

Abstract
Hydrogen sulfide (H2S), independently of any specific transporters, has a number of biological effects on the cardiovascular system. However, until now, the detailed mechanism of H2S was not clear. Recently, a novel post‐translational modification induced by H2S, named S‐sulfhydration, has been proposed. S‐sulfhydration is the chemical modification of specific cysteine residues of target proteins by H2S. There are several methods for detecting S‐sulfhydration, such as the modified biotin switch assay, maleimide assay with fluorescent thiol modifying regents, tag‐switch method and mass spectrometry. H2S induces S‐sulfhydration on enzymes or receptors (such as p66Shc, phospholamban, protein tyrosine phosphatase 1B, mitogen‐activated extracellular signal‐regulated kinase 1 and ATP synthase subunit α), transcription factors (such as specific protein‐1, kelch‐like ECH‐associating protein 1, NF‐κB and interferon regulatory factor‐1), and ion channels (such as voltage‐activated Ca2+ channels, transient receptor potential channels and ATP‐sensitive K+ channels) in the cardiovascular system. Although significant progress has been achieved in delineating the role of protein S‐sulfhydration by H2S in the cardiovascular system, more proteins with detailed cysteine sites of S‐sulfhydration as well as physiological function need to be investigated in further studies. This review mainly summarizes the role and possible mechanism of S‐sulfhydration in the cardiovascular system. The S‐sulfhydrated proteins may be potential novel targets for therapeutic intervention and drug design in the cardiovascular system, which may accelerate the development and application of H2S‐related drugs in the future.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviation
- ATP5A1
ATP synthase subunit α
- CBS
cystathionine‐β‐synthase
- CSE
cystathionine‐γ‐lyase
- eNOS
endothelial NOS
- ER
endoplasmic reticulum
- IRF‐1
interferon regulatory factor‐1
- KATP
ATP‐sensitive K+
- Keap1
kelch‐like ECH‐associating protein 1
- KLF5
krüppel‐like factor 5
- MEK1
mitogen‐activated extracellular signal‐regulated kinase 1
- MMTS
methyl methanethiosulfonate
- MSBT
methylsulfonybenzothiazole
- NF‐κB
nuclear factor κB
- Nrf2
nuclear factor E2‐related factor 2
- PTEN
phosphatase and tensin homologue
- PTP1B
protein tyrosine phosphatase 1B
- SHR
spontaneously hypertensive rats
- SP‐1
specific protein‐1
- SUR2B
sulphonylurea 2B
- TFAM
mitochondrial transcription factor A
- TRP
transient receptor potential
- VEGFR
VEGF receptor
- WT
wild type
Introduction
Hydrogen sulfide (H2S) is a colourless, flammable, water‐soluble gas with a characteristic smell of rotten eggs. H2S was previously regarded as a toxic gas and environmental hazard. However, recent publications have revealed that H2S is synthesized in mammalian tissues, and freely travels through cell membranes. H2S acts independently of any specific transporters, and it has a number of biological effects on various systems (Meng et al., 2015a; Cao and Bian, 2016; Feliers et al., 2016; Ianaro et al., 2016; Katsouda et al., 2016; Cheng et al., 2016b). Nowadays, H2S is regarded as the third endogenous gasotransmitter followed by NO and CO (Hine et al., 2015).
H2S is produced, via the cysteine biosynthesis pathway, by three vital enzymes in mammalian species; these are cystathionine‐γ‐lyase (CSE), cystathionine‐β‐synthase (CBS) and 3‐mercaptopyruvate sulfurtransferase (MPST), and they have different distribution patterns in different tissues (Table 1). CSE is the main enzyme for H2S production in the cardiovascular system (Wang, 2012; Wallace and Wang, 2015). Previous research found that H2S has the potential to produce vasoconstriction or vasodilatation, angiogenesis, smooth muscle growth or apoptosis, cardioprotection and other effects (Mani et al., 2013; Tsikas and Cooper, 2014; Ping et al., 2015; Dunn et al., 2016; Katsouda et al., 2016; Marino et al., 2016). Our studies have suggested that H2S attenuates myocardial hypertrophy and fibrosis in spontaneously hypertensive rats (SHR) (Meng et al., 2015a,c; Meng et al., 2016), inhibits atherosclerotic plaque formation and inflammation in the aorta of apolipoprotein E−/− mice fed a high fat diet (Liu et al., 2013), suppresses oxidative stress and apoptosis in myocardial ischaemia and reperfusion injury (Meng et al., 2015b), augments mitochondrial function and the anti‐oxidative capacity of endothelial cells (Xie et al., 2016a), and activates nuclear factor E2‐related factor 2 (Nrf2) to alleviate diabetes‐accelerated atherosclerosis both in vitro and in vivo (Xie et al., 2016b). H2S also enhances the antioxidant activity, and regulates NO formation and kinase activity to maintain the homeostasis of the cardiovascular system (Liu et al., 2015; Chen et al., 2016; Shimizu et al., 2016; Cheng et al., 2016a). However, until now, the detailed mechanism of H2S has not been clear. Recently, more and more researchers revealed that some of the above effects could be attributed to a novel post‐translational modification induced by H2S, named S‐sulfhydration (Paul and Snyder, 2015a; Sen, 2017).
Table 1.
Distribution of CBS, CSE and MPST
| Names | Distribution |
|---|---|
| CBS | brain, astrocytes, liver |
| CSE | cardiovascular system, respiratory system |
| liver, kidney, uterus, placenta, pancreatic islets | |
| MPST | CNS |
| aortic endothelium and smooth muscles |
MPST, 3‐mercaptopyruvate sulfurtransferase
S‐sulfhydration is a chemical modification on specific cysteine residues of target proteins by H2S. In the presence of H2S, the free thiol groups of cysteine residues with a low pKa become covalently converted into a persulfides group (Mustafa et al., 2009; Paul and Snyder, 2015a). S‐sulfhydration can be induced by H2S on cysteine sulfenic acids (Cys‐SOH) or cysteine disulfides (─S─S) (Figure 1 A‐B), or by polysulfides on cysteine thiol (Cys‐SH, Figure 1 C). It is important to note that H2S also induces S‐sulfhydration on cysteine thiols in oxidation conditions (Figure 1D,E). Similar to S‐nitrosation, protein S‐sulfhydration was reversed by the thioredoxin system (Paul and Snyder, 2015b; Wedmann et al., 2016), which was closely correlated with cardiovascular diseases. This review will focus on the role of protein S‐sulfhydration by H2S in the cardiovascular system.
Figure 1.
