

Abstract
Background and Purpose
Vascular inflammation, including the expression of inflammatory cytokines in endothelial cells, plays a critical role in hyperhomocysteinaemia‐associated vascular diseases. Cathepsin V, specifically expressed in humans, is involved in vascular diseases through its elastolytic and collagenolytic activities. The aim of this study was to determine the effects of cathepsin V on l‐homocysteine‐induced vascular inflammation.
Experimental Approach
A high methionine diet‐induced hyperhomocysteinaemic mouse model was used to assess cathepsin V expression and vascular inflammation. Cultures of HUVECs were challenged with l‐homocysteine and the cathepsin L/V inhibitor SID to assess the pro‐inflammatory effects of cathepsin V. Transfection and antisense techniques were utilized to investigate the effects of cathepsin V on the dual‐specificity protein phosphatases (DUSPs) and MAPK pathways.
Key Results
Cathepsin L (human cathepsin V homologous) was increased in the thoracic aorta endothelial cells of hyperhomocysteinaemic mice; l‐homocysteine promoted cathepsin V expression in HUVECs. SID suppressed the activity of cathepsin V and reversed the up‐regulation of inflammatory cytokines (IL‐6, IL‐8 and TNF‐α), adhesion and chemotaxis of leukocytes and vascular inflammation induced by l‐homocysteine in vivo and in vitro. Increased cathepsin V promoted the degradation of DUSP6 and DUSP7, phosphorylation and subsequent nuclear translocation of ERK1/2, phosphorylation of STAT1 and expression of IL‐6, IL‐8 and TNF‐α.
Conclusions and Implications
This study has identified a novel mechanism, which shows that l‐homocysteine‐induced upregulation of cathepsin V mediates vascular endothelial inflammation under high homocysteine condition partly via ERK1/2/STAT1 pathway. This mechanism could represent a potential therapeutic target in hyperaemia‐associated vascular diseases.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- 5‐AZA
5‐aza‐2′‐deoxycytidine
- DUSP
dual‐specificity protein phosphatase
- ELK1
Ets‐like transcription factor‐1
- Hcy
homocysteine
- SAH
S‐(5′‐adenosyl)‐l‐homocysteine
- SID
N‐[(1,1‐dimethylethoxy)carbonyl]‐l‐tryptophan‐2‐[[[2‐[(2‐ethylphenyl)amino]‐2‐oxoethyl]thio]carbonyl]hydrazide
Introduction
Vascular inflammation is an early and vital event in the development of vascular disease, including hypertension (Poddar et al., 2001; Kamat et al., 2013; Han et al., 2015) and atherosclerosis (Libby, 2012). In the pathological process of vascular inflammation, vascular endothelial cells release inflammatory cytokines, vasoactive substances and proteases, which results in leukocyte infiltration, extracellular matrix degradation, smooth muscle cell proliferation and migration (Li et al., 2012a; Jung et al., 2014; Gulati, 2016). Moreover, inflammatory cytokines can induce the expression of cell adhesion molecules, and the release of vasoactive substances and proteases in vascular endothelial cells (Li et al., 2012a; Donato et al., 2015; Luo et al., 2016). The release of inflammatory cytokines from vascular endothelial cells is the basis of vascular inflammation.
Indeed, homocysteine (Hcy) induces vascular inflammation through the release of inflammatory cytokines from vascular endothelial cells, such as IL‐6, IL‐8 and TNF‐α (Kamat et al., 2013; Han et al., 2015; Li et al., 2016). Homocysteine is an intermediate product formed during the metabolism of methionine and increased levels are seen in cells with a folate deficiency (Chou et al., 2013) or a polymorphism of the methylenetetrahydrofolate reductase, cystathione‐β‐synthase and methionine synthase genes (Nienaber‐Rousseau et al., 2013). Folic acid supplementation, only in part, reduces the incidence of cardiovascular events (Huo et al., 2015). Thus, further studies are needed to elucidate the mechanism of homocysteine‐induced vascular inflammation, so as to identify new intervention targets.
Cathepsins are lysosomal cysteine proteases and belong to the papain family of proteases that comprises 11 members, while endothelial cells mainly express cathepsin K, B, S, L and V (Reichenbach et al., 2013; Platt and Shockey, 2016). The elastolytic and collagenolytic activities of cathepsins are involved in vascular remodelling and inflammation (Du et al., 2013). Cathepsins are released by endothelial cells that sense and respond to TNF‐α (Keegan et al., 2012). The serum levels of cathepsins are associated with inflammatory cytokine levels in patients with cardiovascular diseases (Li et al., 2012b). However, it was recently reported that cathepsin V, which is specifically expressed in humans (Zhou et al., 2015), increases the expression of the inflammatory cytokine TNF‐α in macrophages (Pribis et al., 2015). The actual relationship between inflammatory cytokines and cathepsin V in vascular endothelial cells and potential mechanisms through which they effect each other has not been elucidated.
Homocysteine induces the release of inflammatory cytokines from vascular endothelial cells, and cathepsin V is associated with the expression of inflammatory cytokines. Therefore, we hypothesized that l‐homocysteine‐induced upregulation of cathepsin V promotes the expression of inflammatory cytokines in endothelial cells to induce vascular inflammation under high homocysteine conditions. We accidentally discovered that cathepsin L (human cathepsin V homologous) was increased in the thoracic aorta endothelial cells of hyperhomocysteinaemic mice, and the cathepsin V mRNA level is increased significantly more than other members of the cathepsin family following stimulation of HUVECs with l‐homocysteine (Supporting Information Figure S1). Moreover, our preliminary studies revealed that cathepsin V dramatically promoted the expression of inflammatory cytokines in HUVECs.
The phosphorylation and subsequent nuclear translocation of MAPKs, including ERK1/2, p38 and JNK, are increased on stimulation with homocysteine (Zanin et al., 2015), and dual‐specificity protein phosphotases (DUSPs) can recognize and inactivate MAPKs (Owens and Keyse, 2007). It has also been shown that the Ets‐like transcription factor‐1, ELK1 (Robert et al., 2005), STAT1 (Frau et al., 2013) and STAT3 (Li et al., 2012a) are involved in homocysteine‐induced expression of inflammatory cytokines. We have investigated the effects of cathepsin V on these signalling pathways. Homocysteine has also been shown to increase intracellular S‐adenosylhomocysteine (SAH) levels, which is a major endogenous inhibitor of DNA methyltransferase (DNMT), thereby promoting the transcription of the target gene (Lin et al., 2014). We compared the changes in expression of cathepsin V induced in HUVECs incubated separately with either l‐homocysteine, SAH (endogenous DNMT inhibitor) or 5‐aza‐2′‐deoxycytidine (5‐AZA) (exogenous DNMT inhibitor) to assess the role of the DNMT pathway in l‐homocysteine‐induced expression of cathepsin V. We also evaluated the DNA methylation status of cathepsin V following l‐homocysteine stimulation in HUVECs by bisulfite sequencing PCR (BSP).
