

Abstract
Background and Purpose
Atherosclerosis results from a maladaptive inflammatory response initiated by the intramural retention of LDL in susceptible areas of the arterial vasculature. The ω‐3 polyunsaturated fatty acids (ω‐3) have protective effects in atherosclerosis; however, their molecular mechanism is still largely unknown. The present study used a metabolomic approach to reveal the atheroprotective metabolites of ω‐3 and investigate the underlying mechanisms.
Experimental Approach
We evaluated the development of atherosclerosis in LDL receptor‐deficient mice (LDLR−/−) fed a Western‐type diet (WTD) plus ω‐3 and also LDLR−/− and fat‐1 transgenic (LDLR−/− ‐fat‐1tg) mice fed a WTD. The profiles of ω‐3 in the plasma were screened by LC–MS/MS using unbiased systematic metabolomics analysis. We also studied the effect of metabolites of eicosapentaenoic acid (EPA) on endothelial activation in vitro.
Key Results
The ω‐3 diet and fat‐1 transgene decreased monocyte infiltration, inhibited the expression of pro‐inflammatory genes and significantly attenuated atherosclerotic plaque formation and enhanced plaque stability in LDLR−/− mice. The content of 18‐hydroxy‐eicosapentaenoic acid (18‐HEPE) and 17,18‐epoxy‐eicosatetraenoic acid (17,18‐EEQ), from the cytochrome P450 pathway of EPA, was significantly higher in plasma from both ω‐3‐treated LDLR−/− and LDLR−/− ‐fat‐1tg mice as compared with WTD‐fed LDLR−/− mice. In vitro in endothelial cells, 18‐HEPE or 17,18‐EEQ decreased inflammatory gene expression induced by TNFα via NF‐κB signalling and thereby inhibited monocyte adhesion to endothelial cells.
Conclusions and Implications
EPA protected against the development of atherosclerosis in atheroprone mice via the metabolites 18‐HEPE and/or 17,18‐EEQ, which reduced endothelial activation. These compounds may have therapeutic implications in atherosclerosis.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- 1‐ABT
1‐aminobenzotriazole
- ARA
arachidonic acid
- CCL
chemokine C‐C motif ligand
- CD
chow diet
- CYP
cytochrome P450
- DHA
docosahexarnoic acid
- EC
endothelial cell
- EEQ
epoxyeicosatetraenoic acid
- EPA
eicosapentaenoic acid
- HEPE
hydroxy‐eicosapentaenoic acid
- ICAM‐1
intercellular adhesion molecule 1
- LDLR
LDR receptor
- LOX
lipoxygenase
- MSPPOH
N‐methylsulfonyl‐6‐(2‐propargyloxyphenyl)hexanamide
- NDGA
nordihydroguaiaretic acid
- PUFA
polyunsaturated fatty acid
- VCAM‐1
vascular cell adhesion molecule 1
- VIP
variable importance for prediction
- WTD
Western‐type diet
- αSMA
α‐smooth muscle cell actin
- ω‐3
ω‐3 polyunsaturated fatty acids
- ω‐6
ω‐6 polyunsaturated fatty acids
Introduction
Atherosclerosis is a life‐threatening disease triggered by an increase in the blood concentration of LDL cholesterol and the development of inflammatory networks. Endothelial cells (EC), participating in the intial step of atherosclerosis, are activated by several inflammatory stimuli, which up‐regulate the expression of many inflammatory factors such as E‐selectin and vascular cell adhesion molecule 1 (VCAM‐1) (Ross, 1999). The blood monocytes are captured by activated EC and enter the arterial wall, thereafter differentiating to macrophages. Some of these macrophages with inflammatory markers are expressed on the surface and along with cholesterol are involved in the progressive occlusion of the arterial lumen (Libby, 2012). Suppressing the pro‐inflammatory state of EC could prevent the formation of atherosclerosis.
Arachidonic acid (ARA) (Lopez‐Vicario et al.), a major ω‐6 polyunsaturated fatty acid (ω‐6 PUFA), is catalysed by three major enzymatic pathways – COX, lipoxygenase (LOX) and cytochrome P450 (CYP) – to various metabolites (Gross et al., 2005). Many of these metabolites are pro‐inflammatory and trigger cardiovascular diseases such as hypertension, atherosclerosis and heart failure (Takase et al., 1996; Dwyer et al., 2004; Sacerdoti et al., 2015). In contrast, ω‐3 PUFA (ω‐3), especially eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), described as the ‘Eskimo factor’, have caught the attention of basic and clinical research scientists since the 1940s because of the rare coronary heart disease and cardiovascular events in Greenland Eskimos, who consume a diet rich in seafood (Lavie et al., 2009). EPA and DHA share the same enzymatic system with ARA for transformation into a number of metabolites such as hydroxy‐eicosapentaenoic acid (HEPE) and epoxyeicosatetraenoic acid (EEQ) (Mozurkewich et al., 2016). A number of studies report that a ω‐3 diet can attenuate EC activation, monocyte adhesion and infiltration into the subendothelial space of the arterial wall and reduce atherosclerotic lesions (De Caterina et al., 1994; Collie‐Duguid and Wahle, 1996; De Caterina and Libby, 1996; Hughes et al., 1996; Miles et al., 2000).
Unlike ω‐6 PUFA, ω‐3 PUFA are of prime importance for their anti‐inflammatory effect, but they can only be derived from the diet because mammals lack the endogenous enzymes for ω‐3 desaturation (Spychalla et al., 1997). The Caenorhabditis elegans fat‐1 gene encodes an ω‐3 fatty acid desaturase and converts ω‐6 PUFA to ω‐3 PUFA by adding a double bond at the ω‐3 position. When introduced into mice (Kang et al., 2004), the fat‐1 gene increased endogenous levels of EPA/DHA and reduced the development of atherosclerosis in transgenic mice (fat‐1tg) (Wang et al., 2004; Wan et al., 2010). Hence, EPA and DHA may have an atheroprotective effect. However, the possible underlying mechanisms remain unknown, and whether EPA/DHA themselves or another metabolite(s) have an atheroprotective role is still debatable.
In this study, the potency of metabolites as candidates that mediate the anti‐atherosclerotic effects of ω‐3 were explored. We used an established unbiased systematic metabolomics analysis (Zhang et al., 2015) to screen the changes in both ω‐6 and ω‐3 metabolic profiles in LDL receptor deficient mice and fat‐1tg mice fed a diet supplemented with ω‐3. Among these metabolites, ω‐3‐enriched 18‐HEPE and 17,18‐EEQ were shown to reduce the development of atherosclerosis by decreasing endothelial activation and monocyte adhesion, and these effects were mediated by inhibiting NF‐κB signalling.
Methods
Animal experiments
LDL receptor deficient (LDLR−/−) mice were obtained from the Experimental Animal Centre of Military Medical Science Academy (Beijing, China). Fat‐1tg mice were provided by Dr Alan Zhao from Nanjing Medical University (Wei et al., 2010). Fat‐1tg mice were crossed with LDLR−/− mice to obtain LDLR−/−‐fat‐1tg mice. Six‐week‐old male LDLR−/− mice (18–20 g) were randomized into three groups, which were fed different diets for 6 weeks: a chow diet (CD; MD12016); Western‐type diet (WTD; MD12017, containing 40 kcal% fat, 1.25% cholesterol and 0.5% cholic acid) and WTD supplemented with ω‐3 [WTD + ω‐3; MD12017A, MD12017 mixed with 3% deep‐sea fish oil (Nutrifynn Caps, PA, USA)]. In another set of experiments, LDLR−/− and LDLR−/−‐fat‐1tg mice were fed a WTD for 6 weeks. The investigators were not blinded to the experimental groups. The food intake was monitored throughout the feeding period. All mice were housed in a specific‐pathogen‐free, temperature‐controlled environment in individually ventilated cages with wood shavings as bedding (3–6 per cage), with 12 h light/dark cycles and received food and water ad libitum. At the end of the experiment, mice were killed by exsanguination after being anaesthetized with isoflurane. Plasma samples were collected with butylated hydroxytoluene as an antioxidant and EDTA as an anticoagulant.