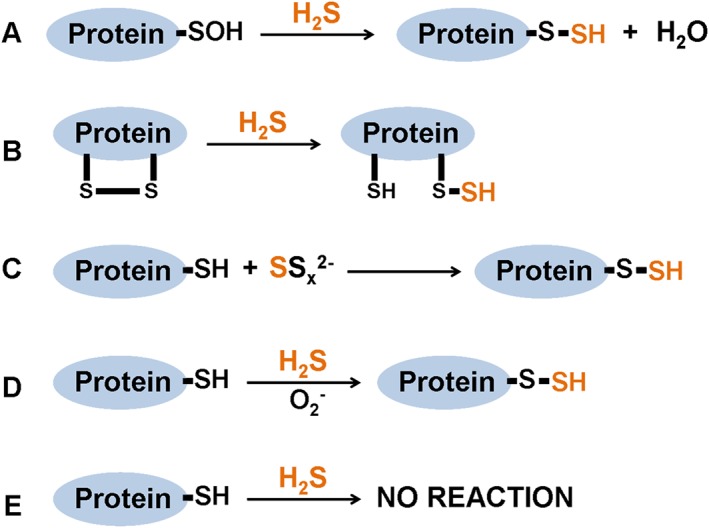
Reaction mechanisms for S‐sulfhydration formation. S‐sulfhydration can be induced by H2S on cysteine sulfenic acids (Cys‐SOH, A) or cysteine disulfides (−S‐S, B), or by polysulfides on cysteine thiols (Cys‐SH, C). H2S induces S‐sulfhydration on cysteine thiols in oxidation conditions (D‐E).
Detection of S‐sulfhydration
Nowadays, it is very difficult to distinguish the persulfides group from the thiol group because of their similar reactivity. The biotin switch used previously for nitrosylation measurement has been modified to detect S‐sulfhydration; this has been named as the ‘modified biotin switch assay’. An alkylating agent S‐methyl methanethiosulfonate (MMTS) was used to block thiol in proteins. The persulfides group was conjugated with N‐[6‐(biotinamido)hexyl]‐3′‐(2′‐pyridyldithio)propionamide (biotin‐HPDP). The biotinylated protein was then immunoprecipitated and analysed by western blotting, which represents the level of protein S‐sulfhydration (Mustafa et al., 2009). However, the thiol and persulfides showed similar reactivity to MMTS, and the selectivity was not good. The basal S‐sulfhydrated proteins account for as much as 25%, most of which might be a false positive (Pan and Carroll, 2013).
S‐sulfhydration was also able to be measured with a maleimide assay with fluorescent thiol modifying regents. Fluorescent maleimide acts on both modified and unmodified sulfhydryl groups. DTT reduces only the modified cysteines, and the decreased fluorescent intensity representing S‐sulfhydration is detected by SDS‐ PAGE (Sen et al., 2012). Unfortunately, the propensity to determine both nitrosylation and sulfenic acids with the malemide assay weakened its credibility for detecting S‐sulfhydration (Reisz et al., 2013; Paul and Snyder, 2015b).
Zhang et al. proposed a novel measurement selective for S‐sulfhydration, which was called ‘tag‐switch method’ (Zhang et al., 2014). Methylsulfonybenzothiazole (MSBT) was used to blocked thiols. Then a reagent containing nucleophile and biotin labelled only persulfides, while there was no binding with the blocked thiol groups. Finally, MSBT labelled persulfides representing S‐sulfhydration were conjugated with streptavidin and visualized by western blots (Zhang et al., 2014; Park et al., 2015). However, the method without higher sensitivity posed a problem for S‐sulfhydration measurement. To increase sensitivity, Wedmann et al. proposed an improved tag‐switch method with new cyanoacetic acid derivatives such as fluorescent BODIPY moiety or the Cy3‐dye (Wedmann et al., 2016).
Mass spectrometry (MS) analysis was also used to filter and identify the protein S‐sulfhydration. After protein samples were blocked with MSBT, only persulfide adducts reacted with CN‐biotin to form biotin‐labelled adducts. Then, these biotin‐labelled proteins were broken into peptides for MS (Park et al., 2015). By comparing the findings with the protein database, the names of the S‐sulfhydrated proteins were identified and specific cysteine sites of protein were ascertained. However, it was very difficult to block the protein samples completely, and, therefore, it was easy to produce false positive results (Gao et al., 2015; Park et al., 2015).
Altogether, as yet, there is no ideal method to detect S‐sulfhydration. More specific probes for the unique identification of S‐sulfhydration are urgently required. The current methods combined with mass spectrometry might be beneficial for qualitative and quantitative detection of protein S‐sulfhydration.
H2S induced protein S‐sulfhydration
Proteins with cysteine residues have the potential to be S‐sulfhydrated. Mustafa et al. found that about 10–25% of proteins extracted from liver, including GAPDH, β‐tubulin and actin, were S‐sulfhydrated in physiological conditions, which suggests that protein S‐sulfhydration might be a common form of post‐translational modification (Mustafa et al., 2009). LC‐MS/MS analysis exhibited that GAPDH was S‐sulfhydrated at Cys150. Moreover, NaHS increased GAPDH activity as high as seven fold, which was absent after C150S mutation (Mustafa et al., 2009). Mir et al. subsequently verified that GAPDH S‐sulfhydration at Cys150 promoted its binding to the E3 ligase protein in brain (Mir et al., 2014). However, one latest study failed to reproduce the conclusion on BL21 (DE3) Escherichia coli containing a pET15b vector expressing the wild‐type (WT) His‐tagged human GAPDH. They found that Cys156 or Cys247, but not Cys152 (which refers to Cys150 in the liver of mice on GAPDH with the normal residue numbering system), was S‐sulfhydrated by sulfide or polysulfide. And there was no increase in GAPDH activity after S‐sulfhydration. In contrast, polysulfides decreased GAPDH activity to about half of the control (Jarosz et al., 2015). Actually, not all cysteine‐rich proteins are able to be S‐sulfhydrated. The VEGFR‐2 contains several cysteine residues, and Cys1045 to Cys1024 serves as the molecular switch of vascular smooth muscle cell migration. However, there is no evidence that VEGFR‐2 is able to be S‐sulfhydrated by H2S (Tao et al., 2013). These discrepancies might be caused by the micro environment or the chemical structures of the proteins, and the different pathlogical states or specific characteristics of the cells or tissues. Generally, S‐sulfhydration can alter the functions of a wide range of cellular proteins. The following is a summary of S‐sulfhydrated proteins (Table 2) induced by H2S in the cardiovascular system.
Table 2.
Categories and functions of protein S‐sulfhydration in the cardiovascular system
| Categories | S‐sulfhydrated proteins | Functions in cardiovascular system |
|---|---|---|
| Enzymes/receptors | ATP5A1 | ATP synthase activity |
| MEK1 | Repair DNA damage | |
| PLN | Myocardial relaxation | |
| PPARγ | Adipogenesis | |
| PTP1B | Restore ER stress homeostasis | |
| p66Shc | Anti‐oxidative stress | |
| Transcription factors | IRF‐1 | Mitochondrial biogenesis |
| Keap1 | Anti‐oxidative stress | |
| p65 | Anti‐apoptosis and anti‐inflammation | |
| SP‐1 | Anti‐myocardial hypertrophy | |
| Endothelial phenotypes regulation | ||
| Ion channels | Kir6.1 | Vasodilatation |
| TRPV4 | Vasodilatation |
PLN, phospholamban
H2S induced S‐sulfhydration on enzymes or receptors in the cardiovascular system
NaHS induced S‐sulfhydration in both cytosolic and membrane proteins of myocardium from isolated working frog hearts or langendorff‐perfused rat hearts. Phospholamban, which is involved in myocardial relaxation through its modulation of intracellular calcium cycling, was identified to be S‐sulfhydrated with the immunoprecipitation and modified biotin switch methods (Mazza et al., 2013). This might be one of the selective post‐translational modifications that maintains cardiac homeostasis.
