

Abstract
A dietary influence on cancer progression has been evident for many decades, and dietary fatty acids, particularly long chain mono- and polyunsaturated fatty acids, have been shown to play significant roles in influencing growth of a variety of human cancers. The discovery of the family of cell-surface free-fatty acid receptors, which include the long-chain fatty acid receptors FFA1 and FFA4, suggest that many of the effects of dietary fats could be receptor-mediated. FFA4 is ubiquitously expressed and has recently been shown to modulate a variety of important anti-inflammatory and metabolic processes. Since FFA4 is currently an attractive drug target for treatment of metabolic disorders such as diabetes and obesity, understanding its role in cancer progression is critical towards the drug discovery process. In this research update, the current body of knowledge on the role of this receptor in regulating cancer cell proliferation, migration, and invasion, as well as in vivo tumorigenesis is reviewed.
Keywords: FFA4, GPR120, free-fatty acids, cancer
Graphical Abstract
1. Introduction
A dietary role has long been implicated in the etiology of cell proliferation, migration, and tumorigenesis in human cancers. An assortment of previous epidemiological and clinical studies have reported potential correlations between dietary fat intake and some cancers, and biochemical characterization in cell-based studies strongly support this linkage. Together, previous studies suggest that dietary factors can influence 30% of all cancers in Western nations (1). Dietary considerations, which can include ingestion of vitamins, grains, fiber, fruits, vegetables, meats, fats, alcohol, and sugars, have been linked to both progression of cancers, as in the case of meats, fats, alcohol, and sugars; as well as prevention of cancers, as in the case of grains, fiber, fruits, fats, and vegetables. Dietary nutrients have been suggested to influence overall cancer risk and progression of nearly all organ-based cancers including, colorectal (2–4), ovarian (5–8), prostate (9–10), lung (11–13), breast (14–16), and pancreatic (17–19) cancers (20). It is now generally accepted that lifestyle, including diet, physical activity, and body weight are important contributors to cancer epidemiology.
According to the World Health Organization, the average global intake of fat has increased by 20 g per person over the last half-century (21). However, not all fats are the same and it is now well established that saturated and unsaturated fats, as well as trans fats, can act in opposing ways to influence human health, including promotion of cancers. In this manner, fatty acids are indispensable dietary substances that have vital roles as energy sources, in maintenance of cell membrane structure and function, as modulators of transcription and gene expression, as well as in regulation of intracellular signaling cascades. Free fatty acids (FFA) are organic compounds with variable linear chain lengths of 6–32 carbons and hydrophilic heads that contain a carboxylic acid. Two important considerations linking FFA structure to health implications are the chain length and the degree of saturation of the carbon chain with hydrogens. Whereas saturated fatty acids, having carbon chains that are fully saturated, have largely been linked to reduced health outcomes; monounsaturated or polyunsaturated fatty acids (MUFA or PUFA, respectively), containing either a single or multiple carbon-carbon double bonds, respectively, have generally been linked to health benefits (22).
Historically, the biologic effects of FFAs were thought to occur via their intracellular metabolism to products that modulate biological function. In the mid-2000’s, the discovery of a family of G protein-coupled receptors (GPCRs) that recognize, and are activated by variable FFAs, led to the realization that many aspects of FFA function are facilitated by these cell-surface localized receptors. This family of FFA receptors includes FFA2 and FFA3, which are agonized by short-chained fatty acids, and FFA1 and FFA4, which are agonized by medium-to-long- chained FFA (23). While FFA1-3 are more closely related, sharing nearly 40% sequence similarity, FFA1 and FFA4 are phylogenetically dissimilar, with only 10% sequence similarity. Yet, FFA1 and FFA4 are both agonized by the same grouping of endogenous fatty acid agonists, which include omega-3 PUFA, such as α-linolenic acid (ALA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), omega-6 PUFA such as linoleic acid, omega-9 MUFA such as oleic acid (OA), and saturated FA such as palmitic and myristic acids (24). Since FFA1 and FFA4 modulate responses to a wide-variety of fats that have been traditionally linked to both beneficial and detrimental health outcomes, these receptors likely mediate similarly divergent outcomes in the context of human cancers. FFA4 is expressed ubiquitously in humans and is densely localized to the lung, gastrointestinal tract, tongue, adipose, skeletal muscle, liver, bone, and macrophages, where it regulates a variety of activities including hormone secretion, taste perception, glucose uptake, and profound reduction in inflammation (reviewed in 23–25). Alternative splicing of exon 3 of the human FFA4 gene gives rise to two distinct isoforms of FFA4 protein, a short isoform (FFA4-S) that contains 361 amino acids and a long isoform (FFA4-L) that contains an additional 16 amino acids in the third intracellular loop (24,26–27). Notably, the longer isoform is not expressed in rodents or non-human primates, having been shown to exist solely in humans (28), where it is highly limited in its expression, to date, only being detected in the colon and in human-derived colon cancer cell lines (29–30).
Agonism of FFA4-S has been shown to preferentially couple to Gαq/11 proteins to facilitate increases in intracellular Ca2+, and presumably diacylglycerol, which activate the protein kinase C (PKC) signaling cascade (23–25). Interestingly, when ectopically expressed in clonal cell lines, the long isoform is incapable of inducing agonist-mediated Ca2+ signals (31). Additionally, there have been reports of tissue-specific coupling of FFA4 to Gαi/o and Gαs proteins in pancreatic delta-cells, gastric ghrelin-secreting cells, and intestinal L-cells (24, 32–34). Following agonism by endogenous or synthetic agonists, FFA4-S is rapidly phosphorylated at its C-terminus by G protein-receptor kinase 6 (GRK6), an effect that leads to robust interaction with the critical scaffolding protein β-arrestin-2 (26, 35–36). This effect has proven to be critical towards the physiological significance of FFA4, as β-arrestin-2 serves as a signaling hub that regulates a variety of important FFA-signaling outcomes, as reviewed in detail by others (24–26).
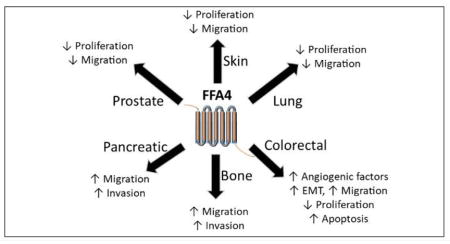
While there have been a variety of recent reviews on the signaling and physiological effects of FFA4, particularly towards its anti-inflammatory effects, the goal of this research update is to present the most recent details on the role of FFA4 in human cancers. The known contributions of FFA4 towards promotion or inhibition of cancer cell proliferation, migration, invasion and tumorigenesis are described based on cancer tissue type below and also summarized in Table 1.
Table 1.
Summary of FFA4 involvement in cancers
| Cancer and Model | Agonist Used | Effect of FFA4 agonism | KD/KO Used | KD/KO Degree | Role of FFA1 | Ref. |
|---|---|---|---|---|---|---|
| Prostate | ||||||
| DU145 Cells | DHA, EPA EPA TUG-891, EPA TUG-891, EPA |
↓ LPA and EGF-induced
Proliferation ↓ LPA and EGF-induced p-ERK1/2, p-FAK, p-p70S6K, CCN ↓ LPA, EGF, and serum-induced proliferation ↓ LPA, and EGF-induced migration |
siRNA (FFA4) | 93% KD after 24 hr 62% KD after 48 hr |
Expressed but no role in proliferation and migration | 41,44 |
| PC-3 Cells | EPA, TUG-891 | ↓ LPA, EGF, and serum-induced Proliferation and migration | siRNA (FFA4) | Not shown | Expressed but no role in proliferation and migration | |
|
| ||||||
| Colorectal | ||||||
| Human CRC tissue | None | Positive correlation with advanced histological staging | None detected | 51 | ||
| HCT116/SW480 | GW9508 | ↑ Expression of angiogenic factors,
↑ EMT and Migration ↑ p-AKT and p-IκBα |
shRNA (FFA4) | 60 – 75% reduction | None detected | 51 |
| HT-29/LOVO | EPA, DHA | ↓Proliferation and ↑
apoptosis ↑ p-YAP and cytosolic sequestration of YAP/TAZ Sensitive to inhibition of PKA |
siRNA (FFA4)/(FFA1) | 25 – 60% reduction | Expressed and contributes to effects | 53 |
| AOM/DSS-induced murine model | Dietary EPA/DHA | ↓ YAP/TAZ and ↑ p-YAP in CRC
tumors Decreased incidence, number, and size of CRC tumors |
Expressed and contributes to effects | 53 | ||
|
| ||||||
| Lung | ||||||
| A549 | GW9508 | ↓ Proliferation, ↓ Migration, ↓ MMP-2 activity | shRNA (FFA1) | Near full | Expressed, ↑ Proliferation and Migration | 63 |
| Rat RLCNR | GW9508 | No effect on proliferation or migration | None | Expressed, ↑ Migration | 63 | |
| Mouse LL/2 | GW9508 | No effect on proliferation or migration | None | Expressed, ↑ Migration | 63 | |
|
| ||||||
| Pancreatic | ||||||
| PANC-1 | GW9508 | ↑ Migration, ↓ MMP-2 activity, ↑ Invasion | shRNA (FFA4)/(FFA1) | Near full for both | Expressed, ↓ Migration | 64 |
| Hamster HPD1NR | GW9508 | No expression detected, no effect | Expressed, ↓ Migration | 64 | ||
| Hamster HPD2NR | GW9508 | ↑ Migration | None detected | 64 | ||
|
| ||||||
| Melanoma | ||||||
| A375 | GW9508 | ↓ Proliferation and Migration | shRNA (FFA4) | > 90% | Expressed, ↑ Proliferation and Migration | 68 |
| G361 | GW9509 | ↓ Proliferation and Migration | None used | Expressed, ↑ Proliferation and Migration | 68 | |
|
| ||||||
| Breast | ||||||
