

Abstract
Despite doxorubicin (Dox) being one of the most widely used chemotherapy agents for breast, blood and lung cancers, its use in colon cancer is limited due to increased drug resistance and severe cardiotoxic side effects that increase mortality associated with its use at high doses. Therefore, better adjuvant therapies are warranted to improve the chemotherapeutic efficacy and to decrease cardiotoxicity. We have recently shown that aldose reductase inhibitor, fidarestat, increases the Dox-induced colon cancer cell death and reduces cardiomyopathy. However, the efficacy of fidarestat in the prevention of Dox-induced endothelial dysfunction, a pathological event critical to cardiovascular complications, is not known. Here, we have examined the effect of fidarestat on Dox-induced endothelial cell toxicity and dysfunction in vitro and in vivo. Incubation of human umbilical vein endothelial cells (HUVECs) with Dox significantly increased the endothelial cell death, and pre-treatment of fidarestat prevented it. Further, fidarestat prevented the Dox-induced oxidative stress, formation of reactive oxygen species (ROS) and activation of Caspase-3 in HUVECs. Fidarestat also prevented Dox-induced monocyte adhesion to HUVECs and expression of ICAM-1 and VCAM-1. Fidarestat pretreatment to HUVECs restored the Dox-induced decrease in the Nitric Oxide (NO)-levels and eNOS expression. Treatment of HUVECs with Dox caused a significant increase in the activation of NF-κB and expression of various inflammatory cytokines and chemokines which were prevented by fidarestat pre-treatment. Most importantly, fidarestat prevented the Dox-induced mouse cardiac cell hypertrophy and expression of eNOS, iNOS, and 3-Nitrotyrosine in the aorta tissues. Further, fidarestat blunted the Dox-induced expression of various inflammatory cytokines and chemokines in vivo. Thus, our results suggest that by preventing Dox-induced endothelial cytotoxicity and dysfunction, AR inhibitors could avert cardiotoxicity associated with anthracycline chemotherapy.
Keywords: Aldose reductase, fidarestat, doxorubicin, endothelial cells, cardiotoxicity
Graphical abstract
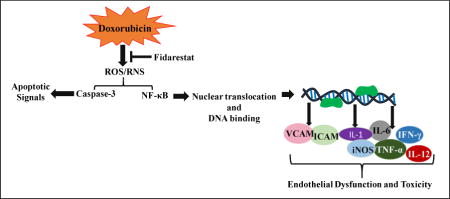
1. Introduction
Doxorubicin (Dox) is one of the highly efficient and low-cost chemotherapeutic drugs used for various types of cancer [1]. However, high doses of Dox, especially in the therapy of colon cancer, is limited due to severe cardiotoxic side effects [2–4]. Dox is known to induce apoptosis in cardiac cells through different mechanisms such as up-regulation of death receptors, pro-apoptotic proteins and reactive oxygen species (ROS) generation [5–7]. Besides being cardiotoxic, Dox-induced toxicity has also been reported in other cell types such as endothelial cells, mesenchymal stem cells, fibroblasts and hepatocytes [8–10]. Since endothelial cells are critical components of the cardiovascular system, their impairment by physical or chemical stress leads to toxic side effects which alter cardiac functions. Further, endothelial dysfunction could lead to cardiovascular complications such as atherosclerosis and restenosis. Anticancer drugs such as Dox, daunorubicin, and bleomycin have been reported to induce damage to endothelial cells and thereby contribute to the cardiotoxic side effects observed with the use of this class of chemotherapeutic drugs [11]. Although widely studied, the mechanisms of Dox-induced toxic side effects are not clearly understood [12, 13] and better preventive strategies are required to control side effects, notably cardiotoxicity.
In an attempt to prevent the toxic side effects of Dox, various antioxidant-based adjuvants and variation in Dox-formulations have gained attention in the recent past but with limited success [14–20]. Our previous studies have demonstrated that aldose reductase (AKR1B1) inhibitor, fidarestat is anti-proliferative and prevents colon cancer growth, metastasis, and angiogenesis in vivo and in vitro models [21–26]. Recently, we have demonstrated that a combination of Dox and fidarestat increases the cytotoxic efficacy of Dox in cultured human colon cancer cells and nude mice models of colon cancer xenografts [27]. Further, our studies also indicate that fidarestat prevents Dox-induced cardiac dysfunctions in mice [27]. However, the mechanism by which fidarestat decreases the cardiotoxic side effects of Dox are not well understood.
In this study, we examined the protective effect of fidarestat on Dox-induced cytotoxicity in human umbilical vein endothelial cells (HUVECs). Specifically, we have examined how fidarestat prevents Dox-induced endothelial dysfunction. Our results demonstrated that fidarestat protected HUVECs from Dox-induced endothelial toxicity in vivo and in vitro by attenuating the oxidative stress-induced signaling events. Fidarestat also prevented the Dox-induced endothelial dysfunction and inhibited the NF-kB-mediated expression of pro-inflammatory cytokines and adhesion molecules. Thus, our results suggest that fidarestat could be used in combination with Dox to prevent the cardiovascular side effects associated with Dox chemotherapy.
2. Materials and Methods
2.1. Materials
Human Endothelial Cell Complete Growth Medium (#1001) was obtained from ScienCell research laboratories. Trypsin/EDTA (#25200–056) was obtained from Invitrogen. Dulbecco’s Phosphate Buffered Saline (PBS) (#21–030-CV) was obtained from Corning Cellgro. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (#M2128) and Doxorubicin Hydrochloride (Dox; #D1515) was obtained from Sigma Aldrich. Antibodies against ICAM-1 (#SC107), VCAM-1 (#SC8304) were obtained from Santa Cruz Biotechnologies. Antibodies against eNOS (#32027), Caspase-3 (#9662), β-Actin (#4970), phospho-NF-κB (#3033) and GAPDH (#14C10) were obtained from Cell Signaling Technologies. Antibodies against iNOS (#ab129372), 4-HNE (#ab46545) and 3-Nitrotyrosine (#ab61392) were obtained from Abcam. Anti-rabbit secondary antibodies (#170–6515) and protein standard for SDS-PAGE (#161–0375) were obtained from Bio-Rad. Fidarestat was a gift from Livwel Therapeutics Inc, CA. USA. The structure of Fidarestat is reported elsewhere [28].
2.2. Cell Culture
Human umbilical vein endothelial cells (HUVECs; #8000) were obtained from ScienCell research laboratories and were cultured in the complete growth medium (#1001) containing 5% FBS, 1X endothelial cell growth supplements (ECGS) and 1X penicillin/streptomycin. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. All experiments were carried out with cells in the passage of 6–8. We have used HUVECs in our study because these cells are the most commonly used and well-characterized cell lines for in vitro studies related to cancer, angiogenesis, and endothelial function. HUVECs show a better response to treatments in vitro and are easy to grow in culture compared to other endothelial cell lines or primary cell lines.
2.3. Cytotoxicity studies
Cells were plated at a density of 3000 cells/well in 96-well plates (n = 8 for each condition) and allowed to adhere overnight. The cells were pretreated with fidarestat (30 μM) overnight followed by Dox (0.1–1 μM) for an additional 24 h. MTT assay was used to determine the cell viability at the end of incubation period. Briefly, 10 μL of 5 mg/mL MTT was added to each well of the 96-well plate and incubated for 2 h. After 2 h, the supernatant was removed, added 100 μL of DMSO, and the absorbance was recorded at 570 nm (BioTek Synergy-2 plate reader). Data is represented either as absorbance values (OD570) or percentage OD compared to control. Caspase3/7 activity was measured by using ApoLive-Glow Multiplex assay kit from Promega (#G6410). HUVECs were pre-treated with fidarestat (30μM) overnight followed by incubation with the indicated concentrations of Dox (0.5μM, 1μM, 2μM) for 24 h. Subsequently, the cells were incubated with Caspase-Glo 3/7 substrate for 30 min, and luminescence was measured using a Synergy2 BioTek plate reader.
