

Abstract
Detection of protein–protein interactions involved in signal transduction in live cells and organisms has a variety of important applications. We report a fluorogenic assay for G protein‐coupled receptor (GPCR)–β‐arrestin interaction that is genetically encoded, generalizes to multiple GPCRs, and features high signal‐to‐noise because fluorescence is absent until its components interact upon GPCR activation. Fluorescence after protease‐activated receptor‐1 activation developed in minutes and required specific serine–threonine residues in the receptor carboxyl tail, consistent with a classical G protein‐coupled receptor kinase dependent β‐arrestin recruitment mechanism. This assay provides a useful complement to other in vivo assays of GPCR activation.
Keywords: protein–protein interaction, G protein coupled receptor, fluorescent reporter, green fluorescent protein
Introduction
The G protein‐coupled receptor (GPCR) superfamily is the largest receptor family in the animal kingdom, with more than 800 GPCRs encoded in the human genome.1 GPCRs share the same topology, consisting of an extracellular N‐terminus, seven membrane‐spanning helices connected by three extracellular and three intracellular loops, and an intracellular C‐terminus. Activated GPCRs act as nucleotide exchange factors for heterotrimeric G proteins, enabling the latter to bind and activate downstream effectors. For example, the GPCR protease‐activated receptor‐1 (PAR1) activates Gq, which in turn activates phospholipase C, phosphoinositide hydrolysis, calcium mobilization, and protein kinase C.2, 3 Activated GPCRs are rapidly phosphorylated by G protein‐coupled receptor kinases (GRKs),4 which enables the GPCRs to bind arrestins, undergo internalization, and trigger arrestin‐dependent signaling. Because GPCRs regulate a host of functions important in physiology and disease and are important drug targets, methods for detecting GPCR activity in cells and tissues have been of intense interest. Examples include detection of GPCR/β‐arrestin interaction as movement of a β‐arrestin–green fluorescent protein (GFP) fusion protein from cytosol to plasma membrane5 and the transcription‐based Tango assay.6, 7 We describe a fluorogenic assay that features a high signal‐to‐noise because fluorescence develops only upon GPCR/β‐arrestin interaction and more rapid response times than the Tango assay, presumably because signal does not require de novo protein synthesis.
Results and Discussion
In this work, to develop a dark‐to‐bright fluorogenic GPCR/β‐arrestin interaction assay, we employed a tripartite GFP that detects protein–protein interaction (PPI) without background fluorescence.8 Previously used bipartite systems, for example, the yellow fluorescent protein Venus‐based protein complementation assay, have significant background fluorescence in the absence of PPI due to intrinsic affinity of the two parts of the split fluorescent protein. The crystal structure of GFP indicates that it is composed of 11 β strands (Supporting Information Movie S1).9, 10 In tripartite GFP, one part contains nine β strands (β1–9); the second part contains the 10th β strand (β10); the third part contains the 11th β strand (β11).8 β1–9 contains three amino acids that form the chromophore,11, 12 and β11 contains the highly conserved Glu222 that catalyzes the chromophore maturation.13 A tripartite GFP was previously demonstrated to detect several PPIs including rapamycin‐induced interaction between FKBP and Frb, which were fused to β11 and β10.8 This system appeared to provide better signal‐to‐noise than the bipartite approach, presumably due to less efficient self‐assembly without proximity of β11 and β10.
To detect GPCR/β‐arrestin interaction, we first designed a positive control by fusing β11 to the transmembrane protein CD4 that is linked to FKBP (i.e., CD4‐FKBP‐β11), and β10 to Frb. HEK293 cells expressing the fusion constructs together with β1–9 and mCherry developed green fluorescence at the plasma membrane upon addition of rapamycin (Supporting Information Fig. S1), suggesting that the tripartite GFP system might be able to detect interaction between a membrane protein and a cytosolic protein, mimicking GPCR and β‐arrestin. Accordingly, we next designed a GPCR/β‐arrestin interaction assay by fusing β11 to C‐terminus of GPCR and β10 to N‐terminus of β‐arrestin. The rationale of this design [Fig. 1(A)] is that β10 and β11 should remain separated until GPCR activation and β‐arrestin brings them into proximity to support stable binding of β1–9, reconstitution of GFP, and development of green fluorescence [Fig. 1(A)]. We dubbed this assay Trio because assembly of a tripartite complex is required.
Figure 1.
