

INTRODUCTION
Genome‐wide association studies (GWAS) and phenome‐wide association studies (PheWAS) have provided powerful methods for investigating the impact of genetic variation on individual drug response and have added extensive knowledge to the understanding of drug targets and effects. We highlight here recent advances in drug development, repurposing, and personalization accelerated by applying GWAS and PheWAS to longitudinal health data information, along with limitations of these methods.
IMPORTANCE OF UNDERSTANDING DRUG TARGETS AND EFFECTS
The challenges facing modern clinical pharmacology can be grouped into two major categories: the efficient development of novel therapeutics and understanding individual variability in response. The development of novel therapeutics is hampered by the problem that despite major advances in the knowledge of disease mechanisms, drug targets, and biomarkers, as well as a continual rise in investment into pharmaceutical research, the number of new drugs approved each year has remained steady.1 Most drugs fail in phase II clinical trials, with 50% of the failures due to lack of efficacy.2, 3 Thus, there is concern that preclinical disease models do not reliably predict efficacy in patients. Human genetics has been proposed as a mechanism to prioritize molecular targets early in the stages of drug discovery towards potentially more efficacious models.4, 5
The problem of variable drug efficacy and susceptibility to side effects has been recognized since the advent of therapeutics. There is now evidence that medication exposure data, outcome data, and genetic information linked together via longitudinal electronic health records (EHRs) can provide a more thorough understanding of drug effects, including response patterns and individuals at risk for potentially rare side effects.6 Knowledge of the genetic mechanisms that drive drug response variation and adverse events can help guide the tailoring of medication therapy. GWAS and PheWAS are modern genetic tools for the exploration of data sets in order to efficiently identify potential targets for novel therapeutics and to provide evidence‐based individualized therapy.
OVERVIEW OF GWAS AND PHEWAS APPROACHES
GWAS is a hypothesis‐generating method to systematically analyze variants across the entire genome (i.e., “genome‐wide”) for association with a phenotype of interest (Figure 1a). Over the past 10 years, the genotyping assays have evolved from early versions assessing hundreds of thousands of single nucleotide polymorphisms (SNPs) to current panels including millions of SNPs.7 At the same time, pipelines for quality control, imputation of genotypes, and statistical analysis for dichotomous, categorical, and continuous traits have matured and become standardized across high‐quality laboratories. The threshold for statistically significant results, given the need to correct for multiple comparisons, has been established at 5 × 10−8. Despite this stringent threshold, use of larger and larger cohorts (including some recent cohorts including more than 700,000 individuals) has enabled identification of many SNPs with statistically significant P‐values.8 GWAS focuses on detection of associations with relatively common variants (e.g., minor allele frequencies 1–5%) so the odds ratios are often small (OR < 1.5). Thus, the major outcome of many GWAS is a better understanding of the genetic architecture of complex traits, but not a set of high effect size variants that would be clinically actionable.7 As described below, examining drug response with GWAS has provided an interesting counterexample to these generalizations, since small numbers can yield signals with large enough effect sizes to be considered for implementation. This may reflect the idea that drug response represents an example of a controlled environmental intervention (drug) interacting with a genome, rather than a multifactorial complex disease state with many potential environmental inputs. Since 2005, over 3,000 GWAS have identified almost 40,000 unique SNP‐trait associations within the GWAS catalog provided by the National Human Genome Research Institute (NHGRI) and European Bioinformatics Institute (EBI).7 The rapid increase in knowledge of common genetic variants in complex diseases has provided significant opportunities for analyzing the association of genetic variation with disease phenotypes and response to therapies.
Figure 1.
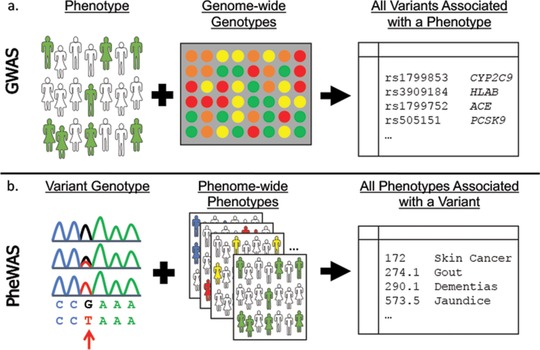
Genome‐wide association studies (GWAS) and phenome‐wide association studies (PheWAS). (a) A GWAS begins with a phenotype of interest and systematically analyzes variants across the entire genome (i.e., “genome‐wide”) for association to the phenotype. GWAS can identify multiple genetic associations to a phenotype in complex or polygenic traits. (b) A PheWAS begins with a genetic variant of interest and systematically analyzes many phenotypes (i.e., “phenome‐wide”) for association to the genotype. PheWAS has the ability to identify pleiotropy, or the finding of multiple independent phenotypes associated with a single genetic variant.
The integration of this wealth of genetic information with phenotypic data by linkage of DNA biobanks with EHRs has led to the development of a reverse genetics approach with EHR‐based genomic studies, termed PheWAS (Figure 1b).6 The first PheWAS study was performed in 2010, in which Denny et al. successfully replicated four known SNP‐disease associations.9 Since then, the use of PheWAS has continued to rise in popularity, with 58 current PubMed indexed publications. PheWAS provides a systematic approach to analyze the many phenotypes potentially associated with a specific genotype, with the ability to identify pleiotropy, or the finding of multiple independent associations with a single genetic association.10 The threshold for statistical significance is less well established for PheWAS; therefore, often a Bonferroni correction is applied in the analyses. However, this is highly stringent, as it assumes independence across all phenotypes, which is unlikely, given that many phenotypes are closely related. Despite this, the use of large cohorts have allowed for PheWAS to not only replicate known findings, but also identify novel associations.
GWAS and PheWAS approaches are complementary, with the ability to replicate and validate the other's findings. Representing the capacity for PheWAS to replicate GWAS, a comprehensive comparison of known GWAS associations within the NHGRI GWAS catalog against the PheWAS method was performed in 2013, showing that 210 of 751 (28%) known SNP‐disease associations were replicated with PheWAS, including 66% of those associations that were adequately powered to detect the association.11 This method also identified 63 potentially novel SNP‐disease associations, again demonstrating pleiotropic effects of the variants. In a pediatric cohort, PheWAS replicated many prior known GWAS associations, including SNPs associated with juvenile rheumatoid arthritis, asthma, autism, and pervasive developmental disorder, and type 1 diabetes.12 Several new SNP‐disease associations were identified within the pediatric population as well, including a cluster of association near the NDFIP1 gene (associated with mental retardation), PLCL1 (developmental delay), and the IL5‐IL13 region (eosinophilic esophagitis).12
Leveraging EHRs for drug‐based genomic and phenomic research
The EHR provides a longitudinal collection of phenotypic information coupled with medication exposures, thus making it an important platform for study of drug effects.6 A broad set of phenotypes collected in an unbiased approach is essential to the PheWAS method.13 To accomplish this, many PheWAS have used phenotypes derived from custom groupings of billing codes, also referred to as phecodes.9 The billing codes used for phecode groupings currently are International Classification of Diseases, Ninth Revision (ICD‐9) codes. ICD codes are published and maintained by the World Health Organization for classification of diseases and services for reimbursement of medical services. While the phecode groupings have been shown to better align with clinical diseases in practice, other methods of phenotype classification also are effective for PheWAS studies.14 Hebbring et al. reported a PheWAS method using individual ICD‐9 codes and parent ICD‐9 three‐digit groups as phenotypes.15 They not only replicated a known association of an HLA class II allele, HLA‐DRB1, with multiple sclerosis, but also replicated associations of HLA‐DRB1 with erythematous conditions and benign neoplasms of the respiratory and intrathoracic organs, found to be significant in a prior study by Denny and colleagues.9, 15 These and other studies highlight the importance of PheWAS techniques for identifying pleiotropic effects.