| MCF-7 | GW9508, TUG-891 | No specific role, likely due to FFA1 | None used | Expressed, ↓ Proliferation and Migration | 81 | |
| MDA-MB-231 | GW9508, TUG-891 | No specific role, likely due to FFA1 | None used | Visually determined | Expressed, ↓ Proliferation and Migration | 81 |
| LA | ↑ Migration and Invasion | shRNA (FFA4) | Expressed, but contribution undetermined | 79 | ||
| EPA, DHA | No effect | KO Mice | Full (genetic KO) | Expressed | 83 | |
|
| ||||||
| Bone | ||||||
| MG-63 | GW9508 | ↑ Migration and Invasion | shRNA (FFA4) | Full | Expressed, ↓ Migration and Invasion | 85 |
| COS1NR | GW9508 | ↑ Migration | shRNA (FFA4) | Not determined | Expressed, but contribution undetermined | 85 |
2. Prostate cancer cell lines
The dietary role of fats, particularly PUFAs, in the in vivo pathogenesis of human prostate carcinomas has proven to be controversial with conflicting results, yet the evidence is decidedly more stout in cell culture based models of prostate cancer. These models typically utilize PC-3 and DU145 cells that are androgen-insensitive and metastatic, as well as LNCaP cells, which are androgen-sensitive but have less migratory capacity. Long-chain omega-3 PUFA such as DHA and EPA have previously been known to reduce androgen-dependent proliferation of these cell types (37–38). In other cases, omega-6 PUFA stimulate proliferation and migration of prostate cancer cell lines, while this effect is specifically inhibited by omega-3 PUFA (39). Mice fed omega-3 PUFA-enriched diets exhibited decreased expression of proliferation-inducing genes, increased prostate apoptosis, and decreased prostatic proliferation (40). Conventionally, the mechanisms of PUFA-mediated prostate cancer inhibition were thought to occur due to intracellular metabolism of PUFA, leading to inhibition of enzymes such as cyclooxygenase and lipoxygenases, which influence cell growth and metastasis. Liu and colleagues were the first to show that both PC-3 and DU145 cell lines express FFA4, and that treatment with DHA or EPA inhibits lysophosphatidic acid (LPA)-induced proliferation in both cell lines (41). Omega-3 PUFA inhibited LPA-mediated signaling to proliferative pathways, where specifically, EPA reduced phosphorylation and subsequent activation of ERK1/2, FAK, and p70S6K, the latter of which acts via AKT signaling, and all of which are intricately involved in LPA-induced prostate cell proliferation (41). Moreover, EPA inhibited transcription and translation of CCN1, an integrin-binding matricellular protein that is critical for prostate cell adhesion, migration, proliferation, and survivability. Interestingly, the effects of omega-3 PUFA were seen to occur on the order of minutes, suggesting a cell-surface receptor mediated mechanism. Indeed, in DU145 cells, the selective synthetic FFA4 agonist TUG-891 inhibited LPA-induced proliferation with an IC50 of 73 nM, compared to a relative potency of 5.7 μM for EPA (41), and these effects were mimicked in PC-3 cells and were also replicated upon use of EGF as the mitogenic stimulus (41). The FFA1/FFA4 agonist GW9508, which displays moderate selectively for FFA1, displayed an IC50 of 950 nM, an effect much higher than its published EC50 of 50 nM for FFA1, and more consistent with its approximately 2 μM potency for FFA4 (42). Ultimately, TUG-891 displayed the highest potential for inhibition of LPA- or serum- induced proliferation of DU145 and PC-3 cells, and this work demonstrated that serum-induced proliferation resumes upon removal of TUG-891, further supporting FFA4 involvement, as the agonist must be continuously present to inhibit proliferation in the face of constant mitogenic sources (41). The same reversal was observed with EPA, which as a fatty acid, has the potential to be readily incorporated in phospholipids within prostate cancer cells to exert non-receptor mediated effects via changes in membrane fluidity and dynamics, as well as generation of intracellularly bioactive omega-3 metabolites (43). Since previous results had shown that omega-3 PUFA can modulate PC-3 cell migration (39), these authors also assessed the role of FFA4 in LPA- and EGF-mediated migration of both DU145 and PC-3 cells. In both cells lines, EPA and TUG-891 treatment significantly reduced cell migration that was induced by either LPA or EGF, to the degree that the mitogen effect was nearly fully inhibited (41). Finally, siRNA mediated knockdown (KD) of FFA4 in DU145 or PC-3 cells had no effect on LPA- or serum- induced migration in the absence of FFA4 agonists. However, FFA4 knockdown eliminated TUG-891 and EPA mediated decreases of both cell proliferation and migration that were observed in scrambled siRNA transfected DU145 and PC-3 cells at 48 and 72 hours. This suggests that FFA4 is chiefly responsible for anti-cancer effects of omega-3 PUFA in these cells (41). Interestingly, the study design involved treatment with siRNA and simultaneous drug (20 μM EPA or 1 μM TUG-891) and mitogen treatment (10 μM LPA or 10 nM EGF), a factor that allows for FFA4 agonists to initiate anti-proliferative and anti-migration effects prior to knockdown of the receptor (i.e., the receptor is still expressed at the beginning of the experiment). Yet, this element was not sufficient to inhibit proliferation over the longer time course of days, and taken together with the reversibility results mentioned previously, suggest that the inhibitory activity of FFA4 agonists requires stable and continued agonism of the receptor to oppose the continuous presence of mitogen.
In a follow up study, the same research group further characterized FFA4 involvement in DU145 and PC-3 cancer cell lines by demonstrating that the anti-proliferative and anti-migration activities of FFA4 require the presence of LPA1 receptor (LPA1R), suggesting a mechanism consistent with negative cross-talk (44). Specifically, while agonism of FFA4 by EPA and TUG-891 inhibited proliferation and migration induced by both LPA and EGF in DU145 and PC-3 cells, siRNA knockdown of LPA1R abrogated the effects of the FFA receptor (44). The effects of EPA and TUG-891 on epidermal growth-factor receptor (EGFR) signaling were also lost upon knockdown of the LPA1R, suggestive of mechanistic cross-talk within a triad between FFA4, LPA1R, and EGFR; whereby agonism of FFA4 directly inhibits LPA1R and as a consequence, inhibits LPA1R transactivation of EGFR (Figure 1). Interestingly, knockdown of β-arrestin-2 completely abrogated LPA- and EGF- induced proliferation and migration in both DU145 and PC-3 cells, but this result also confounds the ability to study the specific role of FFA4/β-arrestin-2 axis in LPA- and EGF-mediated proliferation and migration (44). While this study identified LPA1R as a critical intermediary of FFA4 effects, the direct mechanisms that modulate the FFA4-LPA1R cross-talk in prostate cancer cells remain elusive, albeit, several putative pathways were speculated upon. First, since FFA4 is well known to recruit and interact with β-arrestin-2 (24–26), its agonism may sequester the scaffolding protein away from LPA1R, thereby inhibiting its ability to modulate LPA1R signals (Figure 1). This effect would be similar to that seen in macrophages, where β-arrestin-2 recruitment subsequent to FFA4 agonism sequesters TAB1, inhibiting its downstream interactions with TAK1 and activation of the NF-κB nucleocomplex (24,45). Second, since agonism of FFA4 has been shown to rapidly activate PKC activity and the receptor itself is a substrate for PKC (36), it may be feasible that PKC activity that is induced upon FFA4 agonism facilitates direct heterologous phosphorylation of LPA1R, desensitizing its ability to signal further (Figure 1). Finally, since LPA1R has been shown to form homo- and hetero- dimers with itself and other GPCRs (46), it is conceivable that agonist-induced heterodimerization of FFA4 with LPA1R may facilitate alterations to the signal capabilities of the latter receptor (Figure 1). More research will be needed to decidedly determine the particular intracellular mechanism of action that FFA4 imparts to modulate activities of these two critical prostate mitogenic receptors. Furthermore, a specific role of FFA4 in androgen-dependent prostate cancers has not been examined and there is a lack of information on how the receptor may influence testosterone (T), its conversion by 5-α-reductase to dihydrotestosterone (DHT), the androgen receptor (AR), or downstream effects of AR on androgen response elements and transcriptional activity in these prostate cancers.
Figure 1. Known and putative mechanisms of the role of FFA4 agonism in prostate cancer cells.

Agonism of FFA4 inhibits LPA and EGF induced signaling to ERK1/2, PI3K/AKT, and FAK and inhibits LPA and EGF-mediated cell proliferation and migration in androgen-independent prostate cancer cells. The LPA1 receptor is necessary for this effect and is also partially necessary for the effects of EGF on the EGFR. The proliferative and migration effects of both LPA and EGF are dependent on LPA1 receptor recruitment and interaction with β-arrestin-2. When the LPA1 receptor is unavailable, the effects of FFA4 agonism on both LPA and EGF-events are lost, suggesting that FFA4 activation inhibits LPA1R signaling, which indirectly inhibits EGFR signaling. These known effects are indicated by solid arrows in the figure and are presumed to occur via still-elusive mechanisms that may include the ability of FFA4 agonism to sequester β-arrestin-2 from the LPA1R; to heterologously desensitize LPA1R, perhaps through Gαq/11 signals that lead to PKC-mediated phosphorylation and subsequent internalization; or to inhibit LPA1R via heterodimerization (indicated by dotted lines and question marks). Additionally, the role of FFA4 agonism in modulation of the androgen receptor or in androgen dependent prostate cancers remains unstudied.