Methods
Cell culture
The HUVECs were maintained in DMEM/F12 medium containing 10% v.v‐1 heat‐inactivated FBS. The cells were incubated at 37°C in a humidified atmosphere of 5% CO2. When the cells reached 80% confluence in the culture flask, trypsin–EDTA was used to remove the cells, and the cells were used in experiments or reseeded in a flask. l‐Hcy was prepared at 100 mM in PBS, and SID was prepared at 5 mM in DMSO and was stored at −20°C and reconstituted in the appropriate media before the experiments.
Animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The Animal Ethics Committee of Central South University approved the procedures used for the animal studies. Male C57BL/6 mice (5 weeks old) were supplied by the Hunan Slac Laboratory Animal Company (Changsha, China) and were maintained in our animal facility under controlled temperature (22–24°C) and humidity (50–60%) with a 12 h light/dark cycle and had free access to food (standard rat and mouse food supplied by Beijing KA) and water. The mice were kept for at least 1 week before experimental use and then divided into four groups: control, vehicle (PBS‐DMSO) i.p. injection for 1 month only, once every 2 days; hyperhomocysteinaemia (HHcy), 2% (m.v‐1) methionine/drinking water for 2 months plus vehicle i.p. injection; HHcy + SID, 2% (m.v‐1) methionine/drinking water for 2 months plus SID (once every 2 days, 2.5 mg·kg−1 body weight) i.p. injection for 1 month; and SID, SID infusion only. After the last administration, the mice were anaesthetized and killed with 4% chloral hydrate (400 mg·kg−1 body weight, i.p.), the plasma and thoracic aorta were used for further experiments. This high methionine diet‐induced hyperhomocysteinaemic mice model has been in use for several years (Tan et al., 2006).
Real‐time cellular analysis (RTCA)
RTCA is a non‐invasive, impedance‐based biosensor system that can measure cell viability, migration, growth, spreading and proliferation. Changes in cell morphology and behaviour are continuously monitored in real time using microelectronics located in the wells of the RTCA E‐plates (Stefanowicz‐Hajduk et al., 2016). As cells adhere to the biocompatible gold microelectrodes that line the well bottoms in an E‐plate, they impede the flow of electric current between electrodes. This impedance signal is converted to a specific parameter called cell index. The cell index is an excellent measure of what the cells are actually doing over time: proliferating, spreading, changing shape, dying, responding to specific stimuli and so forth. HUVECs were seeded at a density of 1 × 104 cells per well into an E‐plate 16 (ACEA Biosciences, San Diego, CA, USA) containing 100 μL of medium per well and were monitored on the xCELLigence Real‐Time Cell Analyzer Dual Plate instrument (ACEA Biosciences). When the cells entered the log phase, l‐Hcy was added to the well for at least 48 h and incubated at 37°C in a 5% CO2 atmosphere. The purpose of RTCA in this study was to screen for the appropriate conditions for l‐Hcy intervention, so only three independent experiments were carried out.
Adenovirus infection
Recombinant adenoviruses encoding eGFP (Adv‐Control) and the human cathepsin V (Adv‐Cathepsin V) were purchased from Genechem (Shanghai, China). For cell infection, HUVECs were seeded in six‐well plates or glass bottom cell culture dishes at 37°C overnight. The Adv‐Control (1:50 plaque‐forming unit) or Adv‐Cathepsin V (1:200 plaque‐forming unit) was added to the cells for 48 h. After treatment, cells were harvested for Western blot analysis, or fixed for immunofluorescent staining.
Transfection of siRNA
Control, cathepsin V and STAT1 siRNAs were purchased from Ribobio (Guangzhou, China). These siRNAs were transfected into HUVECs maintained in six‐well plates using Lipo2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). Sixty picomoles of siRNA was diluted in 0.1 mL of DMEM/F12 medium without serum for 5 min, and the Lipo2000 Transfection Reagent (3 μL) was diluted in FBS‐free DMEM/F12 for 5 min. These two reagents were mixed together and incubated at room temperature for 20 min. The cell culture medium was removed from the wells of the six‐well plates, and the mixture of siRNA and Lipo2000 Transfection Reagent was added to each well, and a final volume was added to 1 mL using FBS‐free DMEM/F12 for 5 h. The transfection mixture was removed, and then, 2 mL of DMEM/F12 containing 10% FBS was added to the well. The cells were incubated for 24 h and then were treated with or without 2.0 mM l‐Hcy for another 24 h.
Western blot analysis
After treatment, the cells were washed twice with cold PBS. One hundred microlitres of cell lysis buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X‐100] with 1% 100 mM PMSF and 100 mM sodium orthovanadate was added to each well for 15 min on ice. The lysate was centrifuged at 12 000 g for 15 min, and the protein in the supernatant were collected. The protein concentrations were measured using a bicinchoninic acid assay. Thirty micrograms of each protein lysate was resolved by SDS‐PAGE using 12% gels, and the proteins were then transferred to PVDF membranes (Millipore, Bedford, MA, USA). Nonspecific interactions were blocked with 5% skimmed milk (blocking with 5% BSA for detecting phosphorylation antibody) for 2 h, and the membranes were then probed with the indicated primary antibodies overnight at 4°C. The membranes were incubated with the appropriate secondary antibodies conjugated with peroxidase. The blots were exposed to advansta western bright ECL (Adcansta lnc., Menlo park, CA, USA). Images were collected using ChemiDoc™ MP Imaging System (Bio‐Rad, Hercules, CA, USA). Bands on immunoblots were quantified by densitometry using Image Lab software (version 5.1, Bio‐Rad).
Real‐time PCR analysis
After treatment, the cells were washed twice with cold PBS and were collected by scraping. Total RNA was prepared using TRIzol according to the manufacturer's instructions, and the cDNA was synthesized from 3 μg of total RNA using reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). Real‐time PCR was performed using the SYBR Green Master Mix (Bio‐Rad) and a C1000 Touch thermal cycler (Bio‐Rad). The detector was programmed with the following PCR conditions: 40 cycles for 5 s denaturation at 95°C and 30 s amplification at 60°C. All of the reactions were normalized to the housekeeping gene GAPDH.
Immunohistochemical staining
Thoracic aorta was fixed and sectioned at 5 μm. Following an overnight incubation with an anti‐cathepsin L antibody (1:50, Abcam, Cambridge, UK) or an anti‐CD45 antibody (1:50, Proteintech, Rosemont, IL, USA), the slides were incubated for 1 h with HRP‐conjugated secondary antibodies (1:200, Proteintech). Images were collected using a Nikon eclipse Ci microscope (Nikon, Tokyo, Japan).
Adhesion of monocytes to HUVECs
THP‐1 mononuclear cells were labelled with PKH26 (Sigma, St. Louis, MO, USA) according to manufacturer's protocol. Briefly, THP‐1 mononuclear cells were washed twice with PBS and suspended in diluent ‘C’. Equal volumes of the cell suspension and PKH26 were mixed and incubated at room temperature for 5 min; serum‐containing media were added to the samples to stop the reaction, and it was washed three times with PBS. The PKH26‐labelled THP‐1 cells (5 × 104 cells per well) were incubated with the HUVECs for 30 min in six plates. The adherent monocytes from five randomly selected imaged areas were viewed by fluorescence microscopy (Leica, Germany) and counted using the National Institutes of Health Image J 1.50 software.