Billing codes are not the only source of phenotypes from the EHR. Hebbring et al. have shown that PheWAS can be performed by defining the phenome solely on textual data within clinical documentation.16 For drug effects, a phenome based on billing codes or clinical text alone may not accurately capture drug efficacy or adverse events, nor do they provide the necessary information about drug exposure, including dosing data. One potential method of obtaining this drug exposure and outcome data is to use prospective cohort‐based studies. It has been shown that PheWAS can be used with data obtained through clinical trials, representing a more biased, but targeted, approach at defining a phenome.17, 18
The benefit of leveraging EHRs for both GWAS and PheWAS, as opposed to prospective cohort‐based studies, is the ability to obtain large sample sizes with relatively less time or expense. While the EHR phenome may be incomplete, it includes conditions that are medically relevant, as opposed to clinical trial cohorts in which phenotypes may not represent conditions that necessitate medical attention. Biobanking of genetic data linked to the longitudinal patient data available within the EHR provides an efficient method for aggregating otherwise disparate information. EHR‐based biobanks have the potential to integrate genomic data with medication receipt, laboratory results, or textual data, thus refining both exposures and phenotypes, essential for research on drug effects.
Genomic investigation aids in understanding drug mechanisms
Several features of GWAS suggest its potential for elucidating drug mechanism and identifying relevant novel drug targets. An estimated 21% of published genes within the GWAS catalog are amenable to pharmacological modulation by small molecules.19 Further, the GWAS gene set is enriched with drug targets in comparison to the entire human genome, many of which align with the disease–gene pair identified by GWAS analysis.19
Prior studies support the role of using GWAS to identify alleles that contribute to disease risk and druggable targets. Early GWAS efforts retrospectively identified the genetic basis for drugs already in use for a particular indication. Statins have been used to inhibit 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase and treat hyperlipidemia for decades,20 and GWAS studies in 2008 showed that low‐density lipoprotein (LDL) levels are associated with variation in HMGCR, the gene that encodes HMG‐CoA reductase.21, 22 Further, pharmacogenetics studies have shown that genetic variation in HMGCR is associated with statin efficacy.23, 24 Since then, variants in other genes involved in lipid metabolism but not direct targets for statin action (APOE, LPA, SORT1/CELSR2/PSRC1, and SLCO1B1) have been found in GWAS to be associated with the LDL‐lowering effect of statin therapy.25, 26 A recent GWAS also found that several variants with the LPA locus appear to be associated with coronary heart disease events during statin therapy, independent of the extent of LDL‐cholesterol lowering.27
Other examples of drugs with a mechanism replicated by GWAS are ustekinumab, a monoclonal antibody against interleukin (IL)‐12 used for treatment of psoriasis and inflammatory bowel disease,28, 29 and denosumab, a monoclonal antibody to the receptor activator of nuclear factor‐kappaB ligand (RANKL) for treatment of osteoporosis.30 Metformin has long been used to lower blood glucose levels in individuals with diabetes; however, a 2011 GWAS of 3,920 type 2 diabetes patients clarified the genetic basis for its mechanism with polymorphisms in the ATM gene found to be associated with glycemic control.31 Okada et al. evaluated the role of GWAS in validating the current therapeutic drug targets for rheumatoid arthritis (RA).32 Through a comprehensive genetic study with nearly 100,000 subjects, they found that 18 of 27 currently approved drugs for RA target genes identified as RA risk loci, and also suggest several potential novel therapeutics, some of which had supporting animal studies.32 These early successes fuel enthusiasm for using GWAS to elucidate disease mechanisms and drug targets.19
Early evidence for drug discovery using genomic approaches
In the context of drug development, GWAS advances are relatively recent and are only now being applied to have a potential impact on target discovery. Nevertheless, prior linkage and candidate gene studies have shown that genetics can drive development of novel therapeutics. The development of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors represents this realization. In 2003, it was found that autosomal dominant hypercholesterolemia and an increased incidence of coronary heart disease were associated with gain‐of‐function mutations in the PCSK9 gene.33 Subsequent candidate gene association studies in 2005 and 2006 revealed that PCSK9 loss‐of‐function mutations correlate with reduced levels of LDL cholesterol and a lower incidence of coronary heart disease.34, 35 In 2012, almost 10 years after the first genetic discovery, randomized controlled trials demonstrated that PCSK9‐specific monoclonal antibodies significantly reduce LDL cholesterol levels.36, 37 There are now two US Food and Drug Administration (FDA)‐approved PCSK9 inhibitors. Similar to how candidate gene studies led to the target of PCSK9 and subsequent development of a novel therapy for familial hypercholesterolemia, GWAS and PheWAS hold promise as means to identify novel drug targets. However, the timeline from target to an approved drug is often over 10 years. As findings from GWAS have exponentially increased over the last decade and PheWAS is gaining similar recognition, we anticipate the next decade will show progress toward utilization of that knowledge and drug development.
GWAS for understanding impact of genetic variation on drug efficacy
A considerable amount of variability can exist in a patient's response to drug therapy, including differing efficacy, adverse side effects, and toxicity. A better understanding of the genetic determinants of drug response and mechanism is thought to have potential to individualize drug treatment toward improved efficacy and side effect profiles.38 While candidate gene studies have shown success in identifying genetic variants that contribute to drug response and effects, for many drugs the biological mechanism, metabolic pathways, and potential genetic associations impacting individual response is unknown, limiting the potential for focused gene analysis. In contrast, GWAS are a hypothesis‐free method that can be utilized to determine associations of genetic variation with effects of drug treatment (Figure 2a).
Figure 2.
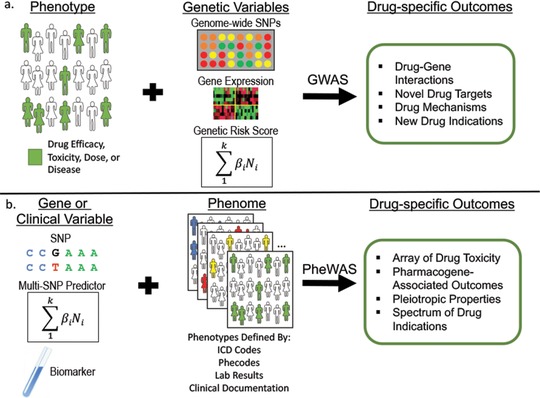
Drug‐specific outcomes identified through genome‐wide association studies (GWAS) and phenome‐wide association studies (PheWAS). (a) Use of drug‐specific phenotypes of interest with genomic predictor variables such as genome‐wide single nucleotide polymorphisms (SNPs), measured or predicted gene expression, or genetic risk scores, can be used in GWAS analysis to gain information for drug mechanisms and discovery. (b) In PheWAS, genetic or clinical variables can be used to search for associations in phenomes curated from different sources of information in the EHR for analysis of drug‐specific associations.