3. Colorectal cancer cell lines and human CRC tissue
Numerous studies have suggested that omega-3 PUFAs, particularly DHA and EPA, can slow the growth of colorectal cancers (CRC) and potentiate the sensitivity to anticancer agents (47–50). However, the evidence pointing to the mechanistic role of FFA4 in modulation of these effects has to date, been conflicting. Wu and colleagues found that FFA4 expression is highly upregulated in human CRC tissue compared to adjacent non-cancerous tissue, while there was no evidence in this report for expression of FFA1 (51). Immunohistochemical analysis of 90 patient sample tissues of malignant CRC with surrounding nonmalignant tissue revealed elevated FFA4 expression in 65 of 90 patient samples, whereas 90 normal tissue samples contained low FFA4 expression, yielding a statistically significant increase of FFA4 expression in CRC tissues with p < 0.001 (51). Moreover, there was a strong clinicopathologic correlation linked to FFA4 expression in human CRC tissues and the expression of the receptor was noted to increase as the clinical stage of cancer advanced, with 100% of stage III histological grade CRCs expressing high levels of FFA4. Additionally, tumor-lymph node-metastasis (TNM) staging demonstrated a positive correlation with high levels of FFA4 expression in 35 out of 40 metastases (p = 0.004) (51). Finally, there was a significant correlation found between human CRC FFA4 expression and body weight, consistent with previous results associating FFA4 expression and obesity (52).
FFA4 expression was also noted to be upregulated in eight human CRC cell lines. Compared to two normal colon cell lines with relative one-fold expression of FFA4, CRC cell lines HCT116 (3.5-fold higher), Colo205 (3-fold), Caco-2 (2.2-fold), HT-29 (2.3-fold), RKO (2.8-fold), DLD-1 (2.9-fold), SW480 (3.2-fold), and SW620 (2.2-fold) all expressed significantly higher levels of FFA4 protein (51). Since the HCT116 and SW480 lines had highest FFA4 expression, they were studied further and noted to lack expression of FFA1 mRNA, allowing for use of GW9508 as a selective FFA4 agonist in these cells. Agonism of FFA4 with GW9508 resulted in enhanced mRNA and protein expression of CRC proangiogenic factors including VEGF, IL-8, and COX-2, and this effect was completely blocked in cells treated with FFA4 shRNA (51). Importantly, reintroduction of FFA4 into the knockdown models was sufficient to restore proangiogenic gene expression, demonstrating that the observed effects were mediated via FFA4. Conditioned media from GW9508-treated CRC cell lines stimulated growth and endothelial branching of human umbilical cord vein endothelial cells (HUVEC) and this response was lost with conditioned media retrieved from HCT116 and SW480 that expressed FFA4 shRNA (51). The effects of FFA4-mediated proangiogenic gene expression were further characterized and shown to result from FFA4-induced activation of PI3K/AKT-NF-κB signaling. This was evidenced by rapid (within 5–10 min) increases in phosphorylation of IκBα and AKT upon GW9508 stimulation, which was blocked by the PI3K inhibitor LY294002. Additionally, increased phosphorylation of IκBα and AKT was not observed upon GW9508 stimulation in the FFA4 knockdown model of HCT 116 and SW480 cells. Pretreatment with either LY294002 or NF-κB inhibitor BAY 11-7082 suppressed the GW9508 induced proangiogenic gene expression noted earlier. Finally, RNA interference of AKT and IκBα eliminated FFA4-mediated proangiogenic gene expression. The proposed CRC signaling pathway is shown in Figure 2, however, the mechanism of signal transduction (i.e., G protein or β-arrestin-2) between FFA4 and PI3K was not investigated. Based on previous studies in adipocytes that show a Gαq/11-dependency of FFA4-signaling to PI3K, it is tempting to speculate that this is the mechanism occurring to link the two proteins in CRC.
Figure 2. Proposed FFA4 signaling in human colorectal cancers.

In human HCT116 and SW480 CRC cells (left), agonism of FFA4 modulates proliferation and cell migration. Agonism of FFA4 activates the PI3K mediated phosphorylation of AKT, which facilitates phosphorylation of IκBα to activate NF-κB. Activation of NF-κB upregulates expression of proangiogenic VEGF, IL-8, and COX-2. In these cells, agonism of FFA4 also increases epithelial-mesenchymal transition (EMT) as evidenced by alterations to EMT markers E-cadherin, N-cadherin, and vimentin. FFA4-induced EMT facilitates cell migration. In these cells, the signal transducer between FFA4 and PI3K remains elusive, as are the intracellular mechanisms of FFA4-mediated EMT and cell migration. On the contrary, in human LOVO and SW480 CRC cells (right), agonism of FFA4 and FFA1 regulates LATS1 mediated phosphorylation of YAP, restricting it to the cytosol and preventing YAP-induced activation of proliferative and survival pathways. Cytosol restricted YAP inhibits proliferation and induces apoptosis. This pathway is blocked by the PKA inhibitor H-89 suggesting a role for canonical PKA/MST1/2-HIPPO signaling.
GW9508 stimulation of FFA4 in SW480 and HCT116 cells also significantly increased chemotactic capacity of the tumor cells in an in vitro transwell chemotaxis assay induced by serum, but failed to promote cell migration upon FFA4 knockdown. Migration was restored upon overexpression of FFA4 in shRNA knockdown cells. Migration of epithelial cells is dependent on loss of cell polarity and adhesion and subsequent gain of mesenchymal function, an effect termed epithelial-mesenchymal transition (EMT), which is characterized biochemically by decreases in the epithelial tumor-suppressing adhesion protein E-cadherin and correlated increases in mesenchymal markers such as vimentin, fibronectin, and N-cadherin. Importantly, treatment of HCT116 and SW480 cells with GW9508 facilitated a significant and time-dependent decrease in E-cadherin that correlated temporally with increased expression of both N-cadherin and vimentin, signifying that FFA4 agonism modulates cell migration via EMT (Figure 2). To assess the effects of FFA4 on angiogenesis and tumor growth in vivo, HCT116 cells expressing scrambled shRNA were used in a nude mouse xenograft paradigm and animals were treated intraperitoneally with 10mg/kg/day of GW9508. Tumors in these mice grew significantly faster from day six than control tumors treated with vehicle, and were four-fold larger at the 30-day period (51). Additionally, mice treated with GW9508 had higher expression of proangiogenic markers that included VEGF, IL-8, and COX-2, as well as its synthetic byproduct PGE2. Importantly, tumors generated from HCT116 cells expressing shRNA for FFA4 grew at the same rate and size in both mice treated with IP injections of GW9508 and vehicle, and lacked upregulation of VEGF, IL-8, and COX-2, demonstrating a specific role for FFA4-mediated angiogenesis in the tumor growth. Upon analyzing whether FFA4 expression correlated with angiogenesis and migration capability of CRC cell lines, it was found that higher FFA4 expression yielded greater proangiogenic gene expression and chemotactic activity. In summary, this work revealed increased FFA4 expression in human CRC tissue, correlating positively with advanced tumor staging, and signaled through the PI3K/AKT-NF-κB pathway to promote proangiogenic gene expression in CRC cell lines. Additionally, higher FFA4 expression induced proangiogenic gene expression and migration capability in CRC cell lines in vivo.
In stark contrast to this study, Zhang and colleagues demonstrated that FFA4, along with FFA1, play roles in suppressing cell proliferation and promoting apoptosis in CRC cells treated with omega-3 PUFA (53). In this study design, in vivo CRC was induced in mice upon treatment with the genotoxic colon carcinogen azoxymethane (AOM), in combination with pro-inflammatory dextran sulfate sodium (DSS), and mice were fed control diets (AIN93) or the same diet supplemented with omega-3 PUFA that included 33% EPA and 23% DHA for 11 weeks. Given this paradigm, AOM/DSS treatment induced tumors in 93% of mice fed control diets and 55% of mice fed the omega-3 supplemented diet (53), although as seen in other studies using similar fats (54), the later animals gained more weight (54). The tumors seen in omega-3 supplemented diet animals were smaller (12.4 versus 25.8 mm) and fewer in number (1.8 versus 4.2) compared to control-diet fed animals (53). To investigate the role of FFA4 in this model, the investigators utilized LOVO and HT-29 CRC cell lines, the latter of which had previously been shown to express over two-fold higher FFA4 as described above (51). Both DHA and EPA exhibited dose- and time-dependent anti-proliferative effects in both cell lines, and moreover, both omega-3 PUFAs elicited significantly higher levels of apoptosis (53).
The transcriptional regulators within the HIPPO-YAP/TAZ pathway play critical roles in cell proliferation and organ growth and development. Dysregulation of HIPPO pathway signals can also elicit initiation, growth, and metastasis of tumors, particularly CRCs (55–57). In its unphosphorylated state, YAP is localized to the nucleus where it can regulate transcription to drive proliferation and survival as well as aberrant cell growth; while phosphorylation on Ser127 facilitates its redistribution to the cytosol, where it remains sequestered and can be proteosomally degraded. GPCRs are known to modulate the HIPPO-YAP/TAZ system via Gαs/cAMP/PKA-dependent phosphorylation of upstream LATS1/2, leading to phosphorylation of YAP/TAZ and inhibition of its nuclear translocation, or alternatively, via signaling to F-actin via Rho GTPases that modulates opposing effects (58–59) (Figure 2). Based on this evidence, the effects of omega-3 PUFA on FFA4-linked HIPPO signaling was investigated and it was found that both DHA and EPA facilitate the dose- and time- dependent phosphorylation of YAP in both LOVO and HT-29 CRC cells (53). Moreover, PUFA treatment modulated cytosolic retention, rather than nuclear translocation, of YAP/TAZ in both CRC cell lines. Further study on this pathway revealed that indeed, the anti-proliferative and apoptotic effects of DHA and EPA were mediated via YAP through this canonical HIPPO pathway. Importantly, this study found that the effects of DHA and EPA were modulated via both FFA1 and FFA4 in these CRC cell lines, and that the PKA inhibitor H-89 abrogates these effects, suggesting that they are mediated via FFA1/FFA4 coupling to the Gαs/cAMP/PKA cascade (53). Finally, the research group investigated the effects of omega-3 enriched diets on YAP/TAZ in vivo using the AOM/DSS CRC model and found that YAP/TAZ were increased in CRC tissue compared to normal tissue, and that YAP and TAZ were significantly reduced in CRC tissues from animals fed omega-3 enriched diets. As seen in CRC cell lines, phosphorylated YAP was also increased in tumors of animals fed omega-3 enriched diets and these animals also expressed lower levels of YAP targeted genes related to proliferation (53).