Protein tyrosine phosphatase 1B (PTP1B) plays a vital role in endoplasmic reticulum (ER) stress and is regarded as a potential target for therapeutic intervention in obesity‐induced cardiomyopathy (Kandadi et al., 2015) and septic shock‐induced cardiovascular dysfunction (Coquerel et al., 2014). Krishnan et al. found that H2S induced PTP1B S‐sulfhydration at Cys215 to inhibit its activity, which facilitated phosphorylation and activation of protein kinase‐like ER kinase, and promoted restoration of ER homeostasis. All of these effects were unavailable in CSE‐deleted HeLa cells (Krishnan et al., 2011). This suggests that H2S regulates ER stress via S‐sulfhydration to inactivate PTP1B, which might be a novel mechanism for the protective effect of H2S in the cardiovascular system.
Recently, H2S was shown to directly S‐sulfhydrate PPARγ. S‐sulfhydration of PPAR γ at Cys139 increased nuclear PPARγ accumulation, enhanced DNA binding activity to promoter of the PPARγ response element, and promoted adipogenesis gene expression in adipocytes, which were blocked by DTT or Cys139 mutation of PPARγ (Cai et al., 2016). As far as we know, PPAR is a dominant factor in blood lipid and glucose metabolism. S‐Sulfhydration of PPARγ might be a novel target for diabetes, obesity, hyperlipidaemia and related complications of the cardiovascular system.
H2S also increased mitogen‐activated extracellular signal‐regulated kinase 1 (MEK1) S‐sulfhydration in both human endothelial cells and human fibroblasts, while there was lower S‐sulfhydration of MEK1 in CSE−/− mice. S‐sulfhydrated MEK1 facilitated ERK1/2 phosphorylation, which subsequently transfers into the nucleus to activate PARP‐1 and to repair DNA damage. Mutation of Cys341 on MEK1 inhibited ERK1/2 phosphorylation and PARP‐1 activation, and failed to mediate DNA damage repair (Zhao et al., 2014). Xie et al. found that both exogenous H2S supplement and CBS overexpression increased p66Shc S‐sulfhydration at Cys59, which decreased the association of PKCβII with p66Shc and attenuated ROS production. However, H2S failed to induce mitochondrial translocation of p66Shc, decrease ROS and protect H2O2‐induced cell senescence if mutation of p66Shc at Cys59 (Xie et al., 2014, 2016c). H2S concentration‐dependently increased S‐sulfhydration of ATP synthase subunit α (ATP5A1) at Cys244 and Cys294 in HEK293 cells. Double mutation of C244S/C294S significantly attenuated ATP synthase activity. And there was lower S‐sulfhydration and weaker activity of ATP5A1 in CSE−/− mice. This suggests that S‐sulfhydration of ATP5A1 might be beneficial for the maintenance of ATP synthase homeostasis in mitochondrial energy disposal as well as antioxidant activity and redox signalling (Módis et al., 2016). Most of the findings revealed that S‐sulfhydration was the main post‐translational modification induced by H2S and played an important role in ROS production and redox signalling in the cardiovascular system.
Besides the cardiovascular system, several enzymes are also S‐sulfhydrated by H2S in the nervous system and endocrine system. NaHS augmented S‐sulfhydration of hippocampal protein phosphatase type 2A, PKA, PKC, and calcium/calmodulin‐dependent protein kinase II (CAMKII) to active postsynaptic signal pathways (Li et al., 2016). H2S also increased the S‐sulfhydration of glucose‐6‐phosphatase and fructose‐1,6‐bisphosphatase to promote gluconeogenesis in primary hepatocytes (Untereiner et al., 2016). S‐sulfhydration and the activity of parkin, a neuroprotective ubiquitin E3 ligase, was up‐regulated after H2S administration, which was attenuated in the brains of patients suffering from Parkinson's disease (Vandiver et al., 2014). However, whether these effects could be extrapolated to the cardiovascular system needs further exploration.
H2S induced S‐sulfhydration on transcription factors in the cardiovascular system
Specific protein‐1 (SP‐1) is an important transcription factor with multi‐functions in the cardiovascular system (Yang et al., 2013b). And there are a total of 11 cysteine residues on SP‐1. Inhibiting H2S production by AOAA (CBS inhibitor) or silencing CBS significantly reduced SP‐1 S‐sulfhydration at Cys68 and Cys755 in HUVECs, which enhanced proteasomal degradation of SP‐1, followed by inhibited SP‐1 binding with VEGFR‐2 or neuropilin‐1 promoter and impaired endothelial tube formation on Matrigel. All of these effects were dramatically restored by exogenous NaHS supplement. This suggests that H2S regulates endothelial key phenotypes such as proliferation and migration via SP‐1 S‐sulfhydration (Saha et al., 2016). Our latest study found that GYY4137, a H2S slow releasing compound, increased S‐sulfhydration on SP‐1 in neonatal rat cardiomyocytes and in myocardium of SHR. There are four residues (Cys659, Cys664, Cys689 and Cys692) in the domain of SP‐1 for binding with krüppel‐like factor 5 (KLF5, a key transcriptional factor involved in myocardial hypertrophy) promoter. And GYY4137 enhanced S‐sulfhydration on SP‐1 if there was an overexpression of WT SP‐1 or SP‐1 had mutations of C659A, C689A and C692A but not C664A in cardiomyocytes. Moreover, GYY4137 failed to attenuate KLF5 promoter activity and mRNA expression, reduce the binding between SP‐1 and KLF5 promoter, decrease the mRNA expression of atrial natriuretic peptide and improve myocardial hypertrophy in angiotensin II‐induced cardiomyocytes if the SP‐1 mutation was C664A (Meng et al., 2016). These findings suggest that S‐sulfhydration at Cys664 is essential for the inhibitory ability of KLF5 transcription and protective effect against myocardial hypertrophy induced by H2S.
