A804598) prevented ATP-mediated induction of renal tubular PAD4. Moreover, our in vivo studies complement the in vitro studies to show that P2X7 receptor activation with BzATP induced renal tubular PAD4 mRNA as well as protein expression in sham-operated mice and further upregulates renal tubular PAD4 in mice subjected to renal IR (Figs. 6 and 7). Our findings support the hypothesis that ATP-mediated induction of renal tubular PAD4 promotes the detrimental renal tubular inflammatory response and death.
It is becoming increasingly clear that the P2X7 receptor plays a role in several kidney pathologies including glomerulonephritis, nephrotoxic nephritis, diabetic nephropathy, as well as renal IR injury (19, 40, 63). Indeed, genetic P2X7 receptor deficiency or P2X7 receptor antagonist treatment attenuated several acute and chronic kidney injury models, including glomerulonephritis and ischemic AKI (51, 63). Moreover, kidney P2X7 receptor expression is significantly increased in obstructive nephropathy as well as renal IR injury. Particularly relevant to our current study, Yan et al. (63) showed that a selective P2X7 receptor antagonist (A438079) provided significant protection against ischemic AKI. Moreover, P2X7 receptor inhibition attenuated the production of proinflammatory chemokines in the kidney after renal IR. In addition, they showed significant induction of renal tubular P2X7 receptor expression after renal IR.
Findings by Yan et al. (63) parallel our previous studies demonstrating genetic PAD4 deletion or pharmacological PAD4 inhibition reduced proinflammatory chemokine induction and protected against ischemic AKI (17, 45). Moreover, their findings are consistent with our current study that shows significant exacerbation of renal tubular necrosis and inflammation with a selective P2X7 receptor agonist (BzATP) treatment after ischemic AKI. Taken together, these studies support the hypothesis that P2X7 receptor activation exacerbates ischemic AKI and renal tubular inflammation via induction of renal tubular PAD4. Furthermore, as we show here that a selective P2X7 agonist, BzATP, exacerbated ischemic AKI in PAD4 wild-type mice but did not increase renal injury in PAD4-deficient mice, these results suggest that P2X7 receptor-mediated exacerbation of renal injury after IR is likely via induction of renal tubular PAD4.
Increase in intracellular calcium concentration is an early hallmark sign of cell death by necrosis that occurs after renal IR injury. Since P2X receptors are ATP-gated cation channels that induce calcium flux and since calcium is required for PAD4 induction and activation, we tested whether P2X (P2X7 in particular) receptor activation causes calcium influx (measured by Fluo-4 AM fluorescence) in human renal proximal tubule cells. Our studies show that P2X receptor activation by ATP or P2X7 agonist (BzATP) induces calcium influx in HK-2 cells and ATP enhanced calcium influx is attenuated by a specific P2X antagonist (A804598). Renal IR-induced ATP release and ATP-mediated P2X7 receptor activation may contribute to the rapid induction of intracellular calcium in renal tubular cells leading to cell death. Interestingly, calcium is a critical factor in PAD4 activation and function (61). It is possible that in addition to directly increasing the synthesis of renal tubular PAD4 enzyme, P2X7 receptor activation via increased calcium influx may also increase the activity of existing intracellular PAD4.
Our studies do not allow us to conclude whether P2X7 receptor activation in other key cell types (e.g., infiltrating neutrophils, lymphocytes, or macrophages) involved in generating renal IR injury plays a role in exacerbating ischemic AKI. Interestingly, a previous study by Arandjelovic et al. (3) showed that ATP via acting on P2X7 receptors induces PAD2 expression in mast cells to promote inflammation consistent with our findings that suggest P2X7-mediated renal tubular PAD4 induction. It remains to be determined whether P2X7 receptor-mediated induction of leukocyte and mast cell PAD2 plays a role in the pathogenesis of ischemic AKI.
We show in this study that ATP, presumably released from necrotic renal tubules after IR, induces renal tubular PAD4 to promote the kidney inflammation and injury via P2X7 receptor activation. Indeed, it is now well known that ATP promotes injury and inflammatory responses in many tissues and cell types via P2 receptor activation (51, 56). It is interesting to note that we and others have previously demonstrated several subtypes of renal P1 adenosine receptors (A1, A2a, and A2b subtypes in particular) protect against ischemic as well as septic AKI (16, 34, 35, 42). Therefore, adenosine, generated by sequential dephosphorylation of ATP, appears to counterbalance or place a “break” on the inflammatory and injury-promoting role of ATP released by necrotic cells. Therefore, whereas the inflammatory nucleotide signaling occurs via P2 purinergic receptor activation, subsequent resolution of tissue inflammation and injury appears to occur by P1 purinergic receptors via direct breakdown of ATP (8, 12).
