

Abstract
Previously, we determined the crystal structure of apo‐TpMglB‐2, a d‐glucose‐binding component of a putative ABC transporter from the syphilis spirochete Treponema pallidum. The protein had an unusual topology for this class of proteins, raising the question of whether the d‐glucose‐binding mode would be different in TpMglB‐2. Here, we present the crystal structures of a variant of TpMglB‐2 with and without d‐glucose bound. The structures demonstrate that, despite its aberrant topology, the protein undergoes conformational changes and binds d‐glucose similarly to other Mgl‐type proteins, likely facilitating d‐glucose uptake in T. pallidum.
Keywords: ABC transporter, syphilis, spirochete, glucose‐binding protein, conformational change
Short abstract
Introduction
The ligand‐binding proteins (LBPs) of ABC‐type transporters are a well‐characterized class of bilobed polypeptides1, 2 that often occur in the periplasms of microorganisms. The lobes are connected by a hinge region, and between them is a ligand‐binding cleft; without ligand, these proteins usually adopt an “open” conformation in which the two lobes are relatively far apart. Upon ligand binding, the lobes become closely apposed, often shielding a bound small molecule completely from solvent exposure. This functionality has been likened to a “Venus fly trap.”3 LBPs called “MglB” are known to bind to d‐glucose and d‐galactose,4, 5 and they undergo a measurable conformational change when binding a native hexose,6 which is an integral part of the overall transport mechanism.
Earlier, we determined the crystal structure of apo‐TpMglB‐2,7 a protein predicted to bind to d‐glucose and d‐galactose. The ligandless protein was in an open conformation, as would be expected. When compared to other Mgl‐B‐like proteins, the protein exhibited an unexpected, circularly permuted topology. All efforts to introduce d‐glucose into the crystals failed, but solution studies indicated that d‐glucose and d‐galactose could bind to the protein with an attendant change in conformation. The current studies examine two crystal structures of a variant of TpMglB‐2 that provide further evidence of this conformational change.
Results and Discussion
The crystal structures of apo‐ and holo‐ TpMglB‐2WA
In our previous work, we used isothermal titration calorimetry (ITC) to characterize the interaction of d‐glucose and other hexoses with a wild‐type, recombinant form of TpMglB‐2 (TpMglB‐2R 7). We found that the protein bound glucose with a K D of 1.1 μM. However, it was always necessary to account for a fraction (15%–30%) of TpMglB‐2R that was incompetent to bind to d‐glucose, and the cause of this incompetency was unknown (it could not be attributed to concentration‐determination errors). Numerous attempts to co‐crystallize TpMglB‐2R with d‐glucose failed. In other ITC experiments, we examined the binding of d‐glucose to the W145A variant of TpMglB‐2 (TpMglB‐2WA). This mutation was designed to abrogate d‐glucose binding, as W145 was a putative d‐glucose‐binding residue located in the ligand‐binding cleft. We observed weaker binding (K D = 12.7 μM), but it occurred with a much lower incompetent fraction (ca. 6%). We thus reasoned that this protein might offer a better platform for co‐crystallization with the sugar. Indeed, we obtained two TpMglB‐2WA crystal forms in the presence of d‐glucose that diffracted to d min spacings of 2.08 and 1.47 Å, respectively. We used molecular replacement to determine both crystal structures, and both refined well, yielding models with excellent R‐values and geometries (Table 1).
Table 1.
