

INTRODUCTION
Reverse translation, defined here as the use of clinical observations to drive scientific investigation, has been used in the field of Pediatric Rheumatology for decades. Forced in part by limited resources and rare childhood diseases, clinical questions have driven scientific investigation and have focused basic science research efforts to address the most clinically relevant questions. Here we discuss the application of reverse translational approaches, by us and others, to improve pharmacotherapy in Pediatric Rheumatology.
Pediatric Rheumatology is a field focused on treating autoimmune and autoinflammatory diseases in childhood. With ∼300 pediatric rheumatologists in North America, and entire states without representation, it is appropriate to say that the clinical need in Pediatric Rheumatology is underserved. However, despite the inherent challenges in caring for a rare pediatric patient population, great strides have been made in advancing the field through scientific discovery inspired by the multitude of questions naturally generated while providing routine clinical care. The concept of “reverse translation” is practiced every day, even at times unintentionally, in fields that let their clinical questions guide their research efforts. Furthermore, in subspecialties like pediatric rheumatology, which is based on a strong foundation of clinical diagnosticians, clinical variability becomes a natural driver for scientific discovery.
Effective use of pharmacotherapy is critical to disease management in the treatment of the Pediatric Rheumatology patient population. However, clinical response to drug therapy can be highly variable, resulting in a trial‐and‐error approach to treatment that can have long‐term deleterious consequences for the patient. These observations have fueled scientific investigation into factors impacting drug response and represents a reverse translational approach to improve patient outcomes through the early use of optimal drug therapy. We posit that three factors primarily contribute to the observed variation in drug response (Figure 1). These factors include heterogeneity in the underlying disease that can significantly impact how the patient responds to the particular drug therapy, pharmacokinetic variation resulting in differences in drug exposure, which is a well‐known factor influencing drug response, and finally, pharmacodynamic variation that results in differences in the biochemical and physiological response to drug therapy that ultimately impacts clinical response. We highlight examples for each of these factors from research in the treatment of juvenile idiopathic arthritis (JIA) as they relate to the reverse translational approach to improving pharmacotherapy in these patients, and we contend that the general concepts fueling this work can be applied to any field.
Figure 1.
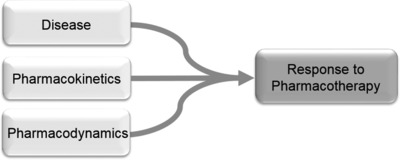
Schematic of factors impacting response to pharmacotherapy. Major factors contributing to the observed variation in drug response among patients includes: heterogeneity in disease, variation in the disposition of the therapeutic agent (i.e., pharmacokinetic variation), and variation in the biochemical and physiological response to the drug (i.e., pharmacodynamic variation).
DISEASE VARIATION AND DRUG RESPONSE
JIA is the most common pediatric autoimmune musculoskeletal condition, estimated to affect 1 in 1,000 children. The prevalence varies considerably depending on the country, due to several factors. JIA has a relatively heterogeneous clinical phenotype, resulting in several classifications based on age, number of joints involved, presence of serologic markers, family history, and the presence of specific systemic signs and symptoms. The currently agreed upon nomenclature for JIA subtypes include: systemic, oligoarticular, polyarticular, enthesitis‐related, psoriatic, and undifferentiated arthritis. JIA is distinct from adult rheumatoid arthritis (RA) in many ways; in fact, only one subtype of JIA (rheumatoid factor positive polyarticular JIA) is clinically and serologically similar to adult RA, but is encountered in less than 10% of all JIA. The systemic JIA (SJIA) subtype is clinically distinct, presenting with daily high spiking fevers, evanescent erythematous rash, organomegaly, and serositis in addition to arthritis. Although it comprises only about 10% of all JIA, it has the highest risk of morbidity and mortality, often secondary to a phenomenon called macrophage activation syndrome (MAS). MAS is characterized by a “cytokine storm” resulting in unremitting fevers, cytopenias, hepatosplenomegaly with hepatic dysfunction, coagulopathy, and CNS involvement such as seizures and change in mental status, and it can be fatal. SJIA has been a challenge to treat, but not only due to its severe clinical course. Throughout the last few decades, when therapeutics were expanded for JIA, it was noted that SJIA does not respond as adequately to methotrexate (MTX) and tumor necrosis factor alpha (TNF‐α) blockade (TNFi), as observed in other JIA subtypes.1
The notable clinical and therapeutic differences in this subtype inspired work that discovered that SJIA is driven by the innate immune system and a distinct group of cytokines compared with nonsystemic JIA and adult RA. Early studies found markedly elevated blood and synovial fluid concentrations of interleukin (IL)‐6, but not TNF‐α, in patients with SJIA compared with nonsystemic JIA and RA.2 In addition, serum from SJIA patients was found to activate peripheral blood mononuclear cells from healthy individuals, resulting in pronounced secretion of IL‐1β and further experimental treatment with the recombinant human IL‐1Ra anakinra induced disease remission in seven of nine SJIA patients.3 Since these landmark discoveries, it has been shown that gene expression profiles in peripheral blood mononuclear cells differ by JIA subtype, and innate immune pathways are uniquely upregulated in SJIA.4 As a result of these discoveries, the treatment paradigm has shifted to the early use of biologic agents targeting IL‐1 and IL‐6 in SJIA. In fact, there is debate now that SJIA should be reclassified as an autoinflammatory condition based on its distinct immunological picture. This is an example of utilizing a clinical disease phenotype and the lackluster response to traditionally successful therapeutics to essentially work backwards to discover a physiologically distinct subtype of childhood arthritis. Furthermore, this discovery has additional important downstream impacts on pharmacotherapeutic decision‐making for the most severe subtype of JIA.