Drug efficacy in particular is often considered to be along a continuum in a patient population. The known genetic variants contributing to statin efficacy discussed above are an indication of the significant clinical and genetic variability that can be seen in a population.25, 26 A high‐yield area of pharmacogenetic investigation utilizing GWAS has been the study of drugs with a narrow therapeutic window and variable efficacious dosing regimens, such as warfarin. While candidate gene studies were used to initially describe the associations of CYP2C9 and VKORC1 with the ability for warfarin to achieve anticoagulation,39, 40, 41, 42 subsequent GWAS have confirmed these findings, showing these to be the strongest genetic predictors of warfarin dose required in individuals of European descent.43, 44 Subsequent GWAS in individuals of African ancestry has also found that in addition to the well‐known CYP2C9*2, CYP2C9*3, and VKORC1 polymorphisms, the CYP2C locus exerts influence by a variant outside of those well established, and this new variant could improve dose prediction in this population.45 Differences in the variants associated with warfarin effect across populations may be due to the differences in mean allele frequencies, ancestry‐specific gene–gene interactions, or population‐specific gene–environment interactions.
Response to clopidogrel therapy is also known to be highly variable. Clopidogrel is a prodrug, and the bioactivation pathway is largely CYP2C19‐dependent.46 Candidate gene studies of cardiovascular events on clopidogrel indicated that CYP2C19 loss‐of‐function variants increased risk.47, 48, 49 Subsequent GWAS in Amish individuals found that the most common loss‐of‐function allele, CYP2C19*2, had the strongest genetic association with the effect of clopidogrel on platelet aggregation; however, this single variant only accounts for ∼12% of the variability in response seen in this population.50 More recently, Zhong et al. identified two novel variants in a Chinese population that were associated with the antiplatelet effect of clopidogrel, as measured by P2Y12‐mediated platelet aggregation, as well as formation of H4, an active metabolite of clopidogrel.51 They estimate that the identified variants, in association with CYP2C19*2, CYP2C19*3 (a variant common in Asian populations), and clinical factors, can improve the predictability of clopidogrel effect to 37.7%.
Another example of GWAS elucidating the genetic underpinnings of variability in drug efficacy is in the use of interferon‐alpha for treatment of hepatitis C infection. A polymorphism adjacent to IL28B has been shown to predict treatment response and viral clearance in individuals on interferon‐alpha for hepatitis C in several GWAS.52, 53, 54 Because the genotype associated with improved response is more common in individuals of Asian and European ancestry than African ancestry populations, this genetic polymorphism may explain the difference in response rates between patients of African and European ancestry.52
These GWAS of statin, clopidogrel, and interferon‐alpha effects all emphasize that variability in drug response is ancestry‐dependent due to the vast difference in distribution of genetic variants, such as CYP2C9, CYPC19, and IL28B, across populations. These findings, along with others, have increased focus towards a personalized approach to disease treatment and encouragement of research efforts from individuals of diverse backgrounds. Studies across ancestries are needed to fully capture the genetic architecture of human traits, including drug response, and ultimately, appropriately implement such variants in clinical practice.
GWAS for understanding impact of genetic variation on drug toxicity
GWAS has been used to determine potential associations of drug toxicities and adverse drug reactions (Figure 2a). The human leukocyte antigen (HLA) variation, in particular, has been associated by GWAS with susceptibility to adverse drug reactions. Drug‐induced liver injury (DILI) is a rare but serious adverse effect secondary to many drug therapies, with increased susceptibility in HLA regions implicated in several studies.55, 56, 57 The first study was in 2008 and focused on ximelagatran, an oral direct thrombin inhibitor that was removed from the market in 2006 due to the development of transaminitis in some patients. In this study, Kindmark et al. performed a GWAS that suggested an association between DILI during use of ximelagatran with HLA class II alleles, which was confirmed with candidate gene studies.58 In 2009, Daly et al. found a strong association of HLA‐B*5701 with DILI following treatment with flucloxacillin.55 Singer et al., in 2010, identified an association of hepatotoxicity after use of lumiracoxib, a selective cyclooxygenase‐2 inhibitor, with common HLA class II haplotypes.56 In 2011, Lucena et al. performed GWAS of 201 cases of DILI after treatment with amoxicillin‐clavulanate compared with 532 controls, finding HLA class I and II SNPs may confer susceptibility to liver injury after this antibiotic treatment.57 Due to the rarity of DILI, the finding of genetic predispositions in GWAS may be limited. Nicoletti et al. recently attempted to overcome this limitation by grouping DILI caused by any drug other than the common causes (flucloxacillin and amoxicillin‐clavulanate) to determine predisposition for DILI.59 They found a strong association of HLA‐A*33:01 with DILI, appearing to be heavily influenced by the effects of terbinafine.59 This demonstrates how novel methods in studies will allow researchers in some cases to overcome the power limitations of GWAS and find rare variants with a large effect size.
The HLA locus has also been implicated in other adverse drug reactions, including skin hypersensitivity. In 2004, Chung et al. reported a strong association in a Han Chinese population between HLA‐B*1502 and Stevens–Johnson syndrome induced by carbamazepine.60 Candidate gene studies were also used to ascertain an association between variation in the HLA region, HLA‐B*5701 (OR = 117), with abacavir skin hypersensitivity, which has since been elucidated both at a structural and mechanistic level.61, 62 Several subsequent GWAS studies across ancestral populations have shown that skin hypersensitivity reactions, ranging from skin rash to severe reactions such as Stevens–Johnson syndrome/Toxic Epidermal Necrolysis, can occur secondary to a wide range of drug therapies. In 2011, Ozeki et al. identified the HLA‐A*3101 allele as a genetic risk factor with a modestly large effect size (OR = 10.8) for carbamazepine‐induced hypersensitivity in a cohort of 53 cases and 882 controls from Japan.63 McCormack et al. shortly after reported the same genetic association with carbamazepine‐induced hypersensitivity reaction in individuals of European descent, finding a large effect size as well (OR = 12.4).64
One important early GWAS example is the study by Link et al., which discovered a single strong association of statin‐induced myopathy in Europeans with an SNP located within SLCO1B1, known to encode an organic anion‐transporting polypeptide that regulates the hepatic uptake of statins (OATP1B1).65 While the variant allele frequency of this significant polymorphism is 0.13 in European populations, carriage of the variant allele resulted in an 18% incidence of myopathy over 5 years, with 60% of cases attributable to the variant allele. Thus, further studies are needed to define the mechanism(s) underlying this “variable penetrance.” A recent study by Mosley et al. evaluated the association of genetic variation with angiotensin‐converting enzyme inhibitor (ACEi)‐induced cough.66 Cough is the most common side effect of ACEi therapy, with epidemiologic variation that suggests a potential genetic predisposition. In GWAS consisting of 1,695 cases of ACEi‐induced cough compared with 5,485 controls, SNPs in KCNIP4 were associated with increased risk for developing cough with ACEi. In recent GWAS, genetic variation has also been implicated as increasing susceptibility to anthracycline‐induced cardiotoxicity and reduced left ventricular function.67, 68 Vancomycin, a commonly used antibiotic, is known to be nephrotoxic, with a GWAS suggesting variation at the chromosome 6q22.31locus could modulate that risk as well.69
PheWAS for understanding drug response variability
PheWAS also has the potential to uncover associations with drug effects, including therapeutic response and side effect profiles (Figure 2b). Neuraz et al. described in 2013 the use of a study population with thiopurine exposure to determine associations with clinical traits after drug exposure to identify adverse events.70 They grouped 442 individuals with thiopurine exposure into three categories based on thiopurine S‐methyltransferase (TPMT) activity, a quantitative trait available from the EHR for patients with clinical TPMT testing. They found that very high TPMT activity was associated with diabetes mellitus and iron‐deficiency anemia. Similarly, they analyzed associations with laboratory data, finding that very high TPMT activity along with thiopurine exposure was associated with an increased incidence of hyperglycemia and anemia by test results. This study shows the ability for PheWAS to identify adverse events potentially associated with drug use, as well as the feasibility of crossvalidation of conventional PheWAS analyses with biological test results.70
Others have noted the potential ability to leverage the identification of pleiotropic effects through PheWAS methods to predict potential adverse events to drug therapy. Diogo et al. analyzed associations of RA‐protecting variants, potential future targets for therapeutics, for additional indications and potential adverse events.71 They first demonstrated that three protein coding variants in tyrosine kinase 2 (TYK2) independently protect against RA. To determine the possibility for TYK2 to be a drug target, they analyzed for associations of TYK2 with any other complex phenotypes. They did not identify any associations meeting statistical significance, suggesting that inhibition of TYK2 may not result in serious adverse events in the treatment of RA. However, the ranking of associations not meeting significance potentially prioritizes adverse events for study in a trial and represents an analytical framework that could show success in the future. This study also highlights the very large populations needed for this study design; among over 20,000 individuals in the PheWAS of one cohort, a total of 2,612 had pneumonia, the potential adverse event most trending toward statistical significance. Using PheWAS to suggest deleterious effects of evolving therapeutics early in the drug development stages could allow resources to target therapeutics with greater potential or can identify patient populations for whom the drug may be contraindicated.