2.4. THP-1 monocyte adhesion assay
In vitro monocyte cell adhesion assay was performed as described earlier with minor modifications [29]. Briefly, HUVECs were seeded in 96-well plates at a density of 3000 cells/well. The cells were pre-treated with fidarestat (30μM) for overnight followed by DOX (1μM). After 24 h, Calcein AM (#C3100MP, Thermo Fisher Scientific) (5μM)-loaded THP-1 cells were added to the HUVEC monolayers in a ratio of 1: 3 (HUVEC : THP-1). After 8h of incubation, the non-adherent THP-1 cells were removed by washing and photographs were taken using an EVOS fluorescence microscope. Quantification of fluorescence intensity was performed using a Synergy2 microplate reader at Ex/Em=495/516.
2.5. Analysis of oxidative stress in HUVECs
Malondialdehyde (MDA), an oxidative stress marker, levels were measured in HUVECs treated with Dox ± fidarestat by using an assay kit (#21044) from Oxis International Inc., following manufacturer’s instructions. Briefly, the cells were scrapped and homogenized in cold PBS containing 5 mM butylated hydroxytoluene (BHT) provided with the kit. The samples were centrifuged at 3000 × g for 10 min at 4 °C, and the supernatant was used for the MDA assay. Total MDA levels (μM) were determined spectrometrically (absorbance 586 nm), and the values are presented as μmoles MDA/μg of protein.
2.6. Quantification of nitric oxide levels
Nitric Oxide (NO) levels were quantified in HUVECs treated with Dox (1μM) ± fidarestat (30μM) for 24 h by using a NO-reactive fluorescent probe DAF-FM Diacetate (#D23844, Thermo Fisher Scientific). After the indicated treatments, the cells were incubated with 5μM of DAF-FM Diacetate for 15 min in HBSS, trypsinized and analyzed using a BD LSRII flow cytometer (Ex/Em:495/515nm). The data were analyzed by Flow Jo software and presented as fold change in Geometric Mean of Fluorescence Intensity (GMFI) compared to unstained control.
2.7. Measurement of ROS in HUVECs
Hydroxyl radical and peroxynitrite sensor dyes, hydroxyphenyl fluorescein (HPF) (#H36004, Thermo Fisher) and CM-H2DCFDA (#C6827, Thermo Fisher), were used to determine the levels of ROS in HUVECs. HUVECs were seeded in 100 mm tissue culture dishes at a density of 50,000 cells/cm2 and treated with 30 μM of fidarestat overnight, followed by treatment with 1 μM Dox for 1h. After the incubation, cells were stained with HPF, and CM-H2DCFDA for 15 min and fluorescence was recorded using a Synergy 2 microplate reader. (Ex/Em: 492-495/517-527 nm).
2.8. Analysis of inflammatory cytokines in HUVECs and mice aorta
Analysis of inflammatory cytokines in the supernatant of HUVEC cell cultures treated with Dox ± fidarestat was performed by using a human cytokine/chemokine magnetic bead multiplex kit from Millipore (#HCYTOMAG-60K) following manufacturer’s instructions. Briefly, after 24h of treatment, the culture media was collected and cleared by centrifugation. The media was concentrated (10x) using a vacuum evaporator, and the protein content was quantified using Bio-Rad protein assay (#500–0006). Equal amount of media was incubated with the pre-mixed beads with agitation on a plate shaker overnight at 2–8 °C. After the incubation, cells were washed with wash buffer using an automated magnetic plate washer (ELx405; Biotek) and incubated with detection antibodies for 1 h at room temperature. The wells containing detection antibodies were counterstained with Streptavidin-Phycoerythrin, incubated for another 30 min, washed, 150 μL of sheath fluid was added and analyzed with a Milliplex analyzer (Millipore). The results are expressed as pg/mL based on the standard curve generated with the standards provided with the kit using Luminex xPONENT software. Similarly as described above, cytokine levels in the mice aorta tissue homogenates were measured using mouse inflammatory multiplex kit from Millipore (#MCYTOMAG-70K).
2.9. Western Blot analysis
After the treatment, the whole cell lysates were prepared in RIPA buffer containing phosphatase and protease inhibitors (#SC-24948; Santa Cruz Biotechnologies). Equal amounts of protein were resolved in 12% SDS-PAGE gels, blotted onto PVDF membranes (#IPVH00010; EMD Millipore) and probed with antibodies for ICAM-1 (1:500), VCAM-1 (1:500), eNOS (1:1000), iNOS (1:1000), 3-Nitrotyrosine (1:1000), Caspase-3 (1:1000), β-Actin (1:1000) phospho-NF-κB(p65) (1:1000) and GAPDH (1:1000). HRP-conjugated anti-rabbit antibodies (#170–6515; 1:1000; Bio-Rad) were used as secondary antibodies. In some cases, the same blots were reprobed with different antibodies or loading controls by stripping with Restore Plus stripping buffer from Thermo Scientific (#46430). Super Signal West Pico Chemiluminescent Substrate (#34080; Thermo Scientific, USA) was used to detect the antigen-antibody complexes. Densitometry was performed using ImageJ software (NIH).
2.10. Preparation of nuclear extracts and NF-κB DNA binding assay
HUVECs were treated with fidarestat (30μM) ± Dox for the indicated time periods (0,15,30,60,120 min) and nuclear extracts were prepared with a nuclear isolation kit from Cayman Chemicals (#10009277). The nuclear isolates were used to determine the NF-κB (p65) transcription factor DNA binding using a kit from Cayman Chemicals (#10007889). Equal amounts of nuclear extracts were loaded onto coated wells followed by the addition of primary and secondary antibodies. The antigen-antibody complex formation was recorded at absorbance 450nm using a Synergy2 microplate reader.
2.11. Animal Studies
All the experiments using mice were performed following the relevant guidelines and protocols approved by Institutional Animal Care and Use Committee (IACUC), UTMB, Galveston. Wild-type C57BL/6J mice (male and female; 6–8 weeks; 19–21g approx weight) obtained from Jackson Labs (#000664) were divided into four groups (n=6). Groups: (1) control mice (no treatment), (2) fidarestat given orally in drinking water (25 mg/ kg). (3) Dox (4 mg/ kg/wk, i.p), and (4) Dox (4 mg/kg/wk, i.p) + fidarestat (25 mg/ kg). Fidarestat dose was selected based on our previous studies [27]. After 3-weeks of treatment, the animals were euthanized by CO2 asphyxiation followed by cervical dislocation, and the tissues were harvested for further analysis.
2.12. Immunohistochemical analysis
Heart tissue sections were fixed with 4% paraformaldehyde and stored in 70% ethanol. The fixed tissues were paraffin-embedded, and sections were stained with antibodies against eNOS, iNOS, 4-HNE, and 3-Nitrotyrosine. Subsequently, the antigen antibody-interactions were visualized using Dako Cytomation LSAB + System-HRP kit (#K0679, Agilent Dako).
2.13. Data availability
The data supporting the findings of this study are available within the article. All other relevant source data are available from the corresponding authors upon request.
2.14. Statistical Analysis
The data were presented as Mean± SD from at least three independent experiments, and the p values were determined using Student t-test for pairwise comparisons using the GraphPad software. p <0.05 was considered as statistically significant.
3. Results
3.1. Effect of fidarestat on Dox-induced HUVEC cell viability
We first examined the effect of Dox on HUVEC viability in the absence and presence of AR inhibitor. Our results shown in the Fig. 1A and 1B suggest that the treatment of HUVEC with Dox led to a dose-dependent decrease in cell viability at 24h and 48h. The decline in the cell viability was more at 48 h as compared to 24 h incubation. Further, pre-treatment of HUVEC with fidarestat prevented the Dox-induced decrease in the cell viability both at 24h and 48h incubation period, suggesting that fidarestat pre-treatment protects HUVECs from Dox-induced cytotoxicity. Since caspase-3 has been shown to be involved in the cytotoxicity leading to apoptosis, we next examined the effect of fidarestat on Dox-induced caspase3/7 activity in HUVECs. The data shown in the Fig. 1D indicate a dose-dependent increase in the activation of caspase-3/7 in the Dox-treated cells and pre-incubation with fidarestat prevented Dox-induced caspase-3/7 activation. These results were further confirmed by measuring the cleaved caspase 3 (17kDa) by immunoblotting. The data shown in the Fig. 1C suggest that treatment of HUVECs with Dox caused a significant increase in the cleaved caspase-3 and pre-treatment with fidarestat prevented it. These results indicate that fidarestat prevents Dox-induced cytotoxicity by blocking the caspase-3-mediated apoptotic signals in HUVECs.
Figure 1. Effect of Dox and Fidarestat on HUVEC viability.