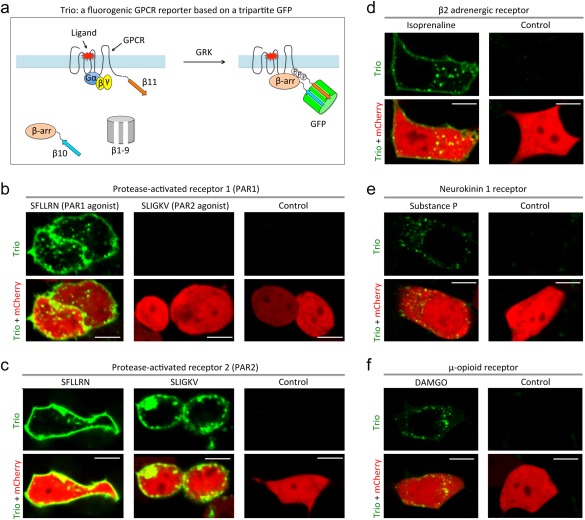
Engineering a fluorogenic assay for detecting GPCR and β‐arrestin interaction. (a) Schematic diagram of the fluorogenic assay Trio. Cells expressing Trio components for PAR1 (b), PAR2 (c), β2‐adrenergic receptor (d), neurokinin‐1 receptor (e), and μ‐opioid receptor (f) were incubated with vehicle (PBS) (Control) or the indicated agonists. Fluorescent images were acquired 60–90 min after agonist addition. Scale, 10 μm. Concentrations of the agonists used were: 100 μM PAR1 agonist; 100 μM PAR2 agonist; 10 μM isoprenaline; 1 μM substance P; 1 μM DAMGO.
As a proof of concept, we first designed a Trio assay to detect the activation of PAR1, the prototypical member of a set of protease‐activated GPCRs14 that mediate important cellular responses to thrombin and other proteases to trigger platelet and endothelial cell activation and other responses important in hemostasis, thrombosis, and inflammation.3 Thrombin cleaves the LDPR/SFLL site in the N‐terminal exodomain of PAR1 to generate a new N‐terminus, which then functions as a tethered peptide agonist and binds intramolecularly to the seven‐transmembrane helix bundle of the receptor and activates G proteins.15 A synthetic peptide SFLLRN that mimics the unmasked (and tethered) N‐terminus is a full agonist for PAR1 activation. Activated PAR1 is phosphorylated by GRKs and binds to β‐arrestins, leading to rapid decoupling from G proteins. The PAR1/β‐arrestin complex then co‐internalizes into endocytic vesicles and is eventually delivered to lysosomes for degradation.
For PAR1/β‐arrestin Trio, we fused β11 to the C‐terminus of PAR1 and β10 to the N‐terminus of β‐arrestin and expressed these fusion proteins in HEK293 cells together with β1–9 and mCherry, the latter as a reference signal for transfection. Green fluorescence was absent in untreated cells. Addition of the PAR1 agonist SFLLRN led to appearance of green fluorescence at the plasma membrane and in intracellular spots [Fig. 1(B)]. No fluorescence was detected after addition of vehicle (i.e., buffer solution) or the PAR2 agonist SLIGKV, which does not activate PAR116 [Fig. 1(B)]. These results suggest that the PAR1 Trio assay detects PAR1 activation at the plasma membrane and internalization to endosomes with the expected pharmacological specificity and a large dynamic range (i.e., from dark to bright fluorescence).
We next developed a Trio assay for another member of the PAR family, PAR2, which can be activated by several trypsin‐like proteases and its tethered ligand peptide SLIGKV as well as the PAR1 agonist SFLLRN.17, 18, 19 Addition of SLIGKV and SFLLRN but not vehicle to HEK293 cells expressing a PAR2‐β11 C‐terminal fusion and the other two Trio components led to development of green fluorescence [Fig. 1(C)], consistent with known PAR2 pharmacology.
Time‐lapse imaging of cells expressing the PAR1 Trio components revealed the appearance of green fluorescence ∼30–50 min after agonist addition [Supporting Information Fig. S3(A) and Movie S2]. The fluorescence was localized to the plasma membrane and vesicles and on filopodia‐like cellular projections. Cells expressing PAR2 Trio components developed green fluorescence ∼30 minutes after agonist addition [Supporting Information Fig. S3(B) and Movie S3]. The fluorescence was mainly localized to vesicles with weak fluorescence on the cell membrane, suggesting rapid endocytosis of PAR2/β‐arrestin complex. Past studies5 suggest that β‐arrestin recruitment can occur within seconds of GPCR activation. Thus, the delay in appearance of fluorescence in the Trio assay likely reflects the time required for ternary complex formation and chromophore maturation.8
To further test whether the Trio assay is generalizable, we generated Trio assays for three more GPCRs: β2 adrenergic receptor (β2AR), neurokinin‐1 receptor (NK1R), and μ‐type opioid receptor (MOR). Addition of the β2AR agonist isoprenaline, the NK1R agonist Subtance P, and the MOR agonist DAMGO triggered appearance of green fluorescence in HEK293 cells expressing the cognate receptor‐β11 C‐terminal fusion proteins and the other Trio components [Fig. 1(D–F)]. Thus, the Trio assay can likely be applied to detect the interaction between many GPCRs and β‐arrestin.