Neutrophil chemotaxis assay
Blood was collected from healthy human volunteers for isolation of neutrophils. All experiments were conducted with the approval of The Third Xiangya Hospital of Central South University Institutional Review Board. Blood was collected into EDTA‐sprayed tubes. The plasma was separated by centrifugation at 2000 g for 10 min, and the neutrophils were isolated using Ficoll‐Hypaque Plus (GE Healthcare Biosciences, Uppsala, Sweden) density gradient. Red blood cell lysis buffer (BD, Franklin Lakes, NJ, USA) was used to remove the residual erythrocytes. The final pellet of neutrophils was resuspended in PBS and labelled with PKH26. Transwell chemotaxis assays were carried out to assess neutrophil recruitment to the endothelial cells using 5 μm transwell (Corning, One Riverfront Plaza, NY, USA). Briefly, HUVECs were seeded into 24‐well plates and were grown until confluent. The cells were then pre‐incubated with or without l‐Hcy or SID for 24 h. The PKH26‐labelled neutrophils (2 × 105 cells per well) were then added to the top chambers above the HUVEC monolayer and allowed to migrate. After 2 h, the cells under the chambers were resuspended and then centrifuged at 1000 g for 5 min in the original well. PKH26‐labelled neutrophils that had migrated to the bottom chamber were viewed by fluorescence microscopy and counted using the National Institutes of Health Image J 1.50 software.
Confocal microscopy for cathepsin V, ERK1/2, p38 and JNK localization
HUVECs were plated at a density of 1 × 104 cells in 15 mm glass bottom cell culture dishes (NEST Biotechnology, Wuxi, China) and were grown at 37°C overnight. The cells were treated with l‐Hcy for 24 h or Adv‐Cathepsin V for 48 h. After treatment, the cells were washed twice with PBS and fixed by incubation in 4% paraformaldehyde for 15 min. The cells were then washed with PBS three times and permeabilized by 0.1% Triton‐X 100 for 20 min at room temperature. The glass bottom cell culture dishes were blocked in 5% BSA for 1 h at room temperature. Antibodies targeting the cathepsin V, ERK1/2, p38 and JNK (1:50) were added to the 1% BSA solution, and cells were separately incubated with these antibodies overnight at 4°C. The cells were washed three times with PBST, according to the source of the primary antibody followed by further incubation with a Cy3‐conjugated goat anti‐mouse antibody or a Cy3‐conjugated goat anti‐rabbit antibody (Keygen Biotech, Nanjing, China, 1:200) for 60 min at room temperature and then washed three times with PBST. For nuclear staining, a DAPI solution (Keygen Biotech) was added at a final concentration of 0.1 μg·mL−1 and incubated for 10 min in the dark. The glass bottom cell culture dishes were subsequently washed three times with PBST. The localization of cathepsin V, ERK1/2, p38 and JNK was imaged using the Olympus FV‐1000 spectral detector confocal system (Olympus Optical Co., Ltd, Japan).
Preparation of nuclear and cytoplasmic extracts
Cytoplasmic and nuclear fractions were isolated using a Nuclear and Cytoplasmic Extraction Kit (CWBIO, Beijing, China) according to the procedure described by the manufacturer. Briefly, harvested cells in suspension were subjected to low‐speed centrifugation (500 g for 3 min) and washed twice in cold PBS. Then 1 mL ‘Nc‐Buffer A’ was added to 1 × 107 cells, vortexed for 15 s and the cells placed on ice for 20 min; 55 μL of ‘Nc‐Buffer B’ was added, the samples were vortexed for 5 s and placed on ice for 1 min. The cell samples were then centrifuged for 15 min at 13 000 g in a microcentrifuge at 4°C and the supernatant (cytosol fraction) transferred to a fresh pre‐chilled 1.5 mL tube; 500 μL ‘Nc‐Buffer C’ was added to the pellet, the sample was vortexed for 5 s and incubated on ice for 10 min. The 5 s vortexing and 10 min incubation procedures were repeated four times, before the sample was centrifuged for 15 min at 13 000 g in a microcentrifuge at 4°C and the supernatant (nuclear extract) transferred to a fresh pre‐chilled 1.5 mL tube. The fractions were stored at −80°C before use.
ELISA
The levels of cathepsin V and chemokines in the supernatants or plasma were measured using ELISA kits according to the manufacturer's instructions. The levels of cathepsin V, IL‐6, IL‐8, CXCL15 and TNF‐α were determined based on the absorbance at 450 nm, measured using EnSpire Multimode Plate Readers (PerkinElmer, Fremont, CA, USA).
Assessment of cathepsin V activity
After treatment, samples from a six‐well plate of HUVECs were digested and washed three times with PBS; the final pellet of HUVECs was resuspended in 100 μL of 25 mM MES, 5 mM DTT, pH 5.5. After 3 cycles of freezing (−80°C, 3 min) and thawing (37°C, 3 min), the cell extracts were centrifuged at 12 000 g for 15 min, and the protein in the supernatant was collected. The protein concentration was determined using coomassie brilliant blue. Eight microlitres of the fluorogenic peptide substrate Z‐LR‐AMC (100 μM) was added to 72 μL of the cell extracts on ice for 20 min, and the activity was assessed using EnSpire Multimode Plate Readers (PerkinElmer). The fluorescence value was standardized to the protein content.
Assessment of plasma levels of homocysteine
The plasma levels of Hcy were measured using an Hcy detection kit (enzymatic cycling assay) on cobas c311 automatic biochemical analyser (Basel, Switzerland).
DNA extraction and BSP
BSP is the most widely used technique to analyse the DNA methylation status in mammals (Tusnády et al., 2005). The extraction of DNA was performed by using a DNA extraction kit (Kangweishiji, Beijing, China); the HUVECs were frozen and then thawed in accordance with the manufacturer's instructions. Then, bisulfite conversion of DNA (1 mg) was manipulated with a BisulFlash DNA Modification Kit (EpiGentek, Farmingdale, NY, USA) according to the manufacturer's instructions. After the bisulfite treatment, the DNA was diluted to concentration of 20 ng·mL−1. Primers sequence of cathepsin V was designed by the MethPrimer, which was as follows: forward, 5′‐GAATGTAATAGATTGGGAAG‐3′ and reverse, 5′‐TCAACAAACAAACCTC TA‐3′. Then, 1 μL DNA sample in 20 μL amplification reaction system, including 10 μL Es Taq MasterMix (Kangweishiji), was amplified with PCR. The PCR amplification programme was as follows: initially the samples were heated to 94°C for 2 min, 35 cycles (including 94°C for 30 s, 50°C for 30 s and 72°C for 30 s for elongation), 72°C for 5 min for elongation. PCR products were purified by gel electrophoresis separation (Kangweishiji). Purified PCR was then linked into PMD 19‐T vectors (Takara, Dalian, China). After the plasmid DNA was extracted, the target segment was amplified with PCR using universal primers of 19‐T vectors: M13F (−47): CGCCAGGGTTTTCCCAGTCACGAC and M13R (−48): AGCGGA TAACAATTTCACACAGGA. The PCR products were purified and sequenced using a 3730XL DNA analyzer (Applied Biosystems, Foster city, CA, USA).