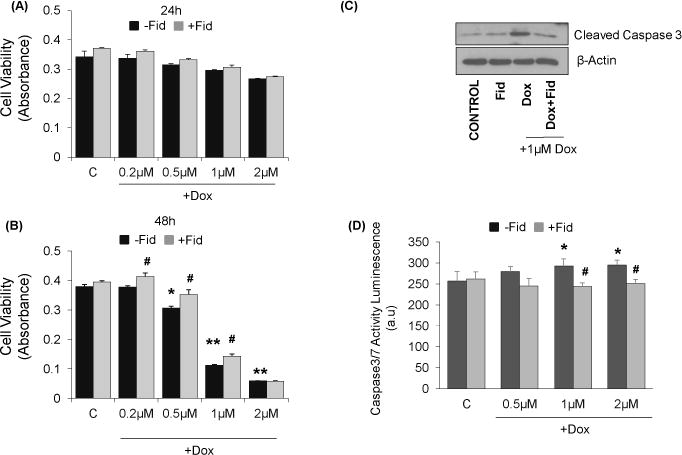
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by treatment with different concentrations of Dox (0μM, 0.2μM, 0.5μM, 1μM, 2μM) for (A) 24h and (B) 48 h. Cell viability was determined by MTT assay (n=6). *p<0.05, **p<0.005 compared to untreated control and #p<0.05 compared to Dox-alone treated cells. (C). Equal amounts of cell lysates were subjected to Western blot analysis using antibodies against Caspase-3 after treatment with Dox-alone (1μM) or in combination with fidarestat. (D) Caspase3/7 activity was measured by using ApoLive-Glow assay kit from Promega following manufacturer’s instructions. Values are mean±SD from at least three independent experiments (n =6 in each group). *p<0.05 vs control; #p<0.05 vs Dox-treated cells. Blots represent one of three independent analysis and antibodies against β-actin was used as loading control.
3.2 Fidarestat prevents Dox-induced oxidative stress, ROS, and peroxynitrite production
Chemotherapeutic drugs are well known to produce ROS and reactive nitrogen species (RNS) which induce oxidative damage in endothelial cells and contribute to drug-induced toxic side effects. Therefore, we next examined the effect of fidarestat on Dox-induced oxidative stress markers. Treatment of HUVEC with Dox increased the CM-H2DCFDA and hydroxyphenyl fluorescein (HPF) fluorescence, which was significantly prevented in HUVEC pre-treated with fidarestat followed by Dox (Fig.2A and 2B). Fidarestat alone did not affect the CM-H2DCFDA or HPF fluorescence. A significant increase in the levels of oxidative stress-induced lipid peroxidation marker, malondialdehyde (MDA) was observed in HUVECs treated with Dox, which was prevented by fidarestat (Fig 2C). Thus, these results suggest that by inhibiting oxidative stress, fidarestat could prevent Dox-induced cytotoxicity in HUVECs.
Figure 2. Effect of Fidarestat on Dox-induced oxidative stress in HUVECs.
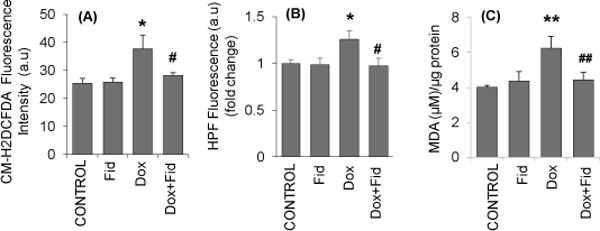
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by Dox (1μM) for 1 h. The cells were stained with 5μM (A) CM-H2DCFDA and (B) Hydroxyphenyl fluorescein (HPF) for 15 min and analyzed by fluorescence plate reader. (C) Growth-arrested HUVECs were pretreated with fidarestat (30μM) for overnight followed by Dox (1μM) for 24 h. The MDA levels were determined by using a MDA assay kit from Oxis International following the manufacturer’s instructions. Data are presented as mean ± SD from three independent experiments (n=5 in each condition). *p<0.05, **p<0.005 compared to untreated control and #p<0.05, ##p<0.005 compared to Dox-alone treated.
3.3 Fidarestat prevents Dox-induced monocyte adhesion and endothelial dysfunction
Chemotherapeutic drugs such as Dox, oxaliplatin and 5-fluorouracil have been reported to induce endothelial dysfunction and are the causative factors for systemic toxicity and in particular cardiotoxicity associated with chemotherapy [30]. However, the role of fidarestat in the prevention of Dox-induced endothelial dysfunction is not known. Therefore, we next examined the effect of fidarestat on Dox-induced monocytes adhesion to HUVECs. The data shown in the Fig. 3A indicate that there was a significant increase in the adhesion of THP-1 monocytes to Dox-treated HUVECs. Fluorescence quantification of Calcein AM loaded fluorescent THP-1 cells on HUVECs showed a substantial increase in fluorescence intensity in Dox-treated cells, which was not observed in cells pretreated with fidarestat followed by Dox (Fig.3B). MTT cell viability assay also showed a significant increase in the absorbance, an indicator of the higher number of attached THP-1 cells, in Dox-treated cells but not in the fidarestat + Dox-treated cells (Fig.3C) suggesting that AR inhibition prevents Dox-induced monocyte adhesion to endothelial cells.
Figure 3. Effect of Fidarestat on Dox-induced monocyte adhesion to HUVECs.
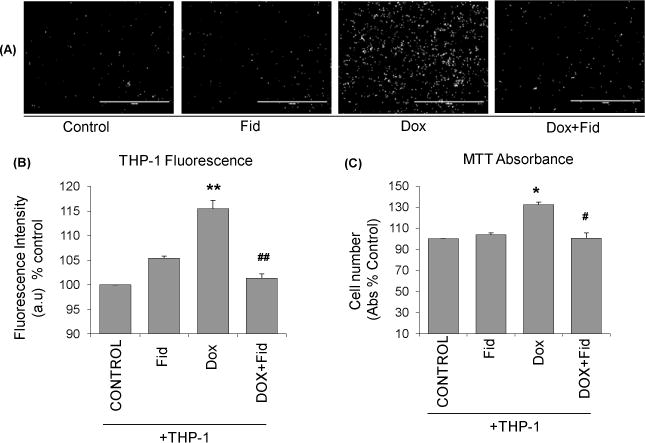
(A) HUVECs were treated with Dox (1μM) alone or Dox (1μM) + fidarestat (30μM) for overnight. Subsequently, Calcein AM -loaded fluorescent THP-1 cells were added and incubated for an additional 8 h. The non-adherent cells were removed by washing, and Calcein AM fluorescence images were captured using an EVOS fluorescence microscope. A representative image from each group is shown (n=6 in each group). Scale bar=1000μm. (B) Quantification of fluorescence intensity in wells was performed using a Synergy2 microplate reader at Ex/Em=495/516. (C) Cell viability was measured using MTT assay. Data are presented as mean ± SD from atleast 3 independent experiments (n=6 in each group). *p<0.05, **p<0.005 compared to untreated control, #p<0.5, ##p<0.005 compared to Dox-alone treated cells.
3.4 Fidarestat prevents Dox-induced expression of adhesion molecules in HUVECs
Since ICAM-1 and VCAM-1 are involved in the adhesion of monocytes to endothelial cells, we next examined the effect of fidarestat on Dox-induced ICAM-1 and VCAM-1 expression in HUVECs. Results shown in the Fig. 4A indicate a significant increase in the expression of ICAM-1 in HUVECs treated with Dox, which was prevented by fidarestat. Similarly, a significant increase in VCAM-1 expression was also observed in Dox-treated cells but not in the fidarestat + Dox-treated cells (Fig. 4B). These results suggest that by preventing the expression of adhesion molecules, fidarestat could inhibit Dox-induced monocytes adhesion to endothelial cells.
Figure 4. Effect of Fidarestat on the Dox-induced expression of adhesion molecules in HUVECs.