Data Collection, Phasing, and Refinement Statistics
| Data set | holo‐TpMglB‐2WA | apo‐TpMglB‐2WA |
|---|---|---|
| PDB accession no. | 6BGC | 6BGD |
| Data collection | ||
| Space group | P1 | C2221 |
| Unit cell dimensions (Å) | ||
| a | 44.257 | 75.834 |
| b | 61.592 | 110.199 |
| c | 70.350 | 89.304 |
| α(°) | 80.2 | 90.0 |
| β (°) | 77.6 | 90.0 |
| γ (°) | 72.7 | 90.0 |
| Resolution (Å) | 39.73–2.08 (2.12–2.08)a | 37.9–1.47 (1.50–1.47) |
| Completeness (%) | 91.8 (91.1) | 95.4 (74.8) |
| Multiplicity | 2.5 (2.3) | 4.4 (4.2) |
| Unique reflections | 36,146 (1816) | 60,862 (2362) |
| R merge b | 0.080 (0.315) | 0.068 (0.386) |
| <I>/σ I | 9.8 (2.1) | 19.7 (2.7) |
| Wilson B (Å2) | 19.8 | 10.4 |
| Refinement | ||
| Resolution (Å) | 39.73–2.08 | 37.9–1.47 |
| No. residues | 736 | 373 |
| No. non‐solvent atoms | 5561 | 2770 |
| No. solvent atoms | 221 | 466 |
| Maximum‐likelihood coordinate error (Å) | 0.22 | 0.1 |
| Average B‐factors | ||
| Non‐solvent (Å2) | 30.15 | 14.91 |
| Ligand | 16.87 | N/A |
| Solvent (Å2) | 25.94 | 28.59 |
| R‐values | ||
| R work c | 0.180 | 0.114 |
| R free d | 0.220 | 0.144 |
| Ramachandran statistics | ||
| Outliers (%) | 0.3 | 0.3 |
| Most favored region (%) | 97.3 | 97.9 |
| r.m.s. deviations | ||
| Bonds (Å) | 0.003 | 0.009 |
| Angles (°) | 0.698 | 1.264 |
Numbers in the parentheses are reported for the highest‐resolution shell of reflections.
where the outer sum (hkl) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
, where F o and F c are observed and calculated structure factor amplitudes, respectively.
R free is calculated using the same formula as R work, but the set hkl is a randomly selected subset (5%) of the total structure factors that are never used in refinement.
Despite the presence of 10 mM d‐glucose, one of the structures (the 1.47‐Å resolution structure) clearly lacked electron density for the ligand. Hereafter, we refer to this as the “apo” form of the protein. Apo‐TpMglB‐2WA crystallized in the same space group as apo‐TpMglB‐2R, with nearly identical unit‐cell dimensions.7 The apo‐TpMglB‐2WA crystal structure [Fig. 1(A)] had no notable secondary‐ or tertiary‐structure differences when compared to apo‐TpMglB‐2R. When superposed,8 the root‐mean‐square deviation of 361 comparable Cα atoms was about 0.5 Å. The only changes of note between the two structures resulted from the engineered change in side‐chain for residue 145. The indole ring of W145 in TpMglB‐2R was missing in apo‐TpMglB‐2WA by design, and the BIS‐TRIS buffer molecule that was observed to stack onto the indole moiety of W145 was absent in the structure of apo‐TpMglB‐2. The open conformation and the lack of any observable electron density for d‐glucose suggest that the crystallization conditions (vide infra) disfavor the binding of the monosaccharide.
Figure 1.
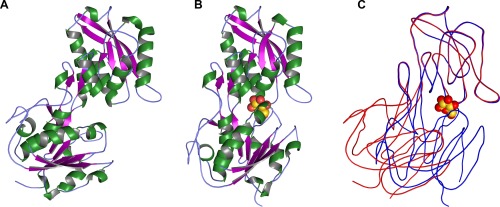
Overall structure and conformational changes in TpMglB‐2WA. (A) The structure of apo‐TpMglB‐2WA. A ribbons‐style representation of the protein is shown, with helices colored green, β‐strands purple, and regions without regular secondary structure light blue. A large cleft exists between the protein's two domains. (B) The holo‐TpMglB‐2WA structure. Atoms of the bound d‐glucose are shown as spheres, with oxygen atoms in red and carbon atoms in gold. (C) Superposition of the two structures. The C‐lobe was used for alignment purposes. The apo‐structure is shown in red, while that of the holo‐structure is in blue. Simplified traces through the mean Cα positions are shown for each respective structure. The position of d‐glucose in the holo‐TpMglB‐2WA structure is shown for reference.