Furthermore, some of the tissue-protective effects of P1 purinergic receptor activation is mediated via activation of highly powerful hypoxia-inducible factor (HIF) transcription factors that may counteract the maladaptive consequences of ATP-mediated P2 purinergic receptor activation (8, 11). Indeed, adenosine formed from breakdown of injurious ATP can activate HIF transcription signaling to protect against ischemia, hypoxia, and inflammatory response via P1 adenosine receptor activation (26).
Our current and previous studies support the acute inflammatory and injurious role of renal tubular PAD4 activation after ischemic AKI. However, the role of chronic PAD4 activation of renal tubules after ischemic injury remains to be determined. Recently published and elegant studies by Martinod et al. (38) suggest that chronic PAD4 activation leads to increased tissue fibrosis in mice. Indeed, they showed that mice deficient in PAD4 had reduced or even absent cardiac and pulmonary fibrosis. It would be interesting to determine in future studies whether chronic PAD4 activation in renal proximal tubules promotes kidney fibrosis after ischemic AKI.
Taken together, our study demonstrates that likely release of ATP by necrotic renal cells after IR induces the synthesis and activity of renal tubular PAD4 via P2X7 receptor activation. We also show here that P2X7 receptor-mediated exacerbation of renal injury and inflammation requires PAD4 as P2X7 receptor failed to exacerbate ischemic AKI in mice genetically deficient in PAD4. Our findings further support the hypothesis that PAD4 inhibition may play a role in preventing the devastating complications of ischemic AKI.
GRANTS
This work was supported by the Dept. of Anesthesiology, Columbia University, and in part by National Institutes of Health Grants DK-058547, GM-067081, and DK-115694 (to H. T. Lee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.R., S.J.H., and H.T.L. conceived and designed research; M.M.R., M.K., H.L., S.J.H., Y.C., and V.D.D. performed experiments; M.M.R., M.K., H.L., S.J.H., Y.C., V.D.D., and H.T.L. analyzed data; M.M.R., M.K., H.L., S.J.H., Y.C., V.D.D., and H.T.L. interpreted results of experiments; M.M.R., M.K., S.J.H., Y.C., and H.T.L. prepared figures; M.M.R. and H.T.L. drafted manuscript; M.M.R., M.K., H.L., S.J.H., Y.C., and H.T.L. edited and revised manuscript; M.M.R., M.K., H.L., S.J.H., Y.C., V.D.D., and H.T.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank K. M. Brown for technical assistance.
REFERENCES
- 1.Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm 2009: 137072, 2009. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzilotti C, Pratesi F, Tommasi C, Migliorini P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev 9: 158–160, 2010. doi: 10.1016/j.autrev.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Arandjelovic S, McKenney KR, Leming SS, Mowen KA. ATP induces protein arginine deiminase 2-dependent citrullination in mast cells through the P2X7 purinergic receptor. J Immunol 189: 4112–4122, 2012. doi: 10.4049/jimmunol.1201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aronson S, Blumenthal R. Perioperative renal dysfunction and cardiovascular anesthesia: concerns and controversies. J Cardiothorac Vasc Anesth 12: 567–586, 1998. doi: 10.1016/S1053-0770(98)90106-9. [DOI] [PubMed] [Google Scholar]
- 5.Bicker KL, Thompson PR. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 99: 155–163, 2013. doi: 10.1002/bip.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnstock G. P2X ion channel receptors and inflammation. Purinergic Signal 12: 59–67, 2016. doi: 10.1007/s11302-015-9493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 8.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol 74: 153–175, 2012. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Greef KE, Ysebaert DK, Ghielli M, Vercauteren S, Nouwen EJ, Eyskens EJ, De Broe ME. Neutrophils and acute ischemia-reperfusion injury. J Nephrol 11: 110–122, 1998. [PubMed] [Google Scholar]
- 10.Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br J Pharmacol 157: 1203–1214, 2009. doi: 10.1111/j.1476-5381.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schönfeld C, Löffler M, Reyes G, Duszenko M, Karhausen J, Robinson A, Westerman KA, Coe IR, Colgan SP. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 202: 1493–1505, 2005. doi: 10.1084/jem.20050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov 13: 852–869, 2014. doi: 10.1038/nrd4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost 97: 738–747, 2007. [PubMed] [Google Scholar]