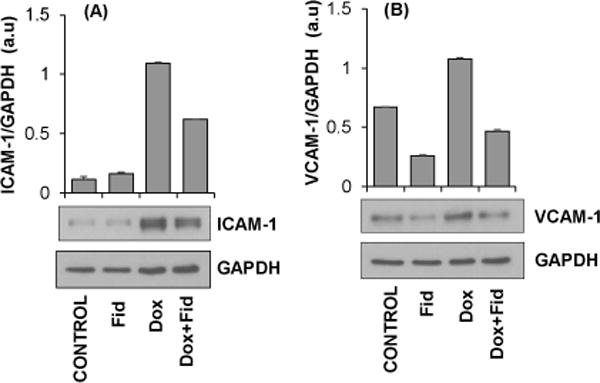
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by Dox (1μM) for an additional 24 h. Equal amounts of cell lysates were subjected to Western blot analysis using antibodies against (A) ICAM-1 and (B) VCAM-1. Blots represent one of three independent analysis and antibodies against GAPDH were used as loading control. Densitometry of blots was performed using Image J software.
3.5 Fidarestat prevents Dox-induced NO, iNOS, and eNOS in HUVECs
Since NO plays a significant role in the endothelial dysfunction, we next examined the effect of fidarestat on Dox-induced NO-levels in HUVECs. Our results shown in the Fig. 5A indicate that treatment of HUVECs with Dox decreased the NO-levels as measured by DAF-FM fluorescence. However, pretreatment with fidarestat significantly restored the Dox-induced decrease in NO levels in HUVECs. Since iNOS and eNOS contribute to NO levels in the cells, we next measured the expression of these proteins in the HUVECs treated with Dox ± fidarestat. Our results shown in the Fig. 5B and 5C indicate that the treatment of HUVECs with Dox decreased the expression of eNOS and increased the iNOS expression. However, pre-treatment of HUVECs with fidarestat prevented the Dox-induced changes in eNOS and iNOS expressions in HUVECs, suggesting that by regulating the expression of iNOS and eNOS, fidarestat restored the Dox-induced endothelial dysfunction.
Figure 5. Effect of Fidarestat on Dox-induced endothelial NO, eNOS and iNOS.
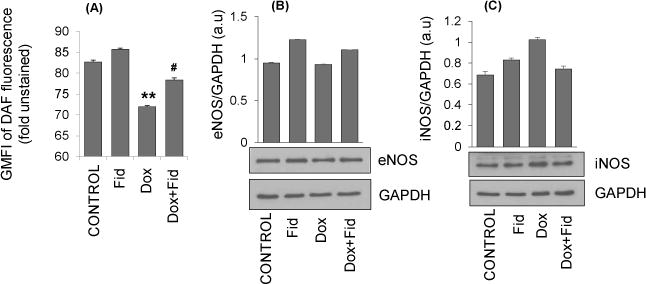
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by Dox (1μM) for an additional 24 h. (A) Cells were stained with 4,5-diaminofluorescein-2 diacetate (DAF-FM) to determine NO-levels and analyzed by flow cytometry. Data are presented as Mean ± SD of GMFI (n=3). **p<0.005 compared to untreated control and #p<0.05 compared to Dox-alone treated. Equal amounts of cell lysates were subjected to Western blot analysis using antibodies against (B) eNOS and (C) iNOS. Blots represent one of three independent analysis and antibodies against GAPDH were used as loading control. Densitometry of blots was performed using Image J software.
3.6 Fidarestat prevents Dox-induced NF-κB activation
Since NF-κB transcription factor is known to transcribe genes related to adhesion molecules, and iNOS and eNOS, we next examined the effect of fidarestat on Dox-induced NF-κB activation in HUVECs. Treatment of HUVECs with Dox caused a time-dependent increase in the nuclear translocation of NF-κB in HUVECs (Fig. 6A). However, pre-treatment with fidarestat blocked the Dox-induced nuclear translocation of NF-κB. Similarly, NF-κB transcription factor binding assay suggests a significant increase in the NF-κB DNA binding activity in Dox-treated HUVECs but not in the Dox plus fidarestat treated cells (Fig.6B).
Figure 6. Effect of Fidarestat on Dox-induced NF-κB activation in HUVECs.
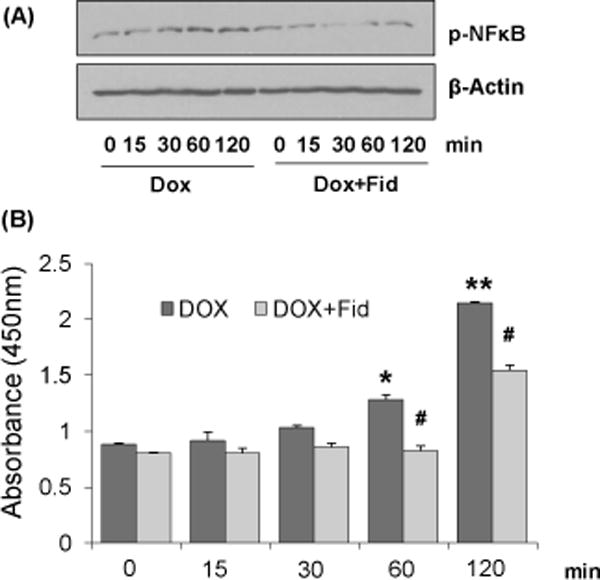
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by Dox (1μM) for the indicated time periods. (A) Equal amounts of nuclear extracts were subjected to Western blot analysis using antibodies against phospho-NF-κB (p65). Blots represent one of three independent analysis and antibodies against β-actin was used as loading control. (B) NF-κB DNA-binding activity was determined spectrophotometrically by using a kit from Cayman Chemical following manufacturer’s instructions. Data are presented as mean ± SD from at least 3 independent experiments (n=5 in each group). *p<0.05, **p<0.005 compared to untreated control (0h) and #p<0.05 compared to Dox-alone treated.
3.7 Fidarestat prevents Dox-induced endothelial toxicity in vivo
To examine the effect of fidarestat on Dox-induced endothelial cytotoxicity in vivo, we examined changes in the expression of endothelial dysfunction markers in mice administered with Dox ± fidarestat. The mice were treated with Dox-alone for 21 days (4 mg kg−1week−1 i.p) in the absence and presence of fidarestat (25 mg kg−1 in drinking water). The levels of endothelial markers were determined in the aorta tissues. A significant decrease in eNOS and increase in iNOS expression was observed in aorta tissues obtained from Dox-treated mice but not in the control or fidarestat alone or Dox+fidarestat-treated mice aortae, suggesting that fidarestat restored Dox-modulated iNOS and eNOS levels in vivo (Fig.7A). Further, a significant increase in the expression of 3-nitrotyrosine was also observed in the aorta tissues of mice treated with Dox, which was not seen in aorta tissues obtained from mice treated with Dox in combination with fidarestat (Fig. 7B). Apart from the aortic tissue, immunohistochemical analysis of formalin-fixed paraffin embedded (FFPE) heart tissue sections of mice treated with Dox showed a significant increase in the 4-hydroxynonenal, 3-nitrotyrosine, iNOS and decrease in eNOS levels (Fig. 8A–D). However, fidarestat restored the Dox-induced alterations in the levels of 4-hydroxynonenal (HNE), 3-nitrotyrosine, iNOS and eNOS in the heart tissues obtained from Dox + fidarestat- treated mice (Fig.8A–D). Hematoxylin and Eosin (H&E) staining of cardiac tissue sections showed indications of hypertrophy in Dox-treated mice, which was prevented in mice treated with Dox + fidarestat (Fig.8E). These results further confirm our in vitro studies that fidarestat prevents Dox-induced endothelial toxicity and dysfunction.
Figure 7. Effect of Fidarestat on Dox-induced eNOS and iNOS levels in mouse aorta.
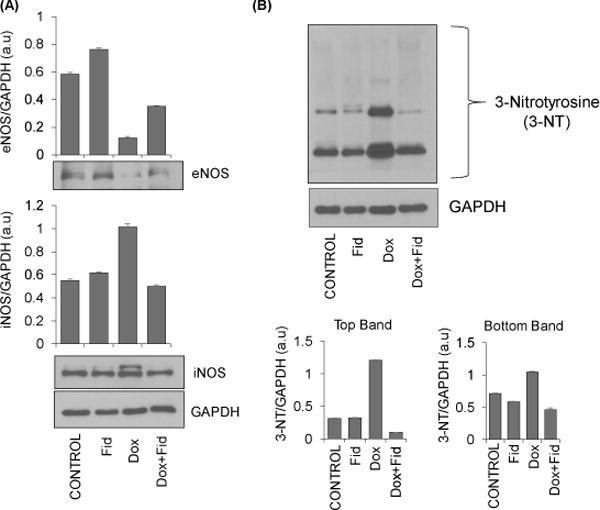
The C57BL/6J mice were treated with Dox (4 mg/kg/week) alone (i.p) or Dox (i.p) in combination with fidarestat in drinking water (25 mg/kg/day) for 21 days. The animals were sacrificed, and aortae were dissected out. Equal amounts of protein from the aorta tissue homogenates were subjected to Western blot analysis using antibodies against (A) eNOS and iNOS, and (B) 3-Nitrotyrosine. Blots represent one of three independent analysis and antibodies against GAPDH were used as loading control. Densitometry of blots was performed using Image J software.