Data analysis
The values are presented as the mean ± SD of the results obtained from five experiments. The significance of the differences between mean values was evaluated with SPSS (version 19, IBM Software Inc., New York, NY, USA) by a one‐way ANOVA followed by the Student–Newman–Keul's test. A P‐value of less than 0.05 was considered significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
HUVECs were purchased from ATCC (Rockville, Maryland, USA); THP‐1 cells were provided by The Third Xiangya Hospital Experimental Medicine Centre of Central South University, passage 10–15 cells were used in the experiments. l‐Homocysteine, SAH and 5‐AZA were purchased from Sigma Chemical (St. Louis, MO, USA). SID 26681509 was obtained from Tocris Bioscience (Bristol, UK). Primary antibodies targeting phospho‐ERK1/2, phospho‐STAT1 and ERK1/2 were purchased from Cell Signaling Technology (Danvers, MA, USA), cathepsin V was purchased from R&D System (Minneapolis, MN, USA), cathepsin L was purchased from Abcam; STAT3 and JNK were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and those primary antibodies targeting STAT1, p38, ELK1, DUSP6, DUSP7 and DUSP9 were purchased from Proteintech (Wuhan, China). Cathepsin V ELISA kit was purchased from R&D System; CXCL15 ELISA kit was purchased from Abcam; and IL‐6, IL‐8 and TNF‐α ELISA kits were purchased from CUSABIO (Wuhan, China).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
l‐Homocysteine up‐regulated endothelial cells the expression of cathepsin V in vivo and in vitro
To assess the effects of l‐Hcy on cathepsin V expression in vivo, we used a high methionine diet‐induced hyperhomocysteinaemic mouse model. Plasma Hcy levels were increased more than 15 μM in hyperhomocysteinaemic mice (Figure 1A). In addition, the expression of cathepsin L (human cathepsin V homologous) in the thoracic arteries was increased approximately two‐ to fourfold (Figure 1B, C) and mainly expressed in the vascular endothelial cells (Figure 1D).
Figure 1.
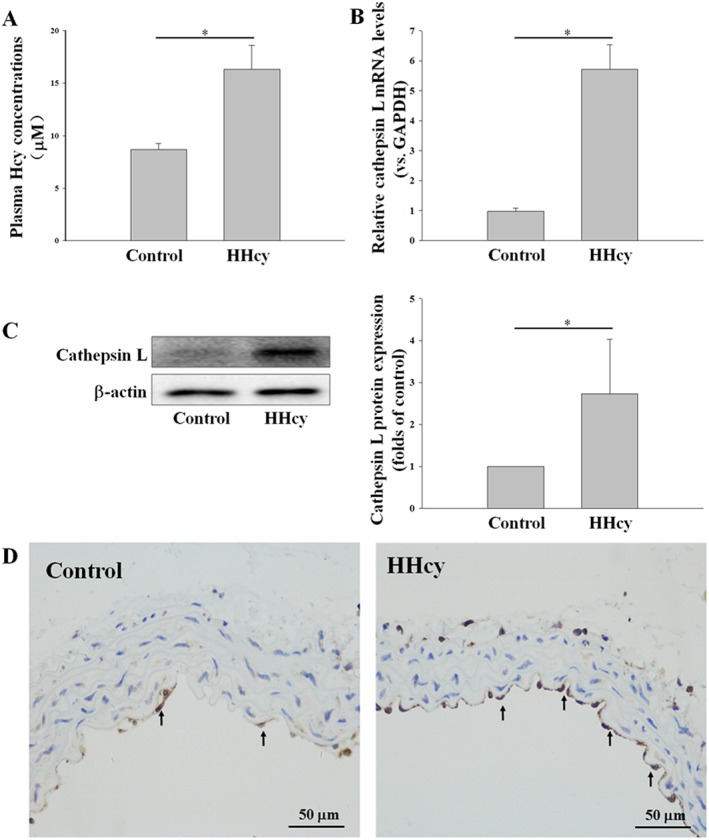
Cathepsin L (human cathepsin V homologous) was increased in the thoracic aorta endothelial cells of hyperhomocysteinaemic mice. The control mice (Control) had free access to water, and the hyperhomocysteinaemic mice (HHcy) were treated with 2% (m·v‐1) methionine/drinking water for 2 months. (A) The plasma Hcy concentrations of control and hyperhomocysteinaemic mice were measured using an automatic biochemical analyser. (B–D) The expression of cathepsin L in thoracic arteries was assessed by real‐time PCR, Western blot and immunohistochemistry. Data represent mean ± SD of five animals per treatment. * P < 0.05.
In order to produce a hyperhomocysteinaemic (HHcy) environment similar to that observed in vivo, we incubated HUVECs with different concentrations of l‐Hcy. l‐Hcy has a temporary inhibitory effect on cell viability/proliferation within 48 h in a concentration‐dependent manner, the status of HUVECs could still be restored after 48 h under l‐Hcy stimulation even when used at a higher concentration (2.0 mM) (Figure 2A). Further investigation found that the mRNA and protein levels of cathepsin V were increased in a dose‐dependent manner under l‐Hcy stimulation (Figure 2C, D), but the supernatant levels were reduced (Figure 2B). It was further demonstrated that cathepsin V was increased in the outside of the nucleus by confocal microscopy (Figure 2E). These results suggested that l‐Hcy significantly increases intracellular cathepsin V expression in vascular endothelial cells.
Figure 2.