The plasma levels of triglycerides, total cholesterol and LDL cholesterol in mice were measured by use of an automated clinical chemistry analyser kit (Biosino Biotech, Beijing, China). Aortic trees were obtained from mice after they were killed and fixed in 4% paraformaldehyde solution for en face staining. Aorta samples for RT‐PCR were snap‐frozen in liquid nitrogen immediately after collection, then stored at −80°C. Samples of aortic roots were embedded in Tissue‐Tec OCT moulds at −80°C after fixing and dehydrating. All procedures involving experimental animals were performed under the principle for replacement, refinement or reduction (the 3Rs) and in accordance with US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85‐23, updated 2011) and were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Tianjin, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Murine atherosclerotic lesion analysis
Aortic trees were fixed and dissected on dark wax, then stained with Oil‐red O to quantify the lesion area by using Image Pro Plus. Cross sections of aortic roots 7 μm thick were stained with Oil‐red O to assess lipid accumulation. Lesion area was determined in haemotoxylin and eosin (H&E)‐stained outflow tract sections. Collagen formation was detected by picrosirius‐red staining (Solarbio, Beijing, China), immunostaining of α‐smooth muscle cell actin (αSMA) for smooth muscle cells (SMC) and F4/80 for macrophages. For immunofluorescence microscopy, aortic root sections were fixed in ice‐cold acetone for 5 min, washed in PBS and blocked with normal goat serum for 1 h at room temperature. Sections were then incubated overnight at 4°C with antibodies for αSMA and F4/80 (both 1:100) in 1% BSA block solution. Sections were then rinsed and incubated with secondary antibody. Images were captured by fluorescence microscopy and analysed by using Image Pro Plus. The vulnerability index of plaques was calculated as (macrophage staining% + lipid staining%)/(SMCs% + collagen fibre%), according to a previous report (Williams et al., 2002).
Cell culture
HUVECs were cultured as described previously (Zhu et al., 1998) at 37°C in a humidified atmosphere containing 5% CO2, and cells between passages 4 and 6 were used for experiments.
Plasmid transfection and luciferase activity assay
HUVECs cultured to sub‐confluence in 6‐well plates were transfected with the firefly luciferase reporter plasmid of NF‐κB containing a TA promoter (pNF‐κB‐TA‐luc, Beyotime Biotechnology, China) along with a β‐galactosidase reporter plasmid (Progema, Madison, WI, USA) for 24 h by using lipofectamine reagent (Invitrogen, Carlsbad, CA, USA) as instructed. Then the cells were treated with of TNFα (0.1 ng·mL−1) and 18‐HEPE (1 μM) or 17,18‐EEQ (1 μM) as indicated for another 24 h. The luciferase activity in cell lysates was measured by using a dual luciferase reporter assay system (Progema, Madison, WI, USA).
Monocyte adhesion
Monocyte adhesion was analysed as described previously but with modifications (Zhu et al., 1998). Briefly, HUVECs were cultured in 6‐well plates, and pretreated with 18‐HEPE (1 μM) or 17,18‐EEQ (1 μM) for 24 h until confluent, then with TNFα (0.1 ng·mL−1) for 6 h. THP1 cells were labelled with BCECF‐AM (Invitrogen), then added to plates at 2 × 106 cells per well. After incubation for 30 min at 37°C, non‐adherent cells were removed by washing three times with PBS. In each sample, the number of stained adhering cells in five random fields was counted under a fluorescence microscope.
Metabolomic analysis
Metabolomic analysis involved LC–MS/MS of metabolites as described previously (Li et al., 2014; Zhang et al., 2015). Briefly, 200 μL plasma samples from mice were spiked with internal standard mixture (5 ng) and extracted by solid‐phase extraction, then cartridges were washed with 2 mL of 5% methanol and vacuum pumped. Analytes were eluted with methanol and evaporated to dryness. The residues were dissolved in 100 μL of 30% acetonitrile. Chromatographic separations involved the use of an UPLC BEH C18 column (1.7 μm, 100 × 2.1 mm i.d.) consisting of ethylene‐bridged hybrid particles (Waters, Milford, MA, USA). Target profiling of PUFA involved the 5500 QTRAP hybrid triple quadruple linear ion‐trap mass spectrometer (AB Sciex, Foster City, CA, USA) equipped with a turbo ion‐spray electrospray ionization source. For unbiased analysis of differences between groups, we used Metaboanalyst 3.0 (http://www.metaboanalyst.ca). Missing values were imputed with half of the minimum positive value, and data were log‐transformed and autoscaled before analysis. Global changes between samples from different groups of mice were compared by non‐parametic tests (P < 0.1) with a fold‐change threshold of 2.
Quantitative RT‐PCR (qPCR)
Real‐time PCR with primers was conducted as previously described (Xue et al., 2015). Total RNA from cells or mouse tissues was isolated by use of RNAiso Plus reagent (Takara Bio, Japan) as instructed and reverse‐transcribed by using the first‐strand cDNA synthesis kit (Thermo Scientific, Rockford, IL, USA). Gene expression was normalized to that of β‐actin. Primers for qPCR are in Supporting Information Table S1.
Western blot analysis
Western blot analysis was performed as described previously (Xue et al., 2015). Cell lysates were resolved by 10% SDS‐PAGE, then transferred to a PVDF membrane, which was incubated with primary antibodies, then horseradish peroxidase‐conjugated secondary antibody. The densities of the protein bands were analysed by using Image J software.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as a mean ± SEM. Statistical analysis involved the use of GraphPad Prism 6 with Student's two‐tailed unpaired t‐test, one‐way ANOVA or two‐way ANOVA with Bonferroni multiple comparison post test, as appropriate. P < 0.05 was considered to be statistically significant.
Materials
Chow diet (MD12016), WTD (MD12017) and WTD supplemented with ω‐3 PUFA (WTD + ω‐3; MD12017A) were from Medicience (Jiangsu, China). The content of EPA and DHA in the WTD + ω‐3 diet was 0.5% (w.w‐1) and 0.35% (w.w‐1) respectively. Standard metabolites of PUFA, including HEPEs and EEQs, and inhibitors of PUFA metabolism [indomethacin (IND), nordihydroguaiaretic acid (NDGA), 1‐aminobenzotriazole (1‐ABT) and N‐methylsulfonyl‐6‐(2‐propargyloxyphenyl)hexanamide (MSPPOH)] were from Cayman Chemical (Ann Arbor, MI). The UPLC BEH C18 column of ethylene‐bridged hybrid particles was from Waters (Milford, MA). The 5500 QTRAP spectrometer was from AB Sciex (Foster City, CA). Triglyceride and cholesterol determination kits were from BioSino Bio‐Technology & Science (Beijing, China). Antibodies against αSMA (Sigma, St. Louis, MO), CD31 (Abcam, Cambridge, MA), MAC‐3 (Santa Cruz Biotechnology, Santa Cruz, CA) and NF‐κB Pathway Sampler Kit #9936 (Cell Signalling Technology, Danvers, MA) were all commercially available.
Nomenclature of targets and ligands
Key protein targets and the ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Supplementation of ω‐3 in WTD reduces the size of plaque formation and enhances plaque stability in LDLR−/− mice
We first examined the efficacy of ω‐3 in WTD‐induced atherosclerosis in LDLR−/− mice. As compared with the chow diet (CD), the WTD induced a marked atherosclerotic lesion in the mice. LDLR−/− mice fed a WTD supplemented with ω‐3 for 6 weeks showed a significantly attenuated atherosclerotic lesion area in the aortic tree and aortic sinus, as measured by en face Oil‐red O staining (Figure 1A) and H&E staining (Figure 1B) respectively. To evaluate the effect of ω‐3 on the nature of the plaques, we further analysed plaque composition in mice. Lipid deposition was lower in atherosclerotic plaques in LDLR−/− mice with ω‐3‐supplemented WTD than control mice (Figure 1C). Furthermore, macrophage infiltration was decreased, as indicated by immunostaining with the macrophage marker MAC‐3 (Figure 1D), in atherosclerotic plaques of ω‐3‐treated mice. In contrast, collagen content was increased, as determined by picrosirius‐red staining (Figure 1E), in plaques of ω‐3‐treated mice. The content of vascular SMC, determined by αSMA immunostaining, was comparable in the ω‐3 and control groups (Figure 1F). As a result, the vulnerability index of the plaque was reduced by the ω‐3 treatment (Figure 1G). Moreover, ω‐3 supplementation significantly reduced the plasma levels of triglycerides, total cholesterol and LDL cholesterol (Figure 1H). Hence, ω‐3 may have potent anti‐atherosclerotic effects either mediated directly or via a reduction in serum lipid levels.