Nrf2 is a vital transcription factor for protection against oxidative stress with kelch‐like ECH‐associated protein 1 (Keap1) as a negative receptor. Guo et al. found that NaHS increased Keap1 S‐sulfhydration to promote dissociation of Keap1/Nrf2 and finally to increase transcription activity of Nrf2, which was involved in the protective effect against ischaemic reperfusion‐induced oxidative stress and cell injury in human gastric epithelial cells (Guo et al., 2014). Yang et al. verified that Keap1 was S‐sulfhydrated in embryonic fibroblasts from WT mice but not CSE‐knockout mice. NaHS S‐sulfhydrated Keap1 at Cys151 to regulate the location, activity and target gene expression of Nrf2 in mouse embryonic fibroblasts. Tramtrack and Bric‐á‐Brac 2 dimerization domain (one functional domain on Keap1) deficiency completely abolished NaHS‐induced Keap1 S‐sulfhydration. The mutation of Cys151 in an intervening region, but not Cys288, failed to enhance Keap1 S‐sulfhydration, promote Nrf2 nuclear translocation or protect against cell senescence. This might be a novel mechanism for preventing from cell ageing by H2S (Yang et al., 2013a). Our latest study found that GYY4137 decreased aortic atherosclerotic plaque formation and ROS levels in aorta of streptozotocin‐induced LDL receptor knockout out mice (LDLr−/−) but not in LDLr−/− and Nrf2−/− double knockout mice. GYY4137 also attenuated foam cell formation and oxidative stress in peritoneal macrophages isolated from WT mice but not Nrf2−/− mice. This suggest that H2S attenuates diabetes‐accelerated atherosclerosis in an Nrf2‐dependent manner. Further study showed that GYY4137 promoted the dissociation of Keap1 from Nrf2 in ox‐LDL and high‐glucose stimulated endothelial cells, which might be attributed to Keap1 S‐sulfhydration at Cys151 and Cys273. We also found that Keap1 mutation of C151A, but not C273A, abolished Keap1/Nrf2 dissociation, Nrf2 nuclear translocation and ROS inhibition induced by GYY4137 administration. It is proposed that protein S‐sulfhydration by H2S might be a novel therapeutic target to prevent diabetes‐accelerated atherosclerosis (Xie et al., 2016b). The latest research found that H2S elevated Keap1 S‐sulfhydration to reduce the association between Keap1 and Nrf2 in the kidneys of rats on a high‐salt diet, followed by decreased collagen deposition and oxidative stress (Huang et al., 2016). All of these results suggest that Keap1 is a key target of H2S in several different cells or tissues. S‐sulfhydration of Keap1 might be a potential target for attenuating oxidative stress and related cardiovascular diseases.
NF‐κB is also a multi‐functional transcription factor. Sen et al. found that TNF‐α enhanced the binding activity between NF‐κB and DNA, followed by increased p65 binding with the promoter of anti‐apoptotic genes in macrophages. However, the anti‐apoptosis effect was abolished in macrophages from CSE−/− mice, which was restored by CSE overexpression or H2S supplement. Further experiments showed that H2S S‐sulfhydrated p65 at the highly conserved Cys38 residue and augmented its association with ribosomal protein S3 as a co‐regulator of NF‐κB to activate the promoter of anti‐apoptotic genes. All these effects were absent after p65‐C38S was transfected (Perkins, 2012; Sen et al., 2012). However, Du et al. found that H2S enhanced p65 S‐sulfhydration in ox‐LDL‐induced macrophages, which was abolished by DTT or p65 mutation at Cys38. Moreover, S‐sulfhydration of p65 by H2S helped to inhibit NF‐κB activation and monocyte chemoattractant protein 1 (MCP‐1 also known as CCL2) generation and suppressed ox‐LDL‐induced inflammation. The various effects of p65 S‐sulfhydration can possibly be attributed to a different condition, which was anti‐apoptosis in physiological but anti‐inflammation in pathological conditions (Du et al., 2014).
Li et al. found that a deficiency in CSE decreased mitochondrial DNA levels and mitochondrial transcription factor A (TFAM) expression in both smooth muscle cells and arteries, which resulted in mitochondrial function disorder. H2S S‐sulfhydrated transcription repressor interferon regulatory factor‐1 (IRF‐1), strengthened the binding between IRF‐1 and DNA methyl transferase 3A promoter to inhibit TFAM promoter methylation. Finally, TFAM methylation was attenuated, while the expressions of TFAM and mitochondrial DNA were increased to restore the mitochondrial biogenesis (Li and Yang, 2015). Bioinformatics analysis found that only Cys53 is located in the DNA‐binding domain of IRF‐1.
H2S induced S‐sulfhydration on ion channels in cardiovascular system
Voltage‐activated Ca2+ channels, one of the most important calcium channels in cardiovascular system, are regulated by H2S (Fukami and Kawabata, 2015; Ping et al., 2015; Zhang et al., 2015). NaHS concentration‐dependently inhibits L‐type calcium currents in cardiomyocytes, which was abolished by DTT. And NaHS decreases the functional free sulfhydryl group in L‐type calcium channel, which provides indirect evidence for S‐sulfhydration of voltage‐activated Ca2+ channels by H2S (Zhang et al., 2012). However, there was no direct evidence that voltage‐activated Ca2+ channels were S‐sulfhydrated by H2S. Whether H2S regulates related subunits or associated cysteine sites is also still unknown.
Transient receptor potential (TRP) channels, as putative pro‐angiogenic Ca2+‐permeable channels, are also modulated by H2S (Munaron et al., 2013; Zhang et al., 2015). Liu et al. found that H2S S‐sulfhydrated TRPV6 at Cys172 and Cys329 sites in bone marrow mesenchymal stem cells. Overexpression of TRPV6 increased Ca2+ influx and activated the PKC/β‐cateine signal pathway to promote osteogenic differentiation. Mutation of TRPV6 at both Cys172 and Cys329, but not only one site alone, resulted in a suppressed PKC/β‐cateine signal pathway and impaired osteogenic differentiation. Moreover, there were also several cysteine residues at other Ca2+ TRP channels, such as TRPV3 and TRPM4, which have the potential to be S‐sulfhydrated by H2S (Liu et al., 2014b). A previous study confirmed that endothelial cells are responsible for endogenous H2S production and H2S‐induced vasodilatation. Naik et al. found that H2S‐induced Ca2+ and K+ influx to dilate vessels was blocked after TRPV4 inhibition. Moreover, S‐sulfhydration of TRPV4 was enhanced after Na2S treatment in aortic endothelial cells (Naik et al., 2016). This suggests that TRPV4 is activated after S‐sulfhydration, which might be the key factor in vasodilatation.