Use of GWAS and PheWAS to identify opportunities for drug repurposing
In addition to elucidating drug mechanism and response variability, GWAS and PheWAS can be used to identify novel treatment methods through drug repurposing (Figure 3).72, 73 Drug repurposing, also termed drug repositioning, is the application of an existing therapeutic drug for new indications. Drug repurposing could significantly speed up the typical >10 years lag time for FDA approval and drug marketing, as preclinical and phase I clinical trials are already complete. While the GWAS gene set is enriched with targets already pursued by drugs that align with the disease–gene pair identified by GWAS analysis, there are also mismatches in which the indication for the drug is not congruent with the associated disease by GWAS, and examples of pleiotropy, where multiple diagnoses are associated with the same genetic signal.19 By comparing known GWAS‐disease associations to the indications of drugs with known gene targets, Sanseau et al. identified 92 individual genes that are targets of drug projects that mapped to a GWAS trait different than their drug indication.19 These instances represent potential drug repurposing opportunities.
Figure 3.
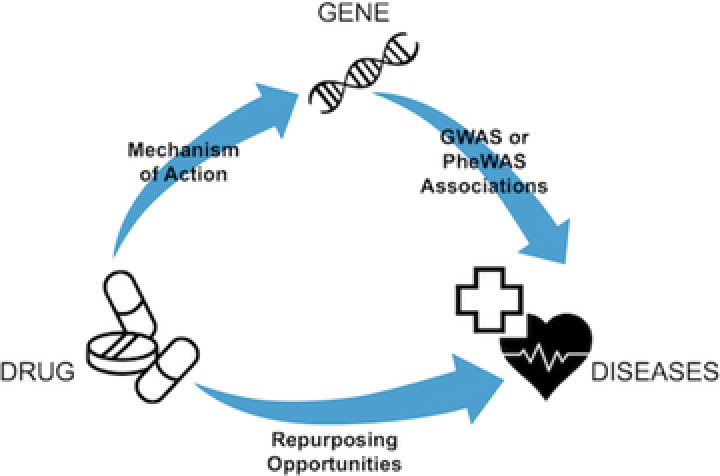
Opportunities for drug repurposing using results of genome‐wide association studies (GWAS) and phenome‐wide association studies (PheWAS). Given a known mechanism of action or genetic target of a currently approved drug, GWAS and PheWAS reveal drug repurposing opportunities through identification of diseases with common genetic associations with the known drug genetic target.
Prior studies have demonstrated the success of this approach, for example, the use of complement inhibitors for the treatment of age‐related macular degeneration (AMD). One of the first GWAS in 2005 found the complement factor H gene to be strongly associated with the risk of AMD.74 At that time, complement inhibitors had been developed for the treatment of sepsis and paroxysmal nocturnal hemoglobinuria.75, 76, 77 This has led to the targeting of factors in the alternative complement pathway in clinical trials with promising findings for reducing the severity of AMD.78 In addition to validating currently approved RA drug therapies, Okada et al. identified several drugs used for other diseases that target biological genes containing RA risk SNPs and thus proposed these as drug repurposing opportunities.32 They found that CDK6 and CDK4, targets of three approved drugs for cancer (palbociclin, capridine, and flavopiridol), include RA risk SNPs, suggesting they should be investigated for efficacy in RA as well.
Analogous to the use of GWAS to identify novel drug uses, the ability of PheWAS to identify pleiotropic effects creates opportunities for drug repurposing (Figure 3).13, 73 As PheWAS can identify diseases that share a common etiology, one can theorize that drugs used to treat one disease may also have efficacy to treat another.16 A hypothesis‐generating study by Rastegar‐Mojarad et al. evaluated the potential for drug repurposing by linking current drug‐targeted genes in DrugBank to the gene–phenotype associations in the PheWAS catalog.73 They validated the disease indications for drugs in 127 cases, but also identified 2,583 that strongly supported potential novel drug–disease associations, available within a cataloged database to the public.79 There are several factors that can influence the ability for a drug–disease identified in PheWAS and poised for drug repurposing to come to fruition. In particular, methods must be developed to narrow the results to candidate drug–disease pairs that are supported in the literature or by mechanistic knowledge. Rastegar‐Mojarad et al. started this approach by cross‐referencing all pairs with the clinical trial registry, noting that incorporation of other biomedical databases could also significantly improve prioritization.73 Recently, Pulley et al. specifically described six genes with pleiotropic effects identified in PheWAS, three of which are currently underway to study repurposing opportunities of drugs with respect to the relevant molecular target.80
Millwood et al. in 2016 used PheWAS methods applied to ICD‐10 codes in the China Kadoorie Biobank to evaluate the potential efficacy of lipoprotein‐associated phospholipase A2 (Lp‐PLA2) inhibitors for the treatment of atherosclerotic disease.81 Loss‐of‐function variants in the PLA2G7 gene is associated with reduced Lp‐PLA2 activity and is relativity common among East Asian populations. Through their PheWAS analysis, they determined there was no association of a loss‐of‐function variant in PLA2G7 with improvement in vascular diseases, such as stroke and coronary events, or nonvascular diseases in a Chinese population. They note that these findings correlate with the lack of efficacy in a 2014 randomized controlled trial with the Lp‐PLA2 inhibitor darapladib.82 Use of PheWAS results such as these in the design of clinical trials could thus help guide study design, saving time and resources.
CHALLENGES OF GWAS AND PHEWAS IN DRUG DISCOVERY, DRUG REPURPOSING, AND PHARMACOGENOMICS
Statistical power in GWAS and PheWAS is determined by the size of the study cohort, the frequency of the variant, and the effect size of the variant. Both methods are limited by a reduced ability to achieve statistical significance given the large number of hypotheses tested.13 While the number of phenotypes tested in PheWAS is relatively small compared with the number of genotypes tested in GWAS, testing of multiple genotypes against a large set of phenotypes exponentially increases the number of statistical tests, requiring smaller and smaller P‐values for statistical significance with Bonferroni correction.
