

Abstract
Background and Purpose
The epithelial sodium channel (ENaC) is expressed in endothelial cells and acts as a negative modulator of vasodilatation. Oxidized LDL (ox‐LDL) is a key pathological factor in endothelial dysfunction. In the present study we examined the role of ENaC in ox‐LDL‐induced endothelial dysfunction and its associated signal transduction pathway.
Experimental Approach
Patch clamp techniques combined with pharmacological approaches were used to examine ENaC activity in the endothelial cells of a split‐open mouse thoracic aorta. Western blot analysis was used to determine ENaC expression in the aorta. The aorta relaxation was measured using a wire myograph assay.
Key Results
Ox‐LDL, but not LDL, significantly increased ENaC activity in the endothelial cells attached to split‐open thoracic aortas, and the increase was inhibited by a lectin‐like ox‐LDL receptor‐1 (LOX‐1) antagonist (κ‐carrageenan), an NADPH oxidase inhibitor (apocynin), and a scavenger of ROS (TEMPOL). Sodium nitroprusside, an NO donor, diminished the ox‐LDL‐mediated activation of ENaC, and this effect was abolished by inhibiting soluble guanylate cyclase (sGC) and PKG. Ox‐LDL reduced the endothelium‐dependent vasodilatation of the aorta pectoralis induced by ACh, and this reduction was partially restored by blocking ENaC.
Conclusion and Implications
Ox‐LDL stimulates ENaC in endothelial cells through LOX‐1 receptor‐mediated activation of NADPH oxidase and accumulation of intracellular ROS. Since the stimulation of ENaC can be reversed by elevating NO, we suggest that both inhibition of ENaC and an elevation of NO may protect the endothelium from ox‐LDL‐induced dysfunction.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- ECs
endothelial cells
- EDR
endothelium‐dependent relaxation
- ENaC
epithelial sodium channel
- eNOS
endothelial NOS
- LOX‐1
lectin‐like oxidized‐LDL receptor‐1
- ox‐LDL
oxidized LDL
- PO
open probability
- PTEN
phosphatase and tensin homolog
- sGC
soluble guanylate cyclise
Introduction
An elevation of plasma oxidized LDL (ox‐LDL) is a hallmark of atherosclerosis (Zeibig et al., 2011) and contributes to endothelial dysfunction (Pandey et al., 2014). This dysfunction is one of the earliest indicators of atherosclerosis and involves numerous changes in endothelial cells, including an up‐regulation of adhesion molecules (Feng et al., 2014), an increase in their proliferation (Sun et al., 2013), a down‐regulation of endothelium‐dependent vasodilatation (Yamamoto et al., 2015) and NO production (Chan et al., 2016), and modifications of their electrophysiological properties (Kuhlmann et al., 2003). Several biological effects of ox‐LDL are mediated via the lectin‐like ox‐LDL receptor‐1 (LOX‐1), the main receptor for ox‐LDL on the endothelial lining of blood vessels (Pirillo et al., 2013). ox‐LDL stimulates LOX‐1 transcription to positively feedback its effects, particularly in endothelial cells directly exposed to the blood and, therefore, greatly elevates ROS in these cells to cause vasoconstriction (Nishimura et al., 2004; Zhou et al., 2016). However, the signalling transduction pathway involved has not been elucidated.
It has been known for a long time that endothelial cells express the epithelial sodium channel (ENaC) (Golestaneh et al., 2001; Jernigan et al., 2008). Interestingly, a slight elevation of extracellular sodium induces the swelling and plasma membrane stiffening of endothelial cells, and these effects are dependent on a functional ENaC (Oberleithner et al., 2004; Oberleithner et al., 2007). Knockdown of the endothelial ENaCα‐subunit leads to a significant softening of the endothelial cortex, suggesting that the ENaC may play a vital role in determining endothelial nanomechanics (Jeggle et al., 2013; Warnock, 2013). In a recent ex vivo study it was shown, using atomic force microscopy, that elevated levels of extracellular sodium can mechanically stiffen endothelial cells of mouse aorta (Korte et al., 2014). For the first time, our patch clamp single‐channel recordings from split‐open artery show that ENaC is functional in endothelial cells. Since ox‐LDL elevates intracellular ROS (Cominacini et al., 2000; Chen et al., 2007; Thum and Borlak, 2008) and ROS can strongly stimulate ENaC in distal nephron principal cells (Ma, 2011; Zhang et al., 2013), ox‐LDL may stimulate ENaC in endothelial cells by elevating intracellular ROS.
More importantly, we have also shown that ENaC controls vascular relaxation by modulating NO levels in endothelial cells (Liu et al., 2015; Zheng et al., 2016). NO is an important signalling molecule and is produced endogenously from l‐arginine in a reaction catalysed by NOSs. Subtle changes in its rate of production may critically affect cellular homeostasis and initiate a variety of cellular signalling processes. ox‐LDL inhibits the expression of endothelial NOS (eNOS) and decreases NO production in endothelial cells (Xu et al., 2015). LOX‐1‐overexpressing mice fed a high‐fat diet show reduced endothelium‐dependent relaxation as a result of decreased NO availability (Eichhorn et al., 2009). We have shown that enhanced ENaC activity can also reduce NO production (Liu et al., 2015; Guo et al., 2016; Zheng et al., 2016), suggesting that ENaC can mediate ox‐LDL‐induced endothelial dysfunction. Our data presented here suggest that ox‐LDL stimulates ENaC in endothelial cells by elevating intracellular ROS and this accounts for the decreased aortic relaxation induced by ox‐LDL.
Methods
Animals
All animal care and experimental procedures were approved by the Harbin Medical University Animal Supervision Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
C57BL/6 mice were purchased from the animal centre of the Second Affiliated Hospital of Harbin Medical University (Harbin, China). A total of 215 male C57BL/6 mice aged 8–10 weeks (24–27 g) were used in the present study. Four to five C57BL/6 mice were housed in a cage and maintained under a 12/12 h light/dark cycle (lights on 0700 h) with the ambient humidity at 50–80% and the temperature at 21 ± 2°C. Food and water were provided ad libitum. Twenty mice were used to assess western blots of α‐ENaC; the aorta pectoralis was isolated from 24 mice and preparations were used to investigate the vasodilatation. 171 C57BL/6 mice were used to record single‐channel activity of ENaC.