The other crystal structure, at a resolution of 2.08 Å, clearly showed evidence of bound d‐glucose (Supplementary Information, Fig. S1), and it is thus termed the “holo” structure. Holo‐TpMglB‐2WA [Fig. 1(B)] adopted a closed conformation upon ligand binding. In the apo‐TpMglB‐2R and apo‐TpMglB‐2WA structures, the two lobes, termed “N” and “C,” were splayed apart, with a solvent‐filled cleft between them. In holo‐TpMglB‐2WA, the lobes were in close apposition, resulting in a net loss of solvent‐exposed surface area (‐1048 Å2). In comparing the two structures, it appeared that closure was accomplished by a rigid‐body movement of lobes about the protein's “hinge” region [Fig. 1(C)]. Some Cα atoms underwent relative motions as high as 17 Å in this conformational change; this is accomplished by a rotation of about 39°, which is consistent with the degree of motion found in MglB from Escherichia coli (EcMglB) upon ligand binding,6 and places the motion of TpMglB‐2 within established ranges for LBPs.9 Analysis using DynDom10 of the domain motions allows for a precise definition of the hinge regions in the primary structure of the protein; they are residues 49–51, 213–216, 270–274, and 283–285. Besides the hinge motion noted in the latter residues, the unique linker region that is present in TpMglB‐2 due to the circular permutation does not undergo significant rearrangements upon binding of d‐glucose.
d‐glucose binding
Buried between the lobes of holo‐TpMglB‐2WA was a single molecule of the β anomer of d‐glucose [Supplementary Information, Fig. S1, Figs. 1(B), 2(A)]. The monosaccharide was essentially solvent‐inaccessible: the calculated solvent accessibilities of the two copies of d‐glucose in the asymmetric unit were 0.5 and 0.1 Å2. As predicted,7, 11 numerous hydrogen bonds were made between cleft‐lining side chains and hydroxyl groups on the sugar. Also, the phenyl ring of F313 stacked on one face of the d‐glucose, as observed in other glucose‐specific LBP structures.5, 6 Of course, one of the residues whose side chain would likely also have stacked on the d‐glucose was missing by design: W145, which was replaced by an alanine in this protein construct. There was electron density in the void created by the loss of W145's indole ring (Supplementary Information, Fig. S2). We found that the density could be well modeled as ethylene glycol (EG), which was present in the cryoprotectant solution at a concentration of 15% (v/v). One hydroxyl group from the EG formed a hydrogen bond with O4 of the bound d‐glucose [Fig. 2(A)]. Despite our ability to observe bound d‐glucose in this structure, there are no overt clues to the lower glucose‐binding incompetency of this protein compared to the wild‐type.
Figure 2.
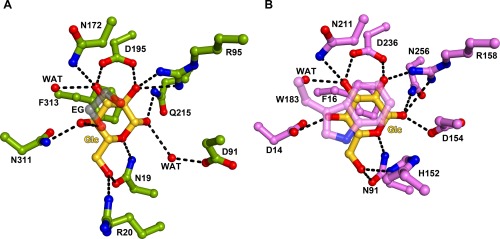
Details of d‐glucose binding. (A) The holo‐TpMglB‐2WA binding site. Residues are shown in stick‐and‐ball format, with oxygen atoms in red, nitrogens in blue, d‐glucose‐derived carbons in gold, protein‐derived carbons in green, and ethylene‐glycol‐derived carbons in gray. Atoms within hydrogen‐bonding distance are connected with dashed black lines. Glc, d‐glucose; EG, ethylene glycol; WAT, water. The molecule of ethylene glycol is shown semi‐transparently to avoid obscuring other atoms in this view. (B) The holo‐EcMglB binding site.6 The color conventions from the previous part are used, except protein‐derived carbons are shown in pink. W183 is also shown semi‐transparently.