PHARMACOKINETIC AND PHARMACODYNAMIC VARIATION AND DRUG RESPONSE
Although MTX continues to be the cornerstone of therapy in the treatment of polyarticular forms of JIA, it is characterized by response failure in ∼30% of patients, and the development of therapy‐limiting toxicities in ∼30% of patients.5 Based on these clinical observations, there have been extensive efforts to use reverse translational approaches to understand the mechanistic basis for the observed variation in drug response. Through an understanding of the mechanisms responsible for the observed variation in response to MTX, it is anticipated that such knowledge will allow for the forward translation of these findings to develop therapeutic strategies to enhance the safety and efficacy of MTX in the treatment of JIA.
Recognizing that pharmacokinetic variation is a well‐known source of clinical response variability, early studies focused on plasma drug level monitoring of MTX. However, these studies failed to find any relationship between plasma MTX levels and either MTX safety or efficacy in JIA,6 and likely reflects the inherent problem of plasma drug level monitoring of MTX in these patients (i.e., once‐weekly dosing with a plasma half‐life of ∼4 h causes MTX to only be transiently present in patient plasma). However, the recognition that MTX is extensively metabolized intracellularly to form polyglutamated metabolites (MTX‐Glun) that are pharmacologically active (Figure 2), and can be measured in circulating erythrocytes, resulted in renewed efforts to evaluate pharmacokinetic variation as a potentially important factor impacting MTX efficacy and toxicity. Although the polyglutamation of MTX was observed initially with high‐dose MTX used for oncologic indications decades ago, early work in RA demonstrated a potential association with response in the low‐dose weekly MTX dosing regimen also adopted by JIA. Subsequent studies have demonstrated that increased erythrocyte accumulation of MTX‐Glun in JIA patients is associated with higher MTX dose and subcutaneous route of administration5 and, importantly, also associated with improved response to MTX.7 Studies have similarly demonstrated that the increased accumulation of erythrocyte MTX‐Glun is associated with the development of gastrointestinal and hepatic toxicities.5 Therefore, optimization of erythrocyte MTX‐Glun levels may represent an approach to fine‐tuning MTX therapy to enhance efficacy and prevent drug‐related toxicities. These observations have driven further basic scientific investigation to identify the basis for the observed variation in intracellular MTX‐Glun formation in patients, with the anticipation that a priori clinical biomarkers can be defined that will promote the safest and most effective use of MTX early in the treatment of these patients.
Figure 2.
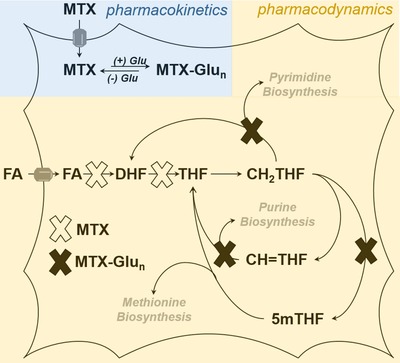
Schematic of intracellular pharmacokinetics and pharmacodynamics of methotrexate (MTX). Following transporter‐mediated uptake, MTX is reversibly metabolized to form a series of polyglutamated metabolites (MTX‐Glun). MTX inhibits the enzymatic conversion of folic acid (FA) to dihydrofolate (DHF), and DHF to tetrahydrofolate (THF), resulting in the depletion of cellular methylene‐THF (CH2THF), methenyl‐THF (CH=THF), and 5‐methyl‐THF (5mTHF). Depletion of the reduced cellular folate pool by MTX and inhibition of folate‐dependent enzymes by MTX‐Glun results in inhibition of purine, pyrimidine, and methionine biosynthesis.