Aorta pectoralis preparation
Briefly, mice (aged 8–10 weeks, 24–27 g body weight) were anaesthetized by an i.p. injection of 0.3% pentobarbital sodium (90 mg·kg−1, P3761; Sigma‐Aldrich, St. Louis, MO, USA) and ventilated with room air using a small animal ventilator. Adequate anaesthesia was characterized by slow but regular breathing and no pedal withdrawal reflex. The aorta pectoralis was removed from each mouse immediately and rinsed with PBS to remove blood and improve the visibility of the tissue. Each aorta pectoralis was dissected from the surrounding fat using watchmaker's forceps in PBS solution and transferred to a 5 × 5 mm cover glass coated with polylysine to immobilize the aorta. The cover glass was then placed in a chamber mounted on an inverted microscope (Nikon, Tokyo, Japan) and incubated in a bathing solution.
In situ patch clamp recording
As described by Climent et al. (2011) and by Liu et al. (2015), in situ patch clamp recordings of ENaC single‐channel currents were performed using intact vascular endothelia. Each dissected aorta pectoralis was placed in a Petri dish containing PSS. They were then placed on a 5 × 5 mm cover glass, coated with l‐polylysine and transferred to a chamber mounted on an inverted Nikon microscope, allowing direct access to the endothelial cell layer. Single‐channel ENaC currents were recorded in a cell‐attached configuration with an Axon Multiclamp 200B amplifier (Axon Instruments, Foster City, CA, USA) at room temperature (22–24°C). Patch pipettes were pulled from borosilicate glass with a Sutter P‐97 horizontal puller, and the resistance of the pipettes ranged from 6 to 10 MΩ when filled with the pipette solution (composition in mM: 135 NaCl, 4.5 KCl, 0.1 EGTA, 5 HEPES and 5 Na‐HEPES; pH 7.2, adjusted with NaOH); for I–V curve measurements, NaCl was substituted with equimolar LiCl to potentiate the single‐channel amplitude of ENaC. The bath solution (composition in mM: 135 NaCl, 4.5 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES and 5 Na‐HEPES; pH 7.2, adjusted with NaOH) was stationary, and the chamber volume was ~0.8 mL. Single‐channel currents were recorded for at least 15 min immediately after gigaseal formation. For most experiments, data were acquired by applying 0 mV to the patch pipettes and sampling at 5 kHz with a low‐pass filter at 1 kHz using Clampex 10.2 software (Molecular Devices, Sunnyvale, CA, USA). Prior to analysis, single‐channel traces were further filtered at 30 Hz. The current–voltage (I–V) relationship was constructed using the single‐channel amplitude measured at the indicated pipette voltages as a function of voltages. Vpipette represents the voltage applied to the electrodes during recordings; therefore, −Vpipette indicates that the intracellular potential deviated from the resting potential of the apical membranes of the endothelial cells. Slope conductance was fit via linear regression using SigmaPlot software (Jandel Scientific, San Diego, CA, USA).
Western blotting
For western blot analysis, samples were homogenized with CelLyticM lysis reagent and a protease inhibitor cocktail (Sigma, Poole, Dorset, UK). Protein concentrations were determined by use of the Bradford assay. Equal amounts of total protein were loaded into SDS‐PAGE wells. GAPDH was used as the internal standard to control for protein quantity. Samples prepared with 5× loading buffer were separated on 10% SDS‐PAGE gels and then transferred to nitrocellulose membranes, which were rinsed with Tris‐buffered saline Tween‐20 (TBS‐T) and then blocked in TBS‐T containing 5% skimmed milk for 1 h at room temperature (22–24°C). Membranes were incubated with primary antibodies against α‐ENaC (NB100–74357; Novus Biologicals, Littleton, CO, USA) and GAPDH (sc‐20357; Santa Cruz Biotechnology, Santa Cruz, California, USA) overnight at 4°C, followed by washing in TBS‐T and incubation with secondary antibodies (1:5000) for another 1 h at room temperature (22–24°C). Membranes were washed with TBS‐T, and blots were detected using an ECL kit (Invitrogen, Carlsbad, CA, USA) and scanned for densitometric analysis (Bio‐Rad, Richmond, CA, USA).
Myograph functional study
Aorta pectoralis muscles were surgically isolated from male C57BL/6 mice and placed in ice‐cold Krebs solution (composition in mM: 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1.2 KH2PO4 and 11 d‐glucose). Aortas were isolated from connective tissues under a dissecting microscope and cut into 2 mm lengths. Each aorta pectoralis preparation was suspended in a wire myograph (Danish Myo Technology, Aarhus, Denmark) and bathed in oxygenated Krebs solution at 37°C. Each ring was stretched to 3 mN and then allowed to equilibrate for 60 min before beginning the experiment. After the 60 min stabilization period, KPSS (containing 60 mM K+) was added to the chambers and washed out with PSS until a reproducible maximal contraction was achieved. EDR induced by ACh (1 nM to 100 μM) was recorded in phenylephrine‐contracted rings in the presence or absence of 10 μg·mL−1 ox‐LDL or 0.5 μM amiloride.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as the mean ± SEM. Statistical analyses were performed with SigmaPlot and SigmaStat software (Jandel Scientific). One‐way ANOVAs (followed by Student–Newman–Keuls post hoc tests) or Student's t‐tests were used where appropriate for statistical analysis. Differences were considered statistically significant at P < 0.05.
Chemicals and reagents
Unless stated otherwise, all chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO, USA). ox‐LDL was purchased from Alfa Aesar (Ward Hill, MA, USA). KT5823, ODQ and apocynin were dissolved separately in a minimum quantity of DMSO and further diluted with the bath solution to achieve the required concentration (the final dilution ratio did exceed 1:1000). The other reagents including ox‐LDL, LDL, amiloride, κ‐carrageenan, TEMPOL and SNP were dissolved in double‐distilled H2O. ox‐LDL, SNP and TEMPOL were protected from light during their preparation and the experiments. All solutions were premade and stored in a −20°C freezer.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
ENaC is expressed and functional in endothelial cells of mouse thoracic aorta
The thoracic aorta was isolated and manually split open to access the apical membrane of endothelial cells. In situ cell‐attached patch clamp experiments were performed to record single‐channel currents of ENaC in the apical membrane of the intact endothelial cells. As shown in Figure 1A, single‐channel currents with slow kinetics and very low amplitude were detected in these cells. The open probability (P O) of this current was significantly reduced by amiloride, a potent blocker of ENaC (P < 0.05; n = 7). To further characterize the currents, current–voltage (I–V) relationships were constructed and fitted via linear regression (Figure 1B, C). The data show that the single‐channel conductance of the currents was ~6.8 pS, which is similar to previously reported single‐channel conductance of ENaC (Ma, 2011). The biophysical features and pharmacological profile of these currents are consistent with those previously reported for ENaC in human dermal endothelial cells (Wang et al., 2009). Western blot was performed using isolated thoracic aorta with or without the endothelial layer. The data show that ENaCα, the major subunit of ENaC, is expressed both in endothelial cells and in smooth muscle cells (Figure 1D).