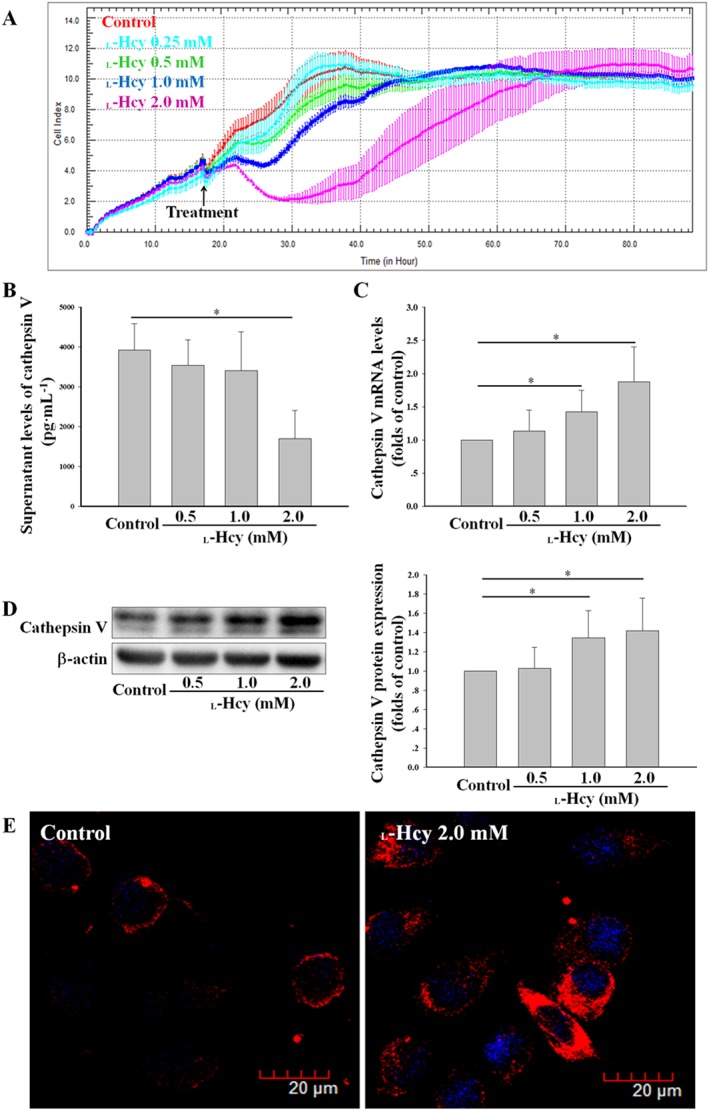
l‐Hcy promoted the expression of cathepsin V in HUVECs. (A) The effects of 0.25–2.0 mM l‐Hcy on HUVECs viability/proliferation were assessed by RTCA. (B) HUVECs were incubated with 0.5–2.0 mM l‐Hcy for 24 h; the supernatant levels of cathepsin V were determined by ELISA. (C–E) The expression of cathepsin V in HUVECs described in (B) was assessed by real‐time PCR, Western blot and confocal microscopy. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
Cathepsin L/V inhibitor SID attenuated vascular inflammation in hyperhomocysteinaemic mice
l‐Hcy increased cathepsin V expression; therefore, the influence of cathepsin V on vascular inflammation was assessed in hyperhomocysteinaemic mice. As a specific inhibitor of cathepsin L/V, SID decreased the number of leukocytes and the expression of IL‐6, CXCL15 (human IL‐8 homologous) and TNF‐α in the thoracic arteries of hyperhomocysteinaemic mice (Figure 3A, B); the plasma levels of these HHcy‐associated inflammatory cytokines were also decreased (Figure 3C). These results indicate that the cathepsin L/V inhibitor SID attenuates vascular inflammation in hyperhomocysteinaemic mice.
Figure 3.
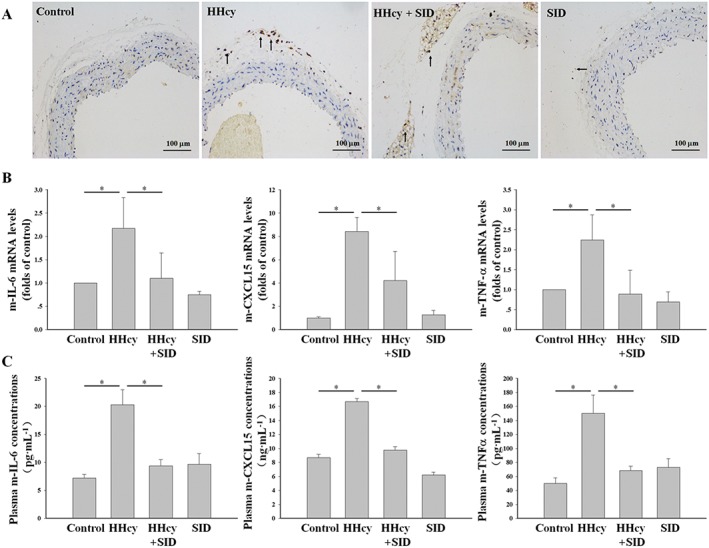
The cathepsin L/V inhibitor SID attenuated vascular inflammation in hyperhomocysteinaemic mice. The control mice and hyperhomocysteinaemic mice were separately treated i.p. with SID (2.5 mg·kg−1 body weight) or vehicle (PBS‐DMSO) for 1 month, administered once every 2 days. (A) Immunohistochemistry staining of the leukocytes (CD45 positive cells) in the thoracic arteries. (B) The mRNA levels of IL‐6, CXCL15 (human IL‐8 homologous) and TNF‐α in the thoracic arteries were assessed by real‐time PCR. (C) The plasma concentrations of IL‐6, CXCL15 and TNF‐α were determined by ELISA. Data represent mean ± SD of five animals per treatment. * P < 0.05.
Cathepsin V mediated the l‐homocysteine‐induced endothelial inflammation in HUVECs
We next wanted to determine the effects of cathepsin V on l‐Hcy‐induced endothelial inflammation. Pretreatment of cells with SID incubation for 24 h had no effect on basal level of cathepsin V activity, but SID distinctly inhibited the cathepsin V activity of HUVECs incubated with 2.0 mM l‐Hcy (Figure 4A). SID also inhibited the monocyte‐endothelial adhesion and neutrophil chemotaxis induced by l‐Hcy (Figure 4B, C). Furthermore, SID reversed the up‐regulation of IL‐6, IL‐8 and TNF‐α induced by l‐Hcy (Figure 4D, E).
Figure 4.
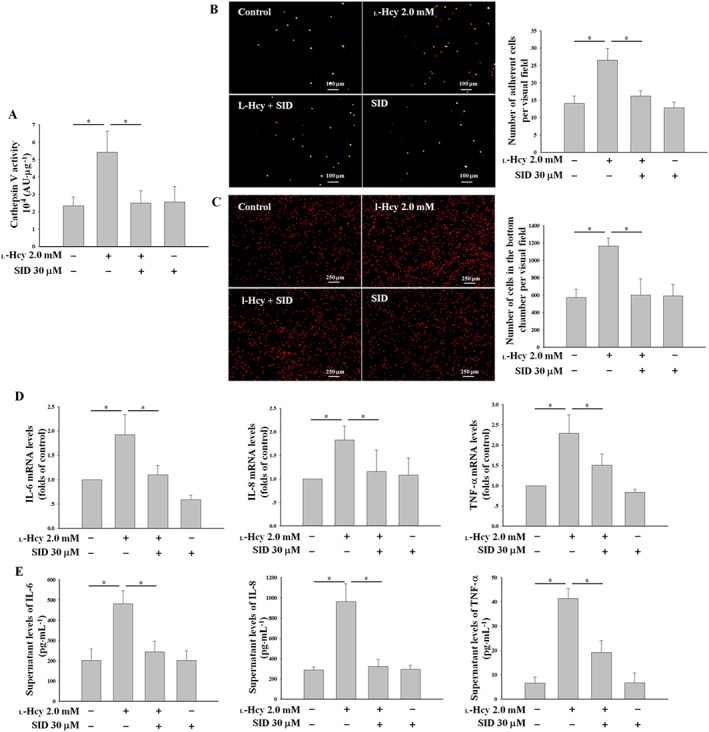
The cathepsin L/V inhibitor SID attenuated l‐Hcy‐induced endothelial inflammation in HUVECs. (A) HUVECs were incubated separately with either 2.0 mM l‐Hcy, 30 μM SID or l‐Hcy + SID for 24 h; the cathepsin V activity of a cell protein extract was analysed. (B) The HUVECs described in (A) were subsequently co‐cultured with THP‐1 cells for 30 min; next, the supernatants and unbound THP‐1 cells were discarded, and adherent THP‐1 cells were counted. (C) The HUVECs were seeded into the bottom chambers of transwell plates overnight and treated with l‐Hcy and SID as described in (A); neutrophils were added to the top chambers and allowed to migrate for 2 h before the number of neutrophils in the bottom wells were counted. (D, E) The mRNA levels and supernatant levels of IL‐6, IL‐8 and TNF‐α were determined separately by real‐time PCR and ELISA. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
In order to further clarify the effects of cathepsin V on endothelial inflammation, the effects of the overexpression of the cathepsin V gene or silencing its gene (mediated by adenovirus infection or siRNA transfection) were investigated. The siRNA‐cathepsin V inhibited the up‐regulation of cathepsin V induced by l‐Hcy (Figure 5A, B) and reversed the up‐regulation of IL‐6, IL‐8 and TNF‐α induced by l‐Hcy (Figure 5C). After infection of Adv‐Cathepsin V, the expression of cathepsin V was up‐regulated (Figure 5D, E), and cathepsin V was located on the outside of the nucleus (Figure 5F). Cathepsin V overexpression also significantly increased the expression of IL‐6, IL‐8 and TNF‐α (Figure 5G). These results indicate that cathepsin V mediates the l‐Hcy‐induced endothelial inflammation in HUVECs.