These two studies are the only ones that delineate a role for FFA4 in CRC and offer markedly contrasting views on the role of the receptor in these cancers. Interestingly, the work of Wu and colleagues (51) did not detect expression of FFA1 in human CRC tissues while the results of Zhang did (53). Further, the results from the former study showed no expression of FFA1 in either CRC cell line used – HCT116 and SW480; while those from the latter show that LOVO and HT-29 CRC cell lines do indeed express FFA1. Hence, there are mixed observations on the expression and role of either long-chain PUFA receptor that can confound the interpretations of data that are derived, particularly given that non-selective FFA1/FFA4 agonists such as EPA, DHA, and GW9508 are used in these studies. The use of selective agonists such as TUG-891, or inclusion of the recently reported FFA4 antagonist AH7614 (60) in future studies should more clearly delineate the role of FFA4 in CRCs. This is especially important given the nature of the described coupling of FFA4 and FFA1 to Gαs rather than the predominately described Gαq/11 effects, raising questions regarding biased-signaling or functional selectivity of one or both receptors in given cell types. The other possible variable in the context of CRCs is the known expression of the FFA4-L isoform in these cells and the possible role that it plays in concert or opposition to those of the shorter isoform. A final point regarding the differences seen in the CRC studies concerns the temporal differences in study design and measure of the outcomes. Much of the work of Zhang and colleagues was done under the context of longer term stimulations with agonists (e.g., 6 hr or more), compared to those done by Wu and colleagues that assessed more rapid measures that occurred on the order of minutes to a few hours. Nonetheless, in vivo tumor progression was markedly progressed by FFA4 in one study, and markedly reduced by FFA4 in the other, so much more work remains to be done to clearly elucidate the cell biology involved.
4. Lung cancer cell lines
The greatest expression of both human and rodent FFA4 transcript has been shown to be localized to the lung (61), yet the physiological role in this tissue has been understudied, as has its involvement in lung cancers. In 2015, a two-staged genome-wide association study of 11,464 lung cancer cases and 12,206 controls from eight independent study datasets identified 30 gene variants, representing 25 independent foci, that were detected in the discovery and subsequently validated in the replication stages of the analysis. The investigators revealed that of these, a variant allele (rs12415204; NG_032670.1:g.9469C>A) of the gene encoding for FFA4 (10q23.33), representing a single nucleotide variation from C to A, had the strongest positive association with a family history of lung cancer, and also had a strong association with increased risk of lung cancer (62). Across the entirety of datasets studied, the FFA4 allele variant had a minor allele frequency ranging from 21–25% and stratified analysis by age, sex, and never-smoking status revealed a potentially increased risk of lung cancer association in females, never-smokers, and those under 50 years of age with the variant FFA4 allele. These data suggest that FFA4 single-nucleotide polymorphisms may increase risk of lung cancer in innately low-risk populations.
Kita and colleagues investigated the roles of FFA4 and FFA1 in rat RLCNR, mouse LL/2 and human A549 lung cancer cells, all three of which were shown to express both long-chain FFA receptor transcripts (63). Given the expression of both FFA4 and FFA1, the study paradigm made use of GW9508 in the absence or presence of the selective FFA1 antagonist GW1100. In cell proliferation assays, no change in growth rate was found upon stimulation with GW9508 alone or in combination with GW1100 in either RLCNR or LL/2 cells. Meanwhile, cell migration was significantly stimulated upon GW9508 treatment alone, and blocked by pretreating with GW1100, suggesting FFA1 mediated effects promoting cell migration. In A549 cells, GW9508 did not induce cell proliferation but GW1100 marginally, yet significantly, inhibited it. Furthermore, GW9508 significantly reduced cell migration in these cells, which was blocked further by GW1100, suggesting that FFA4 plays a role in inhibiting proliferation and migration, while FFA1 induces these effects (63). These results were further reinforced upon FFA1 knockdown in A549 cells, which demonstrated significantly larger GW9508-mediated inhibition of proliferation compared to their control cell line. Furthermore, investigation into the expression and activity of matrix metalloproteinase -2 (MMP-2) or MMP-9, which are known to be associated with migration and angiogenesis in cancer cells, revealed that MMP-2 was equally lowered in both FFA1-knockdown and control cells when treated with GW9508, again suggesting a role for FFA4 as the major contributor to negative regulation of migration in A549 cells (63). In summary, this work demonstrated that FFA1 positivity, while FFA4 negatively regulates tumor progression in lung cancer cells lines.
5. Pancreatic cancer cell lines
On the contrary to results that show that FFA4 agonism inhibits, while FFA1 agonism stimulates, migration of A549 lung cancer cells; in human PANC-1 ductal carcinoma cells, FFA4 agonism promotes, and FFA1 inhibits, malignant properties. These cells were noted to express both long-chain FFA receptors while the hamster pancreatic duct adenocarcinoma-derived HPD1NR and HPD2NR cells expressed only FFA1 and FFA4, respectively (64). Based on the lack of expression of both FFA receptors in the hamster cells, the authors utilized GW9508 as a selective FFA1 or FFA4 agonist in HPD1NR and HPD2NR cells, respectively. One hour of treatment with GW9508 significantly decreased cell migration in HPD1NR cells, while it significantly increased cell migration in HPD2NR cells (64). Treatment of PANC-1 cells, which express both FFA receptors with GW9508 yielded a slight but significant decrease in migration, but a marked increase in migration upon combination of the agonist with the selective FFA1 antagonist GW1100 (64). The effects of GW1100 alone were not shown but collectively, these results demonstrate that FFA1 inhibits, while FFA4 promotes, cell migration in pancreatic cancer cells. To confirm these observations selective knockdown of either FFA4 or FFA1 in PANC-1 cells using shRNA was employed and tumorigenic and invasive properties of these cells were measured, as were MMP-2 and MMP-9 activation. Cell proliferation assays revealed that FFA4-KD cells grew the same as control cells, while FFA1-KD cells showed significantly stunted growth after 72 hours (64). Cell migration was significantly inhibited by GW9508 treatment in FFA4-KD cells compared to control, suggesting that FFA1 is modulating agonist-stimulated inhibition of migration. Meanwhile, in FFA1-KD cells, cell migration was significantly increased, suggesting that FFA4 is modulating agonist-stimulated stimulation of migration (63). These results were confirmed with a wound-healing assay that showed that after 20 hours, FFA4-KD cells migrated to the wound-space to a lesser degree compared to control cells, while FFA1-KD cells migrated to a greater degree compared to control cells. As seen in A549 cells described above, assessment of MMP-2 and MMP-9 activity revealed no changes to MMP-9 but significant reduction of MMP-2 in FFA4-KD cells and significant increases in FFA1-KD cells, suggesting that FFA4 activity regulates increased MMP-2 function. While no effects on tumor growth were reported by this study, a single cell agar based colony formation assay revealed significantly heightened colony diameter from cells expressing FFA1-KD.
6. Melanoma cell lines
There is suggestive epidemiological evidence that omega-3 PUFA can be preventative against melanoma, although the mechanisms of this relationship has not been extensively investigated (65–66). A 2014 study demonstrated that DHA inhibited cell proliferation in vitro in A2058, A375, and SK-Mel 3 human skin melanoma cell lines and also revealed that DHA was efficacious in reducing tumor size in vivo, as tumors from animals fed diets enriched with DHA were 69 and 76% smaller in weight and volume, respectively, compared to those from animals fed diets enriched in omega-6 FA from coconut oil (67). GW9508 significantly reduced proliferation of all three cell lines, however, the authors found only what was described as “negligible” levels of FFA4 transcript in these cells, while FFA1 expression was abundant, suggesting that DHA and GW9508 effects seen here were solely mediated by FFA1.
Following their investigation of FFA4 and FFA1 involvement in pancreatic cancer tumorigenicity, Fukushima and colleagues subsequently studied the roles of both FFA4 and FFA1 in A375 and G361 human skin melanoma cell lines (68). Here, unlike the previous results of Nehra and colleagues (67), in addition to FFA1, FFA4 transcript was noted to be expressed in A375 cells and G361 cells, the latter of which were not used in the previous study (68). GW9508 reduced cell proliferation only in G361 cells at the higher concentration used (10 μM) and only inhibited cell migration in G361 cells. Meanwhile, GW9508 in combination with the FFA1 antagonist GW1100 strongly inhibited cell migration in both melanoma cell lines, suggesting that FFA4 modulates anti-invasive properties in these cells (68). Treatment of both cell lines with tumor promoting agent 12-O-tetradecanolphorbol-13-acetate (TPA) resulted in significantly higher cell migration in A375 and significantly lower migration in G361 cells. Quantitative real-time PCR revealed that acute TPA treatment in A375 cells induced significantly higher FFA1 expression compared to untreated cells, while FFA4 expression did not change. Meanwhile, acute TPA treatment in G361 cells induced significant elevations in both FFA4 and FFA1 transcripts (68). In A375 cells, knockdown of FFA4 decreased cell proliferation and increased cell migration, which was enhanced by GW9508 treatment. Meanwhile, GW9508 increased migration in TPA-primed cells, and this effect was amplified by FFA4-KD, suggesting that FFA4 regulates inhibitory control over cell proliferation and migration. Additionally, TPA-induced robust MMP-9 activation, which was inhibited by FFA4 KD. Together, these results shown that FFA4 and FFA1 work in opposition to respectively inhibit and enhance TPA-induced migration in melanoma cells.