Figure 1.
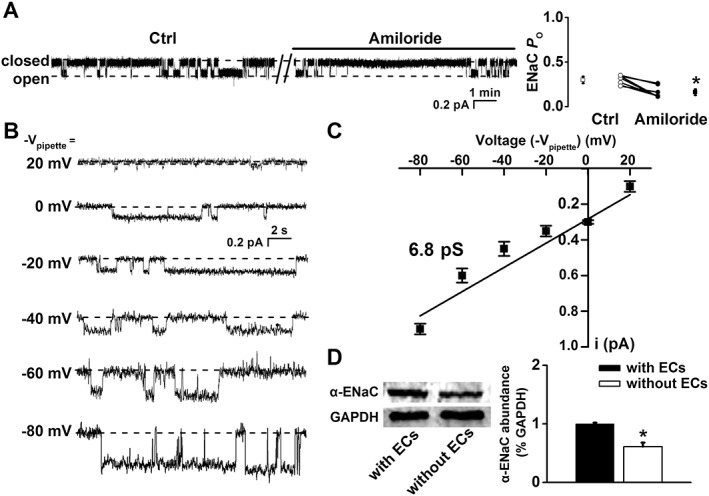
Mouse thoracic aorta expresses functional ENaC. (A) Representative ENaC single‐channel currents recorded in endothelial cells attached to split‐open aorta, before and after application of 0.5 μM amiloride (left). Summarized P O values obtained from the recordings are shown on the right (*P < 0.05; n = 7). (B) Single‐channel currents recorded at the indicated voltages. (C) I–V relationship constructed using single‐channel current amplitude measured as a function of voltages applied to the patch pipette (Vpipette). Single‐channel conductance was determined via linear regression as indicated by the black dashed line (n = 8 for the data point at −Vpipette = 0 mV; n = 7 for the data points at −Vpipette = −20 and −40 mV; n = 6 for the data points at −Vpipette = 20, −60 and −80 mV). (D) Western blot of α‐ENaC from aortic tissue either with or without endothelial cells (ECs) attached (n = 5 for each group; *P < 0.05). Ctrl, control.
ox‐LDL stimulates ENaC in aortic endothelia
Recent studies have shown that ox‐LDL elevates intracellular ROS via its LOX‐1 receptor (Cominacini et al., 2000; Chen et al., 2007; Thum and Borlak, 2008) and ROS can strongly stimulate ENaC (Ma, 2011; Zhang et al., 2013). To determine whether ox‐LDL can alter ENaC activity via this pathway, we performed patch clamp recordings from intact endothelial cells attached to isolated thoracic aorta. Exogenous ox‐LDL was applied to the cells, while LDL was used as a control. ENaC activity was not altered by 10 μg·mL−1 LDL applied to the cells. ENaC P O remained at control levels even when the cells were exposed to LDL for 30 min (0.36 ± 0.03 vs. 0.34 ± 0.03; P > 0.05; n = 7), suggesting that LDL has no acute effect on ENaC activity (Figure 2A, C). However, the application of 10 μg·mL−1 ox‐LDL to the cells significantly elevated ENaC activity; ENaC P O was increased from 0.31 ± 0.03 to 0.74 ± 0.04 (P < 0.05; n = 7) (Figure 2B, D). These results indicate that ox‐LDL, but not LDL, stimulates ENaC activity.
Figure 2.
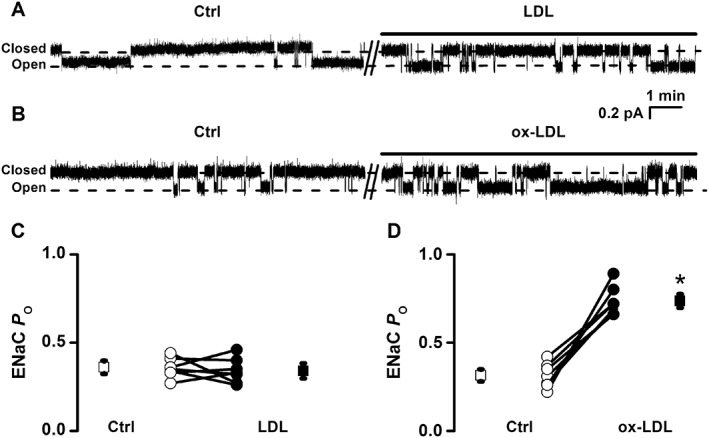
ox‐LDL, but not LDL, stimulates ENaC in endothelial cells attached to a split‐open aorta. (A, B) Representative ENaC single‐channel currents recorded in endothelial cells before and after treatment with either 10 μg·mL−1 LDL (A) or 10 μg·mL−1 ox‐LDL (B). (C, D) Summarized P O values for ENaC before and after the application of LDL or ox‐LDL to the cells are shown in the bottom of the figure (*P < 0.05 vs. respective controls (Ctrl); n = 7).
To investigate whether ox‐LDL stimulates ENaC in a concentration‐dependent manner, endothelial cells attached to the split‐open aorta were either investigated under control conditions or respectively treated with 1, 5, 10, 20, 40 and 50 μg·mL−1 of ox‐LDL for 30 min, prior to the cell‐attached patch clamp examination. The increasing the doses of ox‐LDL gradually elevated ENaC activity, which almost reached a plateau at 50 μg·mL−1 of LDL. ENaC P O was significantly increased by ox‐LDL, from 0.29 ± 0.02 to 0.95 ± 0.02 (50 μg·mL−1) (Figure 3A, B). The calculated mean P O of ENaC was plotted as a function of different doses of ox‐LDL and fitted with Pharmacology Standard Curves Analysis (the solid black line), having an EC50 of 8.60 ± 0.70 μg·mL−1 for ox‐LDL to activate ENaC (Figure 3B; n = 6 for each data point). Therefore, 10 μg·mL−1 ox‐LDL (a dose close to the EC50) was used for the rest of the experiments. We also examined whether prolonged exposure to ox‐LDL affects ENaC activity. Prior to the patch clamp experiments, the split‐open aorta was either kept under control conditions or treated with 10 μg·mL−1 ox‐LDL for 10, 30 or 60 min. As shown in Figure 3C, D, ENaC activity was significantly elevated at 30 min after the treatment and prolonged exposure of the aorta to 10 μg·mL−1 ox‐LDL further increased ENaC activity (P < 0.05, n = 6). These data suggest that ox‐LDL stimulates ENaC in endothelial cells of mouse aorta in a dose‐ and time‐dependent manner.