GWAS and PheWAS for evaluation of drug effects is challenged by small sample sizes, with a subsequent lack of power to detect small or moderately sized effects.83 For example, rare but serious adverse events or drug nonresponders may be associated with rare variants with clinically relevant effect size, but could be potentially missed in traditional GWAS. Due to the rarity of these events in a population, sample size is often much smaller than is typical in GWAS performed for evaluation of disease risk. While the sample size for GWAS in pharmacogenomics studies is typically less than 1,000 individuals, GWAS for common diseases often use thousands of subjects, with meta‐analyses containing even tens of thousands, realized by the pairing of genomic information with EHRs.84 Non‐EHR cohorts often have focused clinical information, lacking drug response trait information. While the EHR can be leveraged to identify drug response traits, the rarity of events necessitates collaboration between biobanks to reach adequate statistical power. Several efforts to encourage data sharing have evolved over the last decade, such as the Electronic Medical Records and Genomics (eMERGE) network, UK Biobank, China Kadoorie Biobank, and Million Veterans Project.
Another challenge facing GWAS and PheWAS is due to the complex architecture of phenotypes with non‐Mendelian inheritance patterns. Disease‐associated alleles, and thus druggable genes, often have a very small effect on the overall risk of the disease, thus variability in drug effects can also only be partially accounted for by an identified genetic variant.4 GWAS and PheWAS are designed and powered to detect associations with common genetic variants in a population, with the majority of these variants having small genetic effects. Thus, while an association may be present between a drug effect and genetic variant, many other environmental and genetic factors are also simultaneously contributing to that variation, resulting in a significant proportion of “missing heritability.” Further, GWAS and PheWAS results are population‐specific, with the majority of large studies being performed in populations of European descent.85, 86 The extent to which these findings can be translated to other populations is unknown, as there are significant differences in linkage disequilibrium and allele frequencies between ancestries.
While GWAS may identify many alleles contributing to disease risk, not all of those alleles or potential gene targets will be disease‐causing or able to be modulated for disease treatment.4 Those genes which harbor causal alleles must be differentiated from the rest in order to narrow the search for potential drug targets. Once a causal allele is identified, it can be difficult to understand the mechanism by which the gene variation contributes to the disease; thus, functional studies are required to fully understand the disease risk attributed to the gene and the potential mechanism of a modifying drug. Another factor that has limited the success of GWAS findings from being translated to marketed therapeutics is the long duration, often over 10 years, before a gene target is translated into an approved marketed drug.19 As previously discussed, drug repurposing is a method to potentially decrease this development time.
Accuracy and scalability of EHR‐based phenotyping for drug response
The use of GWAS and PheWAS to investigate drug efficacy and adverse events relies upon accurate description of the drug response or adverse event. Although EHRs have greatly eased the ability for researchers to identify phenotypes in a population, accurate drug response and side effect phenotypes remain a challenge to assemble in large cohorts.83 EHR‐based GWAS and PheWAS rely on the ability to readily extract structured data from the medical record. For PheWAS, this is often in the form of billing codes, which are unreliably accurate and rarely used for describing drug effects, drug efficacy, and adverse event phenotypes. Thus, manually curated and validated phenotyping algorithms from the EHR must be developed and implemented. While EHRs have allowed for accrual of and access to clinical information, algorithms are necessary to extract this information from the various parts of the EHR, including clinical documents, laboratory data, nursing records, etc. Development and validation of these algorithms can be time‐consuming and require both clinical and technical expertise.
While curation of a single (or few) phenotypes for a GWAS is manageable, this is much more difficult for the thousands of potential phenotypes used in a PheWAS. Phecodes have shown efficacy for PheWAS analyses; however, they do not align precisely with clinical diseases and may not have adequate granularity or specificity for some phenotypes.14, 87 Phecodes currently use ICD‐9‐CM codes as their sole source of information. Efforts are underway to map the codes to ICD‐10, but more important, billing codes alone do not capture all medically relevant phenotypes. For drug effects, while integration of billing codes with other portions of information from the EHR can refine phenotypes and exposures, a significant limitation in obtaining these well‐specified phenotypes from various sources is first the clinical expertise to define the phenotype, followed by the informatics support to extract the information from the EHR.88, 89 Curated phenotypes have been developed for individual diseases, but there is currently no high‐throughput mechanism to produce cases and controls from thousands of detailed phenotype algorithms. New methods are needed to study drug exposures with events at scale, while appropriately assessing the timing of both. Currently, drug response phenotypes are best pursued one‐phenotype‐at‐a‐time.
As we have previously discussed, drug efficacy can vary significantly in a population. However, accurate ascertainment of drug response as a continuous outcome is difficult. For some phenotypes, such as blood pressure reduction or blood glucose control, multiple measurements may allow for more accurate determination of response; however, for the majority of therapeutics there is not a defined scale for response or adverse effect, nor are these measurements made routinely part of clinical care to enable large GWAS studies in EHR cohorts. The recent study by Wells et al. shows how a phenotype along a continuum, left ventricular function by systolic ejection fraction, can be used as the outcome in a GWAS analysis to determine drug side effects, rather than a dichotomous variable, such as the presence or absence of heart failure.68 When feasible, GWAS to measure drug–phenotype associations should use phenotypes defined along a continuum to allow improved accuracy of prediction.
Other limitations of EHR‐based genetics research are secondary to the current confines of EHRs. Due to the decentralization of EHRs, data within the record itself may be incomplete due to the various providers and institutions a patient may visit. Also, EHRs are designed for exchange of clinical information and billing purposes, not specifically for research. Thus, inaccuracies can be introduced by clinical uncertainty or billing errors, and the amount of information available can vary greatly. Further efforts to improve EHR data, centralize information, and allow for phenotype curation from EHR data more efficiently and accurately will greatly facilitate advancement in phenotyping studies.
EMERGING GWAS AND PHEWAS‐RELATED TECHNIQUES FOR PHARMACOGENOMICS
While GWAS methods have provided insight into thousands of variants associated with complex traits, the biological mechanisms underlying the associations remain poorly understood. Gene expression is an intermediate between genetic information and phenotypes and can play an important role in drug response. One proposed method to gain information on biological mechanisms and gene expression is through PrediXcan, a technique that estimates the component of gene expression determined by an individual's genetic profile through use of reference transcriptome data sets and correlates that gene expression with the phenotype of interest.90 PrediXcan can be likened to a limited PheWAS, using imputed gene expression as the PheWAS predictor variable. A major benefit of PrediXcan is its ability to increase power by aggregating the effects of SNPs associated with gene expression. PrediXcan also provides direction of the effect of the genetic variant, for example, increased or decreased gene expression. This is significant for drug discovery and repurposing, as the development of therapeutics that downregulate a gene, and thus gene expression, is often easier to attain than development of drugs that upregulate a gene.90
In addition to potential opportunities with drug development, PrediXcan can provide insights into drug effects. One recent example in which this has been employed is in evaluation of genetically determined expression levels association with chemotherapy‐induced peripheral neuropathy. Dolan et al. analyzed associations with cisplatin‐induced peripheral neuropathy using GWAS and PrediXcan.91 While no SNPs met genome‐wide significance in GWAS, lower expression of RPRD1B, which is predicted by 20 SNPs on chromosome 20 and codes for a protein that regulates transcription of genes involved in the cell cycle, was associated with decreased risk for cisplatin‐induced peripheral neuropathy in PrediXcan (P = 0.0089).91 These recent findings suggest a promising role for PrediXcan methods in the future.