Figure 8. Effect of Fidarestat on Dox-induced eNOS and iNOS and oxidative stress markers in mouse heart.
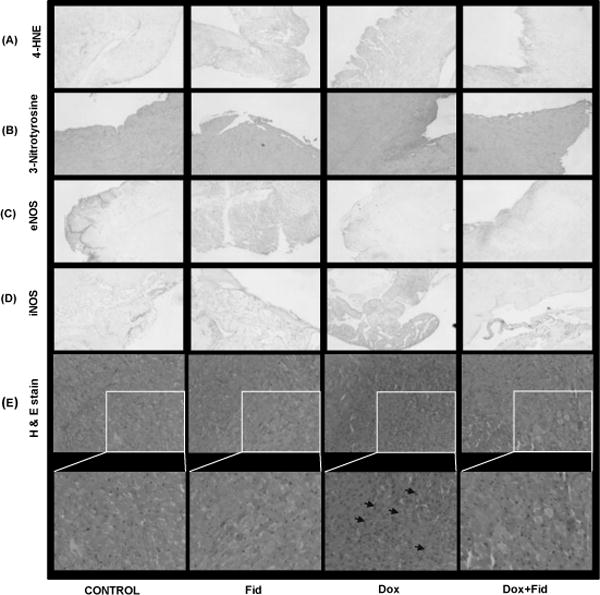
The C57BL/6J mice were treated with either Dox alone (4 mg/kg/week i.p) or Dox (i.p) along-with fidarestat (25 mg/kg/day) in drinking water for 21 days. The animals were sacrificed, heart tissues were dissected out, fixed in formalin and 5 μm sections were cut using a microtome. The sections were stained with (A) 4-HNE, (B) 3-Nitrotyrosine (C) eNOS, (D) iNOS, and (E) Hematoxylin and Eosin. Arrows indicate cellular hyperplasia in Dox-treated heart tissues. A representative image from each group is shown. Magnification 20×.
3.8 Fidarestat prevents Dox-induced inflammatory response in vitro and in vivo
We next examined the effect of fidarestat on the Dox-induced inflammatory response by measuring various inflammatory cytokines and chemokines in the HUVECs and mice aortic tissues. Data shown in the Table-1 indicate a significant increase in inflammatory cytokines such as IL-1α, IL-1β, IL-2, IL-5, IL-8, IL-9, IL-12, IFN-γ, and TNF-α, in cell culture supernatants of HUVECs treated with Dox. However, pre-treatment of HUVECs with fidarestat significantly prevented it. Similarly, a significant increase in inflammatory cytokines such as IL-1α, IL-1β, IL-5, IL-6, IL-7, IL-12, and MIP2 was observed in the aorta tissue homogenates of mice treated with Dox, but not in the Dox+fidarestat- treated mice (Table-2). These results suggest that by preventing NF-κB-mediated expression of various inflammatory markers, adhesion molecules, iNOS and eNOS, fidarestat prevents Dox-induced endothelial toxicity in vitro and in vivo.
Table 1. Effect of Fidarestat on the Dox-induced expression of inflammatory cytokines, chemokines and growth factors in HUVECs.
Growth-arrested HUVECs were pre-treated with fidarestat (30μM) for overnight followed by Dox (1μM) for additional 24 h. The levels of various inflammatory markers were determined in the cell culture media by using human cytokine/chemokine magnetic bead multiplex kit from Millipore following manufacturer’s instructions on a Milliplex system with Luminex xPONENT software. Data presented as mean ± SD from at least three independent experiments (n=3 in each condition).
| Inflammatory | Control | Fid | Dox | Dox+Fid |
|---|---|---|---|---|
|
| ||||
| markers | (pg/mL) | (pg/mL) | (pg/mL) | (pg/mL) |
| EGF | 69.2±0.04 | 89.04±2.1 | 91.2±1.2 | 79.7±0.7# |
| Eotaxin | 4.4±0.9 | 4.1±1.2 | 8.3±0.1 | 4.9±1.4## |
| FGF | 1670.1±94.01 | 1509±18.8 | 2956±444.3* | 2717.01±197.1# |
| IL-5 | 0.19±0.06 | 0.19±0.04 | 0.43±0.02* | 0.24±0.04# |
| GM-CSF | 17.2±0.03 | 18.5±0.67 | 337.2±34.2** | 236.6±2.9# |
| IL-1α | 5.6±0.4 | 3.8±0.1 | 772.4±23.9** | 558.5±23.8## |
| IL-1β | 0.9±0.2 | 0.9±0.1 | 2.6±0.18** | 2.2±0.04 |
| IL-1Ra | 14.01±0.9 | 14.4±1.4 | 27.4±4.6** | 25.3±0.5 |
| IL-10 | 1.09±0.03 | 1.3±0.03 | 3.8±0.1* | 3.1±0.2 |
| IL-12(p40) | 14.2±1.1 | 14.8±0.8 | 28.3±3.8** | 23.8±3.2 |
| IL-12(p70) | 2.02±0.1 | 2.2±0.1 | 4.5±0.4* | 3.1±0.08 |
| IL-2 | 0.8±0.1 | 0.8±0.3 | 1.4±0.2* | 1.2±0.07 |
| IF-α2 | 22.1±2.6 | 19.1±0.6 | 40.5±3.09* | 38.4±1.6 |
| IFN-γ | 5.8±0.5 | 4.2±0.1 | 16.5±1.3** | 13.8±1.1 |
| MCP-3 | 839.1±55.9 | 687.1±14.1 | 7083.8±179.2** | 5737.5±120.1## |
| IL-6 | 525.4±1.8 | 368.6±6.7 | 2540.5±88.3** | 2427.2±109.4# |
| MIP-1α | 15.2±0.4 | 15.5±0.9 | 21.004±1.9* | 18.5±0.3 |
| TGFα | 2.6±0.02 | 2.2±0.1 | 28.05±1.1** | 24.6±0.2 |
| VEGF | 148.5±17.6 | 134.01±3.06 | 293.7±9.6** | 283.3±21.4 |
| TNF-α | 0.3±0.02 | 0.2±0.03 | 1.2±0.2 | 0.9±0.01# |
| PDGF-AABB | 7268.5±131.7 | 7289.06±340 | 5642.06±267.8* | 4877.9±259.6# |
| IL-7 | 10.2±0.2 | 8.8±0.8 | 10.9±1.2* | 9.6±0.9 |
| RANTES | 11.2±0.1 | 9.3±0.2 | 120.3±2.9** | 94.1±1.9## |
| MCP-1 | 6396.9±100.6 | 6666.3±85.2 | 7041.2±7.6* | 6894.6±235.4# |
| MIP-1β | 2.2±0.6 | 2.2±2.1 | 7.04±0.2 | 4.7±1.03 |
p<0.5,
p<0.005 compared to untreated control and
p<0.05,
p<0.005 compared to Dox-alone treated.
Table 2. Effect of Fidarestat on the Dox-induced expression of inflammatory cytokines, chemokines and growth factors in mouse aorta.