Kir6.1, a subunit of the ATP‐sensitive K+ (KATP) channels, was S‐sulfhydrated after CSE overexpression, and this did not occur if CSE was absent or mutated. A further study confirmed that S‐sulfhydrated Kir6.1 at Cys43 attenuated ATP production but elicited more phosphatidylinositol 4,5‐bisphosphate to bind with Kir6.1, which promoted KATP channel activity and improved vasodilatation. Moreover, not only S‐sulfhydration but also vasodilatation induced by H2S was alleviated in Kir6.1‐Cys43 mutants (Mustafa et al., 2011). This might be the key mechanism for H2S to act as an endothelial derived relaxing factor. Kang et al. also found that H2S increased S‐sulfhydration on sulphonylurea 2B (SUR2B) at Cys24 and Cys1455, another subunit of KATP channels complex, to restore smooth muscle contraction (Kang et al., 2015). Liu et al. found that H2S decreased the membrane potential, inhibited the fast inactivation component of the voltage‐dependent potassium channel current in gastric smooth muscle cells and promoted gastric motility, which was suppressed by KV4.3 knockdown. Meanwhile, KV4.3 was S‐sulfhydrated by H2S, which was attenuated by DTT (Liu et al., 2014a). All these data suggested that different cysteine sites might be S‐sulfhydrated in different cells, which have various effects responsible for physiological or pathological process.
Crosstalk with protein S‐sulfhydration and other post‐translational modifications
S‐sulfhydration at cysteine residues usually alters the structure and function of a protein. It is possible that crosstalk occurs with several other post‐translational modification to ultimately regulate a physiological or pathological process. But overall, there are relatively few studies on this type of crosstalk in cardiovascular research.
It was reported that NO elevates S‐nitrosylation on phosphatase and tensin homologue (PTEN) at C83S to attenuate its activity followed by Akt activation. One study found that H2S S‐sulfhydrated PTEN at Cys71 and Cys124 in human neuroblastoma SH‐SY5Y cells (Numajiri et al., 2011). Furthermore, S‐nitrosylation on PTEN increased if H2S production was inhibited by knocking down CBS. Consistent with the reduced S‐sulfhydration, Akt activity increased significantly (Ohno et al., 2015). As far as we know, PTEN is also a key signal molecular in cardiomyocyte apoptosis (Ke et al., 2016), ventricular remodelling (Yang et al., 2016), angiogenesis (Serra et al., 2015) and other cardiovascular diseases. It is thought that protein S‐sulfhydration and S‐nitrosylation of PTEN keep a dynamic balance, and compete with each other to maintain normal function of the protein. PTP1B at Cys215 also underwent S‐nitrosylation and S‐sulfhydration (Chen et al., 2008; Krishnan et al., 2011). S‐sulfhydration of p65, one key submit of NF‐κB, disrupted the S‐nitrosylation of itself to suppress apoptosis in macrophages (Sen et al., 2012). More recently it was verified that the effect of H2S is determined by S‐nitrosylation but not S‐sulfhydration (Sun et al., 2016). However, further studies need to be done on more proteins with simultaneous S‐sulfhydration and S‐nitrosylation to determine the possible physiological significance of each of these processes. Also, it would be beneficial to identify the potential mechanism for the common ‘crosstalk’ between H2S and NO in the cardiovascular system.
Kang et al. reported that H2S increases S‐sulfhydration on SUR2B, one of the key subunits of the KATP channel complex, in mouse colonic smooth muscle cells. Furthermore the peroxynitrite donor SIN‐1 enhances the tyrosine nitration of Kir6.1 (another subunit of th eKATP channel complex). This SIN‐1‐induced up‐regulation of tyrosine nitration on Kir6.1 was restored by NaHS in Chinese hamster ovary cells transfected with Kir6.1 and the SUB2B mutant at C263S but not C24S or C1455S. That is S‐sulfhydration of SUB2B at C24S and C1455S inhibited the nitration of Kir6.1. Moreover, NaHS also reduced the tyrosine nitration of Cav1.2b channels to improve Ca2+‐induced contractions in mouse ileum (Kang et al., 2015). In accord with the previous study, NaHS reduced the activity of L‐Ca2+ channels containing free sulfhydryl groups in cardiomyocytes (Zhang et al., 2012). These results suggest that H2S might directly S‐sulfhydrate L‐Ca2+ channels to regulate Ca2+ homeostasis in the heart. Moreover, H2S S‐sulfhydrates cysteine‐rich proteins to induce several other post‐translational modifications on different proteins. Both of the direct and indirect effects of S‐sulfhydration play a vital role in maintaining the physiological function of proteins in the cardiovascular system.
A significant amount of endogenous H2S anion is generated in rat cardiomyocytes and cardiac fibroblasts. The reduction in H2S production caused by CSE or CBS deletion enhanced the protein S‐guanylation induced by 8‐nitro‐cGMP. Moreover, exogenous NaHS treatment markedly enhanced S‐sulfhydrated 8‐nitro‐cGMP, which attenuated 8‐nitro‐cGMP‐induced H‐Ras activation, but not the activation of the H‐Ras C184S mutant, by H‐Ras S‐guanylation in rat cardiomyocytes and myocardium from a failing heart, due to myocardial infarction (Nishida et al., 2012). As 8‐nitro‐cGMP is a key signalling molecule in cardiovascular system disorders (Akaike et al., 2010), these findings suggest that 8‐nitro‐cGMP S‐sulfhydration by H2S not only inhibits S‐guanylation but also antagonizes the oxidative stress‐induced or electrophile‐mediated cell injury, which might be a novel mechanism of cardioprotection.
At baseline conditions, in endothelial cells isolated from aortae of WT mice, 30% of total endothelial NOS (eNOS) is S‐nitrosylated and 21% is S‐sulfhydrated, and these values are elevated by NO or an H2S donor respectively. Altogether about 5% of total eNOS is S‐sulfhydrated in aortic tissue of WT mice, but S‐sulfhydration is undetectable in CSE knock out mice. Moreover, H2S abolished the S‐nitrosylation of eNOS induced by the NO donor sodium nitroprussiate (SNP), but SNP had no effect on NaHS‐induced S‐sulfhydration of eNOS in endothelial cell lysates. NaHS increases S‐sulfhydration, NO production and eNOS dimerization on WT eNOS and C689S‐eNOS but not C443S‐eNOS. NaHS also increases the phosphorylation of WT‐eNOS or C443G‐eNOS but not S1179A‐eNOS. These results suggest that H2S and NO compete for S‐sulfhydration and S‐nitrosylation at the cysteine residues to regulate the phosphorylation and activity of eNOS. H2S, as a pivotal coordinator, maintains the dynamic homeostasis among several post‐translational modifications of endothelial function (Altaany et al., 2014).
In addition, S‐sulfhydration of p66Shc at Cys59 promoted p66Shc phosphorylation at Ser36 due to an enhanced association with PKCβII and p66Shc (Xie et al., 2014). S‐sulfhydration is involved in the protective effect of the protein‐O‐GlcNAcylation in myocardial ischaemia reperfusion injury (Pagliaro et al., 2011). H2S also induces S‐polythiolation, S‐alkylation, S‐arylation and other post‐translational modifications (Rudolph and Freeman, 2009; Ida et al., 2014). However, whether there is a crosstalk with S‐sulfhydration and the possible effect in the cardiovascular system remains unknown. More knowledge of the crosstalk may speed up our understanding of the role of protein S‐sulfhydration in cardiovascular disease.