Figure 1.
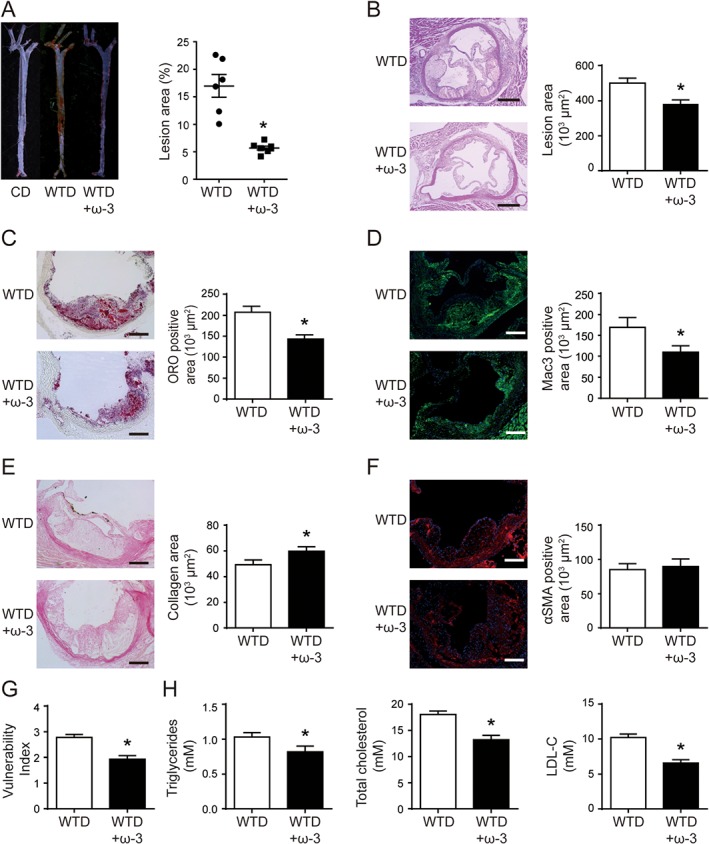
Supplementation of ω‐3 in WTD reduced atherosclerotic plaque formation and increased plaque stability in LDLR−/− mice. LDLR−/− mice were fed a CD or WTD for 6 weeks; one group of LDLR−/− mice were fed the WTD supplemented with ω‐3 (3% wt.wt‐1). (A) Representative aortas from each diet group with Oil‐red O staining (left) and quantification of aortic lesion area (right) by en face staining; each dot represents a single mouse (n = 6 for each group). Horizontal bars represent mean values; whiskers are SEM. (B) Representative photomicrographs of aortic root sections stained with H&E and quantification of lesion area. (C–F) Representative photomicrographs (left) and quantification (right) of aortic root sections stained with Oil‐red O (C), MAC‐3 (D), picrosirius red (E) and αSMA (F) in atherosclerotic plaque. (G) Vulnerability index of plaques. (H) Quantification of plasma levels of triglycerides, total cholesterol and LDL‐C in plasma. (B–H) n = 10 mice in WTD group and n = 9 in WTD + ω‐3 group (one sample in WTD + ω‐3 group was lost during sample preparation). Data are mean ± SEM, *P < 0.05 compared to control.
ω‐3 supplementation of a WTD changed the profiles of PUFA metabolites in LDLR−/− mice
To analyse the profiles of PUFA metabolites, we used LC–MS/MS of plasma samples from the mice. A heat map analysis showed that ω‐3 treatment significantly increased the levels of most ω‐3 metabolites and reduced those of ω‐6 metabolites as compared with controls (Figure 2A and Supporting Information Table S2). The partial least squares discriminant analysis (PLS‐DA) score plot differentiated the profiles by comparison with mice with ω‐3‐supplemented WTD alone (Figure 2B). Variable importance for prediction (VIP) scores were calculated from the scores, and the top 10 most significant metabolites are shown on the Y‐axis (Figure 2C and Table 1). To gain an appreciation of the magnitude and direction of these changes, we plotted a volcano plot (Figure 2D). The red points indicate mediators with expression that reached statistical difference by non‐parametric test (log10 of the P‐value) and changed by at least twofold. These data indicate that HEPEs and EEQs, as metabolites of EPA, may mediate the anti‐atherosclerotic effect of ω‐3 in LDLR−/− mice.
Figure 2.
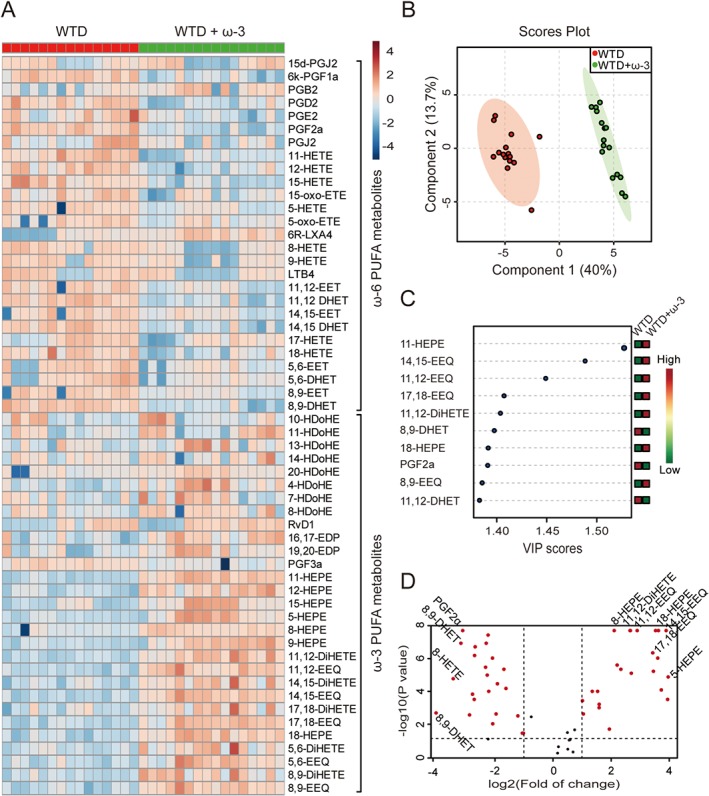
Supplementation of ω‐3 WTD in LDLR−/− mice changed the profiles of PUFA metabolites. Mice were treated as in Figure 1. (A) Heat map showing eicosanoid profiles of ω‐6 and ω‐3 PUFAs in plasma samples from mice with WTD without and with ω‐3. (B) PLS‐DA score plot and (C) features (variables) of top 10 most significant metabolites based on VIP scores from PLS‐DA. The X‐axis shows the correlation scores and the Y‐axis the metabolites. Colour bars show median intensity of variable in the respective group. (D) Volcano plot of PUFA profiling. Red points indicate metabolites with content that both reached statistical difference by non‐parametric test (log10 of the P‐value) and were changed by at least twofold (log2); n = 15 mice in WTD group and n = 16 in WTD + ω‐3 group (one sample in WTD group was missing because of insufficient volume of plasma).
Table 1.