7. Breast Cancer cell lines
Dietary fat intake has been closely related to epidemiological risk of breast cancer development, and higher consumption of dietary omega-3 PUFA from marine sources is associated with a lower incidence of breast cancer (69–73). Additionally, omega-3 PUFAs have been shown to inhibit growth of breast cancer cells in culture and also slow the growth of breast tumor xenografts (74–77). Prior to identification of FFA receptors, the mechanisms of omega-3 PUFA on breast cancer cells were thought to include induction of apoptosis and DNA fragmentation, as well as alterations to AKT and NF-κB signaling (77). These findings were in opposition to those that showed that omega-6 PUFA, such as linoleic acid (LA), induce inflammation, proliferation, EMT, and migration in breast cancer cell lines (78– 81). Initially, it was reported that both FFA4 and FFA1 are expressed in both tumorigenic MCF-7 breast cancer, as well as non-tumorigenic epithelial MCF10A cell lines (78), suggesting that these receptors may modulate the activities of PUFA seen in breast tissue. Previous work has also demonstrated that the omega-9 PUFA oleic acid (OA) induced proliferation, ERK1/2 phosphorylation, and activation of the transcription factor AP-1 in MCF-7 cells, but not in MCF10A cells, in a manner that was dependent on transactivation of EGFR and the non-receptor tyrosine-kinase Src (78). However, this study did not examine the direct role of FFA4 or FFA1 in the effects of OA, and only presumed that one or both of the receptors modulated these activities. Hopkins and colleagues revealed that while FFA1 transcript is present in both MCF-7 and MDA-MB-231 (MDA) cells, FFA4 transcript and protein is only present to a relatively lower degree (82). Experiments measuring the ability of TUG-891 or GW9508 to inhibit LPA-induced proliferation in breast cancer cells demonstrated that both agents inhibited the LPA-effect in a dose dependent manner. However, TUG-891 produced a shallow dose-response, consistent with activity at a non-uniform population of receptors (i.e., agonism of both FFA1 and FFA4), and also required 100-fold higher concentrations than that seen in DU145 prostate cancer cells to fully inhibit the mitogenic effects of LPA (82). Meanwhile, GW9508 demonstrated an IC50 of 16 nM at inhibiting the effect of LPA in MDA cells, consistent with its published EC50 of 50 nM. On the contrary to that seen with TUG891, this effect was 100-fold more potent in breast cancer cells as opposed to DU145, and taken together, these data suggest that FFA1, rather than FFA4, plays the major role in inhibition of LPA-induced proliferation of MDA cells. This hypothesis was consistent with observations that GW9508 also fully inhibited LPA-induced migration in MCF-7 cells (82). Chung and colleagues assessed the in vivo role of FFA4 in mediating dietary PUFA-induced reduction in mammary tumors (83). This group used orthopic injection of Py230 breast cancer cell lines into ovariectomized immune-component mice in order to stimulate diet- or genetic- induced obesity that promotes inflammation and mammary tumors. While omega-3 PUFA inhibited mammary tumor growth through induction of apoptosis, employing an elegant study design that made use of FFA4+-Py230 tumor injection into FFA−/− mice, the authors reveal that the effects seen were completely independent of FFA4 (83).
More recently, Serna-Marquez and colleagues investigated LA-induced migration and invasion of MDA-MB-231 breast cancer cells. In this study, LA was shown to induce AKT-2 phosphorylation and cell invasion and migration in MDA cells. LA-induced cell migration was inhibited by siRNA that selectively targeted FFA4 as well as by treatment of cells with the selective FFA4 antagonist AH7614 (79). Meanwhile, LA-induced invasion was fully and partially blocked by FFA4 siRNA and AH7614, respectively. While the authors speculate that the intracellular mechanisms of these effects could be due to cross-talk with FFA1 or EGFR to activate downstream NF-KB activity and transcription, mechanistic studies on the specific role of FFA4 in this process was not investigated (79).
8. Bone Cancer cell lines
FFA4 has been shown to play a pivotal role in bone development, survival and function, where it suppresses RANKL-induced osteoclast differentiation as well as osteoclastogenesis, and accelerates osteoclast apoptosis and inhibition of bone resorption (84). The same group that investigated the role of FFA1 and FFA4 on lung (63), pancreatic (64), and skin (68) cancer cells has also examined the role of the receptors in bone cancer derived cells. Expression of FFA4 and FFA1 in the MG-63 human bone osteosarcoma cell line was confirmed using RT-PCR, and the team generated MG-63 FFA4-KD cells with near full abrogation of FFA4 transcript expression for use in studies to characterize the role of both receptors. Cell migration of FFA4-KD cells was slightly lower than control cells, however, while GW9508 increased migration of control cells, it robustly inhibited migration in FFA4-KD cells (85). These findings were confirmed in rat COS1NR osteosarcoma cells and suggest that FFA4 modulates migration of osteosarcoma cells (85). The team also investigated the role of FFA4 in highly migratory MG-63 cells that were derived using an approach that utilized only migratory cells that were collected over seven migration cycles. FFA4 transcript expression was shown to be over two-fold higher in these highly migratory MG-63 cells compared to control MG-63 cells, whereas expression of FFA1 was unchanged (85). While the proliferation of highly migratory MG-63 cells was significantly lower than control after 48 hours in culture, cell migration was 200-fold greater in the highly migratory cells compared to control cells. Furthermore, the highly migratory cells showed 100-fold greater invasion through a matrigel extracellular matrix compared to control cells, an effect that was enhanced to nearly 200-fold by treatment with GW9508, which had no effect on invasion in control MG-63 cells (85). Knockdown of FFA4 in highly migratory MG-63 cells showed significant abrogation to cell migration and invasion through the extracellular matrix, which was further reduced by GW9508 treatment. These results show that FFA1 and FFA4 play important, and contrasting, roles in migration and invasion of osteosarcoma cells. Whereas FFA1 seems to negatively regulate migration and invasion, FFA4 positively influences these affects. Intracellular signaling mechanisms weren’t addressed in this study, and no insight into the level of reduction of FFA4 in highly migratory cells was provided. Since these cells exhibited a 2-fold increase in expression of the receptor, the efficacy of FFA4 knockdown would provide a clearer picture into interpretations on the absolute role of agonism of the FFA receptors. Nonetheless, it is clear that FFA4 plays a role in influencing migratory taxis in osteosarcoma cells and will require further study.
9. Chemotherapy resistance
A major barrier to effective cancer treatment is inducible resistance to chemotherapeutics, leading to pharmacotherapeutic failure and cancer relapse and progression. A seminal study in 2011 showed that chemotherapy with platinum-based agents such as cisplatin causes activation of mesenchymal stem cells (MSC) that facilitates chemotherapy resistance (i.e., chemoresistance) (86). In MSC, platinum based chemotherapeutics were shown to induce signaling to cyclooxygenase-1 (COX1) and thromboxane synthase (TXAS) to produce two distinct PUFA, 12-S-hydroxy-5,8,10-heptadecatrienoic acid (12-S-HHT) and hexadeca-4,7,10,13-tetraenoic acid (16:4(n-3)), that in turn, directly mediate platinum-induced chemoresistance at picomolar concentrations (86) (Figure 3). A follow up study by the same research team showed that dietary fish oils, particularly those obtained from mackerel and herring, but also those from salmon and tuna, contained physiologically relevant levels of 16:4(n-3), and that six commercial dietary fish oil supplements were found to contain substantial levels of 16:4(n-3), ranging in concentration from 0.2 to 5.7 μM (87). Importantly, while fish oil monotherapy did not influence tumor size, even picomolar quantities of fish oil induced full cisplatin resistance in tumor bearing mice (87). Finally, ingestion of recommended daily allowances of fish oil in 30 human volunteers facilitated observable levels of plasma 16:4(n-3), which was atypically higher than that found in the oral dose, suggesting metabolism of other fish oil fatty acids to 16:4(n-3) (87). In an elegant and substantial study that sought to further characterize the molecular mechanisms of 12-S-HHT and 16:4(n-3) induced chemotherapy resistance, the same group showed that either splenectomy or clodronate-induced depletion of macrophages prevented the chemoresistance induced by the PUFA, demonstrating that the mechanism specifically involves splenic macrophages (88). Further results showed that 12-S-HHT functions via activation of the low-affinity leukotriene B4 receptor-2 (BLT2) residing on splenic F4/80+/CD11blow macrophages to release lysophosphatidylcholines (LPC) that in turn, alter the ability of platinum-chemotherapeutics to induce DNA damage (88). Notably, the other platinum-induced fatty acid, 16:4(n-3), was shown in this work to not engage BLT2, leaving its specific molecular target unresolved at that time.
Figure 3. Mechanism of FFA4-mediated chemoresistance to the platinum-induced PUFA 16:4(n-3).

Mesenchymal stem cells (MSC) localized to the tumor microenvironment produce 16:4(n-3) PUFA in response to platinum-based chemotherapeutics such as cisplatin via an intracellular signaling mechanism that is dependent on Ca+2, PLA2, thromboxane synthase (TXAS), and cyclooxygenase-1 (COX1) (86). 16:4(n-3) that is released by MSC agonizes FFA4 on splenic macrophages (F4/80+/CD11blow in mice, presumably CD163+ in humans) to activate cytosolic PLA2 via an unresolved pathway that is likely Gαq/11/Ca+2-mediated. FFA4-induced activation of cPLA2 forms the lysophosphatidylcholine LPC(24:1), which directly alters the ability of platinum-based chemotherapeutics to mediate DNA damage responses in cancer cells, leading to chemoresistance to the agents (88–89).