Figure 3.
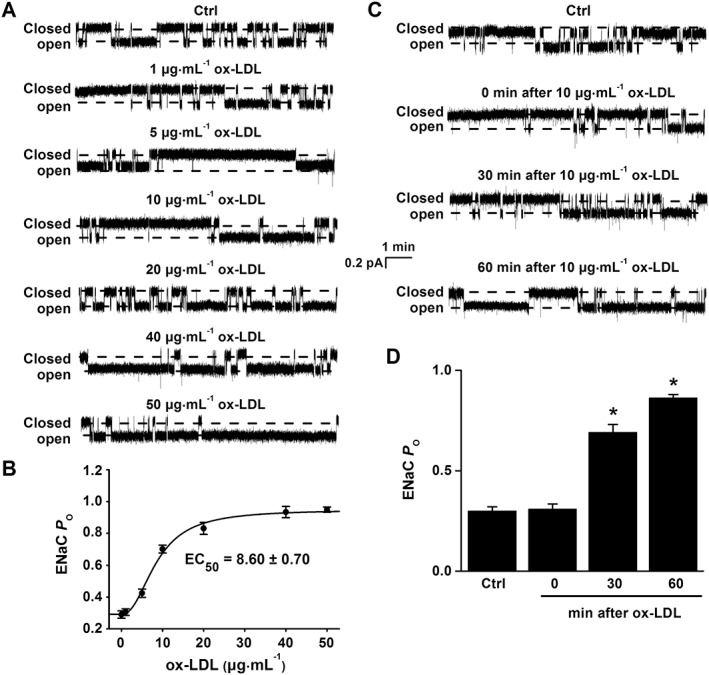
ox‐LDL stimulates ENaC activity in a dose‐ and time‐dependent manner. (A) Representative ENaC single‐channel currents recorded in endothelial cells from a split‐open aorta either under control (Ctrl) conditions or treated with ox‐LDL at concentrations of 1, 5, 10, 20, 40 and 50 μg·mL−1 for 30 min. (B) ENaC P O were plotted as a function of each corresponding concentration of ox‐LDL and fitted with Pharmacology Standard Curves Analysis using SigmaPlot (n = 6 for each data point). (C) Representative ENaC single‐channel currents recorded in endothelial cells from a split‐open aorta either under control conditions or treated with 10 μg·mL−1 ox‐LDL for 0, 30 or 60 min respectively. (D) Summarized ENaC P O under the conditions shown in (C) respectively (*P < 0.05 vs. respective Ctrl; n = 6).
ox‐LDL elevates ENaC activity via LOX‐1/NADPH oxidase/ROS
To determine whether the effect of ox‐LDL on ENaC is due to the binding of ox‐LDL to its receptor LOX‐1, endothelial cells attached to the split‐open aorta were either kept under control conditions or pretreated with 125 μg·mL−1 κ‐carrageenan (a LOX‐1 receptor inhibitor) for 10 min before the cell‐attached recordings were performed. In the presence of κ‐carrageenan, ox‐LDL failed to increase ENaC activity (Figure 4A); ENaC P O remained unchanged, 0.32 ± 0.02 (control) versus 0.35 ± 0.02 (30 min after 10 μg·mL−1 ox‐LDL) (P > 0.05; n = 7). In contrast, κ‐carrageenan itself did not alter ENaC activity. Since the binding of ox‐LDL to LOX‐1 can stimulate NADPH oxidase and rapidly increase intracellular ROS generation in articular chondrocytes (Nishimura et al., 2004), we next examined whether ox‐LDL stimulates ENaC in endothelial cells via a similar pathway. Therefore, the endothelial cells attached to split‐open aorta were either kept under control conditions or treated with either 250 μM TEMPOL (an ROS scavenger) or 100 μM apocynin (an NADPH oxidase inhibitor). After these treatments, ox‐LDL was no longer able to elevate ENaC activity. In the endothelial cells treated with TEMPOL or apocynin, ENaC P O remained at control levels (Figure 4B and C respectively). Although apocynin itself did not alter ENaC activity, TEMPOL tended to decrease ENaC activity. These results suggest that ox‐LDL stimulates ENaC in endothelial cells, at least in part, through its receptor LOX‐1‐mediated generation of intracellular ROS.
Figure 4.
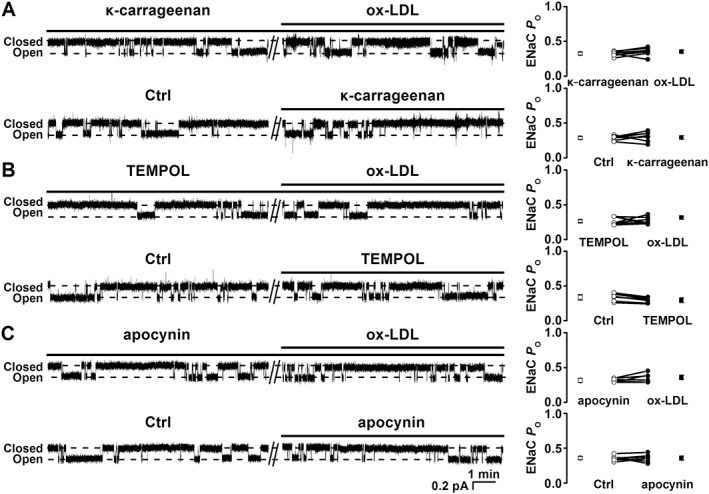
ox‐LDL stimulates ENaC via LOX‐1‐mediated activation of NADPH oxidase. (A) Representative ENaC single‐channel currents recorded in two separate endothelial cells; one was pretreated with 125 μg·mL−1 κ‐carrageenan before being treated with10 μg·mL−1 ox‐LDL (upper trace); the other served as a control (Ctrl) and was treated with 125 μg·mL−1 κ‐carrageenan (lower trace). (B) Representative ENaC single‐channel currents recorded in two separate endothelial cells; one was pretreated with 250 μM TEMPOL before the application of 10 μg·mL−1 ox‐LDL (upper trace); the other served as a Ctrl and was pretreated with 250 μM TEMPOL (lower trace); (C) Representative ENaC single‐channel currents recorded in two separate endothelial cells; one was pretreated with 100 μM apocynin before the application of 10 μg·mL−1 ox‐LDL (upper trace); the other served as a Ctrl and was treated with 100 μM apocynin (lower trace). Summarized ENaC P Os were plotted on the right (n = 7 in each experiment).