Concluding remarks and future perspectives
Over the past few years, significant progress has been achieved in delineating the role of protein S‐sulfhydration by H2S in the cardiovascular system (Figure 2). However, more scientific methods with enhanced sensitivity and specificity to detect S‐sulfhydration are urgently needed. More proteins and detailed cysteine sites of S‐sulfhydration need to be investigated in the cardiovascular system. However, not all of the proteins subjected to S‐sulfhydration have an altered spatial configuration and activity. This might be determined by the location of the S‐sulfhydrated cysteines. If S‐sulfhydrated cysteines are located in the key domain, which is vital to maintain the structure and activity of that protein, protein S‐sulfhydration will alter the protein function and signal transduction. In other words, there might be no significant difference after S‐sulfhydration, which is known as ‘ineffective S‐sulfhydration’. Moreover, the significance of S‐sulfhydration in the cardiovascular system, such as target gene transcription, enzymatic activity and ion channel permeability, are to be investigated in further studies. The exact nature of the crosstalk between S‐sulfhydration and other post‐translational modifications is not yet known and deserves to be better elucidated. In addition, the level of protein S‐sulfhydration is controlled by the thioredoxin system, which suggests that some agents that alter thioredoxin activity or expression will be involved in regulating the intracellular levels of protein S‐sulfhydration and H2S‐mediated biological and pharmacological effects (Wedmann et al., 2016).
Figure 2.
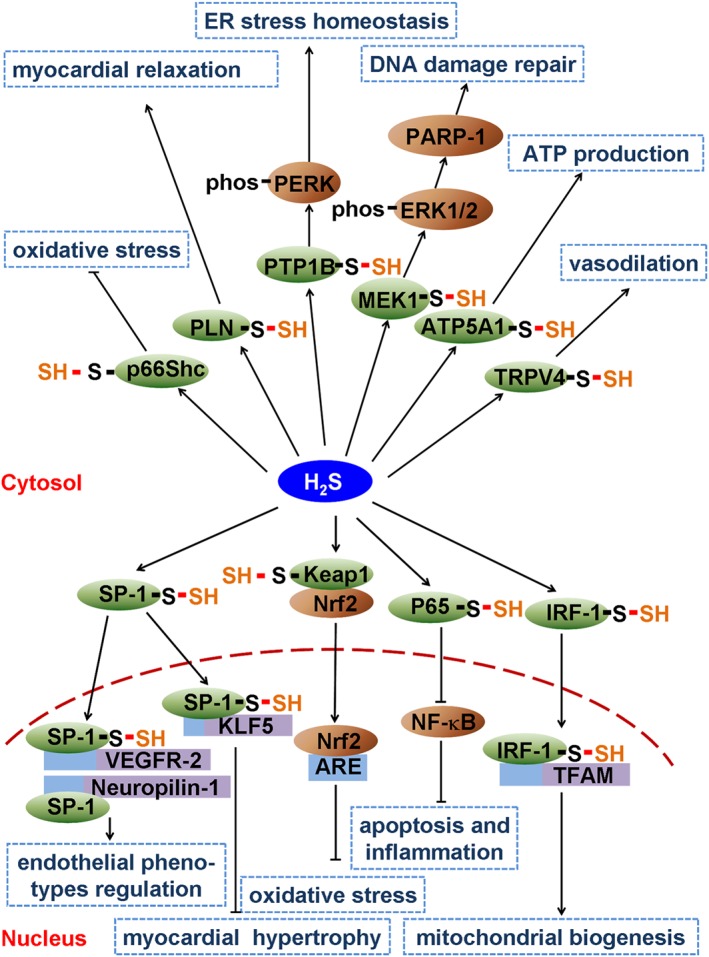
Schematic illustration of possible roles of S‐sulfhydration by H2S in the cardiovascular system. H2S induces S‐sulfhydration on p66Shc to inhibit oxidative stress. S‐sulfhydration on phospholamban (PLN) promotes myocardial relaxation. S‐sulfhydration on protein tyrosine phosphatase 1B (PTP1B) restores endoplasmic reticulum stress homeostatis. H2S also S‐sulfhydrates MEK1 to repair DNA damage. ATP5A1 and transient receptor potential V4 (TRPV4) S‐sulfhydration improves ATP production and vasodilatation respectively. H2S also S‐sulfhydrates SP‐1, Keap1, NF‐κB and IRF‐1 to regulate target gene transcription, which is vital for the regulation of endothelial phenotypes, myocardial hypertrophy, oxidative stress, mitochondrial biogenesis, apoptosis and inflammation.
Protein S‐sulfhydration, as a vital post‐translational modification induced by H2S, is a possible a molecular mechanism for the effects of H2S. Clinically, the relevance of S‐sulfhydration in cardiovascular diseases needs to be studied. More information about S‐sulfhydration will help us to understand how S‐sulfhydration at specific cysteines can have a beneficial effect in various cardiovascular diseases. Moreover, the S‐sulfhydrated proteins may be potential novel targets for therapeutic intervention and drug design in the cardiovascular system, which may accelerate the development and application of H2S‐related drugs in the future.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d,e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant nos. 91639204, 81400203, 81330004, 31371156, 81670209), the Natural Science Foundation of Nantong City (grant no. MS12015015), and the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine.
Meng, G. , Zhao, S. , Xie, L. , Han, Y. , and Ji, Y. (2018) Protein S‐sulfhydration by hydrogen sulfide in cardiovascular system. British Journal of Pharmacology, 175: 1146–1156. doi: 10.1111/bph.13825.
Contributor Information
Yi Han, Email: hanyi79@163.com.
Yong Ji, Email: yongji@njmu.edu.cn.