In early 2017, a collaborative effort between the group that pioneered the above described evidence of platinum-induced PUFA chemotherapy resistance and a group that has contributed to much of the known molecular pharmacology of FFA4, revealed that the molecular target of 16:4(n-3), which affords chemotherapy resistance, is indeed FFA4 (89). In a similar manner as that seen for 12-S-HHT/BLT2, it was shown that 16:4(n-3) agonizes FFA4 on the surface of F4/80+/CD11blow macrophages to modulate this effect. Interestingly and as may be expected, in clonal cell models, 16:4(n-3) activated both Gαq/11 as well as β-arrestin-2 linked signaling to both FFA4 and FFA1, demonstrating that it is an agonist at both long-chain fatty acid receptors (89). In a well-designed model utilizing conditioned media from mouse splenic cells treated with 16:4(n-3), GW9508, or TUG-891, it was shown that tumor xenografts that were markedly reduced in volume upon treatment with cisplatin, were insensitive to the effects of the platinum agent when it was co-administered with the splenic conditioned media (sCM), suggesting an important role for the FFA receptors in chemotherapy resistance in vivo (89). Further studies using the selective antagonists of FFA4 and FFA1, AH7614 and GW1100, respectively, showed that 16:4(n-3)-induced chemoresistance was modulated by FFA4 and not FFA1, as tumor volume was significantly decreased by cisplatin/16:4(n-3) + AH7614 sCM but not by cisplatin/16:4(n-3) + GW1100 sCM (89). These results were confirmed to be FFA4 mediated and specific to 16:4(n-3), as sCM derived from FFA4−/− mice was incapable of inducing 16:4(n-3) chemoresistance, while it had no effect on 12-S-HHT chemoresistance, which as noted previously, occurs via agonism of BLT2. While F4/80−/CD11blow, F4/80+/CD11bhigh, and F4/80−/CD11bhigh macrophage subpopulations did not appear to express FFA4, the effects of 16:4(n-3) were shown to occur via agonism of FFA4 localized to F4/80+/CD11blow splenic macrophages, which expressed FFA4 and FFA1, in addition to the short-chain fatty acid receptors FFA2 and FFA3 (89). Within F4/80+/CD11blow cells, agonism of FFA4 by 16:4(n-3) was shown to cause a rapid phosphorylation and increases in activity of cytosolic phospholipase A2 (cPLA2), which was blocked by the FFA4 antagonist AH7614, and was not reproduced in other macrophages (89). Mechanistically, FFA4-mediated cPLA2 activity facilitated the generation of a 24:1 unsaturated lysophosphatidylcholine (LPC24:1), which directly modulates chemoresistance (88–89) (Figure 3). The specific role of this FFA4-cPLA2-LPC24:1 axis was confirmed using sCM from FFA4−/− mice, pharmacological inhibition of cPLA2, and direct administration of LPC24:1 (89). Finally, the research team demonstrated that human CD163+ splenocytes have heightened expression of FFA4, similar to that seen with orthologous murine F4/80+ cells, and that humans subjected to four hours of platinum-based chemotherapy had significantly higher levels of plasma LPC24:1 compared to those that had undergone non-platinum based therapies, suggesting that this mechanism is directly translatable to human chemoresistance.
10. Concluding Remarks
Since the first study on the role of FFA4 in cancer cells was published by Wu and colleagues in 2013 (51), many other reports have been published, establishing a linkage to either promotion or inhibition of proliferation and migration of prostate, colorectal, lung, pancreatic, skin, and breast cancers, as well as chemoresistance. As described here, many of these cancer cells express both long-chain FFA receptors. Given that many of the initial studies made use of GW9508, an agonist with more selectively for FFA1, but which also agonizes FFA4, interpretation of the effects in these cells is somewhat cloudy. The utilization of siRNA/shRNA-based approaches does shed light on the role of FFA4 in these studies, however, since some of the findings do not report the efficacy of FFA4-knockdown, again, it is difficult to pinpoint a definitive role for FFA4 in the case that FFA1 is present and non-selective pharmacological agents are used. Since many of the studies also present evidence that the two FFA receptors have opposing actions on proliferation, migration, and or invasion, it is conceivable that loss of activity of one receptor via knockdown approaches may dysregulate the opposing balance, particularly when non-selective agonists are used to compare effects in control cells. Further work will be needed using more selective agonists (e.g., TUG-891, cpdA) as well as antagonists, such as AH7614, whose activity was first noted in 2014, to more clearly define the role for FFA4 in cancers. While the studies performed thus far delineate involvement of FFA4 in many of these cancers, the detailed intracellular cascades at play in each cancer type, and likely even each cell type, will also require further study in order to understand the mechanisms of cell signaling that link FFA4 to its anti- or pro- cancerous effects in each given tissue. Since FFA4 is agonized by a variety of medium-to-long-chain fatty acids, including MUFA, omega-3, -6, and -9 PUFA, as well as saturated fats, all of which have historically been described either as “good” or “bad” fats, it is difficult at this point to link the receptor to a definitive benefit or risk towards cancer signaling. Given the possibility and context of these fats inducing agonist-directed functional selectivity via FFA4, further study will be needed to conclusively expound the role of the receptor in these and other cancers, and to elucidate the cellular biology employed by receptor activation.
Acknowledgments
This work was supported by NIH grant DK098730 to NHM.
Abbreviations
- FFA
Free-fatty acid
- PUFA
polyunsaturated fatty acid
- DHA
docosahexaenoic acid
- EPA
Eicosapentaenoic acid
- OA
Oleic acid
- LA
Linoleic acid
- LPA
1-oleoyl lysophosphatidic acid
- LPC
lysophosphatidylcholine
- EGFR
Epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
Footnotes
Chemical compounds studied in this article: GW9508 (CID 11595431), TUG-891 (57522038), AH7614 (233085), GW1100 (11692123), DHA (445580), EPA (446284)
AUTHOR CONTRIBUTIONS: ISS and NHM wrote the manuscript.
CONFLICT OF INTERESTS: Neither author has any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Instit. 1981;66:1191–308. [PubMed] [Google Scholar]
- 2.Lin PH, Aronson W, Freedland SJ. Nutrition, dietary interventions and prostate cancer: the latest evidence. BMC Med. 2015;13(3):1–5. doi: 10.1186/s12916-014-0234-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vargas AJ, Thompson PA. Diet and Nutrient Factors in Colorectal Cancer Risk. Nutr Clin Pract. 2012;27:613–623. doi: 10.1177/0884533612454885. [DOI] [PubMed] [Google Scholar]
- 4.Kunzmann AT, Coleman HG, Huang WY, Kitahara CM, Cantwell MM, Berndt SI. Dietary fiber intake and risk of colorectal cancer and incident and recurrent adenoma in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Am J Clin Nutr. 2015;102(4):881–90. doi: 10.3945/ajcn.115.113282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kushi LH, Mink PJ, Folsom AR, Anderson KE, Zheng W, Lazovich D, Sellers TA. Prospective study of diet and ovarian cancer. Am J Epidemiol. 1999;149(1):21–31. doi: 10.1093/oxfordjournals.aje.a009723. [DOI] [PubMed] [Google Scholar]
- 6.Mommers M, Schouten LJ, Goldbohm RA, van den Brandt PA. Dairy consumption and ovarian cancer risk in the Netherlands Cohort Study on Diet and Cancer. Br J Cancer. 2006;94(1):165–70. doi: 10.1038/sj.bjc.6602890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsson SC, Bergkvist L, Wolk A. Milk and lactose intakes and ovarian cancer risk in the Swedish Mammography Cohort. Am J Clin Nutr. 2004;80(5):1353–57. doi: 10.1093/ajcn/80.5.1353. [DOI] [PubMed] [Google Scholar]
- 8.Larsson SC, Holmberg L, Wolk A. Fruit and vegetable consumption in relation to ovarian cancer incidence: the Swedish Mammography Cohort. Br J Cancer. 2004;90(11):2167–70. doi: 10.1038/sj.bjc.6601872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristal AR, Arnold KB, Neuhouser ML, Goodman P, Platz EA, Albanes D, Thompson IM. Diet, supplement use, and prostate cancer risk: results from the prostate cancer prevention trial. Am J Epidemiol. 2010;172(5):566–77. doi: 10.1093/aje/kwq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballon-Landa E, Parsons JK. Nutrition, physical activity, and lifestyle factors in prostate cancer prevention. Curr Opin Urol. 2018 Jan;28(1):55–61. doi: 10.1097/MOU.0000000000000460. [DOI] [PubMed] [Google Scholar]
- 11.Vieira AR, Abar L, Vingeliene S, Chan DS, Aune D, Navarro-Rosenblatt D, Stevens C, Greenwood D, Norat T. Fruits, vegetables and lung cancer risk: a systematic review and meta-analysis. Ann Oncol. 2016 Jan;27(1):81–96. doi: 10.1093/annonc/mdv381. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Qin S, Zhang T, Song X, Zhang S. The effect of fruit and vegetable intake on the development of lung cancer: a meta-analysis of 32 publications and 20,414 cases. Eur J Clin Nutr. 2015 Nov;69(11):1184–92. doi: 10.1038/ejcn.2015.64. [DOI] [PubMed] [Google Scholar]