- 14.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int 66: 486–491, 2004. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 15.Furlan-Freguia C, Marchese P, Gruber A, Ruggeri ZM, Ruf W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest 121: 2932–2944, 2011. doi: 10.1172/JCI46129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol 289: F369–F376, 2005. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 17.Ham A, Rabadi M, Kim M, Brown KM, Ma Z, D’Agati V, Lee HT. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 307: F1052–F1062, 2014. doi: 10.1152/ajprenal.00243.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis 34: 384–399, 1999. doi: 10.1016/S0272-6386(99)70375-6. [DOI] [PubMed] [Google Scholar]
- 19.Hillman KA, Burnstock G, Unwin RJ. The P2X7 ATP receptor in the kidney: a matter of life or death? Nephron, Exp Nephrol 101: e24–e30, 2005. doi: 10.1159/000086036. [DOI] [PubMed] [Google Scholar]
- 20.Hilmi IA, Damian D, Al-Khafaji A, Planinsic R, Boucek C, Sakai T, Chang CC, Kellum JA. Acute kidney injury following orthotopic liver transplantation: incidence, risk factors, and effects on patient and graft outcomes. Br J Anaesth 114: 919–926, 2015. doi: 10.1093/bja/aeu556. [DOI] [PubMed] [Google Scholar]
- 21.Idzko M, Ferrari D, Riegel AK, Eltzschig HK. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 124: 1029–1037, 2014. doi: 10.1182/blood-2013-09-402560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jablonski P, Howden BO, Rae DA, Birrell CS, Marshall VC, Tange J. An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35: 198–204, 1983. doi: 10.1097/00007890-198303000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11: 88–101, 2015. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 24.Jarvis MF, Khakh BS. ATP-gated P2X cation-channels. Neuropharmacology 56: 208–215, 2009. doi: 10.1016/j.neuropharm.2008.06.067. [DOI] [PubMed] [Google Scholar]
- 25.Jones DR, Lee HT. Perioperative renal protection. Best Pract Res Clin Anaesthesiol 22: 193–208, 2008. doi: 10.1016/j.bpa.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Ju C, Colgan SP, Eltzschig HK. Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (Berl) 94: 613–627, 2016. doi: 10.1007/s00109-016-1408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim M, Kim M, Park SW, Pitson SM, Lee HT. Isoflurane protects human kidney proximal tubule cells against necrosis via sphingosine kinase and sphingosine-1-phosphate generation. Am J Nephrol 31: 353–362, 2010. doi: 10.1159/000298339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D’Agati VD, Lee HT. Selective renal overexpression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 299: F347–F358, 2010. doi: 10.1152/ajprenal.00194.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron, Exp Nephrol 109: e102–e107, 2008. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinsey GR, Okusa MD. Expanding role of T cells in acute kidney injury. Curr Opin Nephrol Hypertens 23: 9–16, 2014. doi: 10.1097/01.mnh.0000436695.29173.de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolachala VL, Bajaj R, Chalasani M, Sitaraman SV. Purinergic receptors in gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol 294: G401–G410, 2008. doi: 10.1152/ajpgi.00454.2007. [DOI] [PubMed] [Google Scholar]
- 32.Kork F, Balzer F, Spies CD, Wernecke KD, Ginde AA, Jankowski J, Eltzschig HK. Minor postoperative increases of creatinine are associated with higher mortality and longer hospital length of stay in surgical patients. Anesthesiology 123: 1301–1311, 2015. doi: 10.1097/ALN.0000000000000891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kusch A, Hoff U, Bubalo G, Zhu Y, Fechner M, Schmidt-Ullrich R, Marko L, Müller DN, Schmidt-Ott KM, Gürgen D, Blum M, Schunck WH, Dragun D. Novel signalling mechanisms and targets in renal ischaemia and reperfusion injury. Acta Physiol (Oxf) 208: 25–40, 2013. doi: 10.1111/apha.12089. [DOI] [PubMed] [Google Scholar]
- 34.Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol 15: 102–111, 2004. doi: 10.1097/01.ASN.0000102474.68613.AE. [DOI] [PubMed] [Google Scholar]
- 35.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol 291: R959–R969, 2006. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 36.Lee YJ, Park SH, Jeung TO, Kim KW, Lee JH, Han HJ. Effect of adenosine triphosphate on phosphate uptake in renal proximal tubule cells: involvement of PKC and p38 MAPK. J Cell Physiol 205: 68–76, 2005. doi: 10.1002/jcp.20367. [DOI] [PubMed] [Google Scholar]
- 37.Leventhal JS, Schröppel B. Toll-like receptors in transplantation: sensing and reacting to injury. Kidney Int 81: 826–832, 2012. doi: 10.1038/ki.2011.498. [DOI] [PubMed] [Google Scholar]
- 38.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med 214: 439–458, 2017. doi: 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330: 362–366, 2010. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 40.Menzies RI, Tam FW, Unwin RJ, Bailey MA. Purinergic signaling in kidney disease. Kidney Int 91: 315–323, 2017. doi: 10.1016/j.kint.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Morandini AC, Savio LE, Coutinho-Silva R. The role of P2X7 receptor in infectious inflammatory diseases and the influence of ectonucleotidases. Biomed J 37: 169–177, 2014. doi: 10.4103/2319-4170.127803. [DOI] [PubMed] [Google Scholar]