ox‐LDL reduces endothelium‐dependent aortic relaxation, which is partially dependent on ENaC activity
We previously showed that an elevation in ENaC activity reduces endothelium‐dependent artery relaxation (Liu et al., 2015). Therefore, we tested whether ENaC mediates ox‐LDL‐induced endothelial dysfunction. The experiments were performed using isolated aorta pectoralis with intact endothelia. As shown in Figure 5A, B, a contraction of the aorta pectoralis was induced by 1 μM phenylephrine; the EDR was induced by ACh at 10−9, 10−8, 10−7, 10−6, 10−5 and 10−4 M respectively. Experiments were then carried out to determine how amiloride (a potent ENaC blocker) and ox‐LDL affect the ACh‐induced relaxation. The data show that the ACh‐induced relaxation of the aorta pectoralis was smaller after pretreatment with 10 μg·mL−1 ox‐LDL. Amiloride did not alter the relaxation, indicating that basal ENaC activity does not affect aortic tone. However, the reduced relaxation induced by ox‐LDL was partially but significantly reversed by blocking ENaC with amiloride. These data suggest that an elevated ENaC activity participates in the decreased endothelium‐dependent aortic relaxation induced by ox‐LDL.
Figure 5.
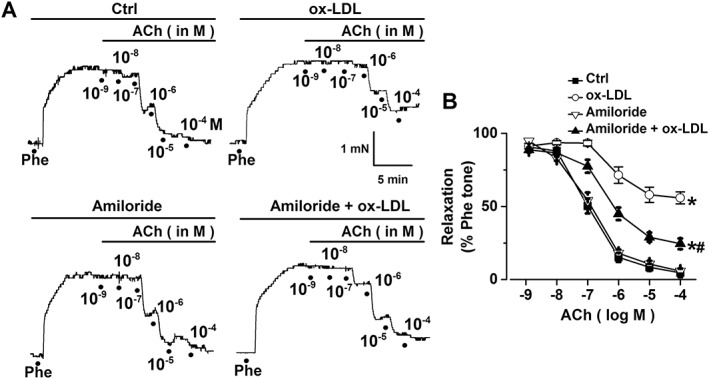
ox‐LDL reduces ACh‐induced aortic relaxation via an ENaC‐dependent pathway. (A) Representative traces of ACh‐induced relaxation of mouse aorta pectoralis either under control (Ctrl) conditions or after treatment with 10 μg·mL−1 ox‐LDL, 0.5 μM amiloride or 0.5 μM amiloride +10 μg·mL−1 ox‐LDL. Aorta pectoralis preparations were treated with 1 μM phenylephrine (Phe) to induce a contraction before examining the ACh‐induced relaxation. (B) Summaries of relaxation induced by ACh at 10−9, 10−8, 10−7, 10−6, 10−5 and 10−4 M under the conditions described in (A) (n = 6 for each data point). The data for 10−4 M ACh were analysed, and showed *P < 0.05 versus Ctrl; #P < 0.05 versus ox‐LDL.
NO reverses ox‐LDL‐induced ENaC activity via soluble guanylate cyclase and PKG
As it is well‐known that NO can cause vascular relaxation, NO may antagonize the effect of ox‐LDL on ENaC activity. Hence, we tested whether NO could reverse ox‐LDL‐induced ENaC activity. Consistently, ENaC P O in endothelial cells attached to the split‐open aorta was markedly increased after application of 10 μg·mL−1 ox‐LDL to the cells, which was from 0.30 ± 0.02 (before) to 0.79 ± 0.03 (30 min after ox‐LDL) (P < 0.05; n = 6); this increase was reversed by 1 mM SNP, an NO donor; ENaC P O was reduced to 0.28 ± 0.03 (P < 0.05; n = 6). Furthermore, ox‐LDL failed to stimulate ENaC when the cells were pretreated with SNP (Figure 6A). Therefore, we concluded that NO can prevent or attenuate activation of ENaC by ox‐LDL. Since NO is known to stimulate soluble guanylate cyclase (sGC) and PKG (Shahidullah and Delamere, 2006), the endothelial cells attached to the split‐open aorta were treated with either 10 μM ODQ (an sGC inhibitor) or 10 μM KT5823 (a PKG inhibitor) for 10 min before the patch clamp experiments. The data show that after inhibition of sGC with ODQ, SNP was no longer able to reverse ox‐LDL‐induced ENaC activity (Figure 6B). Similarly, after inhibition of PKG with KT5823, SNP failed to attenuate ox‐LDL‐induced ENaC activity (Figure 6C). These results suggest that NO reverses the activation of ENaC by ox‐LDL via a pathway associated with sGC/PKG.
Figure 6.
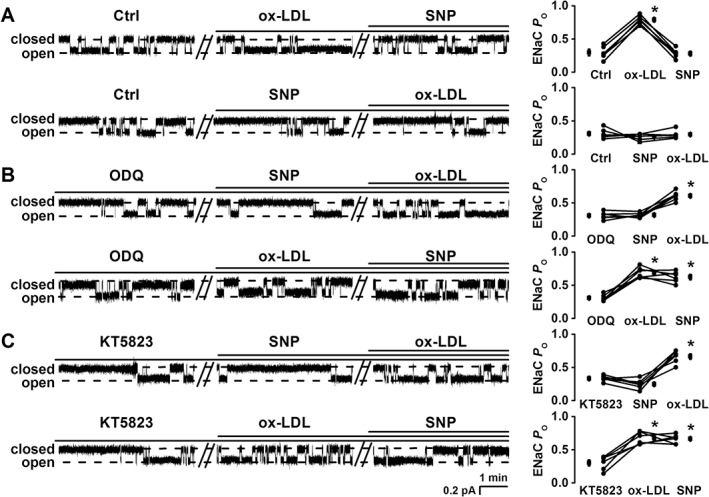
Exogenous NO reverses the ox‐LDL‐induced increase in ENaC activity via sGC/PKG. (A–C) Representative ENaC single‐channel currents recorded in endothelial cells (left) and summarized ENaC P O (right); basal ENaC activity in the same patch served as its own control (Ctrl). (A) SNP (an NO donor; 1 mM) reversed the increased ENaC activity induced by 10 μg·mL−1 ox‐LDL under Ctrl conditions, no matter whether ox‐LDL was applied to the cell first (upper trace) or SNP was applied to the cell first (lower trace); (B, C) SNP (1 mM) no longer reversed the increased ENaC activity induced by 10 μg·mL−1 ox‐LDL in two separate cells; one was pretreated with 10 μM ODQ, an sGC inhibitor (B); the other was pretreated with 10 μM KT5823 (C), no matter whether SNP was applied to the cell first (upper traces in B and C) or ox‐LDL was applied to the cell first (lower traces in B and C). *P < 0.05 vs. either Ctrl or pre‐treament; n = 6 paired experiments.