- 13.Wakai K, Sugawara Y, Tsuji I, Tamakoshi A, Shimazu T, Matsuo K, et al. Risk of lung cancer and consumption of vegetables and fruit in Japanese: A pooled analysis of cohort studies in Japan. Cancer Sci. 2015;106(8):1057–65. doi: 10.1111/cas.12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang YJ, Hou YC, Chen LJ, Wu JH, Wu CC, Chang YJ, Chung KP. Is vegetarian diet associated with a lower risk of breast cancer in Taiwanese women? BMC Public Health. 2017;17(1):800. doi: 10.1186/s12889-017-4819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deschasaux M, Julia C, Kesse-Guyot E, Lécuyer L, Adriouch S, Méjean C, et al. Are self-reported unhealthy food choices associated with an increased risk of breast cancer? Prospective cohort study using the British Food Standards Agency nutrient profiling system. BMJ Open. 2017;7(6):e013718. doi: 10.1136/bmjopen-2016-013718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwasaki M, Tsugane S. Risk factors for breast cancer: epidemiological evidence from Japanese studies. Cancer Sci. 2011;102(9):1607–1614. doi: 10.1111/j.1349-7006.2011.01996.x. [DOI] [PubMed] [Google Scholar]
- 17.Arem H, Reedy J, Sampson J, Jiao L, Hollenbeck AR, Risch H, Mayne ST, Stolzenberg-Solomon RZ. The Healthy Eating Index 2005 and risk for pancreatic cancer in the NIH-AARP study. J Natl Cancer Inst. 2013;105(17):1298–305. doi: 10.1093/jnci/djt185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosetti C, Bravi F, Turati F, Edefonti V, Polesel J, Decarli A, Negri E, et al. Nutrient-based dietary patterns and pancreatic cancer risk. Ann Epidemiol. 2013;23(3):124–8. doi: 10.1016/j.annepidem.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Chan JM, Gong Z, Holly EA, Bracci PM. Dietary patterns and risk of pancreatic cancer in a large population-based case-control study in the San Francisco Bay Area. Nutr Cancer. 2013;65(1):157–64. doi: 10.1080/01635581.2012.725502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohler LN, Garcia DO, Harris RB, Oren E, Roe DJ, Jacobs ET. Adherence to Diet and Physical Activity Cancer Prevention Guidelines and Cancer Outcomes: A Systematic Review. Cancer Epidemiol Biomarkers Prev. 2016;25(7):1018–1028. doi: 10.1158/1055-9965.EPI-16-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. [Accessed 2017 Dec]; http://www.who.int/dietphysicalactivity/publications/trs916/kit/en/
- 22.Connor WE. Importance of n-3 fatty acids in health and disease. Am J Clin Nutr. 2000;71(1 Suppl):171S–5S. doi: 10.1093/ajcn/71.1.171S. [DOI] [PubMed] [Google Scholar]
- 23.Milligan G, Shimpukade B, Ulven T, Hudson BD. Complex Pharmacology of Free Fatty Acid Receptors. Chem Rev. 2017;117(1):67–110. doi: 10.1021/acs.chemrev.6b00056. [DOI] [PubMed] [Google Scholar]
- 24.Moniri NH. Free-fatty acid receptor-4 (GPR120): Cellular and molecular function and its role in metabolic disorders. Biochem Pharmacol. 2016;110–111:1–15. doi: 10.1016/j.bcp.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulven T, Christiansen E. Dietary Fatty Acids and Their Potential for Controlling Metabolic Diseases Through Activation of FFA4/GPR120. Annu Rev Nutr. 2015;35:239–63. doi: 10.1146/annurev-nutr-071714-034410. [DOI] [PubMed] [Google Scholar]
- 26.Burns RN, Moniri NH. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun. 2010;396(4):1030–5. doi: 10.1016/j.bbrc.2010.05.057. [DOI] [PubMed] [Google Scholar]
- 27.Cheshmehkani A, Senatorov IS, Dhuguru J, Ghoneim O, Moniri NH. Free-fatty acid receptor-4 (FFA4) modulates ROS generation and COX-2 expression via the C-terminal β-arrestin phosphosensor in Raw 264.7 macrophages. Biochem Pharmacol. 2017;146:139–150. doi: 10.1016/j.bcp.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore K, Zhang Q, Murgolo N, Hosted T, Duffy R. Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: comparison with human GPR120 splice variants. Comp Biochem Physiol B Biochem Mol Biol. 2009;154(4):419–26. doi: 10.1016/j.cbpb.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Galindo MM, Voigt N, Stein J, van Lengerich J, Raguse JD, Hofmann T, Meyerhof W, Behrens M. G protein-coupled receptors in human fat taste perception. Chem Senses. 2012;37(2):123–39. doi: 10.1093/chemse/bjr069. [DOI] [PubMed] [Google Scholar]
- 30.Kim JM, Lee KP, Park SJ, Kang S, Huang J, Lee JM, Sato K, Chung HY, Okajima F, Im DS. Omega-3 fatty acids induce Ca(2+) mobilization responses in human colon epithelial cell lines endogenously expressing FFA4. Acta Pharmacol Sin. 2015;36(7):813–20. doi: 10.1038/aps.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson SJ, Brown AJ, Holliday ND. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol. 2012;81(5):631–42. doi: 10.1124/mol.111.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone VM, Dhayal S, Brocklehurst KJ, Lenaghan C, Sörhede Winzell M, Hammar M, Xu X, Smith DM, Morgan NG. GPR120 (FFAR4) is preferentially expressed in pancreatic delta cells and regulates somatostatin secretion from murine islets of Langerhans. Diabetologia. 2014;57(6):1182–91. doi: 10.1007/s00125-014-3213-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engelstoft MS, Park WM, Sakata I, Kristensen LV, Husted AS, Osborne-Lawrence S, Piper PK, et al. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol Metab. 2013;2(4):376–92. doi: 10.1016/j.molmet.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsukahara T, Watanabe K, Watanabe T, Yamagami H, Sogawa M, et al. Tumor necrosis factor α decreases glucagon-like peptide-2 expression by up-regulating G-protein-coupled receptor 120 in Crohn disease. Am J Pathol. 2015;185(1):185–96. doi: 10.1016/j.ajpath.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, Milligan G, Tobin AB. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem. 2014;289(26):18451–65. doi: 10.1074/jbc.M114.568816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burns RN, Singh M, Senatorov IS, Moniri NH. Mechanisms of homologous and heterologous phosphorylation of FFA receptor 4 (GPR120): GRK6 and PKC mediate phosphorylation of Thr347, Ser350, and Ser357 in the C-terminal tail. Biochem Pharmacol. 2014;87(4):650–9. doi: 10.1016/j.bcp.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung BH, Mitchell SH, Zhang JS, Young CY. Effects of docosahexaenoic acid and eicosapentaenoic acid on androgen-mediated cell growth and gene expression in LNCaP prostate cancer cells. Carcinogenesis. 2001;22(8):1201–6. doi: 10.1093/carcin/22.8.1201. [DOI] [PubMed] [Google Scholar]
- 38.Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, Cramer SD, et al. Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer. Carcinogenesis. 2012;33(2):404–12. doi: 10.1093/carcin/bgr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown MD, Hart CA, Gazi E, Bagley S, Clarke NW. Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br J Cancer. 2006;94(6):842–53. doi: 10.1038/sj.bjc.6603030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akinsete JA, Ion G, Witte TR, Hardman WE. Consumption of high ω-3 fatty acid diet suppressed prostate tumorigenesis in C3(1) Tag mice. Carcinogenesis. 2012;33(1):140–8. doi: 10.1093/carcin/bgr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Hopkins MM, Zhang Z, Quisenberry CB, Fix LC, Galvan BM, Meier KE. Omega-3 fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J Pharmacol Exp Ther. 2015;352(2):380–94. doi: 10.1124/jpet.114.218974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148(5):619–28. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu Z, Suburu J, Chen H, Chen YQ. Mechanisms of omega-3 polyunsaturated fatty acids in prostate cancer prevention. Biomed Res Int. 2013;2013:824563. doi: 10.1155/2013/824563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hopkins MM, Liu Z, Meier KE. Positive and Negative Cross-Talk between Lysophosphatidic Acid Receptor 1, Free Fatty Acid Receptor 4, and Epidermal Growth Factor Receptor in Human Prostate Cancer Cells. J Pharmacol Exp Ther. 2016;359(1):124–33. doi: 10.1124/jpet.116.233379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–98. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaslavsky A, Singh LS, Tan H, Ding H, Liang Z, Xu Y. Homo- and hetero-dimerization of LPA/S1P receptors, OGR1 and GPR4. Biochim Biophys Acta. 2006;1761(10):1200–12. doi: 10.1016/j.bbalip.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Fan YY, Ran Q, Toyokuni S, Okazaki Y, Callaway ES, Lupton JR, Chapkin RS. Dietary fish oil promotes colonic apoptosis and mitochondrial proton leak in oxidatively stressed mice. Cancer prevention research. 2011;4:1267–1274. doi: 10.1158/1940-6207.CAPR-10-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calviello G, Di Nicuolo F, Gragnoli S, Piccioni E, Serini S, et al. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and –2 and HIF-1alpha induction pathway. Carcinogenesis. 2004;25:2303–2310. doi: 10.1093/carcin/bgh265. [DOI] [PubMed] [Google Scholar]
- 49.Touil Y, Igoudjil W, Corvaisier M, Dessein AF, Vandomme J, Monte D, Stechly L, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clinical cancer research. 2014;20:837–846. doi: 10.1158/1078-0432.CCR-13-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granci V, Cai F, Lecumberri E, Clerc A, Dupertuis YM, Pichard C. Colon cancer cell chemosensitisation by fish oil emulsion involves apoptotic mitochondria pathway. The British journal of nutrition. 2013;109:1188–1195. doi: 10.1017/S000711451200308X. [DOI] [PubMed] [Google Scholar]
- 51.Wu Q, Wang H, Zhao X, Shi Y, Jin M, Wan B, et al. Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene. 2013;32(49):5541–50. doi: 10.1038/onc.2013.264. [DOI] [PubMed] [Google Scholar]
- 52.Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483(7389):350–4. doi: 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]