Profile of lipid mediators in plasma samples in WTD versus WTD + ω‐3 fed mice
| Compound name | FC | log2(FC) | −log10(P) | Concentration (ng·mL−1) |
|---|---|---|---|---|
| 14,15‐EEQ | 59.02 | 5.88 | 8.18 | 0.056 ± 0.012 versus 3.284 ± 0.521 |
| 11‐HEPE | 60.78 | 5.93 | 8.18 | 0.078 ± 0.014 versus 4.752 ± 1.023 |
| 18‐HEPE | 51.32 | 5.68 | 8.18 | 0.200 ± 0.048 versus 10.262 ± 2.701 |
| 11,12‐EEQ | 33.69 | 5.07 | 8.18 | 0.106 ± 0.016 versus 3.570 ± 0.632 |
| 8,9‐EEQ | 26.78 | 4.74 | 8.18 | 0.134 ± 0.032 versus 3.602 ± 0.613 |
| 11,12‐DiHETE | 22.15 | 4.47 | 8.18 | 5.559 ± 1.058 versus 123.1 ± 17.57 |
| 8‐HEPE | 21.09 | 4.40 | 8.18 | 0.251 ± 0.046 versus 4.945 ± 0.992 |
| 17,18‐EEQ | 43.92 | 5.46 | 6.70 | 0.221 ± 0.050 versus 9.722 ± 1.873 |
| 12‐HEPE | 43.92 | 5.46 | 6.35 | 10.851 ± 2.135 versus 605.7 ± 154.4 |
| 14,15‐DiHETE | 15.48 | 3.95 | 5.89 | 0.725 ± 0.123 versus 11.21 ± 1.243 |
LDLR−/−‐fat‐1tg mice had reduced atherosclerotic plaque formation and increased plaque stability
To exclude the lipid‐lowering effect of ω‐3 in this model, we examined the development of atherosclerosis in LDLR−/−‐fat‐1tg mice fed a WTD. Fat‐1tg mice with C. elegans ω‐3 desaturase can produce ω‐3 endogenously from ω‐6. We used a short‐term atherosclerotic study, feeding LDLR−/−‐fat‐1tg and LDLR−/− mice a WTD for 6 weeks. Consistent with ω‐3 supplementation, fat‐1 expression reduced the lesion area of the atherosclerotic plaque and enhanced plaque stability in mice (Figure 3A–G) without altering serum lipid levels (Figure 3H).
Figure 3.
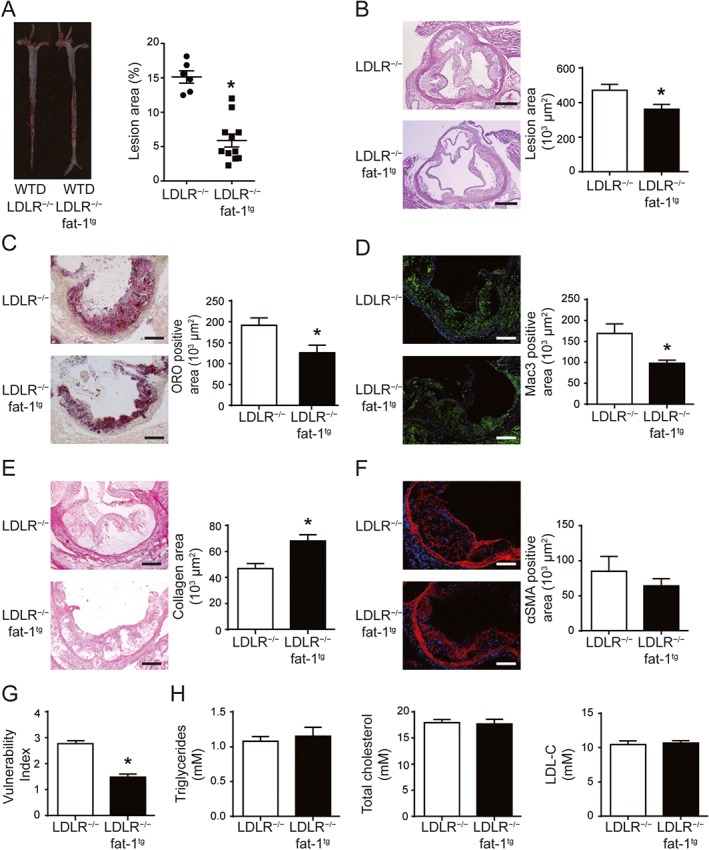
The fat‐1 transgene reduced atherosclerotic plaque formation and increased plaque stability but did not alter plasma levels of triglycerel and cholesterol in LDLR−/−‐fat‐1tg mice. LDLR−/− and LDLR−/−‐fat‐1tg mice were fed a WTD for 6 weeks. (A) Representative aortas from each group with Oil‐red O staining (left) and quantification (right) of aortic lesion area by en face staining; each dot represents a single mouse; n = 6 mice in LDLR−/− group and n = 10 in LDLR−/−‐fat‐1tg group. Horizontal bars represent mean values; whiskers are SEM. (B) Representative photomicrographs of aortic root sections stained with H&E to calculate lesion area. (C–F) Representative photomicrographs (left) and quantification (right) of aortic root sections stained with Oil‐red O (C), picrosirius red (D), MAC‐3 (E) and αSMA (F) in atherosclerotic plaque. Positive area in lesions was quantified. (G) Vulnerability index of plaques. (H) Quantification of plasma levels of triglycerides and total cholesterol in plasma. (B–H) n = 10 in each group. Data are mean ± SEM, *P < 0.05 compared to control.
The fat‐1 transgene altered the profile of PUFA metabolites endogenously in LDLR−/− mice
To assay the profile of PUFA metabolites, we used LC–MS/MS to assay the plasma from LDLR−/−‐fat‐1tg and LDLR−/− mice fed a WTD. Heat map analysis showed that as compared with LDLR−/− mice, LDLR−/−‐fat‐1tg mice showed a modestly increased ω‐3 content and decreased ω‐6 content (Figure 4A and Supporting Information Table S3). The PLS‐DA score plot differentiated LDLR−/−‐fat‐1tg and LDLR−/− mice (Figure 4B), and VIP scores were calculated to reveal the top 10 most significant metabolites (Figure 4C, Table 2) shown in a volcano plot (Figure 4D). The content was greater for metabolites derived from EPA by lipoxygenase (11‐HEPE) and CYP (18‐HEPE, 17,18‐EEQ) than other PUFA compounds.
Figure 4.
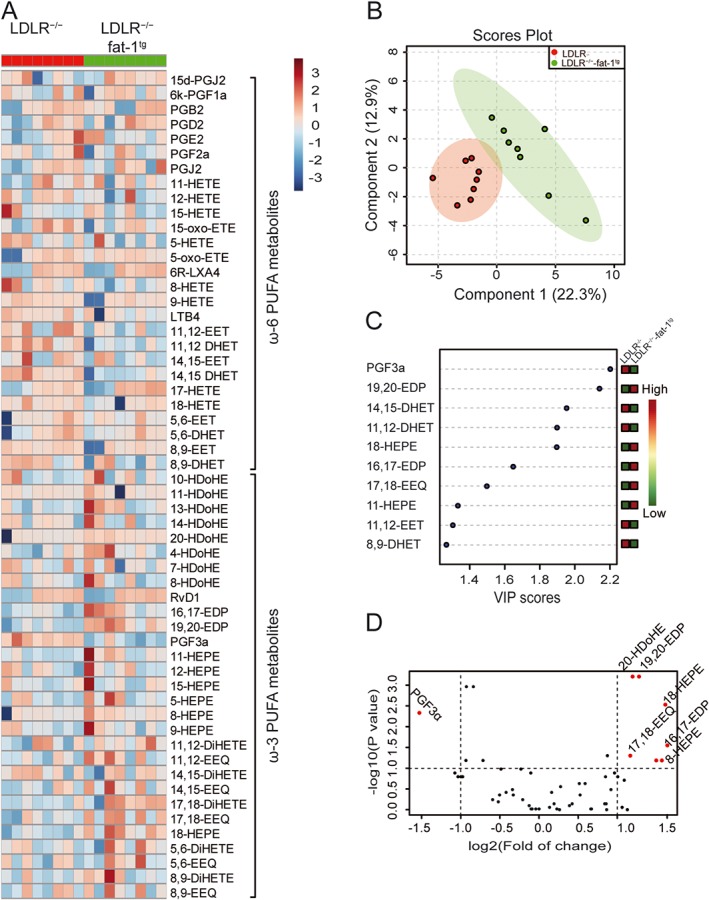
LDLR−/−‐fat‐1tg mice showed endogenously altered profile of PUFA metabolites compared with LDLR−/− mice. Mice were treated as in Figure 3. (A) Heat map showing eicosanoid profile of ω‐6 and ω‐3 PUFAs in plasma samples from LDLR−/− and LDLR−/−‐fat‐1tg mice (n = 8 in each group). PLS‐DA scores plot (B) and features (variables) of top 10 metabolites based on VIP scores (C) from PLS‐DA. The X‐axis shows the correlation scores and the Y‐axis the metabolites. Colour bars show median intensity of variables. (D) Volcano plot of PUFA profiling. Red points indicate metabolites with content that both reached statistical difference by non‐parametric test (log10 of the P‐value) and were changed by at least twofold (log2).