Discussion
The major findings of the present study are: (i) ENaC is expressed in endothelial cells of mouse thoracic aorta and its activity is significantly elevated by ox‐LDL, but not by LDL; (ii) ox‐LDL stimulates ENaC via LOX‐1‐mediated generation of intracellular ROS; (iii) blockade of ENaC significantly attenuates ox‐LDL‐induced decrease in endothelium‐dependent aortic relaxation; and (iv) NO reverses ox‐LDL‐mediated increase in ENaC activity via the sGC/PKG pathway and, probably, also through NO inhibiting oxidative stress generated from NADPH oxidase. Our findings may provide a new therapeutic strategy for treating vascular dysfunction caused by ox‐LDL.
It is well accepted that ox‐LDL is a critical risk factor/biomarker for several concurrent pathophysiological processes, including chronic inflammation, metabolic disorder and oxidative stress, during atherogenesis, and a cause of vasculature dysfunction (Zhang et al., 2016a). Our data show, for the first time, that ENaC is expressed and functional in the endothelial cells of thoracic aorta and mediates the decreased EDR induced by ox‐LDL. The ox‐LDL concentration in the circulation of healthy subjects is normally maintained at a very low level (~0.14 μg·mL−1) (Wang et al., 2007). However, ox‐LDL levels are significantly elevated in patients with hyperlipidaemia (Owens et al., 2012), type 2 diabetes mellitus (Nour Eldin et al., 2014), unstable or stable angina (Neri Serneri et al., 2013) or even coronary heart disease (Holm et al., 2011). Not surprisingly, ox‐LDL stimulates ENaC in aortic endothelial cells in a dose‐ and time‐dependent manner, suggesting that the elevated ENaC activity evoked by the chronically enhanced ox‐LDL levels may contribute to vasculature dysfunction induced by oxidative stress.
Our recent studies demonstrated that elevated ENaC activity accounts for the reduced EDR by decreasing NO levels in endothelial cells from salt‐sensitive rats fed a high‐salt (HS) diet, and that blockade of ENaC by amiloride increased both phosphorylated eNOS and NO and, therefore, prevented the HS diet‐induced loss of vasorelaxation and endothelial dysfunction (Wang et al. 2018). Others have also shown that inhibition of ENaC increases NOS phosphorylation and NO production in small‐diameter rat mesenteric arteries (Perez et al., 2009). Furthermore, in cultured alveolar type II monolayers, NO may inhibit ENaC‐mediated Na+ reabsorption (Guo et al., 1998). In the present study, it was demonstrated that exogenous NO reverses the ox‐LDL‐induced increase in ENaC activity not only via the sGC/PKG pathway but also through inhibition of ROS, since NO can inhibit oxidative stress generated from NADPH oxidase (Shen et al., 2010). Together, these findings suggest that there might be a feedback mechanism between ENaC activity and NO production. This feedback implies that enhanced ENaC activity reduces NO production in endothelial cells and that, conversely, a reduction in NO will relieve its inhibitory effects on ENaC activity. ox‐LDL is a product of chronic oxidative stress; it is also a potent inducer of oxidative stress, as ox‐LDL injections have been shown to increase both bone marrow and blood ROS levels in mice (Zhang et al., 2016b). Importantly, incubation of cultured human aortic endothelial cells with ox‐LDL enhances ROS via the phosphorylation of p66Shc, an important mediator of oxidative stress (Shi et al., 2014). In a previous study, it was revealed that ox‐LDL activates NADPH oxidase (both the gp91 phox and p47 phox subunits) expression and enhances ROS generation via LOX‐1 in human coronary artery endothelial cells (Dandapat et al., 2007). Therefore, these findings suggest that there might be another positive feedback mechanism between ox‐LDL and ROS to elevate both ox‐LDL and ROS.
In the present study, it was found that ox‐LDL also stimulates ENaC. Therefore, a strong elevation of both ox‐LDL and ROS due to the positive feedback mechanism should significantly enhance ENaC activity. The results described above provide a rationale for the hypothesis that ox‐LDL probably stimulates endothelial ENaC through an ox‐LDL/LOX‐1‐mediated accumulation of intracellular ROS. Our data show that the stimulating effects of ox‐LDL on endothelial ENaC activity in the aorta were respectively abolished by TEMPOL (an ROS scavenger) and apocynin (an NADPH oxidase inhibitor), as well as by a LOX‐1 antagonist. Together, these data suggest that ox‐LDL regulates endothelial ENaC activity through its receptor, LOX‐1,‐mediated activation of NADPH oxidase.
In the present study, we did not explore the mechanisms by which ROS stimulates ENaC. However, in our previous studies we demonstrated that ROS stimulates ENaC via PI3K/PTEN‐mediated increase in phosphatidylinositol 3,4,5‐trisphosphate (PI(3,4,5)P3) at the apical membrane in distal nephron cells (Ma, 2011; Zhang et al., 2013). Therefore, it is possible that the ox‐LDL‐induced elevation of ROS might also increase PI(3,4,5)P3 to stimulate ENaC in endothelial cells. Finally, there is accumulating evidence demonstrating that ox‐LDL impairs ACh‐mediated vasorelaxation (Valente et al., 2014). Furthermore, we also demonstrated that blockade of ENaC attenuates the impaired EDR in aorta treated with ox‐LDL, suggesting a direct involvement of ENaC in the regulation of endothelial and vascular function. Amiloride is known to inhibit the sodium–hydrogen exchanger (NHE). However, the binding affinity of amiloride to ENaC is ~100‐fold higher than that to NHE (Weinbrenner et al., 2003). Hence, it is unlikely that the concentration of amiloride (0.5 μM) used in the present study also affected the NHE.