The C57BL/6J mice were treated with Dox (4 mg/kg/week i.p) in the absence and presence of fidarestat (25 mg/kg in drinking water) for 21 days. The animals were sacrificed, and aortae were dissected out. The tissue lysates in each group of animals were pooled together, and equal amounts of protein in the aorta homogenates were subjected to Multiplex cytokine analysis to determine various inflammatory markers. A mouse cytokine/chemokine magnetic bead multiplex kit from Millipore was used to obtain the data following manufacturer’s instructions on a Milliplex system with Luminex xPONENT software. Data presented as mean ± SD (n=3).
| Inflammatory | Control | Fid | Dox | Dox+Fid |
|---|---|---|---|---|
|
| ||||
| markers | (pg/mL) | (pg/mL) | (pg/mL) | (pg/mL) |
| G-CSF | 1.6±0.2 | 1.9±0.05 | 3.2±0.2* | 4.7±0.2 |
| GM-CSF | 8.6±0.05 | 18.2±2.5 | 12.1±3.5 | 8.8±0.2## |
| IFN-γ | 1.6±0.1 | 5.7±1.9 | 8.8±0.4* | 8.4±0.1 |
| IL-15 | 27.9±1.07 | 51.6±9.9 | 71.3±5.7* | 73.6±0.4 |
| IL-1α | 62.5±1.9 | 88.9±17.8 | 128.7±5.4** | 80.5±18.6## |
| IL-1β | 19.9±2.6 | 21.7±0.8 | 62.5±2.6** | 7.2±0.4## |
| IL-4 | 16.6±0.2 | 13.6±0.04 | 10.4±0.2* | 16.07±0.1 |
| IL-5 | 1.7±0.3 | ND | 2.5±0.3** | 0.9±0.4## |
| IL-6 | 13.6±0.1 | 8.3±0.5 | 37.2±1.7** | 22.4±5.02## |
| IL-7 | 3.6±0.2 | 6.1±0.9 | 8.6±0.9 | 5.5±2.1 |
| IL-9 | 389.9±25.6 | 301.03±14.9 | 171.3±21.7** | 354.7±9.5 |
| IL-10 | 8.09±0.05 | 8.04±1.02 | 7.03±1.01 | 8.5±0.4 |
| IL-12(p40) | 2.2±1.9 | 5.02±1.8 | 15.6±4.3** | 3.6±0.4# |
| IP-10 | 63.9±0.5 | 106.4±3.3 | 99.4±2.8 | 37.2±1.3## |
| KC | 33.04±0.4 | 25.2±1.7 | 16.5±2.06 | 18.8±0.6 |
| MCP-1 | 14.7±0.8 | 31.06±0.6 | 22.9±1.6 | 14.8±1.06# |
| MIP-1α | 43.3±3.05 | 47.8±0.9 | 23.1±2.1* | 28.7±0.2 |
| MIP-2 | 60.05±3.1 | 68.7±5.5 | 135.1±2.7** | 74.9±0.6## |
| RANTES | 5.2±0.06 | 5.5±0.03 | 4.1±0.2 | 5.3±0.8 |
| TNF-α | 3.8±0.1 | 4.3±0.2 | 1.9±0.5 | 2.7±0.6 |
p<0.5,
p<0.005 compared to untreated control and
p<0.05,
p<0.005 compared to Dox-alone treated.
ND= not detectable.
Discussion
Vascular endothelial cells play a major role in maintaining homeostasis of the vascular system. Endothelium provides a physical barrier between the vessel wall and lumen. The endothelium secretes a number of cell signaling mediators such as inflammatory cytokines, adhesion molecules, NO and prostaglandins that regulate platelet aggregation, coagulation, fibrinolysis, and vascular tone [31–33]. Further, the endothelium plays an important role in the microvascular dysfunction that could lead to cardiovascular complications [34, 35]. We have recently shown that AR inhibitor, fidarestat, increases Dox-induced colon cancer cell death as well as prevents cardiomyopathy [27]. However, it is not known how AR inhibitor prevents Dox-induced cardiac toxicity. In this study, we demonstrate that AR inhibitor, fidarestat prevents Dox-induced endothelial cell death and dysfunction. Specifically, we found that fidarestat prevents Dox-induced endothelial cell death by inhibiting the activation of caspase-3 and prevents endothelial dysfunction by blocking the NF-κB-mediated pro-inflammatory signals as well as altering the cellular ROS/NO levels and eNOS/iNOS expressions.
Dox-induced endothelial dysfunction is one of the major contributors to cardiotoxic side-effects of Dox, possibly due to excessive ROS production. Excessive production of several species of ROS including superoxide anion (O2−) peroxynitrite anion (ONOO−), and hydroxyl radical (HO•) and subsequent activation of signaling cascades have been implicated in the toxic effects of oxidative stress [36]. Thus, understanding the impact of Dox on the oxidative stress-associated signaling pathways should be helpful in designing effective cardio-protective strategies to combat oxidative stress-induced toxic side-effects [37]. Our results demonstrate that fidarestat protects HUVECs from Dox-induced toxicity, which in part, can be attributed to attenuation of Dox-induced oxidative stress signals in HUVECs. Our results show that fidarestat prevents Dox-induced oxidative stress, the formation of ROS and peroxynitrites. Oxidative stress has been reported to be the dominant contributor and effector of Dox-induced cell death in vitro and in vivo [36]. Further, oxidative stress-generated superoxide anions (O2−) react with NO to form peroxynitrite which is known to cause cell death/apoptosis by the induction of lipid peroxidation [38, 39]. Besides peroxynitrite formation, which has many toxic side effects, decreased availability of NO causes endothelial dysfunction and affects vascular cell function [40, 41]. We have observed that fidarestat significantly decreases the Dox-induced peroxynitrite formation, caspase-3 cleavage and lipid peroxidation in HUVECs. This indicates that cytoprotective functions of fidarestat may be mediated through inhibition of oxidative stress-associated signaling pathways.
Adhesion of neutrophils to endothelial cells, loss of endothelial cell permeability, and overexpression of adhesion molecules such as ICAM-1, VCAM-1, and E-selectin have been shown to be associated with endothelial dysfunction [42, 43]. Our results show that Dox-induced induction of adhesion molecules such as ICAM-1 and VCAM-1 is significantly prevented in HUVECs pre-treated with fidarestat. Further, a significant decrease in the Dox-induced increase in the adhesion of THP-1 cells to HUVECs was also observed in Dox + fidarestat-treated cells demonstrating the ability of fidarestat to protect HUVECs from Dox-induced endothelial dysfunction. Furthermore, a Dox-induced decrease in eNOS and increase in iNOS were also significantly prevented in HUVECs pre-treated with fidarestat indicating the protective functions of fidarestat could be mediated through regulation of cellular NO levels. Similar results were also observed in aorta tissue lysates of mice treated with Dox, where fidarestat restores Dox-induced decrease in eNOS and increase in iNOS proteins. Few studies indicate that the reduction in eNOS leads to the decreased bioavailability of NO in endothelial cells which contributes to endothelial dysfunction [44, 45]. Strategies that increase the bioavailability of NO in endothelial cells have been shown to contribute to endothelial protection [46–48]. Thus, fidarestat could protect endothelial cell function by preventing decrease in eNOS and restoring the NO bioavailability.
Cytokines, chemokines and growth factors play a significant role in the pathophysiology of many human diseases including cancer [49–52]. Elevated levels of pro-inflammatory cytokines have been reported during chemotherapy of cancer patients [52–55]. Maintaining a proper balance of pro- and anti-inflammatory cytokines is an important factor necessary to overcome the systemic side effects and toxicities associated with chemotherapy [56, 57]. Since inflammatory cytokines play a major role in maintaining endothelial function; we measured various inflammatory cytokines in HUVECs and mice aortic tissues. HUVECs treated with Dox and aorta tissues obtained from Dox-treated mice showed a significant increase in the inflammatory cytokine (IL-1α, IL-1β, IL-2, IL-5, IL-8, IL-9, IL-12, IFN-γ, and TNFα) expressions. However, such an increase in inflammatory cytokines was not observed in HUVECs treated with Dox+fidarestat and also in aortic tissues obtained from Dox + fidarestat -treated mice (Table-1 and Table-2). These results indicate that fidarestat through its potent anti-inflammatory actions protects endothelial dysfunction in vitro and in vivo. Our findings are in agreement with other investigators, who have shown an increase in the expression of inflammatory cytokines and chemokines in Dox-treated mice [58, 59]. Several other investigators have also demonstrated that the prevention of Dox-induced cytokine release might be one of the potential targets to prevent Dox-induced cardiotoxicity [60–62]. In one of our recently published reports, we have demonstrated cardio-protective and chemo-adjuvant functions of fidarestat in combination with Dox. Further, we have found that Dox-induced secretion of several inflammatory cytokines was prevented in mice treated with Dox+fidarestat. Apart from protection from Dox-induced inflammatory effects, we have shown that fidarestat exerts anti-inflammatory effects in different cell types in response to various oxidant stimuli [27, 63].