Lastly, we applied Trio to investigate requirements for PAR1/β‐arrestin interaction.
Serine and threonine residues in the cytoplasmic C‐terminal tail of PAR1 are phosphorylated after receptor activation, and these residues are required for normal receptor uncoupling or internalization.20, 21, 22 The requirement for specific potential phosphorylation sites for PAR1‐β‐arrestin interactions has not been examined directly. We first mutated Ser/Thr to Ala at three regions of the cytoplasmic tail [Fig. 2(A)]. Mutant 1 (M1) contains such mutations between residues 380 and 389; M2 contains Ser/Thr → Ala mutations between 390 and 405; and M3 has Ser/Thr → Ala mutations for residues from 406 to the C‐terminus . Fluorescence imaging using PAR1 Trio indicated that the M1 and the M2 mutants interacted with β‐arrestin like WT while the M3 mutant did not [Fig. 2(B)]. Accordingly, we generated four more mutants with Ser/Thr → Ala mutations within the region between 406 and the C‐terminus [Fig. 2(A)]: M4 with a S411A mutation; M5 with a T415A mutation; M6 with S417A/S418A mutations, and M7 with a S423A mutation. Fluorescence imaging using Trio assay revealed that M7 interacted with β‐arrestin like WT. In contrast, the other three mutants showed reduced interaction with β‐arrestin based on the green fluorescence normalized by mCherry's red fluorescence [Fig. 2(B)]. All of these mutants triggered calcium signaling (see below), and appending the C‐terminal tail of arginine vasopressin receptor 2 (AVPR2) to the M3 mutant of PAR1 (named M3‐AVPR2) rescued M3–β‐arrestin interaction. Thus, these receptor mutants were expressed and functional. These results suggest that S411/T415/S417/S418/S423 appeared to play necessary roles in interaction of PAR1 with β‐arrestin.
Figure 2.
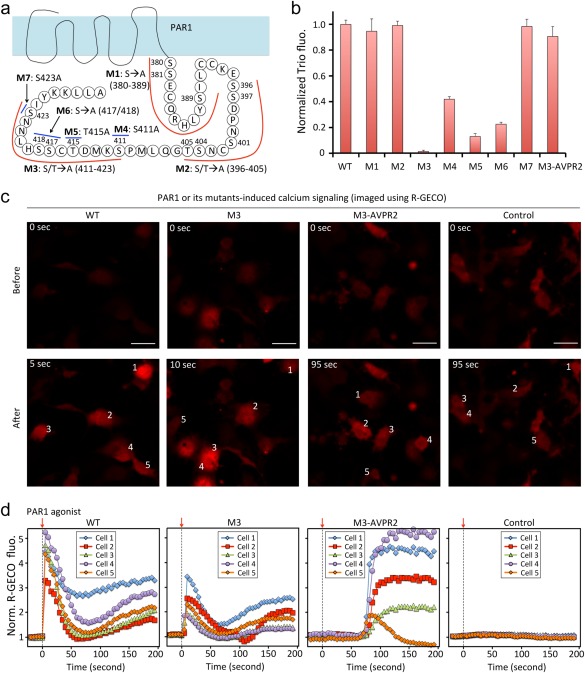
Trio identifies key residues in PAR1 and β‐arrestin interaction. (a) Schematic diagram of PAR1 and its mutants. (b) Normalized Trio fluorescence of PAR1 and mutants. Fluorescence images (c) and quantitative analysis (d) of calcium signaling upon addition of PAR1 agonist to COS7 cells expressing exogenous PAR1 and mutants. Scale, 50 μm.