Comparisons to EcMglB
This binding of d‐glucose to TpMglB‐2WA was very similar to that observed in structures of this ligand binding to EcMglB [Fig. 2(B)]. In that latter structure,5, 6 the ligand is sandwiched between two hydrophobic residues (F16 and W183), with an extensive network of hydrogen bonds between other side chains and the equatorially arranged hydroxyl groups of the d‐glucose. Most of the contacts between the ligand and protein are conserved in the TpMglB‐2WA structure, with a few exceptions. In EcMglB, H152 makes contact with the O6 oxygen atom of d‐glucose; this contact is absent in the d‐glucose/TpMglB‐2WA structure, despite the structural conservation of the histidine (H89 in the latter protein). Also, the side chain of D154 in EcMglB made a direct hydrogen bond to bound d‐glucose, but the homologous residue in TpMglB‐2WA, D91, contacted the sugar by a water‐mediated interaction [Fig. 2(A)]. Intriguingly, this direct interaction in EcMglB confers no steric or geometric advantages to binding the β anomer, despite the facts that only this anomer has been observed in crystal structures12 and it binds preferentially in solution.6 However, in TpMglB‐2, the water that makes contact to the anomeric OH group is rigidly held in place by hydrogen bonds to a carboxylate oxygen atom of D91 and the main‐chain oxygen atom of N19; this position favors binding of the β anomer, as the α‐anomeric OH group would have a poor interaction with the water (specifically, a hydrogen‐bonding angle of about 79°). The preferences of both TpMglB‐2R and EcMglB for d‐glucose over the epimeric sugar d‐galactose have been noted;7, 13 whether the mechanisms of these preferences are the same is unknown, as a structure of d‐galactose bound to TpMglB‐2 is not available, and the discrimination appears to be subtly distributed around the monosaccharide‐binding site in EcMglB.12 The observed differences in the binding sites could account for the apparently lower affinity of wild‐type TpMglB‐2R for d‐glucose compared to EcMglB (1.1 vs. 0.2 μM, respectively7, 13). However, a definitive pronouncement is not possible because of the widely divergent pHs (7.4 vs. 6.0, respectively) used in the affinity measurements.
It is unclear to what degree these unanticipated changes were inherent in the TpMglB‐2 binding site, given the W145A mutation present in the studied protein. As stated above, removal of the indole side chain of W145 leaves a void that is filled with EG in the present structure. The EG does not directly contact D91, and its closest distance to H89 is 4.1 Å (a possible van der Waals contact). The differences in the binding sites do not substantially affect the respective positions of the bound d‐glucoses (Supporting Information, Fig. S3). Thus, while it is conceivable that the EG is affecting the position of H89, a full characterization of the structural features of binding ultimately will depend on obtaining a wild‐type holo‐TpMglB‐2R structure.