Techniques such as PrediXcan, which aim to increase the power of GWAS methods, may overcome some of the limitations for GWAS to identify associations with rare variants or small effects. Further, although analysis of GWAS data often uses stringent thresholds for statistical significance, there is likely information that can be gleaned from associations with P‐values that fail to meet the 5 × 10−8 threshold. Some have proposed analyzing GWAS data using a multiple‐locus‐based approach, drawing on protein pathway‐ or domain‐based data to develop a candidate gene data set, which can then be integrated with known drug–gene target sets to identify potential drug repurposing opportunities.72, 92 This has been suggested for a wide range of complex diseases, including type 1 and 2 diabetes, bipolar disorder, Crohn's disease, hypertension, coronary artery disease, and RA.72, 93
Although the initial applications of the PheWAS methodology have focused on identification of phenotypes that are associated with single SNPs, recent approaches have involved a search for associations with aggregated genetic information or other phenotypic data.94 These advances also aim to overcome the power and effect size limitations of traditional PheWAS studies. Use of a set of SNPs as the input for a PheWAS can be one way to increase the effect size in PheWAS. The set of SNPs can be used to generate a genetic risk score derived from GWAS data and weighted based on an individual SNP effect size. Krapohl et al. used genetic risk scores of thousands of SNPs derived from GWAS of psychiatric traits to determine associations with phenotypes.95 They also demonstrate the use of a limited phenome, consisting only of behavioral phenotypes only, which can be used to yield greater power.
Similarly, methods for the joint testing of multiple correlated traits can be performed to increase the power in a PheWAS analysis.96 As many phenotypes are known to be correlated, the Bonferroni correction often applied to PheWAS is likely overly‐conservative, resulting in significant associations being missed. Performing the analysis on an a priori grouping of correlated traits could increase likelihood of finding associations. Any significant association could then be more closely analyzed individually, decreasing the number of tests performed in a single PheWAS compared with analysis using the entire phenome.96
It is the curation of the EHR phenome that enables PheWAS, and the technique is not limited to the study of genetic effects. PheWAS methods can also be used to investigate the association of other factors, such as laboratory parameters or comorbidities, with human traits, an analysis that can be termed a phenotype‐only PheWAS. Using this approach, Warner et al. demonstrated that elevated white blood counts (WBC) in an intensive care unit are associated with diagnoses of Clostridium difficile infection and bacterial sepsis.97 This study also takes advantage of the nonbinary features of many clinical traits, such as continuous laboratory measurements, to show the varying WBC across the phenome. Limiting dichotomization of these features, which could lead to loss of significant information and ability to find associations, will be important in future PheWAS.
Phenotype‐only PheWAS can also be used to describe features associated with a disease process, as shown recently in the description of features associated with systemic loxoscelism.98 In another study, Liao et al. used the predictor in a PheWAS as the presence of autoantibodies among a cohort of patients with RA, and determined a significant association between several different epitopes and comorbidities.99 A similar approach was used by Doss et al. to define subgroups of RA patients based on serology for rheumatoid factor, finding that seronegative RA was associated with fibromyalgia and seropositive RA was associated with chronic airway obstruction.100 In addition to demonstrating the use of nongenetic information for PheWAS analyses, these studies show the ability for PheWAS to identify subtypes within diseases; for example, associations with other diseases, severity of disease, variable phenotypic manifestations of disease, or differing response to therapeutics. Outside of clinical phenotypes as predictors in PheWAS, another opportunity for the future is to apply PheWAS to PrediXcan, in which predictors of gene expression can be used to identify traits associated with predicted increased or decreased expression of a gene. Each of these developing techniques have the potential to add insight on subgroups of diseases that respond to medication therapy differently, including patient populations with the development of adverse effects or lack of efficacy.
While the potential for evolution of PheWAS techniques are vast, the goal will remain the same—to improve the ability for PheWAS to identify novel associations by increasing power and improving predictive capacity.
CONCLUSION
GWAS and PheWAS not only provide insight into the biology of diseases, but also provide opportunities for drug targeting, development, and identification of populations at risk for drug‐related adverse events. Further investigations using current and future methods will provide the linkages between disease–gene associations, cellular mechanisms, and therapeutic approaches. GWAS and PheWAS pharmacogenomic studies with larger sample sizes, facilitated by multiinstitutional collaboration and consistent phenotyping through utilization of EHRs, can allow future studies to achieve greater power to identify small to moderate genetic effects on drug response. Techniques such as genetic risk scores to analyze all risk genes, including those with small and large effect size in a population, will further facilitate greater accuracy in prediction of response to drug therapy.
Conflict of Interest
S.L.V. discloses participating as an invited speaker at Merck.
Acknowledgments
J.R. Robinson receives salary and tuition support by the T15 LM007450 training grant from the NIH National Library of Medicine. S.L. Van Driest is supported by an Innovation in Regulatory Science Award from the Burroughs Wellcome Foundation (1015006) and a Clinical Scientist Development Award from the Doris Duke Foundation, and has been supported by the NIH National Center for Advancing Translational Sciences KL2 TR000446. This work is also supported by NIH funding through R01 LM0106085 and P50 GM115305.
Author Contributions
J.R.R. reviewed the relative literature for data collection and analysis and wrote the article. S.L.V. designed the format of the review, added significant data for inclusion, and provided critical review of the article. J.C.D. and D.M.R. both added significant data for inclusion and provided critical review of the article. All authors approved the final article.