Overall, the majority of data presented here were obtained from ex vivo experiments, which have limitations as regards their physiological relevance in vivo. Nevertheless, these findings provide the first direct evidence that the mouse thoracic aorta expresses a functional ENaC and that ox‐LDL, but not LDL, stimulates ENaC in endothelial cells. The stimulating effects of ox‐LDL on endothelial ENaC were significantly inhibited by the NO donor and amiloride. Thus, we suggest that blockade of ENaC or the elevation of NO may be useful approaches to prevent ox‐LDL‐induced vascular dysfunction.
Author contributions
Z.‐R.Z. conceived and designed the experiments; Z.‐R.Z. and H.‐P.M. revised the manuscript; C.L. and Q.‐S.W. collected and analysed the data and drafted the manuscript; X.Y., N.N., Q.‐Q.H., B.‐L.Z., X.C., C.‐J.Y., B.‐L.S. and M.‐M.W. collected the data; all authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was generously supported by grants from Key Project of Chinese National Program for Fundamental Research and Development (973 Program 2014CB542401 to Z.Z.), National Natural Science Foundation of China (nos. 91639202 and 81320108002 to Z.Z.) and by Nn10 plan from Harbin Medical University Cancer Hospital. This work was also partially supported by a grant from National Institute of Health (NIH) (R01 DK100582 to H.‐P.M.). We gratefully thank Dr Yu Huang from the Institute of Vascular Medicine of the Chinese University of Hong Kong for the suggestion with the experimental design.
Liang, C. , Wang, Q.‐S. , Yang, X. , Niu, N. , Hu, Q.‐Q. , Zhang, B.‐L. , Wu, M.‐M. , Yu, C.‐J. , Chen, X. , Song, B.‐L. , Zhang, Z.‐R. , and Ma, H.‐P. (2018) Oxidized low‐density lipoprotein stimulates epithelial sodium channels in endothelial cells of mouse thoracic aorta. British Journal of Pharmacology, 175: 1318–1328. doi: 10.1111/bph.13853.
References
- Alexander SP, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Hung CH, Shih JY, Chu PM, Cheng YH, Tsai YJ et al. (2016). Baicalein is an available anti‐atherosclerotic compound through modulation of nitric oxide‐related mechanism under oxLDL exposure. Oncotarget 7: 42881–42891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XP, Xun KL, Wu Q, Zhang TT, Shi JS, Du GH (2007). Oxidized low density lipoprotein receptor‐1 mediates oxidized low density lipoprotein‐induced apoptosis in human umbilical vein endothelial cells: role of reactive oxygen species. Vascul Pharmacol 47: 1–9. [DOI] [PubMed] [Google Scholar]
- Climent B, Zsiros E, Stankevicius E, de la Villa P, Panyi G, Simonsen U et al. (2011). Intact rat superior mesenteric artery endothelium is an electrical syncytium and expresses strong inward rectifier K+ conductance. Biochem Biophys Res Commun 410: 501–507. [DOI] [PubMed] [Google Scholar]
- Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M et al. (2000). Oxidized low density lipoprotein (ox‐LDL) binding to ox‐LDL receptor‐1 in endothelial cells induces the activation of NF‐kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem 275: 12633–12638. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandapat A, Hu C, Sun L, Mehta JL (2007). Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX‐1‐dependent redox‐sensitive pathway. Arterioscler Thromb Vasc Biol 27: 2435–2442. [DOI] [PubMed] [Google Scholar]
- Eichhorn B, Muller G, Leuner A, Sawamura T, Ravens U, Morawietz H (2009). Impaired vascular function in small resistance arteries of LOX‐1 overexpressing mice on high‐fat diet. Cardiovasc Res 82: 493–502. [DOI] [PubMed] [Google Scholar]
- Feng Y, Cai ZR, Tang Y, Hu G, Lu J, He D et al. (2014). TLR4/NF‐kappaB signaling pathway‐mediated and oxLDL‐induced up‐regulation of LOX‐1, MCP‐1, and VCAM‐1 expressions in human umbilical vein endothelial cells. Genet Mol Res 13: 680–695. [DOI] [PubMed] [Google Scholar]
- Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M (2001). Mineralocorticoid receptor‐mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun 280: 1300–1306. [DOI] [PubMed] [Google Scholar]
- Guo D, Liang S, Wang S, Tang C, Yao B, Wan W et al. (2016). Role of epithelial Na+ channels in endothelial function. J Cell Sci 129: 290–297. [DOI] [PubMed] [Google Scholar]
- Guo Y, DuVall MD, Crow JP, Matalon S (1998). Nitric oxide inhibits Na+ absorption across cultured alveolar type II monolayers. Am J Physiol 274 (3 Pt 1): L369–L377. [DOI] [PubMed] [Google Scholar]
- Holm S, Ueland T, Dahl TB, Michelsen AE, Skjelland M, Russell D et al. (2011). Fatty acid binding protein 4 is associated with carotid atherosclerosis and outcome in patients with acute ischemic stroke. PLoS One 6: e28785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggle P, Callies C, Tarjus A, Fassot C, Fels J, Oberleithner H et al. (2013). Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 61: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, LaMarca B, Speed J, Galmiche L, Granger JP, Drummond HA (2008). Dietary salt enhances benzamil‐sensitive component of myogenic constriction in mesenteric arteries. Am J Physiol Heart Circ Physiol 294: H409–H420. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte S, Strater AS, Druppel V, Oberleithner H, Jeggle P, Grossmann C et al. (2014). Feedforward activation of endothelial ENaC by high sodium. FASEB J 28: 4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann CR, Schafer M, Li F, Sawamura T, Tillmanns H, Waldecker B et al. (2003). Modulation of endothelial Ca(2+)‐activated K(+) channels by oxidized LDL and its contribution to endothelial proliferation. Cardiovasc Res 60: 626–634. [DOI] [PubMed] [Google Scholar]
- Liu HB, Zhang J, Sun YY, Li XY, Jiang S, Liu MY et al. (2015). Dietary salt regulates epithelial sodium channels in rat endothelial cells: adaptation of vasculature to salt. Br J Pharmacol 172: 5634–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HP (2011). Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3‐kinase‐dependent pathway. J Biol Chem 286: 32444–32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri Serneri GG, Coppo M, Bandinelli M, Paoletti P, Toscano T, Micalizzi E et al. (2013). Exaggerated myocardial oxLDL amount and LOX‐1 receptor over‐expression associated with coronary microvessel inflammation in unstable angina. Atherosclerosis 226: 476–482. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Akagi M, Yoshida K, Hayakawa S, Sawamura T, Munakata H et al. (2004). Oxidized low‐density lipoprotein (ox‐LDL) binding to lectin‐like ox‐LDL receptor‐1 (LOX‐1) in cultured bovine articular chondrocytes increases production of intracellular reactive oxygen species (ROS) resulting in the activation of NF‐kappaB. Osteoarthritis Cartilage 12: 568–576. [DOI] [PubMed] [Google Scholar]