Conclusions
Our results provide strong structural evidence that TpMglB‐2, despite its deviant topology, undergoes a conformational change akin to other LBPs upon binding to d‐glucose. These structures buttress the hydrodynamic evidence of ligand‐induced conformational changes obtained with the wild‐type protein.7 Given the likely importance of d‐glucose as both carbon and energy sources for T. pallidum,14 it is not surprising to find that TpMglB‐2 probably does not deviate from the basic mechanisms of nutrient transport by ABC transporters, despite its topological distinctness. The hypothesis that TpMglB‐2 acts as the d‐glucose‐binding component of an ABC transporter is further supported by the existence of genes for other presumptive components of the transporter in T. pallidum, namely tp0685 (mglA; the ATP‐binding component) and tp0686 (mglC; the permease).15
Materials and Methods
Protein purification
The procedure for expression and purification of the recombinant W145A variant protein of TpMglB‐2 (TpMglB‐2WA) was essentially as previously described.7 The purified protein was dialyzed overnight at 4°C against 1 L of 10 mM phosphate buffer, pH 7.4 containing 100 mM NaCl (Buffer A). The dialyzed protein was incubated with 10 mM d‐glucose overnight before crystallization. When necessary, the protein was concentrated using an Amicon centrifugal filter device (10‐kDa molecular mass cutoff; Millipore, Danvers, MA). Concentrations were determined spectrophotometrically using extinction coefficients calculated by the ProtParam utility of ExPASy.16
Protein crystallization and cryoprotection
Crystallization was initiated by mixing equal volumes (0.3 μL) of the purified protein and the precipitant solutions equilibrated against 100 μL of crystallization solution in a sitting‐drop configuration. For the holo‐TpMglB‐2 crystals, the protein concentration was 27 mg/mL in Buffer A containing 10 mM d‐glucose. Crystals appeared in approximately five days with a well solution containing 200 mM NaCl, 100 mM 2‐(N‐morpholino)ethanesulfonic acid (MES) pH 6.0, 20% (w/v) polyethylene glycol 6000 (PEG 6000). Prior to flash cooling in liquid nitrogen, crystals were cryo‐protected by incubating them in crystallization solution containing 20% (v/v) EG.
For apo‐MglB‐2WA, the protein was prepared as described above, except that the concentration was 18 mg/mL. For crystallization, the protein solution (0.35 μL) was mixed with an equal volume of the crystallization solution (0.2 M sodium citrate, 20% (w/v) PEG 3350) and incubated at room temperature. Prior to flash‐cooling in liquid nitrogen, the crystals were cryo‐protected by transferring them into a solution containing 0.2 M sodium citrate, 0.1 M NaCl, 20% PEG 3350, 10 mM d‐glucose, and 25% (v/v) EG.
X‐ray structure determination
All X‐ray diffraction experiments were conducted at beamline 19‐ID of the Advanced Photon Source at Argonne National Laboratories under direction of the Structural Biology Center. Crystals of holo‐TpMglB‐2WA diffracted X‐rays to a d min spacing of 2.08 Å, and they had the symmetry of space group P1. Two reciprocal‐space lattices were clearly present in the diffraction data, although the crystals showed no outward appearance of epitaxy or twinning. We considered this to be a special case of non‐merohedral twinning, with minimal overlap between the two visible lattices. Using HKL3000,17 we were able to index only the dominant lattice, with data reduction and scaling proceeding without any special provisions in the software. Phases for the holo‐TpMglB‐2WA data were determined by molecular replacement. We used the wild‐type apo‐TpMglB‐2R model (denuded of all waters and ligands) as the search model, but we split it into the N and C domains and performed separate searches in anticipation of the domain closure on bound d‐glucose. The linkers between the two domains were excised for this purpose. Phaser18 located two copies of each domain in the asymmetric unit with high confidence (final log‐likelihood gain = 5116 and translation function Z‐score = 25.3). After a round of rigid‐body, simulated‐annealing, positional, and B‐factor refinement in PHENIX,19 difference electron density for d‐glucose and the missing linkers was apparent. After adding these moieties to the model, refinement in PHENIX proceeded using positional, TLS, and individual B‐factor protocols. Between rounds of refinement, the model was adjusted in Coot.20
Crystals of apo‐TpMglB‐2WA diffracted X‐rays to a d min spacing of 1.47 Å and had the symmetry of space group C2221. No twinning was observed in the data from these crystals, and the data were indexed, reduced, merged, and scaled in HKL3000. Again, molecular replacement was used to obtain phases for the data. We used the wild‐type apo‐TpMglB‐2, denuded of all waters and ligands, as the search model. Phaser found one molecule of apo‐TpMglB‐2 in the asymmetric unit, with a final log‐likelihood gain of 4394 and translation‐function Z‐score of 49.7. No evidence of a bound ligand was observed in difference electron‐density maps, despite the inclusion of d‐glucose in the crystallization and cryoprotection media. The model was refined using the positional and anisotropic B‐factor refinement protocols in PHENIX. Occupancies of alternative conformations for amino‐acid residues were also refined. In the refinements of both apo‐ and holo‐TpMglB‐2WA, the weights for the stereochemical and B‐factor restraints were refined in the late stages of refinement. All structure figures were rendered in PyMOL (Schrödinger, LLC), and that program was also used for the surface‐area calculations.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
Some results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE‐AC02–06CH11357.