Table 2.
Profile of lipid mediators in plasma samples in LDLR−/− versus LDLR−/−fat‐1tg mice fed a WTD for 6 weeks
| Compound name | FC | log2(FC) | −log10(P) | Concentration (ng·mL−1) |
|---|---|---|---|---|
| 19,20‐EDP | 3.60 | 1.85 | 3.21 | 1.779 ± 0.461 versus 6.403 ± 1.892 |
| 20‐HDoHE | 3.60 | 1.85 | 3.21 | 1.407 ± 0.397 versus 5.072 ± 1.814 |
| 18‐HEPE | 3.78 | 1.92 | 2.52 | 0.179 ± 0.052 versus 0.674 ± 0.194 |
| PGF3a | 0.48 | −1.05 | 2.33 | 0.670 ± 0.133 versus 0.324 ± 0.078 |
| 16,17‐EDP | 5.83 | 2.54 | 1.55 | 0.530 ± 0.191 versus 3.091 ± 1.659 |
| 17,18‐EEQ | 3.41 | 1.77 | 1.30 | 0.049 ± 0.043 versus 0.530 ± 0.179 |
| 8‐HEPE | 6.79 | 2.76 | 1.19 | 0.067 ± 0.060 versus 1.463 ± 1.055 |
| 11‐HEPE | 6.85 | 2.78 | 1.19 | 0.075 ± 0.018 versus 0.512 ± 0.380 |
ω‐3 PUFA decreased the expression of inflammatory genes in the mouse aorta
To analyse the inflammatory response in the mouse aorta, we assayed the gene expression of pro‐inflammatory factors including macrophage markers (CD68, F4/80); endothelial activation markers intercellular adhesion molecule 1 (ICAM‐1), VCAM‐1, E‐selectin and P‐selectin; interleukins; and CC chemokines. The expression of CD68, F4/80, ICAM‐1, VCAM‐1, IL‐1β and TNFα was reduced in aortas of mice fed the ω‐3‐supplemented WTD (Figure 5A–H), which suggests that the ability of ω‐3 to attenuate macrophage infiltration and endothelial activation might mediate its anti‐atherosclerotic effect.
Figure 5.
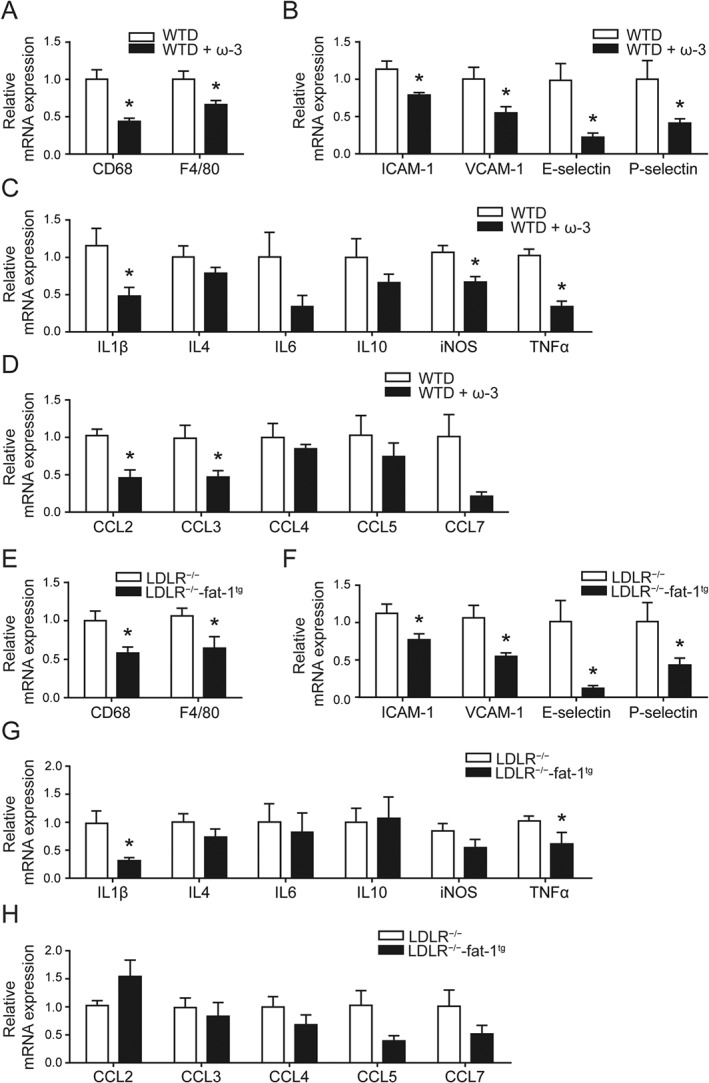
Increased the levels of ω‐3 metabolites in plasma reduced the expression of pro‐inflammatory genes in the mouse aorta. (A–D) Mice were treated as in Figure 1; (E–H) Mice were treated as in Figure 3. Quantification of mRNA expression of macrophage markers (A, E), endothelial activation markers (B, F), interleukins (ILs) and CC chemokines (C, D, G, H) in the aorta in mice; n = 6 in each group. Data are mean ± SEM, *P < 0.05 compared to control.
Macrophages isolated from fat‐1tg mice did not show anti‐inflammatory properties
To determine the mechanism responsible for the anti‐atherosclerotic effect of ω‐3 supplementation and fat‐1 overexpression, macrophages from LDLR−/− or LDLR−/−‐fat‐1tg mice were isolated and treated with LPS. The macrophage expression of inflammatory factors such as inducible NOS (iNOS), TNFα, IL‐1β, IL‐6 and chemokine C‐C motif ligand 2 (CCL2) in different genotypes was increased by LPS compared to the PBS control but was comparable in macrophages from LDLR−/− and LDLR−/−‐fat‐1tg mice (Supporting Information Figure S1). Therefore, altered PUFA metabolism induced by an overexpression of fat‐1 in macrophages may not contribute to the anti‐inflammatory response in vessels.
18‐HEPE and 17,18‐EEQ inhibited TNFα‐induced endothelial activation and monocyte adhesion
EC activation along with monocyte adhesion is considered the initial step of atherosclerosis. To further explore the protective effect of ω‐3 on atherogenesis, we determined whether ω‐3 could decrease EC activation in response to inflammatory stimuli. We pretreated HUVECs with EPA or ARA for 24 h before exposure to TNFα. EPA but not ARA inhibited the TNFα‐induced expression of VCAM‐1 in HUVECs (Figure 6A). The anti‐inflammatory effect of EPA was reversed on pretreatment with 1‐ABT, a general CYP inhibitor, but not indomethacin or NDGA, inhibitors of the two other pathways, COX or LOX respectively (Figure 6B). Hence, the effect of EPA might depend on metabolites from the CYP pathway, whose content was markedly increased in plasma of mice fed ω‐3.
Figure 6.