References
- Akaike T, Fujii S, Sawa T, Ihara H (2010). Cell signaling mediated by nitrated cyclic guanine nucleotide. Nitric Oxide 23: 166–174. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The concise guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaany Z, Ju Y, Yang G, Wang R (2014). The coordination of S‐sulfhydration, S‐nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal 7: ra87. [DOI] [PubMed] [Google Scholar]
- Cai J, Shi X, Wang H, Fan J, Feng Y, Lin X et al. (2016). Cystathionine γ lyase‐hydrogen sulfide increases peroxisome proliferator‐activated receptor γ activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochim Biophys Acta 1861: 419–429. [DOI] [PubMed] [Google Scholar]
- Cao X, Bian JS (2016). The role of hydrogen sulfide in renal system. Front Pharmacol 7: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Chen Q, Zhu XJ, Kong DS, Wu L, Shao JJ et al. (2016). Diallyl trisulfide protects against ethanol‐induced oxidative stress and apoptosis via a hydrogen sulfide‐mediated mechanism. Int Immunopharmacol 36: 23–30. [DOI] [PubMed] [Google Scholar]
- Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH et al. (2008). Cysteine S‐nitrosylation protects protein‐tyrosine phosphatase 1B against oxidation‐induced permanent inactivation. J Biol Chem 283: 35265–35272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YS, Dai DZ, Dai Y, Zhu DD, Liu BC (2016a). Exogenous hydrogen sulphide ameliorates diabetic cardiomyopathy in rats by reversing disordered calcium‐handling system in sarcoplasmic reticulum. J Pharm Pharmacol 68: 379–388. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Garikipati VN, Nickoloff E, Wang C, Polhemus DJ, Zhou J et al. (2016b). Restoration of hydrogen sulfide production in diabetic mice improves reparative function of bone marrow cells. Circulation 134: 1467–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquerel D, Neviere R, Delile E, Mulder P, Marechal X, Montaigne D et al. (2014). Gene deletion of protein tyrosine phosphatase 1B protects against sepsis‐induced cardiovascular dysfunction and mortality. Arterioscler Thromb Vasc Biol 34: 1032–1044. [DOI] [PubMed] [Google Scholar]
- Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M et al. (2014). Hydrogen sulfide suppresses oxidized low‐density lipoprotein (ox‐LDL)‐stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF‐κB) pathway. J Biol Chem 289: 9741–9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WR, Alexander SP, Ralevic V, Roberts RE (2016). Effects of hydrogen sulphide in smooth muscle. Pharmacol Ther 158: 101–113. [DOI] [PubMed] [Google Scholar]
- Feliers D, Lee HJ, Kasinath BS (2016). Hydrogen sulfide in renal physiology and disease. Antioxid Redox Signal 25: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami K, Kawabata A (2015). Hydrogen sulfide and neuronal differentiation: focus on Ca2+ channels. Nitric Oxide 46: 50–54. [DOI] [PubMed] [Google Scholar]
- Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M et al. (2015). Quantitative H2S‐mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 4: e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Liang F, Shah Masood W, Yan X (2014). Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s‐sulfhydration, MAPK dependent anti‐apoptosis and NF‐κB dependent anti‐inflammation pathway. Eur J Pharmacol 725: 70–78. [DOI] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L et al. (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Shen Z, Liu J, Huang Y, Chen S, Yu W et al. (2016). Hydrogen sulfide inhibits high‐salt diet‐induced renal oxidative stress and kidney Injury in Dahl rats. Oxid Med Cell Longev 2016 2807490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianaro A, Cirino G, Wallace JL (2016). Hydrogen sulfide‐releasing anti‐inflammatory drugs for chemoprevention and treatment of cancer. Pharmacol Res 111: 652–658. [DOI] [PubMed] [Google Scholar]
- Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y et al. (2014). Reactive cysteine persulfides and S‐polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A 111: 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz AP, Wei W, Gauld JW, Auld J, Özcan F, Aslan M et al. (2015). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) is inactivated by S‐sulfuration in vitro. Free Radic Biol Med 89: 512–521. [DOI] [PubMed] [Google Scholar]
- Kandadi MR, Panzhinskiy E, Roe ND, Nair S, Hu D, Sun A (2015). Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet‐induced obesity: role of AMPK‐dependent autophagy. Biochim Biophys Acta 1852: 299–309. [DOI] [PubMed] [Google Scholar]
- Kang M, Hashimoto A, Gade A, Akbarali HI (2015). Interaction between hydrogen sulfide‐induced sulfhydration and tyrosine nitration in the KATP channel complex. Am J Physiol Gastrointest Liver Physiol 308: G532–G539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouda A, Bibli SI, Pyriochou A, Szabo C, Papapetropoulos A (2016). Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol Res 113: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke ZP, Xu P, Shi Y, Gao AM (2016). MicroRNA‐93 inhibits ischemia‐reperfusion induced cardiomyocyte apoptosis by targeting PTEN. Oncotarget 7: 28796–28805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK (2011). H2S‐Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal 4: ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yang G (2015). Hydrogen sulfide maintains mitochondrial DNA replication via demethylation of TFAM. Antioxid Redox Signal 23: 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Zhou J, Zhang H, Luo Y, Long LH, Hu ZL et al. (2016). Hydrogen sulfide promotes surface insertion of hippocampal AMPA receptor gluR1 subunit via phosphorylating at serine‐831/serine‐845 sites through a sulfhydration‐dependent mechanism. CNS Neurosci Ther 22: 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DH, Huang X, Meng XM, Zhang CM, Lu HL, Kim YC et al. (2014a). Exogenous H2S enhances mice gastric smooth muscle tension through S‐sulfhydration of KV 4.3, mediating the inhibition of the voltage‐dependent potassium current. Neurogastroenterol Motil 26: 1705–1716. [DOI] [PubMed] [Google Scholar]
- Liu MH, Lin XL, Zhang Y, He J, Tan TP, Wu SJ et al. (2015). Hydrogen sulfide attenuates doxorubicin‐induced cardiotoxicity by inhibiting reactive oxygen species‐activated extracellular signal‐regulated kinase 1/2 in H9c2 cardiac myocytes. Mol Med Rep 12: 6841–6848. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yang R, Liu X, Zhou Y, Qu C, Kikuiri T et al. (2014b). Hydrogen sulfide maintains mesenchymal stem cell function and bone homeostasis via regulation of Ca(2+) channel sulfhydration. Cell Stem Cell 15: 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Han Y, Li L, Lu H, Meng G, Li X et al. (2013). The hydrogen sulfide donor, GYY4137, exhibits anti‐atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. Br J Pharmacol 169: 1795–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC et al. (2013). Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127: 2523–2534. [DOI] [PubMed] [Google Scholar]
- Marino A, Martelli A, Citi V, Fu M, Wang R, Calderone V et al. (2016). The novel H2S donor 4‐carboxy‐phenyl isothiocyanate inhibits mast cell degranulation and renin release by decreasing intracellular calcium. Br J Pharmacol 173: 3222–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza R, Pasqua T, Cerra MC, Angelone T, Gattuso A (2013). Akt/eNOS signaling and PLN S‐sulfhydration are involved in H2S ‐dependent cardiac effects in frog and rat. Am J Physiol Regul Integr Comp Physiol 305: R443–R451. [DOI] [PubMed] [Google Scholar]
- Meng G, Ma Y, Xie L, Ferro A, Ji Y (2015a). Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br J Pharmacol 172: 5501–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Wang J, Xiao Y, Bai W, Xie L, Shan L et al. (2015b). GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J Biomed Res 29: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Zhu J, Xiao Y, Huang Z, Zhang Y, Tang X et al. (2015c). Hydrogen sulfide donor GYY4137 protects against myocardial fibrosis. Oxid Med Cell Longev 2015: 691070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Xiao Y, Ma Y, Tang X, Xie L, Liu J et al. (2016). Hydrogen sulfide regulates krüppel‐like factor 5 transcription activity via specificity protein 1 S‐sulfhydration at Cys664 to prevent myocardial hypertrophy. J Am Heart Assoc 5: e004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir S, Sen T, Sen N (2014). Cytokine‐induced GAPDH sulfhydration affects PSD95 degradation and memory. Mol Cell 56: 786–795. [DOI] [PubMed] [Google Scholar]