Figure 5.
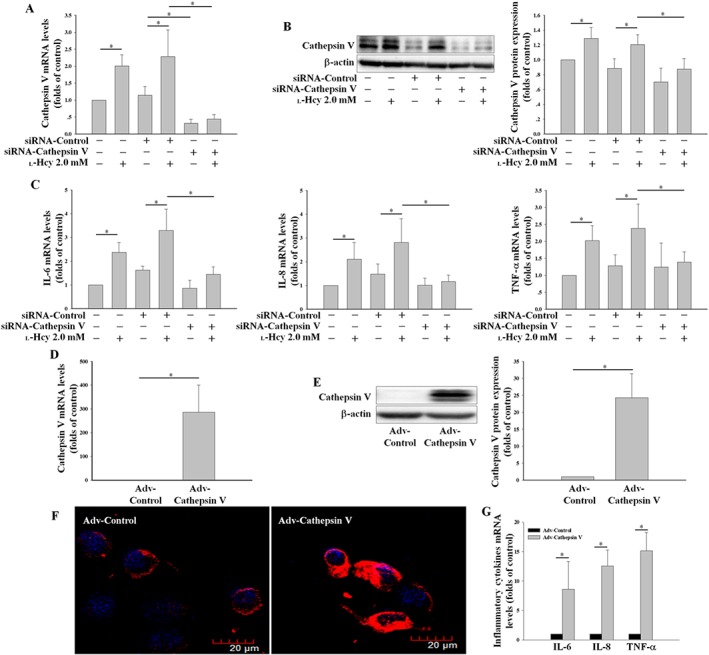
Cathepsin V mediates the l‐Hcy‐induced up‐regulation of inflammatory cytokines. (A, B) HUVECs were transfected with control (siRNA‐Control) or cathepsin V siRNA (siRNA‐Cathepsin V) for 24 h and then co‐incubated with l‐Hcy 2.0 mM for 24 h; the cathepsin V expression was assessed by real‐time PCR and Western blot. (C) The effects of siRNA‐Cathepsin V on the mRNA levels of IL‐6, IL‐8 and TNF‐α were determined by real‐time PCR. (D–F) HUVECs were transfected with eGFP (Adv‐Control) or eGFP‐Cathepsin V (Adv‐Cathepsin V) by adenovirus infection for 48 h; the expression of cathepsin V was assessed by real‐time PCR, Western blot and confocal microscopy. (G) The effects of Adv‐Cathepsin V on the mRNA levels of IL‐6, IL‐8 and TNF‐α were determined by real‐time PCR. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
ERK1/2/STAT1 pathway is involved in cathepsin V‐induced inflammatory cytokine expression
Considering that inflammatory cytokines are regulated by cathepsin V at the transcriptional level whereas cathepsin V is mainly expressed in the outside of nucleus, there must be a nuclear translocation mechanism to translate the cathepsin V signal to the nucleus. We evaluated the mechanisms underlying cathepsin V‐induced inflammatory cytokine expression by investigating the expression of MAPKs. The overexpression of cathepsin V down‐regulated the expression of p38 and JNK, but had no effect on ERK1/2 (Figure 6A). As shown in Figure 6B, ERK1/2 was translocated to the nucleus in the cathepsin V overexpression group. This translocation of ERK1/2 was also determined by nucleocytoplasmic separation analysis; ERK1/2 was increased in the nucleus and decreased in the cytoplasm (Figure 6D). We also investigated the downstream targets of phospho‐ERK1/2, including the transcription factors ELK1, STAT1 and STAT3. When the phosphorylation of ERK1/2 was increased in the cathepsin V overexpression group, the phosphorylation of STAT1 was increased, but the total protein of STAT3 and ELK1 was significantly reduced (Figure 6C). Transfection of siRNA‐STAT1 inhibited the expression of STAT1 (Figure 6E, F) and reversed the up‐regulation of IL‐6 and TNF‐α, but not IL‐8, induced by l‐Hcy (Figure 6G). As shown in Figure 6I, the phosphorylation of ERK1/2 and STAT1 was increased in the thoracic arteries of hyperhomocysteinaemic mice; the cathepsin V inhibitor SID reversed this increased phosphorylation of ERK1/2 and STAT1, which is similar to the results obtained in vitro. These results demonstrate that cathepsin V augments IL‐6 and TNF‐α expression partly via the ERK1/2/STAT1 pathway in vitro and in vivo.
Figure 6.