References
- 1. Scannell, J.W. , Blanckley, A. , Boldon, H. & Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Disc. 3, 191–200 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Disc. 8, 711–715 (2004). [DOI] [PubMed] [Google Scholar]
- 3. Arrowsmith, J. & Miller, P. Trial watch: phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Disc. 8, 569 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Plenge, R.M. , Scolnick, E.M. & Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Disc. 8, 581–594 (2013). [DOI] [PubMed] [Google Scholar]
- 5. Nelson, M.R. et al The support of human genetic evidence for approved drug indications. Nat. Genet. 8, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Denny, J.C. Surveying recent themes in translational bioinformatics: big data in ehrs, omics for drugs, and personal genomics. Yearb. Med. Inform. 199–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hindorff, L.A. et al Potential etiologic and functional implications of genome‐wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 23, 9362–9367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marouli, E. et al Rare and low‐frequency coding variants alter human adult height. Nature 7640, 186–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Denny, J.C. et al PheWAS: demonstrating the feasibility of a phenome‐wide scan to discover gene‐disease associations. Bioinformatics (Oxford, UK) 9, 1205–1210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hebbring, S.J. The challenges, advantages and future of phenome‐wide association studies. Immunology 2, 157–165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denny, J.C. et al Systematic comparison of phenome‐wide association study of electronic medical record data and genome‐wide association study data. Nat. Biotechnol. 12, 1102–1110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Namjou, B. et al Phenome‐wide association study (PheWAS) in EMR‐linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5‐IL13 to Eosinophilic Esophagitis. Front. Genet. 401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Denny, J.C. , Bastarache, L. & Roden, D.M. Phenome‐wide association studies as a tool to advance precision medicine. Annu. Rev. Genom. Hum. Genet. 353–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei, W.Q. et al Evaluating phecodes, clinical classification software, and ICD‐9‐CM codes for phenome‐wide association studies in the electronic health record. PLoS One 7, e0175508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hebbring, S.J. , Schrodi, S.J. , Ye, Z. , Zhou, Z. , Page, D. & Brilliant, M.H. A PheWAS approach in studying HLA‐DRB1*1501. Genes Immun. 3, 187–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hebbring, S.J. , Rastegar‐Mojarad, M. , Ye, Z. , Mayer, J. , Jacobson, C. & Lin, S. Application of clinical text data for phenome‐wide association studies (PheWASs). Bioinformatics (Oxford, UK) 12, 1981–1987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moore, C.B. et al Phenome‐wide association study relating pretreatment laboratory parameters with human genetic variants in AIDS clinical trials group protocols. Open Forum Infect. Dis. 1, ofu113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hall, M.A. et al Detection of pleiotropy through a Phenome‐wide association study (PheWAS) of epidemiologic data as part of the Environmental Architecture for Genes Linked to Environment (EAGLE) study. PLoS Genet. 12, e1004678 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanseau, P. et al Use of genome‐wide association studies for drug repositioning. Nat. Biotechnol. 4, 317–320 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Mabuchi, H. et al Effect of an inhibitor of 3‐hydroxy‐3‐methyglutaryl coenzyme A reductase on serum lipoproteins and ubiquinone‐10‐levels in patients with familial hypercholesterolemia. N. Engl. J. Med. 9, 478–482 (1981). [DOI] [PubMed] [Google Scholar]
- 21. Kathiresan, S. et al Six new loci associated with blood low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2, 189–197 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burkhardt, R. et al Common SNPs in HMGCR in micronesians and whites associated with LDL‐cholesterol levels affect alternative splicing of exon13. Arterioscler. Thromb. Vasc. Biol. 11, 2078–2084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chasman, D.I. , Posada, D. , Subrahmanyan, L. , Cook, N.R. , Stanton, V.P. Jr. & Ridker, P.M. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA 23, 2821–2827 (2004). [DOI] [PubMed] [Google Scholar]
- 24. Krauss, R.M. et al Variation in the 3‐hydroxyl‐3‐methylglutaryl coenzyme a reductase gene is associated with racial differences in low‐density lipoprotein cholesterol response to simvastatin treatment. Circulation 12, 1537–1544 (2008). [DOI] [PubMed] [Google Scholar]
- 25. Chasman, D.I. , Giulianini, F. , MacFadyen, J. , Barratt, B.J. , Nyberg, F. & Ridker, P.M. Genetic determinants of statin‐induced low‐density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2, 257–264 (2012). [DOI] [PubMed] [Google Scholar]
- 26. Postmus, I. et al Pharmacogenetic meta‐analysis of genome‐wide association studies of LDL cholesterol response to statins. Nat. Commun. 5068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei, W. et al LPA variants are associated with residual cardiovascular risk in patients receiving statins. American Heart Association Scientific Sessions (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jostins, L. et al Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 7422, 119–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsoi, L.C. et al Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 12, 1341–1348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Estrada, K. et al Genome‐wide meta‐analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 5, 491–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou, K. et al Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 2, 117–120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okada, Y. et al Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 7488, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abifadel, M. et al Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2, 154–156 (2003). [DOI] [PubMed] [Google Scholar]
- 34. Cohen, J. , Pertsemlidis, A. , Kotowski, I.K. , Graham, R. , Garcia, C.K. & Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2, 161–165 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Cohen, J.C. , Boerwinkle, E. , Mosley, T.H. Jr. & Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 12, 1264–1272 (2006). [DOI] [PubMed] [Google Scholar]
- 36. Stein, E.A. et al Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low‐density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet 9836, 29–36 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Stein, E.A. et al Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 12, 1108–1118 (2012). [DOI] [PubMed] [Google Scholar]
- 38. Roden, D.M. , Wilke, R.A. , Kroemer, H.K. & Stein, C.M. Pharmacogenomics: the genetics of variable drug responses. Circulation 15, 1661–1670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rost, S. et al Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 6974, 537–541 (2004). [DOI] [PubMed] [Google Scholar]
- 40. Rieder, M.J. et al Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 22, 2285–2293 (2005). [DOI] [PubMed] [Google Scholar]
- 41. Lindh, J.D. , Lundgren, S. , Holm, L. , Alfredsson, L. & Rane, A. Several‐fold increase in risk of overanticoagulation by CYP2C9 mutations. Clin. Pharmacol. Ther. 5, 540–550 (2005). [DOI] [PubMed] [Google Scholar]
- 42. Wadelius, M. et al Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenom. J. 4, 262–270 (2005). [DOI] [PubMed] [Google Scholar]
- 43. Cooper, G.M. et al A genome‐wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 4, 1022–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takeuchi, F. et al A genome‐wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 3, e1000433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perera, M.A. et al Genetic variants associated with warfarin dose in African‐American individuals: a genome‐wide association study. Lancet 9894, 790–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hulot, J.S. et al Cytochrome P450 2C19 loss‐of‐function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 7, 2244–2247 (2006). [DOI] [PubMed] [Google Scholar]
- 47. Kim, K.A. , Park, P.W. , Hong, S.J. & Park, J.Y. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin. Pharmacol. Ther. 2, 236–242 (2008). [DOI] [PubMed] [Google Scholar]
- 48. Mega, J.L. et al Cytochrome p‐450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 4, 354–362 (2009). [DOI] [PubMed] [Google Scholar]
- 49. Simon, T. et al Genetic determinants of response to clopidogrel and cardiovascular events. N. Engl. J. Med. 4, 363–375 (2009). [DOI] [PubMed] [Google Scholar]
- 50. Shuldiner, A.R. et al Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 8, 849–857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhong, W.P. et al Genomewide association study identifies novel genetic loci that modify antiplatelet effects and pharmacokinetics of clopidogrel. Clin. Pharmacol. Ther. 6, 791–802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ge, D. et al Genetic variation in IL28B predicts hepatitis C treatment‐induced viral clearance. Nature 7262, 399–401 (2009). [DOI] [PubMed] [Google Scholar]