- 42.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. A(2A) adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol 279: F809–F818, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 266–280, 2012. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SW, Kim M, Kim M, D’Agati VD, Lee HT. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate 1 receptor activation. Kidney Int 80: 1315–1327, 2011. doi: 10.1038/ki.2011.281. [DOI] [PubMed] [Google Scholar]
- 45.Rabadi M, Kim M, D’Agati V, Lee HT. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am J Physiol Renal Physiol 311: F437–F449, 2016. doi: 10.1152/ajprenal.00254.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol 3: 360, 2012. doi: 10.3389/fimmu.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol 22: 416–425, 2011. doi: 10.1681/ASN.2010040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulze-Lohoff E, Hugo C, Rost S, Arnold S, Gruber A, Brüne B, Sterzel RB. Extracellular ATP causes apoptosis and necrosis of cultured mesangial cells via P2Z/P2X7 receptors. Am J Physiol Renal Physiol 275: F962–F971, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Schwiebert EM, Zsembery A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim Biophys Acta 1615: 7–32, 2003. doi: 10.1016/S0005-2736(03)00210-4. [DOI] [PubMed] [Google Scholar]
- 50.Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem 114: 810–819, 2010. doi: 10.1111/j.1471-4159.2010.06809.x. [DOI] [PubMed] [Google Scholar]
- 51.Solini A, Usuelli V, Fiorina P. The dark side of extracellular ATP in kidney diseases. J Am Soc Nephrol 26: 1007–1016, 2015. doi: 10.1681/ASN.2014070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song JH, Kim M, Park SW, Chen SW, Pitson SM, Lee HT. Isoflurane via TGF-beta1 release increases caveolae formation and organizes sphingosine kinase signaling in renal proximal tubules. Am J Physiol Renal Physiol 298: F1041–F1050, 2010. doi: 10.1152/ajprenal.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefano L, Rössler OG, Griesemer D, Hoth M, Thiel G. P2X(7) receptor stimulation upregulates Egr-1 biosynthesis involving a cytosolic Ca(2+) rise, transactivation of the EGF receptor and phosphorylation of ERK and Elk-1. J Cell Physiol 213: 36–44, 2007. doi: 10.1002/jcp.21085. [DOI] [PubMed] [Google Scholar]
- 54.Sun S, Xia S, Ji Y, Kersten S, Qi L. The ATP-P2X7 signaling axis is dispensable for obesity-associated inflammasome activation in adipose tissue. Diabetes 61: 1471–1478, 2012. doi: 10.2337/db11-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trabanelli S, Ocadlíková D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, Idzko M, Di Virgilio F, Ferrari D, Lemoli RM. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol 189: 1303–1310, 2012. doi: 10.4049/jimmunol.1103800. [DOI] [PubMed] [Google Scholar]
- 56.Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal 2: pe6, 2009. doi: 10.1126/scisignal.256pe6. [DOI] [PubMed] [Google Scholar]
- 57.Tsao HK, Chiu PH, Sun SH. PKC-dependent ERK phosphorylation is essential for P2X7 receptor-mediated neuronal differentiation of neural progenitor cells. Cell Death Dis 4: e751, 2013. doi: 10.1038/cddis.2013.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Beers JJ, Zendman AJ, Raijmakers R, Stammen-Vogelzangs J, Pruijn GJ. Peptidylarginine deiminase expression and activity in PAD2 knock-out and PAD4-low mice. Biochimie 95: 299–308, 2013. doi: 10.1016/j.biochi.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 59.Vergani A, Tezza S, Fotino C, Visner G, Pileggi A, Chandraker A, Fiorina P. The purinergic system in allotransplantation. Am J Transplant 14: 507–514, 2014. doi: 10.1111/ajt.12567. [DOI] [PubMed] [Google Scholar]
- 60.Vinay P, Gougoux A, Lemieux G. Isolation of a pure suspension of rat proximal tubules. Am J Physiol Renal Fluid Electrolyte Physiol 241: F403–F411, 1981. [DOI] [PubMed] [Google Scholar]
- 61.Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta 1829: 1126–1135, 2013. doi: 10.1016/j.bbagrm.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wildeman E, Pires MM. Facile fluorescence-based detection of PAD4-mediated citrullination. ChemBioChem 14: 963–967, 2013. doi: 10.1002/cbic.201300173. [DOI] [PubMed] [Google Scholar]
- 63.Yan Y, Bai J, Zhou X, Tang J, Jiang C, Tolbert E, Bayliss G, Gong R, Zhao TC, Zhuang S. P2X7 receptor inhibition protects against ischemic acute kidney injury in mice. Am J Physiol Cell Physiol 308: C463–C472, 2015. doi: 10.1152/ajpcell.00245.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]