To investigate consequences of these mutations in the downstream signaling of PAR1, we conducted calcium imaging as a measure of PAR1‐triggered Gαq and PLC activation and calcium mobilization. We exogenously expressed wild‐type PAR1 and its mutants in COS7 cells because these cells do not contain endogenous PAR1. To image calcium signaling, we utilized the genetically encoded red fluorescent calcium reporter R‐GECO. Addition of PAR1 agonist to COS7 cells transfected with empty vector did not trigger calcium signaling. Addition of PAR1 agonist to cells expressing wild‐type PAR1 rapidly led to calcium transients as expected [<5 sec, Fig. 2(C,D)]. While the M3 mutant PAR1 did not show interaction with β‐arrestin, addition of PAR1 agonist to cells expressing M3 did trigger calcium mobilization, albeit slightly delayed compared to wild‐type PAR1. The M3‐AVPR2 mutant with rescued β‐arrestin interaction behaved similarly. Taken together, these data are consistent with the classical model of GPCR activation triggering phosphorylation‐independent G protein activation and phosphorylation‐dependent arrestin‐binding and support the notion that the Trio system reads out phosphorylation site‐dependent arresting binding as expected.
In summary, we have developed a GPCR/β‐arrestin interaction assay, Trio, based on the tripartite GFP. Trio is not fluorescent before GPCR/β‐arrestin interaction and produces green fluorescence when the two proteins interact upon GPCR activation and phosphorylation. Using Trio, we have identified residues necessary for recruiting β‐arrestin upon PAR1 activation, which demonstrates that Trio is useful to investigate structural basis of GPCR/β‐arrestin interaction and raises the possibility that this system might be useful for probing other PPIs of activated GPCRs. Although the fluorescence signal is slower than Förster resonance energy transfer (FRET)‐based reporters due to the chromophore maturation time, GFP‐based protein complementation assays are known to provide a larger dynamic range than FRET‐based reporters in detecting PPIs. Trio appears to provide still better signal to noise compared to the two‐part split GFP‐based assay with no background fluorescence in the absence of stimulated PPIs. Because of the importance in detecting GPCR/β‐arrestin interaction as shown by the transcription‐based Tango and PRESTO‐Tango assays,6, 7 the tripartite GFP‐based Trio will be another valuable tool for the study of GPCRs in cell‐based assays.
Materials and Methods
DNA constructs
All plasmid constructs were created by standard molecular biology techniques and confirmed by exhaustively sequencing the cloned fragments. DNA of the tripartite GFP was synthesized.
Mammalian cell cultures
The HEK293T/17 (ATCC CRL‐11268) was obtained from ATCC. Cells were passaged in Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), non‐essential amino acids, penicillin (100 units/mL), and streptomycin (100 μg/mL). All culture supplies were obtained from the UCSF Cell Culture Facility. HEK293T/17 cells were transiently transfected with the GPCR Trio reporter with the calcium phosphate method. Cells were grown in 35 mm glass bottom microwell (14 mm) dishes (MatTek Corporation). Transfection was performed when cells were cultured to ∼50% confluence. For each transfection, 4.3 μg of plasmid DNA was mixed with 71 μL of 1X Hanks' Balanced Salts buffer (HBS) and 4.3 μL of 2.5M CaCl2. Cells were imaged 24 h after transient transfection. The COS7 cells were transfected with PAR1 or its mutants along with the calcium reporter R‐GECO.
Confocal microscopy
For fluorescence imaging of the Trio assay in cultured mammalian cells, transfected HEK293T/17 cells were imaged in 35 mm glass bottom microwell dishes on a Nikon Eclipse Ti inverted microscope equipped with a Yokogawa CSU‐W1 confocal scanner unit (Andor), a digital CMOS camera ORCA‐Flash4.0 (Hamamatsu), a ASI MS‐2000 XYZ automated stage (Applied Scientific Instrumentation), and a Nikon Plan Apo λ 20X air (N.A. 0.75) objective. Laser inputs were provided by an Integrated Laser Engine (Spectral Applied Research) equipped with laser lines (Coherent) 488 nm (6.3 mW) for GFP imaging and 561 nm (3.5 mW) for mCherry imaging. The confocal scanning unit was equipped with the following emission filters: 525/50‐nm for GFP imaging and 610/60‐nm for mCherry imaging. For imaging Trio based reporters in mammalian cells, images were acquired with an exposure time of 100 ms for both GFP and mCherry. Image acquisition was controlled by the NIS‐Elements Ar Microscope Imaging Software (Nikon). Images were processed using NIS‐Elements and ImageJ (NIH).
Imaging in cultured mammalian cells
HEK293T/17 cells or COS7 cells transiently transfected with the Trio constructs were imaged in 35 mm glass bottom dishes ∼24 h after transfection. For time‐lapse imaging of the Trio upon agonist addition, cells at ∼24 h after transient transfection with the reporter were grown in 35 mm glass bottom dishes to ∼90% confluence. Time‐lapse microscopy was performed using the confocal microscope described above with the aid of an environmental control unit incubation chamber (InVivo Scientific), which was maintained at 37°C and 5% CO2. To activate GPCRs, appropriate agonists were added to cells.