- 53. Suppiah, V. et al IL28B is associated with response to chronic hepatitis C interferon‐alpha and ribavirin therapy. Nat. Genet. 10, 1100–1104 (2009). [DOI] [PubMed] [Google Scholar]
- 54. Tanaka, Y. et al Genome‐wide association of IL28B with response to pegylated interferon‐alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 10, 1105–1109 (2009). [DOI] [PubMed] [Google Scholar]
- 55. Daly, A.K. et al HLA‐B*5701 genotype is a major determinant of drug‐induced liver injury due to flucloxacillin. Nat. Genet. 7, 816–819 (2009). [DOI] [PubMed] [Google Scholar]
- 56. Singer, J.B. et al A genome‐wide study identifies HLA alleles associated with lumiracoxib‐related liver injury. Nat. Genet. 8, 711–714 (2010). [DOI] [PubMed] [Google Scholar]
- 57. Lucena, M.I. et al Susceptibility to amoxicillin‐clavulanate‐induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 1, 338–347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kindmark, A. et al Genome‐wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenom. J. 3, 186–195 (2008). [DOI] [PubMed] [Google Scholar]
- 59. Nicoletti, P. et al Association of liver injury from specific drugs, or groups of drugs, with polymorphisms in hla and other genes in a genome‐wide association study. Gastroenterology 5, 1078–1089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chung, W.H. et al Medical genetics: a marker for Stevens‐Johnson syndrome. Nature 6982, 486 (2004). [DOI] [PubMed] [Google Scholar]
- 61. Mallal, S. et al Association between presence of HLA‐B*5701, HLA‐DR7, and HLA‐DQ3 and hypersensitivity to HIV‐1 reverse‐transcriptase inhibitor abacavir. Lancet 9308, 727–732 (2002). [DOI] [PubMed] [Google Scholar]
- 62. Illing, P.T. et al Immune self‐reactivity triggered by drug‐modified HLA‐peptide repertoire. Nature 7404, 554–558 (2012). [DOI] [PubMed] [Google Scholar]
- 63. Ozeki, T. et al Genome‐wide association study identifies HLA‐A*3101 allele as a genetic risk factor for carbamazepine‐induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Gen. 5, 1034–1041 (2011). [DOI] [PubMed] [Google Scholar]
- 64. McCormack, M. et al HLA‐A*3101 and carbamazepine‐induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 12, 1134–1143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Link, E. et al SLCO1B1 variants and statin‐induced myopathy–a genomewide study. N. Engl. J. Med. 8, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 66. Mosley, J.D. et al A genome‐wide association study identifies variants in KCNIP4 associated with ACE inhibitor‐induced cough. Pharmacogenom. J. 3, 231–237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Aminkeng, F. et al A coding variant in RARG confers susceptibility to anthracycline‐induced cardiotoxicity in childhood cancer. Nat. Genet. 9, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wells, Q.S. et al Genome‐wide association and pathway analysis of left ventricular function after anthracycline exposure in adults. Pharmacogenet. Genom. 7, 247–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Van Driest, S.L. et al Genome‐Wide association study of serum creatinine levels during vancomycin therapy. PLoS One 6, e0127791 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neuraz, A. et al Phenome‐wide association studies on a quantitative trait: application to TPMT enzyme activity and thiopurine therapy in pharmacogenomics. PLoS Comp. Biol. 12, e1003405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Diogo, D. et al TYK2 protein‐coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non‐autoimmune complex traits. PLoS One 4, e0122271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Grover, M.P. et al Identification of novel therapeutics for complex diseases from genome‐wide association data. BMC Med. Genom. S8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rastegar‐Mojarad, M. , Ye, Z. , Kolesar, J.M. , Hebbring, S.J. & Lin, S.M. Opportunities for drug repositioning from phenome‐wide association studies. Nat. Biotechnol. 4, 342–345 (2015). [DOI] [PubMed] [Google Scholar]
- 74. Klein, R.J. et al Complement factor H polymorphism in age‐related macular degeneration. Science 5720, 385–389 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nurnberger, W. , Gobel, U. , Stannigel, H. , Eisele, B. , Janssen, A. & Delvos, U. C1‐inhibitor concentrate for sepsis‐related capillary leak syndrome. Lancet 8799, 990 (1992). [DOI] [PubMed] [Google Scholar]
- 76. Hack, C.E. et al C1‐esterase inhibitor substitution in sepsis. Lancet 8789, 378 (1992). [DOI] [PubMed] [Google Scholar]
- 77. Hillmen, P. et al Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 6, 552–559 (2004). [DOI] [PubMed] [Google Scholar]
- 78. Yaspan, B.L. et al Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age‐related macular degeneration. Sci. Transl. Med. 395 (2017). [DOI] [PubMed] [Google Scholar]
- 79. Moosavinasab, S. et al ‘RE:fine drugs’: an interactive dashboard to access drug repurposing opportunities. Database (Oxford, UK) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pulley, J.M. et al Accelerating precision drug development and drug repurposing by leveraging human genetics. Assay Drug Dev. Technol. 3, 113–119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Millwood, I.Y. et al A phenome‐wide association study of a lipoprotein‐associated phospholipase A2 loss‐of‐function variant in 90 000 Chinese adults. Int. J. Epidemiol. 5, 1588–1599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. White, H.D. et al Darapladib for preventing ischemic events in stable coronary heart disease. N. Engl. J. Med. 18, 1702–1711 (2014). [DOI] [PubMed] [Google Scholar]
- 83. Daly, A.K. Genome‐wide association studies in pharmacogenomics. Nat. Rev. Genet. 4, 241–246 (2010). [DOI] [PubMed] [Google Scholar]
- 84. Bowton, E. et al Biobanks and electronic medical records: enabling cost‐effective research. Sci. Transl. Med. 234, 234cm233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rosenberg, N.A. , Huang, L. , Jewett, E.M. , Szpiech, Z.A. , Jankovic, I. & Boehnke, M. Genome‐wide association studies in diverse populations. Nat. Rev. Genet. 5, 356–366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Popejoy, A.B. & Fullerton, S.M. Genomics is failing on diversity. Nature 7624, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Leader, J.B. et al Contrasting association results between existing PheWAS phenotype definition methods and five validated electronic phenotypes. AMIA Annu. Symp. Proc. 824–832 (2015). [PMC free article] [PubMed] [Google Scholar]
- 88. Wei, W.Q. & Denny, J.C. Extracting research‐quality phenotypes from electronic health records to support precision medicine. Genom. Med. 1, 41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pathak, J. , Kho, A.N. & Denny, J.C. Electronic health records‐driven phenotyping: challenges, recent advances, and perspectives. J. Am. Med. Inform. Assoc. e2, e206‐211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gamazon, E.R. et al A gene‐based association method for mapping traits using reference transcriptome data. Nat. Genet. 9, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dolan, M.E. et al Clinical and genome‐wide analysis of cisplatin‐induced peripheral neuropathy in survivors of adult‐onset cancer. Clin. Cancer Res. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ballouz, S. et al Candidate disease gene prediction using Gentrepid: application to a genome‐wide association study on coronary artery disease. Mol. Genet. Genom. Med. 1, 44–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Grover, M.P. et al Novel therapeutics for coronary artery disease from genome‐wide association study data. BMC Med. Genom. S1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Roden, D.M. Phenome‐wide association studies: a new method for functional genomics in humans. J. Physiol. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Krapohl, E. et al Phenome‐wide analysis of genome‐wide polygenic scores. Mol. Psychiatry 9, 1188–1193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bush, W.S. , Oetjens, M.T. & Crawford, D.C. Unravelling the human genome‐phenome relationship using phenome‐wide association studies. Nat. Rev. Genet. 3, 129–145 (2016). [DOI] [PubMed] [Google Scholar]
- 97. Warner, J.L. & Alterovitz, G. Phenome based analysis as a means for discovering context dependent clinical reference ranges. AMIA Annu. Symp. Proc. 1441–1449 (2012). [PMC free article] [PubMed] [Google Scholar]
- 98. Robinson, J.R. , Kennedy, V.E. , Doss, Y. , Bastarache, L. , Denny, J. & Warner, J.L. Defining the complex phenotype of severe systemic loxoscelism using a large electronic health record cohort. PLoS One 4, e0174941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liao, K.P. et al Phenome‐Wide association study of autoantibodies to citrullinated and noncitrullinated epitopes in rheumatoid arthritis. Arthritis Rheumatol. (Hoboken, NJ). 4, 742–749. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Doss, J. , Mo, H. , Carroll, R.J. , Crofford, L.J. & Denny, J.C. Phenome‐wide association study of rheumatoid arthritis subgroups identifies association between seronegative disease and fibromyalgia. Arthritis Rheumatol. (Hoboken, NJ). 2, 291–300, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]