In contrast to the well‐accepted role of pharmacokinetic variability as a major source of variation in drug response, the role of pharmacodynamic variation is not as well established nor understood. In the case of MTX, the most direct and measureable pharmacodynamic effect is the systemic depletion of the biologically active forms of folate resulting from inhibition of dihydrofolate reductase, including tetrahydrofolate (THF), methylene‐THF (CH2‐THF), methenyl‐THF (CH=THF), and 5‐methyl‐THF (5mTHF) (Figure 2). Together, the inhibition of folate‐dependent biochemical pathways through the depletion of intracellular folates and the direct inhibition of folate‐dependent enzymes by MTX‐Glun results in inhibition of one‐carbon metabolism represented as inhibition of purine, pyrimidine, and methionine biosynthesis. Although MTX has been demonstrated to cause marked and variable perturbations in folate homeostasis in JIA patients,8 no studies have evaluated whether this observed pharmacodynamic variation is related to or contributes to the observed variation in response to MTX, and is the focus of ongoing investigation in our laboratory.
More recent efforts in our lab have focused on downstream pharmacodynamic measures of response to MTX therapy in JIA to understand the observed variation in drug response. These studies have been based on the recognition that cytokines represent mediators of disease activity in JIA, are responsive to drug therapy, and are the target of newer therapies. Most notably, recent work by our group has found that patients with elevated plasma TNF‐α levels at the time of MTX initiation are more likely to fail MTX therapy and require the addition of a TNFi in the first 3 months of treatment.9 These finding suggest that disease activity associated with marked elevations in TNF‐α, may benefit from the early addition of TNFi therapy. Similarly, we have found that elevations in plasma levels of the adipocytokine and rate‐limiting enzyme in NAD biosynthesis, nicotinamide phosphoribosyltransferase (NAMPT), is associated with poor response to MTX.10 Using a reverse translational approach, we used this clinical research observation to conduct a mechanism‐based in vitro study on the effect of NAMPT on the pharmacological activity of MTX, and were able to demonstrate that reductions in the enzymatic activity of NAMPT promotes pharmacological response to MTX. This suggests the potential prognostic value of NAMPT level monitoring in patients as a predictor of response to MTX therapy, as well as the potential use of NAMPT inhibitors as a method to increase response to MTX.
In summary, reverse translation can be used effectively to drive discovery even in pediatric subspecialties where resources are scarce, such as Pediatric Rheumatology. It is a conceptual approach that can be applied readily in the clinical and research setting to address problems of high clinical significance, such as variability in drug response and the optimization of drug therapy to improve clinical outcomes, as described here. In the case of observed variation in response to pharmacotherapy, we propose that heterogeneity in disease, as well as variation in pharmacokinetic and pharmacodynamic parameters, represent the primary factors driving variation in drug response, and reverse translational studies evaluating these factors can be applied universally and adopted in any therapeutic area.
Conflict of Interest
The author declared no conflicts of interest.
References
- 1. Russo, R.A. & Katsicas, M.M. Clinical remission in patients with systemic juvenile idiopathic arthritis treated with anti‐tumor necrosis factor agents. J. Rheumatol. 36, 1078–1082 (2009). [DOI] [PubMed] [Google Scholar]
- 2. de Benedetti, F. et al Correlation of serum interleukin‐6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 34, 1158–1163 (1991). [DOI] [PubMed] [Google Scholar]
- 3. Pascual, V. et al Role of interleukin‐1 (IL‐1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL‐1 blockade. J. Exp. Med. 201, 1479–1486 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barnes, M.G. et al Subtype‐specific peripheral blood gene expression profiles in recent‐onset juvenile idiopathic arthritis. Arthritis Rheum. 60, 2102–2112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becker, M.L. et al The effect of genotype on methotrexate polyglutamate variability in juvenile idiopathic arthritis and association with drug response. Arthritis Rheum. 63, 276–285 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Ravelli, A. et al Plasma levels after oral methotrexate in children with juvenile rheumatoid arthritis. J. Rheumatol. 20, 1573–1577 (1993). [PubMed] [Google Scholar]
- 7. Calasan, M.B. et al Methotrexate polyglutamates in erythrocytes are associated with lower disease activity in juvenile idiopathic arthritis patients. Ann. Rheum. Dis. 74, 402–407 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Funk, R.S. et al Folate depletion and increased glutamation in juvenile idiopathic arthritis patients treated with methotrexate. Arthritis Rheumatol. 66, 3476–3485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Funk, R.S. , Chan, M.A. & Becker, M.L. Cytokine biomarkers of disease activity and therapeutic response after initiating methotrexate therapy in patients with juvenile idiopathic arthritis. Pharmacotherapy 37, 700–711 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Funk, R.S. et al Nicotinamide phosphoribosyltransferase attenuates methotrexate response in juvenile idiopathic arthritis and in vitro. Clin. Transl. Sci. 9, 149–157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]