NF-κB is an important mediator of inflammatory responses and mediates cellular gene expression in response to external and internal stimuli [64, 65]. Our results demonstrate that fidarestat prevents Dox-induced nuclear translocation of NF-κB and its DNA binding (Fig.6). Activation of NF-κB is reported to promote pro-apoptotic functions in endothelial cells and cardiomyocytes, whereas it has been reported to exert anti-apoptotic functions in cancer cells [66, 67]. Thus, inhibition of NF-κB activation by fidarestat and subsequent inhibition of expression of target genes and inflammatory cytokines would contribute to the cardioprotective functions of fidarestat. We have shown earlier that AR inhibition prevents activation of NF-κB signals by blocking the phosphorylation of PKC and PI3K [22,68]. Specifically, we have shown that AR- reduced lipid aldehydes and glutathione-lipid aldehydes mediate oxidative stress signals leading to activation of transcription factors such as NF-κB and AP1 [68]. Our current results suggest that AR inhibitor, fidarestat, by inhibiting Dox-induced oxidative stress signals (ROS/peroxynitrite/MDA/NO/caspase-3) and inflammatory signals (NF-κB/ICAM-1/VCAM-1/iNOS/cytokines) prevents endothelial death and dysfunction.
In conclusion, our results provide insights into the mechanisms of how AR inhibition prevents endothelial toxicity associated with Dox. Our study demonstrates that AR inhibitor, fidarestat, prevents Dox-induced endothelial cell death and dysfunction by inhibiting the Dox-induced oxidative stress as well as inflammatory responses in vitro and in vivo. These findings will help in paving ways to develop AR inhibitors such as fidarestat for adjuvant chemotherapy to increase the cytotoxic efficacy of chemotherapeutic drugs towards cancer cells and to decrease the cardiotoxic side effects.
Acknowledgments
Supported by NIH grants CA129383 and DK 104786.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: Authors declare no Conflict of Interest.
Author Contributions: H.S. performed all experiments, data analysis, and wrote the manuscript. P.B.P. helped H.S. in studies using animal models, read the manuscript and provided input. K.S. helped tissue culture, edited the manuscript and provided input. A.S. edited and provided the input on the manuscript. S.K.S designed the study along with K.V.R and edited the manuscript. K.V.R. designed the experimental plan, interpreted the data and prepared the final draft of the manuscript. K.V.R. is final approval and overall responsibility for the published work.
References
- 1.Hortobagyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs. 1997;54(Suppl 4):1–7. doi: 10.2165/00003495-199700544-00003. [DOI] [PubMed] [Google Scholar]
- 2.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Seminars in oncology. 1992;19(6):670–86. [PubMed] [Google Scholar]
- 3.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. The New England journal of medicine. 1998;339(13):900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 4.Jain D. Cardiotoxicity of doxorubicin and other anthracycline derivatives. Journal of nuclear cardiology : official publication of the American Society of Nuclear Cardiology. 2000;7(1):53–62. doi: 10.1067/mnc.2000.103324. [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. The Journal of biological chemistry. 2004;279(24):25535–43. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- 6.Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Scientific reports. 2017;7:44735. doi: 10.1038/srep44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Archivum immunologiae et therapiae experimentalis. 2009;57(6):435–445. doi: 10.1007/s00005-009-0051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cappetta D, Rossi F, Piegari E, Quaini F, Berrino L, Urbanek K, et al. Doxorubicin targets multiple players: A new view of an old problem. Pharmacological research. 2017 doi: 10.1016/j.phrs.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Oliveira MS, Carvalho JL, Campos ACDA, Gomes DA, de Goes AM, Melo MM. Doxorubicin has in vivo toxicological effects on ex vivo cultured mesenchymal stem cells. Toxicology Letters. 2014;224(3):380–386. doi: 10.1016/j.toxlet.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 10.King PD, Perry MC. Hepatotoxicity of chemotherapy. The oncologist. 2001;6(2):162–76. doi: 10.1634/theoncologist.6-2-162. [DOI] [PubMed] [Google Scholar]
- 11.Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochimica et biophysica acta. 2006;1760(2):267–71. doi: 10.1016/j.bbagen.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Wouters KA, Kremer LC, Miller TL, Herman EH, Lipshultz SE. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. British journal of haematology. 2005;131(5):561–78. doi: 10.1111/j.1365-2141.2005.05759.x. [DOI] [PubMed] [Google Scholar]
- 13.Torres VM, Simic VD. Doxorubicin-Induced Oxidative Injury of Cardiomyocytes - Do We Have Right Strategies for Prevention? In: Fiuza M, editor. Cardiotoxicity of Oncologic Treatments. InTech; Rijeka: 2012. p. Ch 5. [Google Scholar]
- 14.Jeyaseelan R, Poizat C, Wu HY, Kedes L. Molecular mechanisms of doxorubicin-induced cardiomyopathy. Selective suppression of Reiske iron-sulfur protein, ADP/ATP translocase, and phosphofructokinase genes is associated with ATP depletion in rat cardiomyocytes. The Journal of biological chemistry. 1997;272(9):5828–32. doi: 10.1074/jbc.272.9.5828. [DOI] [PubMed] [Google Scholar]
- 15.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. Journal of molecular and cellular cardiology. 2012;52(6):1213–25. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Gu J, Hu W, Zhang DD. Resveratrol, a polyphenol phytoalexin, protects against doxorubicin-induced cardiotoxicity. Journal of cellular and molecular medicine. 2015;19(10):2324–8. doi: 10.1111/jcmm.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quach B, Birk A, Szeto H. Mechanism of preventing doxorubicin-induced mitochondrial toxicity with cardiolipin-targeted peptide, SS-31 (966.1) The FASEB Journal. 2014;28(1 Supplement) [Google Scholar]
- 18.Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Experimental Hematology & Oncology. 2012;1:10–10. doi: 10.1186/2162-3619-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Razavi-Azarkhiavi K, Iranshahy M, Sahebkar A, Shirani K, Karimi G. The Protective Role of Phenolic Compounds Against Doxorubicin-induced Cardiotoxicity: A Comprehensive Review. Nutrition and cancer. 2016;68(6):892–917. doi: 10.1080/01635581.2016.1187280. [DOI] [PubMed] [Google Scholar]
- 20.Granados-Principal S, Quiles JL, Ramirez-Tortosa CL, Sanchez-Rovira P, Ramirez-Tortosa MC. New advances in molecular mechanisms and the prevention of adriamycin toxicity by antioxidant nutrients. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2010;48(6):1425–38. doi: 10.1016/j.fct.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Saxena A, Tammali R, Ramana KV, Srivastava SK. Aldose Reductase Inhibition Prevents Colon Cancer Growth by Restoring Phosphatase and Tensin Homolog Through Modulation of miR-21 and FOXO3a. Antioxidants & Redox Signaling. 2013;18(11):1249–1262. doi: 10.1089/ars.2012.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tammali R, Srivastava SK, Ramana KV. Targeting Aldose Reductase for the Treatment of Cancer. Current cancer drug targets. 2011;11(5):560–571. doi: 10.2174/156800911795655958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tammali R, Saxena A, Srivastava SK, Ramana KV. Aldose Reductase Inhibition Prevents Hypoxia-induced Increase in Hypoxia-inducible Factor-1α (HIF-1α) and Vascular Endothelial Growth Factor (VEGF) by Regulating 26 S Proteasome-mediated Protein Degradation in Human Colon Cancer Cells. The Journal of biological chemistry. 2011;286(27):24089–24100. doi: 10.1074/jbc.M111.219733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tammali R, Reddy ABM, Saxena A, Rychahou PG, Evers BM, Qiu S, et al. Inhibition of aldose reductase prevents colon cancer metastasis. Carcinogenesis. 2011;32(8):1259–1267. doi: 10.1093/carcin/bgr102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tammali R, Reddy ABM, Srivastava SK, Ramana KV. Inhibition of Aldose Reductase Prevents Angiogenesis in vitro and in vivo. Angiogenesis. 2011;14(2):209–221. doi: 10.1007/s10456-011-9206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shoeb M, Ramana KV, Srivastava SK. Aldose Reductase Inhibition Enhances TRAIL-Induced Human Colon Cancer Cell Apoptosis through AKT/FOXO3a-dependent upregulation of Death Receptors. Free radical biology & medicine. 2013;63:280–290. doi: 10.1016/j.freeradbiomed.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonowal H, Pal PB, Wen JJ, Awasthi S, Ramana KV, Srivastava SK. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. Scientific reports. 2017;7(1):3182. doi: 10.1038/s41598-017-03284-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuno K, Kato N, Makino M, Suzuki T, Shindo M. Continuous Inhibition of Excessive Polyol Pathway Flux in Peripheral Nerves by Aldose Reductase Inhibitor Fidarestat Leads to Improvement of Diabetic Neuropathy. Journal of Diabetes and its Complications. 1999;13(3):141–150. doi: 10.1016/s1056-8727(99)00038-0. [DOI] [PubMed] [Google Scholar]