References
- 1. Davidson AL, Maloney PC (2007) ABC transporters: how small machines do a big job. Trends Microbiol 15:448–455. [DOI] [PubMed] [Google Scholar]
- 2. Quiocho FA, Ledvina PS (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol 20:17–25. [DOI] [PubMed] [Google Scholar]
- 3. Mao B, Pear MR, McCammon JA, Quiocho FA (1982) Hinge‐bending in L‐Arabinose‐ binding protein: the “Venus‐flytrap” model. J Biol Chem 257:1131–1133. [PubMed] [Google Scholar]
- 4. Vyas MN, Jacobson BL, Quiocho FA (1989) The calcium‐binding site in the galactose chemoreceptor protein. Crystallographic and metal‐binding studies. J Biol Chem 264:20817–20821. [PubMed] [Google Scholar]
- 5. Vyas NK, Vyas MN, Quiocho FA (1988) Sugar and signal‐transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 242:1290–1295. [DOI] [PubMed] [Google Scholar]
- 6. Borrok MJ, Kiessling LL, Forest KT (2007) Conformational changes of glucose/galactose‐binding protein illuminated by open, unliganded, and ultra‐high‐resolution ligand‐bound structures. Protein Sci 16:1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brautigam CA, Deka RK, Liu WZ, Norgard MV (2016) The Tp0684 (MglB‐2) lipoprotein of Treponema pallidum: a glucose‐binding protein with divergent topology. PLoS One 11:e0161022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 9. Trakhanov S, Vyas NK, Luecke H, Kristensen DM, Ma J, Quiocho FA (2005) Ligand‐free and ‐bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: Trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44:6597–6608. [DOI] [PubMed] [Google Scholar]
- 10. Hayward S, Berendsen HJC (1998) Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins Struct Funct Genet 30:144–154. [PubMed] [Google Scholar]
- 11. Deka RK, Goldberg MS, Hagman KE, Norgard MV (2004) The Tp38 (TpMg1B‐2) lipoprotein binds glucose in a manner consistent with receptor function in Treponema pallidum . J Bacteriol 186:2303–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vyas MN, Vyas NK, Quiocho FA (1994) Crystallographic analysis of the epimeric and anomeric specificity of the periplasmic transport/chemosensory protein receptor for D‐glucose and D‐galactose. Biochemistry 33:4762–4768. [DOI] [PubMed] [Google Scholar]
- 13. Zukin RS, Strange PG, Heavey LR, Koshland DE (1977) Properties of the galactose binding protein of Salmonella typhimurium and Escherichia coli . Biochemistry 16:381–386. [DOI] [PubMed] [Google Scholar]
- 14. Radolf JD, Deka RK, Anand A, Smajs D, Norgard MV, Yang XF (2016) Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat Rev Microbiol 14:744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, Sodergren E, Hardham JM, McLeod MP, Salzberg S, Peterson J, Khalak H, Richardson D, Howell JK, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton MD, Fujii C, Garland S, Hatch B, Horst K, Roberts K, Sandusky M, Weidman J, Smith HO, Venter JC (1998) Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375–388. [DOI] [PubMed] [Google Scholar]
- 16. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server In: Walker JM, Ed. (2005) The proteomics protocols handbook. Totowa, NJ: Humana Press, pp 571–607. [Google Scholar]
- 17. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Cryst 62:859–866. [DOI] [PubMed] [Google Scholar]
- 18. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure determination. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Emsley P, Cowtan K (2004) Coot: Model‐building tools for molecular graphics. Acta Cryst 60:2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information