- Nour Eldin EE, Almarzouki A, Assiri AM, Elsheikh OM, Mohamed BE, Babakr AT (2014). Oxidized low density lipoprotein and total antioxidant capacity in type‐2 diabetic and impaired glucose tolerance Saudi men. Diabetol Metab Syndr 6: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberleithner H, Ludwig T, Riethmuller C, Hillebrand U, Albermann L, Schafer C et al. (2004). Human endothelium: target for aldosterone. Hypertension 43: 952–956. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Riethmuller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M (2007). Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A 104: 16281–16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens AP 3rd, Passam FH, Antoniak S, Marshall SM, McDaniel AL, Rudel L et al. (2012). Monocyte tissue factor‐dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J Clin Invest 122: 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Bhunia A, Oh YJ, Chang F, Bergman Y, Kim JH et al. (2014). OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ Res 115: 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FR, Venegas F, Gonzalez M, Andres S, Vallejos C, Riquelme G et al. (2009). Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3‐kinase/Akt in small‐diameter mesenteric arteries. Hypertension 53: 1000–1007. [DOI] [PubMed] [Google Scholar]
- Pirillo A, Norata GD, Catapano AL (2013). LOX‐1, OxLDL, and atherosclerosis. Mediators Inflamm 2013: 152786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Delamere NA (2006). NO donors inhibit Na,K‐ATPase activity by a protein kinase G‐dependent mechanism in the nonpigmented ciliary epithelium of the porcine eye. Br J Pharmacol 148: 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Gao L, Hsu YT, Bledsoe G, Hagiwara M, Chao L et al. (2010). Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt–eNOS signaling. Am J Physiol Heart Circ Physiol 299: H1419–H1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Luscher TF, Camici GG (2014). Dual role of endothelial nitric oxide synthase in oxidized LDL‐induced, p66Shc‐mediated oxidative stress in cultured human endothelial cells. PLoS One 9: e107787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Chen D, Cao L, Zhang R, Zhou J, Chen H et al. (2013). MiR‐490‐3p modulates the proliferation of vascular smooth muscle cells induced by ox‐LDL through targeting PAPP‐A. Cardiovasc Res 100: 272–279. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J (2008). LOX‐1 receptor blockade abrogates oxLDL‐induced oxidative DNA damage and prevents activation of the transcriptional repressor Oct‐1 in human coronary arterial endothelium. J Biol Chem 283: 19456–19464. [DOI] [PubMed] [Google Scholar]
- Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B (2014). OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med 70: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Meng F, Mohan S, Champaneri B, Gu Y (2009). Functional ENaC channels expressed in endothelial cells: a new candidate for mediating shear force. Microcirculation 16: 276–287. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fang X, Wang S, Feng Y, Yin J (2007). Relation between plasma oxLDL antibodies and oxLDL in the circulation. Inflammation 30: 7–13. [DOI] [PubMed] [Google Scholar]
- Wang ZR, Liu HB, Sun YY, Hu QQ, Li YX, Zheng WW et al. (2018). Dietary salt blunts vasodilation by stimulating epithelial sodium channels in endothelial cells from salt‐sensitive Dahl rats. Br J Pharmacol 175: 1305–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DG (2013). The amiloride‐sensitive endothelial sodium channel and vascular tone. Hypertension 61: 952–954. [DOI] [PubMed] [Google Scholar]
- Weinbrenner T, Cladellas M, Isabel Covas M, Fito M, Tomas M, Senti M et al. (2003). High oxidative stress in patients with stable coronary heart disease. Atherosclerosis 168: 99–106. [DOI] [PubMed] [Google Scholar]
- Xu L, Wang S, Li B, Sun A, Zou Y, Ge J (2015). A protective role of ciglitazone in ox‐LDL‐induced rat microvascular endothelial cells via modulating PPARgamma‐dependent AMPK/eNOS pathway. J Cell Mol Med 19: 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Kakino A, Takeshita H, Hayashi N, Li L, Nakano A et al. (2015). Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin‐like oxLDL receptor. FASEB J 29: 3342–3356. [DOI] [PubMed] [Google Scholar]
- Zeibig S, Li Z, Wagner S, Holthoff HP, Ungerer M, Bultmann A et al. (2011). Effect of the oxLDL binding protein Fc‐CD68 on plaque extension and vulnerability in atherosclerosis. Circ Res 108: 695–703. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chen S, Liu H, Zhang B, Zhao Y, Ma K et al. (2013). Hydrogen sulfide prevents hydrogen peroxide‐induced activation of epithelial sodium channel through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS One 8: e64304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jia YH, Zhao XS, Zhou FH, Pan YY, Wan Q et al. (2016a). Trichosanatine alleviates oxidized low‐density lipoprotein induced endothelial cells injury via inhibiting the LOX‐1/p38 MAPK pathway. Am J Transl Res 8: 5455–5464. [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Chen L, Si Z, Bu H, Narasimhulu CA, Song X et al. (2016b). Probucol protects endothelial progenitor cells against oxidized low‐density lipoprotein via suppression of reactive oxygen species formation in vivo. Cell Physiol Biochem 39: 89–101. [DOI] [PubMed] [Google Scholar]
- Zheng WW, Li XY, Liu HB, Wang ZR, Hu QQ, Li YX et al. (2016). AMP‐activated protein kinase attenuates high salt‐induced activation of epithelial sodium channels (ENaC) in human umbilical vein endothelial cells. Oxid Med Cell Longev 2016 1531392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Cao XQ, Liu ZH, Cao YJ, Liu CF, Zhang YL et al. (2016). Rapamycin inhibits oxidized low density lipoprotein uptake in human umbilical vein endothelial cells via mTOR/NF‐kappaB/LOX‐1 pathway. PLoS One 11: e0146777. [DOI] [PMC free article] [PubMed] [Google Scholar]