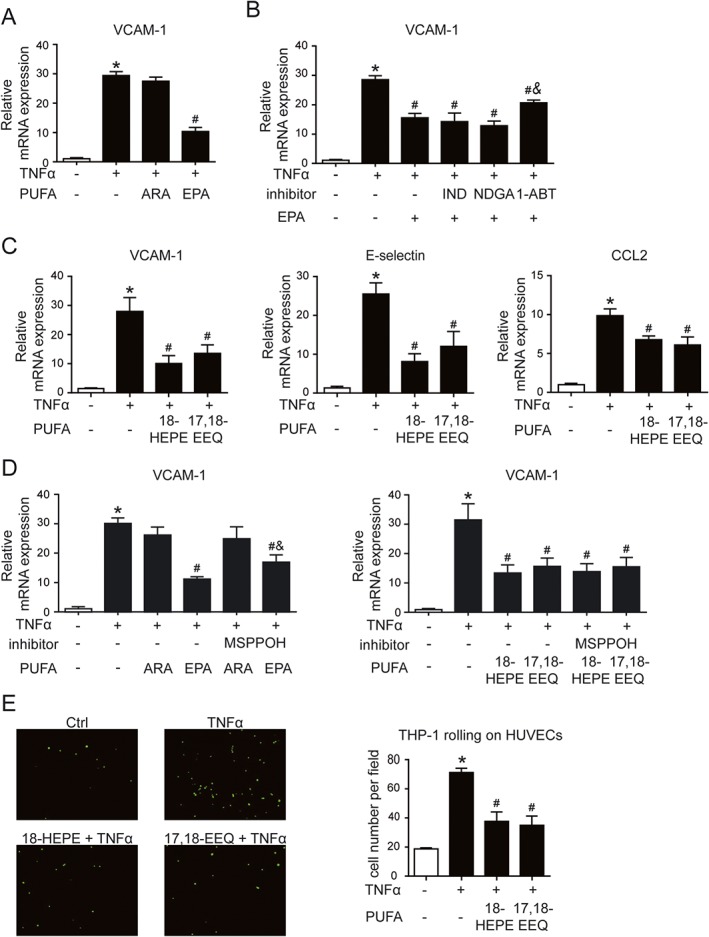
18‐HEPE and 17,18‐EEQ derived from EPA reduced endothelial activation and monocyte adhesion. (A) HUVECs were pretreated with ARA (100 μM) or EPA (100 μM) for 24 h, then exposed to TNFα (0.1 ng·mL−1) for an additional 6 h. (B) HUVECs were pretreated with EPA (100 μM) with or without 1‐ABT (5 μM), indomethacin (IND; 10 μM) or NDGA (10 μM) for 24 h, then with TNFα (0.1 ng·mL−1) for 6 h. (C, D) HUVECs were pretreated with ARA (100 μM), EPA (100 μM), 18‐HEPE (1 μM) or 17,18‐EEQ (1 μM) with or without MSPPOH (5 μM) for 24 h, then with TNFα (0.1 ng·mL−1) for 6 h. Cells were then analysed by qPCR for mRNA levels of VCAM1, E‐selectin or CCL2. (E) HUVECs in 6‐well plates were pretreated with 18‐HEPE (1 μM) or 17,18‐EEQ (1 μM) for 24 h, then with TNFα (0.1 ng·mL−1) for 6 h. BCECF‐AM‐labelled THP1 cells were added to the EC monolayer and incubated for 30 min. Fluorescence microscopy of the attached THP1 cells (green). The ratio of adherent THP1 cells to total ECs was measured. Data are mean ± SEM from five independent experiments. *P < 0.05 versus Ctrl; # P < 0.05 versus TNFα; & P < 0.05 versus TNFα+EPA.
To verify this metabolomic finding in vivo, we further tested the effect of 18‐HEPE and 17,18‐EEQ, the metabolites of EPA by CYP epoxygenase. The TNFα‐induced mRNA levels of pro‐inflammatory factors such as VCAM‐1, E‐selectin and CCL2 were significantly decreased with 18‐HEPE or 17,18‐EEQ treatment in HUVECs (Figure 6C). Moreover, a CYP epoxygenase inhibitor, MSPPOH, reduced the effect of EPA on VCAM‐1 expression but failed to reverse the effect of 18‐HEPE or 17,18‐EEQ (Figure 6D), which suggests the anti‐inflammatory effect of EPA is mediated via the CYP‐mediated metabolites. Correspondingly, the increased monocyte adhesion with TNFα treatment was greatly reduced by pretreatment with both 18‐HEPE and 17,18‐EEQ (Figure 6E). Thus, our data suggest that EPA attenuated EC activation via its metabolites 18‐HEPE and 17,18‐EEQ.
Anti‐inflammatory effect of 18‐HEPE and 17,18‐EEQ in ECs was via an inhibitory action on the NF‐κB pathway
The NF‐κB pathway plays an important role in regulating inflammatory responses in various cells (Tabruyn et al., 2009). For instance, it promotes EC activation and vessel inflammation (Pan et al., 2011). To determine the mechanism of the anti‐inflammatory effect of 18‐HEPE and 17,18‐EEQ, we examined the TNFα‐activated NF‐κB pathway. TNFα‐induced NF‐κB‐dependent luciferase activity was attenuated by 18‐HEPE and 17,18‐EEQ pretreatment in ECs (Figure 7A). The protein level of phosphorylated p65, a subunit of NF‐κB, was increased 3.5‐fold by TNFα in HUVECs as compared with the PBS control. Pretreatment with 18‐HEPE and 17,18‐EEQ significantly reversed the phosphorylation of p65 (Figure 7B). Furthermore, the phosphorylation of IKKα, the upstream kinase of NF‐κB, but not its total protein level, was decreased by pretreatment with 18‐HEPE and 17,18‐EEQ, as was TNFα‐stimulated IκBα degradation (Figure 7C). Thus, our data suggest that 18‐HEPE and 17,18‐EEQ attenuated EC activation via an effect on the NF‐κB signalling cascade.
Figure 7.
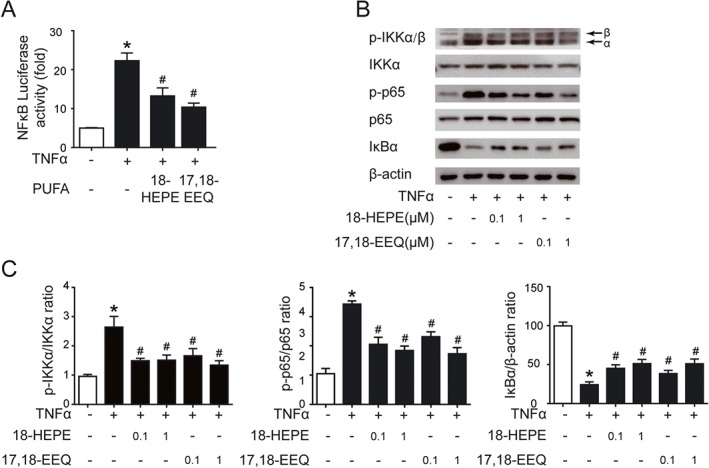
Anti‐inflammatory effect of 18‐HEPE and 17,18‐EEQ in ECs was through inhibition of the NF‐κB pathway. (A) HUVECs cultured to sub‐confluence in 6‐well plates were transfected with pNF‐κB‐TA‐luc, and a β‐galactosidase plasmid for 24 h, treated with TNFα (0.1 ng·mL−1) and 18‐HEPE (1 μM) or 17,18‐EEQ (1 μM) as indicated for another 24 h. Luciferase activity was measured in the cell lysates by a dual luciferase reporter assay system. (B, C) HUVEC were treated with different concentrations (0.1, 1 μM) of 18‐HEPE or 17,18‐EEQ for 1 h, then stimulated with TNFα (0.1 ng·mL−1) for 0.5 h. Western blot analysis of protein expression of IKKα, phospho‐IKKα/β (p‐IKKα/β), p65, phospho‐p65 (p‐p65), IκBα and β‐actin. The mean values of the control group were set to 1. The values of other groups were normalized to control group values, represented as fold or % of control values. Data are mean ± SEM from five independent experiments. *P < 0.05 versus control; # P < 0.05 versus TNFα.
Discussion
Endothelial activation is the initial step in atherogenesis; many metabolites of ARA are pro‐inflammatory and play an important role in this process. However, ω‐3 PUFAs, in contrast, are known to prevent the development of atherosclerosis via their anti‐hypercholesterolaemic and anti‐inflammatory effects, some by regulating endothelial function (Balestrieri et al., 1996; Dessi et al., 2013). Previous studies mainly focused on the protective effect of major components of ω‐3, EPA and DHA, on endothelial function and atherosclerosis, but the effect of the metabolites of ω‐3 still remained largely unknown. In this study, we studied the profiles of ω‐6 and ω‐3 and their metabolites in plasma from atheroprone mice, screening specific metabolites by using unbiased systematic LC–MS/MS. Using pharmacological and genetic models, we identified 18‐HEPE and 17,18‐EEQ as the effective metabolites of ω‐3 that mediate the anti‐atherosclerotic effect by inhibiting NF‐κB activation in ECs.