- Módis K, Ju Y, Ahmad A, Untereiner AA, Altaany Z, Wu L et al. (2016). S‐Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol Res 113: 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munaron L, Avanzato D, Moccia F, Mancardi D (2013). Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 53: 77–84. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK et al. (2009). H2S signals through protein S‐sulfhydration. Sci Signal 2: ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al. (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik JS, Osmond JM, Walker BR, Kanagy NL (2016). Hydrogen sulfide‐induced vasodilation mediated by endothelial TRPV4 channels. Am J Physiol Heart Circ Physiol 311: H1437–H1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, Ihara H et al. (2012). Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat Chem Biol 8: 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W et al. (2011). On‐off system for PI3‐kinase‐Akt signaling through S‐nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc Natl Acad Sci U S A 108: 10349–10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Okuda K, Uehara T (2015). Endogenous S‐sulfhydration of PTEN helps protect against modification by nitric oxide. Biochem Biophys Res Commun 456: 245–249. [DOI] [PubMed] [Google Scholar]
- Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C (2011). Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal 14: 833–850. [DOI] [PubMed] [Google Scholar]
- Pan J, Carroll KS (2013). Persulfide reactivity in the detection of protein s‐sulfhydration. ACS Chem Biol 8: 1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CM, Macinkovic I, Filipovic MR, Xian M (2015). Use of the "tag‐switch" method for the detection of protein S‐sulfhydration. Methods Enzymol 555: 39–56. [DOI] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2015a). H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci 40: 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2015b). Protein sulfhydration. Methods Enzymol 555: 79–90. [DOI] [PubMed] [Google Scholar]
- Perkins ND (2012). Cysteine 38 holds the key to NF‐κB activation. Mol Cell 45: 1–3. [DOI] [PubMed] [Google Scholar]
- Ping NN, Li S, Mi YN, Cao L, Cao YX (2015). Hydrogen sulphide induces vasoconstriction of rat coronary artery via activation of Ca(2+) influx. Acta Physiol (Oxf) 214: 88–96. [DOI] [PubMed] [Google Scholar]
- Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM (2013). Thiol‐blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J 280: 6150–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph TK, Freeman BA (2009). Transduction of redox signaling by electrophile‐protein reactions. Sci Signal 2: re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Chakraborty PK, Xiong X, Dwivedi SK, Mustafi SB, Leigh NR et al. (2016). Cystathionine β‐synthase regulates endothelial function via protein S‐sulfhydration. FASEB J 30: 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N (2017). Functional and molecular insights of hydrogen sulfide signaling and protein sulfhydration. J Mol Biol 429: 543–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R et al. (2012). Hydrogen sulfide‐linked sulfhydration of NF‐κB mediates its antiapoptotic actions. Mol Cell 45: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra H, Chivite I, Angulo‐Urarte A, Soler A, Sutherland JD, Arruabarrena‐Aristorena A et al. (2015). PTEN mediates Notch‐dependent stalk cell arrest in angiogenesis. Nat Commun 6: 7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Nicholson CK, Lambert JP, Barr LA, Kuek N, Herszenhaut D et al. (2016). Sodium sulfide attenuates ischemic‐induced heart failure by enhancing proteasomal function in an Nrf2‐dependent manner. Circ Heart Fail 9: e002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Aponte AM, Menazza S, Gucek M, Steenbergen C, Murphy E (2016). Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S‐sulfhydration vs. S‐nitrosylation. Cardiovasc Res 110: 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao BB, Liu SY, Zhang CC, Fu W, Cai WJ, Wang Y et al. (2013). VEGFR2 functions as an H2S‐targeting receptor protein kinase with its novel Cys1045‐Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal 19: 448–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsikas D, Cooper AJ (2014). Letter by Tsikas and Cooper regarding article, “dysregulation of hydrogen sulfide (H2S) producing enzyme cystathionine γ‐lyase (CSE) contributes to maternal hypertension and placental abnormalities in preeclampsia”. Circulation 129: e516. [DOI] [PubMed] [Google Scholar]
- Untereiner AA, Wang R, Ju Y, Wu L (2016). Decreased gluconeogenesis in the absence of cystathionine gamma‐lyase and the underlying mechanisms. Antioxid Redox Signal 24: 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM et al. (2014). Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 4: 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Wang R (2015). Hydrogen sulfide‐based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14: 329–345. [DOI] [PubMed] [Google Scholar]
- Wang R (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92: 791–896. [DOI] [PubMed] [Google Scholar]
- Wedmann R, Onderka C, Wei S, Szijártó IA, Miljkovic JL, Mitrovic A et al. (2016). Improved tag‐switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem Sci 7: 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Feng H, Li S, Meng G, Liu S, Tang X et al. (2016a). SIRT3 mediates the antioxidant effect of hydrogen sulfide in endothelial cells. Antioxid Redox Signal 24: 329–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y et al. (2016b). Hydrogen sulfide induces keap1 S‐sulfhydration and suppresses diabetes‐accelerated atherosclerosis via Nrf2 activation. Diabetes 65: 3171–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZZ, Liu Y, Bian JS (2016c). Hydrogen sulfide and cellular redox homeostasis. Oxid Med Cell Longev 2016 6043038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZZ, Shi MM, Xie L, Wu ZY, Li G, Hua F et al. (2014). Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal 21: 2531–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S et al. (2013a). Hydrogen sulfide protects against cellular senescence via S‐sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18: 1906–1919. [DOI] [PubMed] [Google Scholar]
- Yang P, Zhang Y, Pang J, Zhang S, Yu Q, He L et al. (2013b). Loss of Jak2 impairs endothelial function by attenuating Raf‐1/MEK1/Sp‐1 signaling along with altered eNOS activities. Am J Pathol 183: 617–625. [DOI] [PubMed] [Google Scholar]
- Yang X, Qin Y, Shao S, Yu Y, Zhang C, Dong H et al. (2016). MicroRNA‐214 inhibits left ventricular remodeling in an acute myocardial infarction rat model by suppressing cellular apoptosis via the phosphatase and tensin homolog (PTEN). Int Heart J 57: 247–250. [DOI] [PubMed] [Google Scholar]
- Zhang D, Macinkovic I, Devarie‐Baez NO, Pan J, Park CM, Carroll KS et al. (2014). Detection of protein S‐sulfhydration by a tag‐switch technique. Angew Chem Int Ed Engl 53: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Sun Y, Tsai H, Tang C, Jin H, Du J (2012). Hydrogen sulfide inhibits L‐type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PLoS One 7: e37073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xu C, Yang G, Wu L, Wang R (2015). Interaction of H2S with calcium permeable channels and transporters. Oxid Med Cell Longev 2015: 323269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Ju Y, Li S, Altaany Z, Wang R, Yang G (2014). S‐sulfhydration of MEK1 leads to PARP‐1 activation and DNA damage repair. EMBO Rep 15: 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]