- 29.Ramana KV, Bhatnagar A, Srivastava SK. Inhibition of aldose reductase attenuates TNF-alpha-induced expression of adhesion molecules in endothelial cells. FASEB Journal. 2004;18(11):1209–18. doi: 10.1096/fj.04-1650com. [DOI] [PubMed] [Google Scholar]
- 30.Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos MA, et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer treatment reviews. 2012;38(5):473–83. doi: 10.1016/j.ctrv.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Sandoo A, van Zanten JJCSV, Metsios GS, Carroll D, Kitas GD. The Endothelium and Its Role in Regulating Vascular Tone. The Open Cardiovascular Medicine Journal. 2010;4:302–312. doi: 10.2174/1874192401004010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 33.Michiels C. Endothelial cell functions. Journal of cellular physiology. 2003;196(3):430–43. doi: 10.1002/jcp.10333. [DOI] [PubMed] [Google Scholar]
- 34.Rubanyi GM. The role of endothelium in cardiovascular homeostasis and diseases. Journal of cardiovascular pharmacology. 1993;22(Suppl 4):S1–14. doi: 10.1097/00005344-199322004-00002. [DOI] [PubMed] [Google Scholar]
- 35.Hadi HAR, Carr CS, Al Suwaidi J. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vascular Health and Risk Management. 2005;1(3):183–198. [PMC free article] [PubMed] [Google Scholar]
- 36.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. Journal of molecular and cellular cardiology. 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circulation research. 2015;116(3):531–49. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay P, Rajesh M, Batkai S, Kashiwaya Y, Hasko G, Liaudet L, et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. American journal of physiology. Heart and circulatory physiology. 2009;296(5):H1466–83. doi: 10.1152/ajpheart.00795.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng S, Kruger A, Schmidt A, Metzger A, Yan T, Gödtel-Armbrust U, et al. Differential roles of nitric oxide synthase isozymes in cardiotoxicity and mortality following chronic doxorubicin treatment in mice. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2009;380(1):25–34. doi: 10.1007/s00210-009-0407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huie RE, Padmaja S. The reaction of no with superoxide. Free radical research communications. 1993;18(4):195–9. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 41.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. The American journal of physiology. 1996;271(5 Pt 1):C1424–37. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 42.Liao JK. Linking endothelial dysfunction with endothelial cell activation. The Journal of clinical investigation. 2013;123(2):540–1. doi: 10.1172/JCI66843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mestas J, Ley K. Monocyte-Endothelial Cell Interactions in the Development of Atherosclerosis. Trends in cardiovascular medicine. 2008;18(6):228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siragusa M, Fleming I. The eNOS signalosome and its link to endothelial dysfunction. Pflugers Archiv: European journal of physiology. 2016;468(7):1125–37. doi: 10.1007/s00424-016-1839-0. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Vanhoutte PM, Leung SWS. Vascular nitric oxide: Beyond eNOS. Journal of Pharmacological Sciences. 2015;129(2):83–94. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Current vascular pharmacology. 2012;10(1):4–18. doi: 10.2174/157016112798829760. [DOI] [PubMed] [Google Scholar]
- 47.Cannon RO., 3rd Role of nitric oxide in cardiovascular disease: focus on the endothelium. Clinical chemistry. 1998;44(8 Pt 2):1809–19. [PubMed] [Google Scholar]
- 48.Craige SM, Kroller-Schon S, Li C, Kant S, Cai S, Chen K, Contractor MM, et al. PGC-1alpha dictates endothelial function through regulation of eNOS expression. Scientific reports. 2016;6:38210. doi: 10.1038/srep38210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2014;1843(11):2563–2582. doi: 10.1016/j.bbamcr.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 50.Klampfer L. Cytokines, inflammation and colon cancer. Current cancer drug targets. 2011;11(4):451–464. doi: 10.2174/156800911795538066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow MT, Luster AD. Chemokines in Cancer. Cancer immunology research. 2014;2(12):1125–1131. doi: 10.1158/2326-6066.CIR-14-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nature reviews Cancer. 2008;8(11):887–899. doi: 10.1038/nrc2507. [DOI] [PubMed] [Google Scholar]
- 53.Cheung YT, Ng T, Shwe M, Ho HK, Foo KM, Cham MT, Lee JA, et al. Association of proinflammatory cytokines and chemotherapy-associated cognitive impairment in breast cancer patients: a multi-centered, prospective, cohort study() Annals of Oncology. 2015;26(7):1446–1451. doi: 10.1093/annonc/mdv206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith AK, Conneely KN, Pace TWW, Mister D, Felger JC, Kilaru V, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain, Behavior, and Immunity. 2014;38:227–236. doi: 10.1016/j.bbi.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vyas D, Laput G, Vyas AK. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets and therapy. 2014;7:1015–1023. doi: 10.2147/OTT.S60114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine. 2004;25(3):94–102. doi: 10.1016/j.cyto.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nature reviews Cancer. 2008;8(11):887–99. doi: 10.1038/nrc2507. [DOI] [PubMed] [Google Scholar]
- 58.Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the NLRP3 inflammasome. Cancer biology & therapy. 2011;11(12):1008–16. doi: 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pecoraro M, Del Pizzo M, Marzocco S, Sorrentino R, Ciccarelli M, Iaccarino G, et al. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicology and Applied Pharmacology. 2016;293(Supplement C):44–52. doi: 10.1016/j.taap.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 60.Abou El Hassan MAI, Verheul HMW, Jorna AS, Schalkwijk C, van Bezu J, van der Vijgh WJF, et al. The new cardioprotector Monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. British Journal of Cancer. 2003;89(2):357–362. doi: 10.1038/sj.bjc.6601022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruynzeel AME, Abou El Hassan MA, Schalkwijk C, Berkhof J, Bast A, Niessen HWM, et al. Anti-inflammatory agents and monoHER protect against DOX-induced cardiotoxicity and accumulation of CML in mice. British Journal of Cancer. 2007;96(6):937–943. doi: 10.1038/sj.bjc.6603640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elsea CR, Roberts DA, Druker BJ, Wood LJ. Inhibition of p38 MAPK Suppresses Inflammatory Cytokine Induction by Etoposide, 5-Fluorouracil, and Doxorubicin without Affecting Tumoricidal Activity. PLoS ONE. 2008;3(6):e2355. doi: 10.1371/journal.pone.0002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramana KV, Willis MS, White MD, Horton JW, DiMaio JM, Srivastava D, et al. Endotoxin-induced cardiomyopathy and systemic inflammation in mice is prevented by aldose reductase inhibition. Circulation. 2006;114(17):1838–46. doi: 10.1161/CIRCULATIONAHA.106.630830. [DOI] [PubMed] [Google Scholar]
- 64.Lawrence T. The Nuclear Factor NF-κB Pathway in Inflammation. Cold Spring Harbor Perspectives in Biology. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. International immunopharmacology. 2002;2(11):1509–20. doi: 10.1016/s1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 66.Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochemical Journal. 2002;367(Pt 3):729–740. doi: 10.1042/BJ20020752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Bakly WM, Louka ML, El-Halawany AM, Schaalan MF. 6-gingerol ameliorated doxorubicin-induced cardiotoxicity: role of nuclear factor kappa B and protein glycation. Cancer chemotherapy and pharmacology. 2012;70(6):833–41. doi: 10.1007/s00280-012-1975-y. [DOI] [PubMed] [Google Scholar]
- 68.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, et al. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. The Journal of biological chemistry. 2006;281(26):17652–60. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]