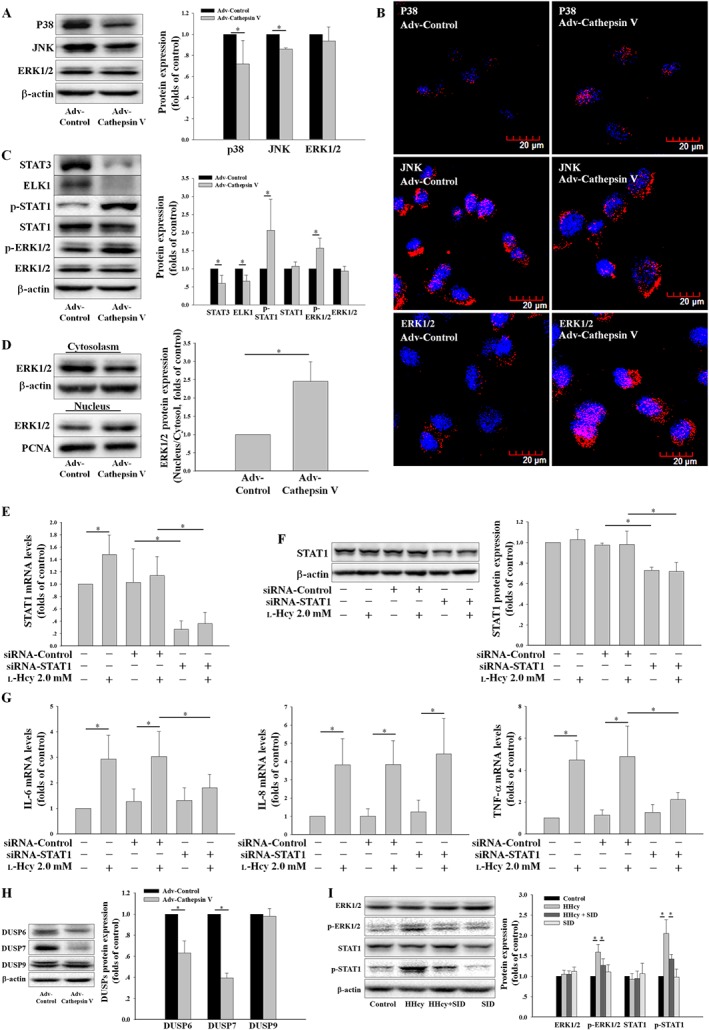
The ERK1/2/STAT1 pathway is involved in cathepsin V‐induced inflammatory cytokine expression. (A, B) The protein expression and localization of ERK1/2, p38 and JNK induced by Adv‐Cathepsin V were assessed by Western blot and confocal microscopy. (C) The protein expression of STAT3, ELK1, STAT1, phosphor‐STAT1 (p‐STAT1), ERK1/2 and phosphor‐ERK1/2 (p‐ERK1/2) were determined by Western blot. (D) The nuclear translocation of ERK1/2 was detected by Western blot. (E, F) HUVECs were transfected with control (siRNA‐Control) or STAT1 siRNA (siRNA‐STAT1) for 24 h and then co‐incubated with 2.0 mM l‐Hcy for 24 h, the STAT1 expression was assessed by real‐time PCR and Western blot. (G) HUVECs described in (E) were used to assay the mRNA levels of IL‐6, IL‐8 and TNF‐α by real‐time PCR. (H) The protein expression of DUSP6, DUSP7 and DUSP9 induced by Adv‐Cathepsin V were assessed by Western blot. (I) Control mice and hyperhomocysteinaemic mice were separately treated with SID or vehicle, the protein expression of ERK1/2, p‐ERK1/2, STAT1 and p‐STAT1 in the thoracic arteries were assessed by Western blot. The data shown are the mean ± SD of five independent experiments or five animals per treatment. * P < 0.05.
DUSPs inactivate ERK1/2 by dephosphorylating both the phosphoserine/threonine and phosphotyrosine residues. Since cathepsin V activity is dependent upon low pH environment in lysosome and its basic function is to hydrolyze protein, cathepsin V mediates ERK1/2 phosphorylation possibly through degradation of certain protein phosphatases. DUSP6, DUSP7 and DUSP9 only exist in the cytosol and inactivate ERK1/2 in vitro and in vivo whereas they display little or no activity towards either p38 or JNK (Owens and Keyse, 2007). The overexpression of cathepsin V down‐regulated the expression of DUSP6 and DUSP7 in the cytosol, but had no effect on DUSP9 (Figure 6H). Our results demonstrate that cathepsin V mediates ERK1/2 phosphorylation partly via the degradation of DUSP6 and DUSP7.
DNA demethylation pathway may be involved in l‐homocysteine‐induced cathepsin V expression
In addition, we preliminarily investigated the potential mechanisms underlying the l‐Hcy‐induced expression of cathepsin V. DNMT is a key enzyme mediating the epigenetic regulation of gene expression, and DNA hypermethylation in promoter regions is often associated with the down‐regulation or silencing of transcription (Xu et al., 2014). Hcy can induce an increase in SAH, which is a major endogenous inhibitor of DNMT (Lin et al., 2014). We compared the changes in cathepsin V expression in HUVECs incubated separately with either l‐Hcy, SAH (endogenous DNMT inhibitor) or 5‐AZA (exogenous DNMT inhibitor); both SAH and 5‐AZA up‐regulated cathepsin V expression, and l‐Hcy had a similar effect (Figure 7A, B). We also evaluated the DNA methylation of cathepsin V following l‐Hcy stimulation in HUVECs by BSP. As shown in our results (Figure 7C), methylation of the promoter region of cathepsin V was decreased following l‐Hcy stimulation. These results indicate that the DNA demethylation pathway may be involved in the expression of cathepsin V induced by l‐Hcy.
Figure 7.
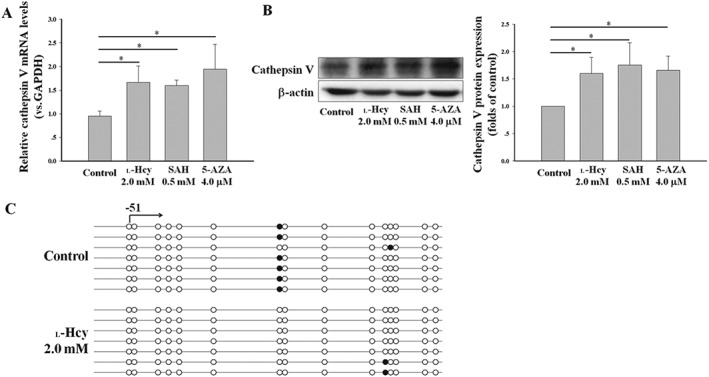
DNA demethylation pathway may be involved in l‐Hcy‐induced cathepsin V expression. (A, B) HUVECs were incubated separately with either 2.0 mM l‐Hcy, 0.5 mM SAH or 4.0 μM 5‐AZA for 24 h, the expression of cathepsin V was analysed by realtime‐PCR and Western blot. (C) Methylation profile of cathepsin V in HUVECs. Each line represents an individual clone allele, and each circle represents a CpG site. Blank circle, non‐methylated; solid circle, methylated. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
Discussion
The major new findings of this study were that l‐Hcy stimulated intracellular cathepsin V expression in endothelial cells probably via the DNA demethylation pathway, and this increased expression of cathepsin V promotes the vascular inflammation partly through the ERK1/2/STAT1 pathway under high homocysteine conditions.
It is well recognized that vascular inflammation is the basis for the development of cardiovascular diseases (Libby, 2012). Furthermore, it has been demonstrated that Hcy‐induced vascular inflammation is an important player in the pathophysiology of hyperhomocysteinaemia‐associated hypertension and atherosclerosis (Familtseva et al., 2016; Platt and Shockey, 2016). However, it was not clear which factors are responsible for vascular inflammation in hyperhomocysteinaemia. It has been reported that cathepsin V plays an important role in vascular inflammation partly because of its potent elastolytic and collagenolytic activities (Du et al., 2013). In the current study, we found that cathepsin L (human cathepsin V homologous) was mainly expressed in vascular endothelial cells rather than smooth muscle cells in the thoracic arteries of hyperhomocysteinaemic mice. Moreover, l‐Hcy also significantly promoted intracellular cathepsin V expression in endothelial cells. These results indicate that intracellular cathepsin V may be involved in l‐Hcy‐induced vascular inflammation in endothelial cells.