- 53.Zhang K, Hu Z, Qi H, Shi Z, Chang Y, Yao Q, Cui H, Zheng L, Han Y, Han X, Zhang Z, Chen T, Hong W. G-protein-coupled receptors mediate ω-3 PUFAs-inhibited colorectal cancer by activating the Hippo pathway. Oncotarget. 2016;7(36):58315–58330. doi: 10.18632/oncotarget.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheshmehkani A, Senatorov IS, Kandi P, Singh M, Britt A, Hayslett R, Moniri NH. Fish oil and flax seed oil supplemented diets increase FFAR4 expression in the rat colon. Inflamm Res. 2015;64(10):809–815. doi: 10.1007/s00011-015-0864-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ou C, Sun Z, Li S, Li G, Li X, Ma J. Dual roles of yes-associated protein (YAP) in colorectal cancer. Oncotarget. 2017;8(43):75727–75741. doi: 10.18632/oncotarget.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avruch J, Zhou D, Bardeesy N. YAP oncogene overexpression supercharges colon cancer proliferation. Cell Cycle. 2012;11(6):1090–6. doi: 10.4161/cc.11.6.19453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu FX, Meng Z, Plouffe SW, Guan KL. Hippo pathway regulation of gastrointestinal tissues. Annu Rev Physiol. 2015;77:201–27. doi: 10.1146/annurev-physiol-021014-071733. [DOI] [PubMed] [Google Scholar]
- 58.Zhou X, Wang Z, Huang W, Lei QY. G protein-coupled receptors: bridging the gap from the extracellular signals to the Hippo pathway. Acta Biochim Biophys Sin. 2015;47(1):10–5. doi: 10.1093/abbs/gmu108. [DOI] [PubMed] [Google Scholar]
- 59.Sebio A, Lenz HJ. Molecular Pathways: Hippo Signaling, a Critical Tumor Suppressor. Clin Cancer Res. 2015;21(22):5002–7. doi: 10.1158/1078-0432.CCR-15-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sparks SM, Chen G, Collins JL, Danger D, Dock ST, Jayawickreme C, et al. Identification of diarylsulfonamides as agonists of the free fatty acid receptor 4 (FFA4/GPR120) Bioorg Med Chem Lett. 2014;24(14):3100–3. doi: 10.1016/j.bmcl.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11(1):90–4. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 62.Poirier 1, Brennan P, McKay JD, Spitz MR, Bickeböller H, Risch A, et al. Informed genome-wide association analysis with family history as a secondary phenotype identifies novel loci of lung cancer. Genet Epidemiol. 2015;39(3):197–206. doi: 10.1002/gepi.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kita T, Kadochi Y, Takahashi K, Fukushima K, Yamasaki E, Uemoto T, et al. Diverse effects of G-protein-coupled free fatty acid receptors on the regulation of cellular functions in lung cancer cells. Exp Cell Res. 2016;342(2):193–9. doi: 10.1016/j.yexcr.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 64.Fukushima K, Yamasaki E, Ishii S, Tomimatsu A, Takahashi K, Hirane M, et al. Different roles of GPR120 and GPR40 in the acquisition of malignant properties in pancreatic cancer cells. Biochem Biophys Res Commun. 2015;465(3):512–5. doi: 10.1016/j.bbrc.2015.08.050. [DOI] [PubMed] [Google Scholar]
- 65.Serini S, Fasano E, Celleno L, Cittadini A, Calviello G. Potential of long-chain n-3 polyunsaturated fatty acids in melanoma prevention. Nutr Rev. 2014;72(4):255–66. doi: 10.1111/nure.12093. [DOI] [PubMed] [Google Scholar]
- 66.Noel SE, Stoneham AC, Olsen CM, Rhodes LE, Green AC. Consumption of omega-3 fatty acids and the risk of skin cancers: a systematic review and meta-analysis. Int J Cancer. 2014 Jul 1;135(1):149–56. doi: 10.1002/ijc.28630. [DOI] [PubMed] [Google Scholar]
- 67.Nehra D, Pan AH, Le HD, Fallon EM, Carlson SJ, Kalish BT, Puder M. Docosahexaenoic acid, G protein-coupled receptors, and melanoma: is G protein-coupled receptor 40 a potential therapeutic target? Surg Res. 2014;188(2):451–8. doi: 10.1016/j.jss.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fukushima K, Takahashi K, Fukushima N, Honoki K, Tsujiuchi T. Different effects of GPR120 and GPR40 on cellular functions stimulated by 12-O-tetradecanoylphorbol-13-acetate in melanoma cells. Biochem Biophys Res Commun. 2016;475(1):25–30. doi: 10.1016/j.bbrc.2016.05.023. [DOI] [PubMed] [Google Scholar]
- 69.Kim EH, Willett WC, Colditz GA, Hankinson SE, Stampfer MJ, Hunter DJ, et al. Dietary fat and risk of postmenopausal breast cancer in a 20-year follow-up. Am J Epidemiol. 2006;164:990–7. doi: 10.1093/aje/kwj309. [DOI] [PubMed] [Google Scholar]
- 70.Park SY, Kolonel LN, Henderson BE, Wilkens LR. Dietary fat and breast cancer in postmenopausal women according to ethnicity and hormone receptor status: the Multiethnic Cohort Study. Cancer Prev Res. 2012;5:216–28. doi: 10.1158/1940-6207.CAPR-11-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saadatian-Elahi M, Norat T, Goudable J, Riboli E. Biomarkers of dietary fatty acid intake and the risk of breast cancer: a meta-analysis. Int J Cancer. 2004;111:584–91. doi: 10.1002/ijc.20284. [DOI] [PubMed] [Google Scholar]
- 72.MacLean CH, Newberry SJ, Mojica WA, Khanna P, Issa AM, Suttorp MJ, et al. Effects of omega-3 fatty acids on cancer risk: a systematic review. JAMA. 2006;295:403–15. doi: 10.1001/jama.295.4.403. [DOI] [PubMed] [Google Scholar]
- 73.Zheng JS, Hu XJ, Zhao YM, Yang J, Li D. Intake of fish and marine n-3 polyunsaturated fatty acids and risk of breast cancer: meta-analysis of data from 21 independent prospective cohort studies. BMJ. 2013 Jun 27;346:f3706. doi: 10.1136/bmj.f3706. [DOI] [PubMed] [Google Scholar]
- 74.Karmali RA, Marsh J, Ruchs C. Effect of ω-3 fatty acids on growth of a rat mammary tumor. J Natl Cancer Inst. 1984;73:457–461. doi: 10.1093/jnci/73.2.457. [DOI] [PubMed] [Google Scholar]
- 75.Sauer LA, Dauchy RT, Blask DE, Krause JA, Davidson LK, Dauchy EM. Eicosapenaenoic acid suppresses cell proliferation in MCF-7 human breast cancer xenografts in nude rats via a pertussis toxin-sensitive signal transduction pathway. J Nutr. 2005;135:2124–2129. doi: 10.1093/jn/135.9.2124. [DOI] [PubMed] [Google Scholar]
- 76.Jiang W, Zhu Z, McGinley JN, el Bayouny K, Manni A, Thompson JH. Identification of a molecular signature underlying inhibition of mammary carcinoma growth by dietary n-3 fatty acids. Cancer Res. 2012;72:3795–3806. doi: 10.1158/0008-5472.CAN-12-1047. [DOI] [PubMed] [Google Scholar]
- 77.Schley PD, Jijon HB, Robinson LE, Field CJ. Mechanisms of ω-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat. 2005;92:187–195. doi: 10.1007/s10549-005-2415-z. [DOI] [PubMed] [Google Scholar]
- 78.Soto-Guzman A, Robledo T, Lopez-Perez M, Salazar EP. Oleic acid induces ERK1/2 activation and AP-1 DNA binding activity through a mechanism involving Src kinase and EGFR transactivation in breast cancer cells. Mol Cell Endocrinol. 2008;294(1–2):81–91. doi: 10.1016/j.mce.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 79.Serna-Marquez N, Diaz-Aragon R, Reyes-Uribe E, Cortes-Reynosa P, Salazar EP. Linoleic acid induces migration and invasion through FFAR4- and PI3K-/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Med Oncol. 2017;34(6):111. doi: 10.1007/s12032-017-0969-3. [DOI] [PubMed] [Google Scholar]
- 80.Yonezawa T, Haga S, Kobayashi Y, Katoh K, Obara Y. Unsaturated fatty acids promote proliferation via ERK1/2 and Akt pathway in bovine mammary epithelial cells. Biochem Biophys Res Commun. 2008;367:729–35. doi: 10.1016/j.bbrc.2007.12.190. [DOI] [PubMed] [Google Scholar]
- 81.Reyes N, Reyes I, Tiwari R, Geliebter J. Effect of linoleic acid on proliferation and gene expression in the breast cancer cell line T47D. Cancer Lett. 2004;209:25–35. doi: 10.1016/j.canlet.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 82.Hopkins MM, Zhang Z, Liu Z, Meier KE. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J Clin Med. 2016;5(2) doi: 10.3390/jcm5020016. pii: E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chung H, Lee YS, Mayoral R, Oh DY, Siu JT, Webster NJ, et al. Omega-3 fatty acids reduce obesity-induced tumor progression independent of GPR120 in a mouse model of postmenopausal breast cancer. Oncogene. 2015;34(27):3504–13. doi: 10.1038/onc.2014.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim HJ, Yoon HJ, Kim BK, Kang WY, Seong SJ, Lim MS, et al. G Protein-Coupled Receptor 120 Signaling Negatively Regulates Osteoclast Differentiation, Survival, and Function. J Cell Physiol. 2016;231(4):844–51. doi: 10.1002/jcp.25133. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi K, Fukushima K, Onishi Y, Node Y, Inui K, Fukushima N, Honoki K, Tsujiuchi T. Different effects of G-protein-coupled receptor 120 (GPR120) and GPR40 on cell motile activity of highly migratory osteosarcoma cells. Biochem Biophys Res Commun. 2017;484(3):675–680. doi: 10.1016/j.bbrc.2017.01.175. [DOI] [PubMed] [Google Scholar]
- 86.Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20(3):370–83. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 87.Daenen LG, Cirkel GA, Houthuijzen JM, Gerrits J, Oosterom I, Roodhart JM, et al. Increased Plasma Levels of Chemoresistance-Inducing Fatty Acid 16:4(n-3) After Consumption of Fish and Fish Oil. AMA Oncol. 2015;1(3):350–8. doi: 10.1001/jamaoncol.2015.0388. [DOI] [PubMed] [Google Scholar]
- 88.Houthuijzen JM, Daenen LG, Roodhart JM, Oosterom I, van Jaarsveld MT, Govaert KM, et al. Lysophospholipids secreted by splenic macrophages induce chemotherapy resistance via interference with the DNA damage response. Nat Commun. 2014;5:5275. doi: 10.1038/ncomms6275. [DOI] [PubMed] [Google Scholar]
- 89.Houthuijzen JM, Oosterom I, Hudson BD, Hirasawa A, Daenen LGM, McLean CM, et al. Fatty acid 16:4(n-3) stimulates a GPR120-induced signaling cascade in splenic macrophages to promote chemotherapy resistance. FASB J. 2017;31(5):2195–2209. doi: 10.1096/fj.201601248R. [DOI] [PMC free article] [PubMed] [Google Scholar]