AUTHOR CONTRIBUTIONS
X.S. conceived the project. Q.Z. conducted the experiments. Y.Z. and S.R.C. constructed the MOR Trio vector. Q.Z., X.S., S.R.C. wrote the manuscript.
Supporting information
Supporting Information Movie S1
Supporting Information Movie S2
Supporting Information Movie S3
Supplementary Material
Competing Financial Interests: None declared.
References
- 1. Lefkowitz RJ (2013) A brief history of G‐protein coupled receptors. Angew Chem Int Ed 52:6366–6378. [DOI] [PubMed] [Google Scholar]
- 2. Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK (2012) High‐resolution crystal structure of human protease‐activated receptor 1. Nature 492:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coughlin SR (2000) Thrombin signalling and protease‐activated receptors. Nature 407:258–264. [DOI] [PubMed] [Google Scholar]
- 4. Pitcher JA, Freedman NJ, Lefkowitz RJ (1998) G protein‐coupled receptor kinases. Annu Rev Biochem 67:653–692. [DOI] [PubMed] [Google Scholar]
- 5. Barak LS, Ferguson SS, Zhang J, Caron MG (1997) A beta‐arrestin/green fluorescent protein biosensor for detecting G protein‐coupled receptor activation. J Biol Chem 272:27497–27500. [DOI] [PubMed] [Google Scholar]
- 6. Kroeze WK, Sassano MF, Huang X‐P, Lansu K, McCorvy JD, Giguere PM, Sciaky N, Roth BI (2015) PRESTO‐Tango as an open‐source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol 22:362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ (2008) The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cabantous S, Nguyen HB, Pedelacq J‐D, Koraichi F, Chaudhary A, Ganguly K, Lockard MA, Favre G, Terwilliger TC, Waldo GS (2013) A new protein‐protein interaction sensor based on tripartite split‐GFP association. Sci Rep 3:2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang F, Moss LG, Phillips GN (1996) The molecular structure of green fluorescent protein. Nat Biotechnol 14:1246–1251. [DOI] [PubMed] [Google Scholar]
- 10. Orm M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- 11. Tsien RY (2009) Constructing and exploiting the fluorescent protein paintbox. Angew Chem Int Ed Engl 48:5612–5626. [DOI] [PubMed] [Google Scholar]
- 12. Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67:509–544. [DOI] [PubMed] [Google Scholar]
- 13. Sniegowski JA, Lappe JW, Patel HN, Huffman HA, Wachter RM (2005) Base catalysis of chromophore formation in Arg96 and Glu222 variants of green fluorescent protein. J Biol Chem 280:26248–26255. [DOI] [PubMed] [Google Scholar]
- 14. Vu TK, Hung DT, Wheaton VI, Coughlin SR (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64:1057–1068. [DOI] [PubMed] [Google Scholar]
- 15. Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR (1991) Domains specifying thrombin‐receptor interaction. Nature 353:674–677. [DOI] [PubMed] [Google Scholar]
- 16. Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM (1996) Ligand cross‐reactivity within the protease‐activated receptor family. J Biol Chem 271:16466–16471. [DOI] [PubMed] [Google Scholar]
- 17. Santulli RJ, Derian CK, Darrow AL, Tomko KA, Eckardt AJ, Seiberg M, Scarborough RM, Andrade‐Gordon P (1995) Evidence for the presence of a protease‐activated receptor distinct from the thrombin receptor in human keratinocytes. Proc Natl Acad Sci USA 92:9151–9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nystedt S, Emilsson K, Wahlestedt C, Sundelin J (1994) Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA 91:9208–9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coughlin SR (1994) Protease‐activated receptors start a family. Proc Natl Acad Sci USA 91:9200–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, Coughlin SR (1994) Inhibition of thrombin receptor signaling by a G‐protein coupled receptor kinase—functional specificity among G‐protein coupled receptor kinases. J Biol Chem 269:1125–1130. [PubMed] [Google Scholar]
- 21. Tiruppathi C, Yan W, Sandoval R, Naqvi T, Pronin AN, Benovic JL, Malik AB (2000) G protein‐coupled receptor kinase‐5 regulates thrombin‐activated signaling in endothelial cells. Proc Natl Acad Sci USA 97:7440–7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hammes SR, Shapiro MJ, Coughlin SR (1999) Shutoff and agonist‐triggered internalization of protease‐activated receptor 1 can be separated by mutation of putative phosphorylation sites in the cytoplasmic tail. Biochemistry 38:9308–9316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Movie S1
Supporting Information Movie S2
Supporting Information Movie S3
Supplementary Material