Fish oil and/or ω‐3 are well‐known efficient lipid‐lowering agents in both humans and in animal models. In this study, we generated the atherosclerosis model in LDLR−/− mice by a 6‐week atheroprone diet involving ω‐3 supplementation of a WTD. In line with previous studies (Chang et al., 2009), ω‐3 treatment markedly lowered plasma LDL cholesterol level. Furthermore, monocyte infiltration (marked by CD68 and F4/80) was decreased, with the formation of fewer plaque lesions and reduced stabilization. ApoE−/−‐fat‐1tg mice showed no significant differences in plasma total or LDL cholesterol levels (Wan et al., 2010). To exclude the cholesterol‐lowering effect on atherosclerotic lesion formation, we used fat‐1tg mice to further study the role of specific metabolites in local vessels. WTD‐fed LDLR−/−‐fat‐1tg mice showed reduced lesion size and less monocyte infiltration but nearly equal cholesterol concentration. Consistent with a previous report of apoE−/−‐fat‐1tg mice (Wan et al., 2010), our findings suggest that ω‐3 directly protects against the development of atherosclerosis and this effect is, at least in part, independent of plasma lipid levels.
The mechanism through which ω‐3 supplementation but not fat‐1 transgenic modifications reduce plasma cholesterol level has not yet been clarified. Possibly, the effect in transgenic mice is compensated for by other changes during atherosclerosis development. Furthermore, ω‐3 supplementation produced a more robust and tight anti‐atherosclerotic phenotype, with higher fold changes in the levels of 18‐HEPE (51.32 vs. 3.78) and 17,18‐EEQ (43.92 vs. 3.41) in our study. Moderately high concentrations of 18‐HEPE and 17,18‐EEQ may induce a more anti‐inflammatory than lipid‐lowering effect.
ω‐3 PUFAs are reported to be direct ligands for the retinoid X receptor, interact with the FFA4 receptor (GPR120) or change the ω‐6/ω‐3 ratio in transducing signalling to regulate inflammatory cascades (Lengqvist et al., 2004; Oh et al., 2010; Wan et al., 2010). In addition, the production of potent anti‐inflammatory metabolites was considered an important factor to achieve the lowest levels of inflammation with ω‐3 treatment. However, because of limited methods to detect the metabolite profile, the function of ω‐3 metabolites was not clear. In our study, we used our latest established LC–MS/MS method and identified 56 metabolites with significantly changed expression in mouse plasma. Among the 56 metabolites in the COX, LOX and CYP pathways of ω‐6 and ω‐3, the levels of two specific EPA metabolites, 18‐HEPE and 17,18‐EEQ, belonging to the CYP pathway, were significantly enhanced with both ω‐3 treatment and fat‐1 overexpression in vivo, which suggests that they might participate in the anti‐atherosclerotic effect of ω‐3. 18‐HEPE and 17,18‐EEQ can be detected and quantified in a stable state in mouse and human plasma (Wishart et al., 2009). In the circulation, 18‐HEPE is converted to epoxide intermediates via transcellular biosynthesis by sequential actions of a leukocyte 5‐lipoxygenase‐like reaction, which leads to the formation of RvE1. 17,18‐EEQ is hydrolyzed to 17,18‐DiHETE by soluble epoxide hydrolase. However, the real half‐life of the activity of both metabolites has not been reported. In addition, the mRNA levels of monocyte/macrophage markers were significantly decreased in mice with both ω‐3 treatment and fat‐1 overexpression as compared with controls, which implies that monocyte infiltration, a critical trigger for atherosclerosis, was reduced with ω‐3 supplementation. EC activation is the main cause of monocyte adhesion and infiltration. We also found that EPA attenuated the TNFα‐induced expression of inflammatory factors, which was blocked by a CYP epoxygenase inhibitor. Finally, the effective metabolites 18‐HEPE and 17,18‐EEQ reversed EC activation and subsequent monocyte adhesion via the NFκB pathway.
EETs, the metabolites of ARA generated by the CYP2C and 2J epoxygenase family, possess cardioprotective functions (Spector et al., 2004). A clinical study showed that carriers of CYP2C9 mutant alleles exhibit diminished metabolic capacity and are at increased risk of developing atherosclerosis (Ercan et al., 2008). The G‐50T polymorphism in CYP2J2 may be an important risk factor for the development of coronary heart disease events in African‐Americans, whereas cigarette smoking may modify the relationship between the I264M and K399R polymorphisms in CYP2C8 and the coronary heart disease risk in Caucasians (Lee et al., 2007). The overexpression of CYP2J2 in apoE−/− mice protected against WTD‐induced atherosclerosis by increasing the production of EETs, but any change in EEQ levels was not mentioned (Liu et al., 2016). In our previous studies, we demonstrated that the development of atherosclerosis can be attenuated by reducing the anti‐inflammatory effect of EET by soluble epoxide hydrolase (sEH) inhibition or sEH deficiency in bone marrow‐derived cells such as monocytes and macrophages, which suggests that the CYP metabolic pathway is important for maintaining vessel homeostasis (Li et al., 2016). ω‐3 shares the same enzymes as ARA to produce EEQ. sEH inhibition was found to increase the level of 17,18‐EEQ, which modulates inflammation and autophagy in obese adipose tissue and liver (Lopez‐Vicario et al., 2015). However, in our study, ω‐3 supplementation failed to suppress the inflammatory response to LPS in macrophages. EETs may be more efficient than EEQs in reducing the inflammatory response in macrophages, with ECs showing greater sensitivity to EEQs. These findings have revealed a new mechanism of the anti‐atherosclerotic effect of ω‐3 treatment. Of note, this study was based on a vertebrate animal study; these results need to be validated in atherosclerotic samples from humans given ω‐3 supplements.
In conclusion, we used LC–MS/MS to screen the metabolite profile of ω‐3 PUFA in atheroprone diet‐treated mouse models with ω‐3 supplementation or fat‐1 expression and identified 18‐HEPE and 17,18‐EEQ as the main increased metabolites of ω‐3. In addition, 18‐HEPE and 17,18‐EEQ attenuated EC activation via the NF‐κB signalling pathway. Our study suggests potential molecular targets for treating atherosclerosis and that ω‐3 combined with sEH inhibition might have an enhanced anti‐atherosclerotic effect.
Author contributions
Y.L., D.L., X.Z. and Y.Z. conceived and designed the experiments; Y.L., X.F., D.L., X.Z., J.H., J.H., L.P. and C.Y. performed the experiments, data collection and analysis; Y.W. and F.X. were responsible for supplying the cells and materials; Y.L., D.L., D.A. and Y.Z. drafted, edited and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 LPS induced the proinflammatory gene expression in macrophages from in LDLR−/− and LDLR−/−fat‐1tg mice. Macrophages were isolated from LDLR−/− and LDLR−/−‐fat‐1tg mice. Proinflammatory gene expression were measured of the macrophages after lipopolysaccharide stimulation (1 ng ml‐1) for 4 h. n = 5 for each group. Data are mean ± SEM.
Table S1 List of oligonucleotide primer pairs used in qPCR.
Table S2 Comparison of PUFA metabolites in plasma from LDLR−/− with different diets.
Table S3 Comparison of PUFA metabolites in plasma from LDLR−/− and LDLR−/−‐fat‐1tg mice with WTD.
Acknowledgements
This work was supported in part by grants from the Ministry of Science and Technology of China [2016YFC0903000] to Y.Z. and the National Natural Science Foundation of China [81420108003 to Y.Z.; 81322006; 81370396 to D.A.; 91539108 to X.Z.].
Liu, Y. , Fang, X. , Zhang, X. , Huang, J. , He, J. , Peng, L. , Ye, C. , Wang, Y. , Xue, F. , Ai, D. , Li, D. , and Zhu, Y. (2018) Metabolic profiling of murine plasma reveals eicosapentaenoic acid metabolites protecting against endothelial activation and atherosclerosis. British Journal of Pharmacology, 175: 1190–1204. doi: 10.1111/bph.13971.
Contributor Information
Dan Li, Email: yidan_cpu@126.com.
Yi Zhu, Email: zhuyi@tmu.edu.cn.
References
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrieri GP, Maffi V, Sleiman I, Spandrio S, Di Stefano O, Salvi A et al (1996). Fish oil supplementation in patients with heterozygous familial hypercholesterolemia. Recenti Prog Med 87: 102–105. [PubMed] [Google Scholar]
- Chang CL, Seo T, Matsuzaki M, Worgall TS, Deckelbaum RJ (2009). n‐3 fatty acids reduce arterial LDL‐cholesterol delivery and arterial lipoprotein lipase levels and lipase distribution. Arterioscler Thromb Vasc Biol 29: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collie‐Duguid ES, Wahle KW (1996). Inhibitory effect of fish oil N‐3 polyunsaturated fatty acids on the expression of endothelial cell adhesion molecules. Biochem Biophys Res Commun 220: 969–974. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Caterina R, Cybulsky MI, Clinton SK, Gimbrone MA Jr, Libby P (1994). The omega‐3 fatty acid docosahexaenoate reduces cytokine‐induced expression of proatherogenic and proinflammatory proteins in human endothelial cells. Arterioscler Thromb 14: 1829–1836. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Libby P (1996). Control of endothelial leukocyte adhesion molecules by fatty acids. Lipids 31 (Suppl): S57–S63. [DOI] [PubMed] [Google Scholar]
- Dessi M, Noce A, Bertucci P, Manca di Villahermosa S, Zenobi R, Castagnola V et al (2013). Atherosclerosis, dyslipidemia, and inflammation: the significant role of polyunsaturated Fatty acids. ISRN Inflamm 2013: 191823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R et al (2004). Arachidonate 5‐lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med 350: 29–37. [DOI] [PubMed] [Google Scholar]
- Ercan B, Ayaz L, Cicek D, Tamer L (2008). Role of CYP2C9 and CYP2C19 polymorphisms in patients with atherosclerosis. Cell Biochem Funct 26: 309–313. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Falck JR, Gross ER, Isbell M, Moore J, Nithipatikom K (2005). Cytochrome P450 and arachidonic acid metabolites: role in myocardial ischemia/reperfusion injury revisited. Cardiovasc Res 68: 18–25. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Southon S, Pinder AC (1996). (n‐3) Polyunsaturated fatty acids modulate the expression of functionally associated molecules on human monocytes in vitro. J Nutr 126: 603–610. [DOI] [PubMed] [Google Scholar]
- Kang JX, Wang J, Wu L, Kang ZB (2004). Transgenic mice: fat‐1 mice convert n‐6 to n‐3 fatty acids. Nature 427: 504. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie CJ, Milani RV, Mehra MR, Ventura HO (2009). Omega‐3 polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardiol 54: 585–594. [DOI] [PubMed] [Google Scholar]
- Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC (2007). CYP2J2 and CYP2C8 polymorphisms and coronary heart disease risk: the Atherosclerosis Risk in Communities (ARIC) study. Pharmacogenet Genomics 17: 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengqvist J, Mata De Urquiza A, Bergman AC, Willson TM, Sjovall J, Perlmann T et al (2004). Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor alpha ligand‐binding domain. Mol Cell Proteomics 3: 692–703. [DOI] [PubMed] [Google Scholar]
- Li D, Liu Y, Zhang X, Lv H, Pang W, Sun X et al (2016). Inhibition of soluble epoxide hydrolase alleviated atherosclerosis by reducing monocyte infiltration in Ldlr(−/−) mice. J Mol Cell Cardiol 98: 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D et al (2014). Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One 9: e94092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P (2012). Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wang T, He X, Liu X, Wang B, Liu Y et al (2016). CYP2J2 overexpression increases EETs and protects against HFD‐induced atherosclerosis in ApoE−/− mice. J Cardiovasc Pharmacol 67: 491–502. [DOI] [PubMed] [Google Scholar]
- Lopez‐Vicario C, Alcaraz‐Quiles J, Garcia‐Alonso V, Rius B, Hwang SH, Titos E et al (2015). Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega‐3 epoxides. Proc Natl Acad Sci U S A 112: 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles EA, Wallace FA, Calder PC (2000). Dietary fish oil reduces intercellular adhesion molecule 1 and scavenger receptor expression on murine macrophages. Atherosclerosis 152: 43–50. [DOI] [PubMed] [Google Scholar]
- Mozurkewich EL, Greenwood M, Clinton C, Berman D, Romero V, Djuric Z et al (2016). Pathway markers for pro‐resolving lipid mediators in maternal and umbilical cord blood: a secondary analysis of the mothers, omega‐3, and mental health study. Front Pharmacol 7: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W et al (2010). GPR120 is an omega‐3 fatty acid receptor mediating potent anti‐inflammatory and insulin‐sensitizing effects. Cell 142: 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan LL, Liu XH, Gong QH, Wu D, Zhu YZ (2011). Hydrogen sulfide attenuated tumor necrosis factor‐alpha‐induced inflammatory signaling and dysfunction in vascular endothelial cells. PLoS One 6: e19766. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ross R (1999). Mechanisms of disease – atherosclerosis – an inflammatory disease. N Engl J Med 340: 115–126. [DOI] [PubMed] [Google Scholar]
- Sacerdoti D, Pesce P, Di Pascoli M, Brocco S, Cecchetto L, Bolognesi M (2015). Arachidonic acid metabolites and endothelial dysfunction of portal hypertension. Prostaglandins Other Lipid Mediat 120: 80–90. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL (2004). Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43: 55–90. [DOI] [PubMed] [Google Scholar]
- Spychalla JP, Kinney AJ, Browse J (1997). Identification of an animal omega‐3 fatty acid desaturase by heterologous expression in Arabidopsis. Proc Natl Acad Sci U S A 94: 1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabruyn SP, Memet S, Ave P, Verhaeghe C, Mayo KH, Struman I et al (2009). NF‐kappaB activation in endothelial cells is critical for the activity of angiostatic agents. Mol Cancer Ther 8: 2645–2654. [DOI] [PubMed] [Google Scholar]
- Takase B, Maruyama T, Kurita A, Uehata A, Nishioka T, Mizuno K et al (1996). Arachidonic acid metabolites in acute myocardial infarction. Angiology 47: 649–661. [DOI] [PubMed] [Google Scholar]
- Wan JB, Huang LL, Rong R, Tan R, Wang J, Kang JX (2010). Endogenously decreasing tissue n‐6/n‐3 fatty acid ratio reduces atherosclerotic lesions in apolipoprotein E‐deficient mice by inhibiting systemic and vascular inflammation. Arterioscler Thromb Vasc Biol 30: 2487–2494. [DOI] [PubMed] [Google Scholar]
- Wang HH, Hung TM, Wei J, Chiang AN (2004). Fish oil increases antioxidant enzyme activities in macrophages and reduces atherosclerotic lesions in apoE‐knockout mice. Cardiovasc Res 61: 169–176. [DOI] [PubMed] [Google Scholar]
- Wei D, Li J, Shen M, Jia W, Chen N, Chen T et al (2010). Cellular production of n‐3 PUFAs and reduction of n‐6‐to‐n‐3 ratios in the pancreatic beta‐cells and islets enhance insulin secretion and confer protection against cytokine‐induced cell death. Diabetes 59: 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams H, Johnson JL, Carson KG, Jackson CL (2002). Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 22: 788–792. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B et al (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res 37 (Database issue): D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue SS, He JL, Zhang X, Liu YJ, Xue FX, Wang CJ et al (2015). Metabolomic analysis revealed the role of DNA methylation in the balance of arachidonic acid metabolism and endothelial activation. Biochim Biophys Acta 1851: 1317–1326. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yang N, Ai D, Zhu Y (2015). Systematic metabolomic analysis of eicosanoids after omega‐3 polyunsaturated fatty acid supplementation by a highly specific liquid chromatography‐tandem mass spectrometry‐based method. J Proteome Res 14: 1843–1853. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Lin JH, Liao HL, Friedli O Jr, Verna L, Marten NW et al (1998). LDL induces transcription factor activator protein‐1 in human endothelial cells. Arterioscler Thromb Vasc Biol 18: 473–480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 LPS induced the proinflammatory gene expression in macrophages from in LDLR−/− and LDLR−/−fat‐1tg mice. Macrophages were isolated from LDLR−/− and LDLR−/−‐fat‐1tg mice. Proinflammatory gene expression were measured of the macrophages after lipopolysaccharide stimulation (1 ng ml‐1) for 4 h. n = 5 for each group. Data are mean ± SEM.
Table S1 List of oligonucleotide primer pairs used in qPCR.
Table S2 Comparison of PUFA metabolites in plasma from LDLR−/− with different diets.
Table S3 Comparison of PUFA metabolites in plasma from LDLR−/− and LDLR−/−‐fat‐1tg mice with WTD.