Our experimental observations revealed that the supernatant level of cathepsin V was decreased in a dose‐dependent manner following l‐Hcy stimulation (Figure 2B), and the release of inflammatory cytokines was increased (Figure 5C), which indeed suggests that l‐Hcy inhibited cathepsin V secretion and promoted inflammatory cytokine release. Consistent with this notion, it has been reported that cathepsin V concentrations are reduced and inflammatory cytokine level increased in the bronchoalveolar lavage fluid (BALF) of Besnier–Boeck–Schaumann (BBS) patients (Naumnik et al., 2015).
Cathepsins are lysosomal cysteine proteases and belong to the papain family of proteases that is composed of 11 members, while endothelial cells mainly express cathepsins K, B, S, L and V (Platt and Shockey, 2016). It has reported that human cathepsin V, but not human cathepsin L, is the analogue of mouse cathepsin L (Yasuda et al., 2004). Urbich et al. (2005) have demonstrated that cathepsin L is highly expressed in endothelial progenitor cells (EPCs) as opposed to matured endothelial cells and is essential for matrix degradation and invasion by EPC in vitro. It also has been demonstrated that plasma cathepsin L levels are significantly higher in abdominal aortic aneurysm patients than in controls, whereas cathepsins K and V levels are lower (Lv et al., 2013). These findings suggested that cathepsin V may have a different function from other members of the cathepsin family. The rationale for our focus on cathepsin V in the present study was based on our preliminary findings that that cathepsin V mRNA level was increased the most when compared with cathepsins B and S (Figure S1), whereas the detailed roles of cathepsins B and S in endothelial cells clearly warrant investigation in the future.
Cathepsin V is specifically expressed in humans (Zhou et al., 2015), indicating that cathepsin V has certain special functions for humans. Surprisingly, this study found that intracellular cathepsin V played a pivotal role in promoting the expression and release of inflammatory cytokines. As reported, cathepsin K knockout reduces macrophage contents and the expression of inflammatory cytokines in the lesion of injury‐induced neointimal hyperplasia (Hu et al., 2014). Indeed, an elevated expression of inflammatory cytokines (e.g. IL‐6, IL‐8 and TNF‐α) induced by Hcy is described as a typical feature of the inflammatory response of endothelial cells (Poddar et al., 2001; Kamat et al., 2013; Han et al., 2015).
Inhibition of cathepsin S reduces TGFβ1‐induced Smad2 and Smad3 activation in mice subjected to myocardial infarction (Chen et al., 2013). Cathepsin K contributes to Notch1 processing and activates downstream signalling, thereby promoting the ischaemia‐induced neovascularization (Jiang et al., 2014). There is increasing evidence that cathepsins modulate cardiovascular remodelling and regeneration by affecting the activity of several intracellular signalling pathways. Our data indicate that inflammatory cytokines are regulated by cathepsin V at the transcriptional level, but cathepsin V is mainly expressed outside of the nucleus. Therefore, there must surely be a nuclear translocation mechanism to translate the cathepsin V signal to the nucleus. We demonstrated that an increased expression of cathepsin V promotes the phosphorylation and subsequent nuclear translocation of ERK1/2. As reported previously, cathepsin S enhances the VEGF/ERK1/2 signalling pathway (Li et al., 2015). We also found that cathepsin V up‐regulated the expression of IL‐6 and TNF‐α, but not that of IL‐8, via the ERK1/2/STAT1 pathway. The mechanism by which cathepsin V up‐regulates IL‐8 remains to be investigated further.
In physiological conditions, ERK1/2 is in a un‐phosphorylated state in the cytosol, but is translocated to the nucleus upon phosphorylation. ERK1/2 phosphorylation is maintained by a balance between MAP kinase and ERK kinase (MEK1/2) activation and is negatively controlled by compounds such as the DUSPs. DUSPs inactivate ERK1/2 by dephosphorylating both the phosphoserine/threonine and phosphotyrosine residues. Since cathepsin V activity is dependent on a low pH environment in lysosomes and its basic function is to hydrolyze protein, cathepsin V mediates ERK1/2 phosphorylation possibly through degradation of certain protein phosphotases. DUSP6, DUSP7 and DUSP9 only exist in the cytosol and inactivate ERK1/2 in vitro and in vivo whereas they displaying little or no activity towards either p38 or JNK (Owens and Keyse, 2007). Our results demonstrated that cathepsin V mediates ERK1/2 phosphorylation partly via the degradation of DUSP6 and DUSP7, but, not DUSP9. Phospho‐ERK1/2 can phosphorylate and activate its downstream signalling molecules (such as transcription factor STAT1) in the nucleus, and phospho‐STAT1 will promote the transcription of target genes (such as IL‐6 and TNF‐α).
A genome‐wide analysis revealed that the hypomethylation of chromosomal DNA predominates in atherosclerotic plaques (Aavik et al., 2015). DNMT is a key enzyme mediating the epigenetic regulation of gene expression, and DNA hypomethylation in the promoter regions is often associated with an up‐regulation of transcription (Xu et al., 2014). In the methionine cycle, which connects Hcy metabolism and the transmethylation reaction of DNA, SAH is hydrolyzed to form Hcy; therefore, increased levels of Hcy inhibit the decomposition of SAH, resulting in the accumulation of SAH. Mild to moderate increases in d‐ and l‐Hcy (30–1000 μM) induce an increase in SAH, which is a major endogenous inhibitor of DNMT (Lin et al., 2014); increased levels of SAH promote genomic demethylation (Han et al., 2014). Thus, these findings indicate that SAH is involved in l‐Hcy‐induced DNA demethylation. In the present study, we demonstrated that l‐Hcy had a similar effect to SAH (endogenous DNMT inhibitor) or 5‐AZA (exogenous DNMT inhibitor), which promote the transcription of cathepsin V, an observation that is consistent with published results showing that 5‐AZA treatment enhances the mRNA expression and attenuates DNA methylation of cathepsin V (Arai et al., 2008). Furthermore, we also demonstrated, by BSP, that the methylation of promoter regions of cathepsin V was decreased under l‐Hcy stimulation in HUVECs.
Although the concentrations of Hcy were only 15–30 μM in hyperhomocysteinaemic mice, we incubated cells with 2.0 mM l‐Hcy in vitro. This concentration was chosen for the following reasons: (i) it is reported that the release of IL‐8 from HUVECs is increased after a 24 h incubation with 1.0 mM–10.0 mM Hcy (Elordieta and Calleja, 2005); (ii) from our results, it was shown that the status of HUVECs could be restored after 48 h when stimulated with l‐Hcy 2.0 mM indicating that this concentration has no direct cytotoxic effect on the HUVECs; (iii) cathepsin V mRNA levels were increased in a dose‐dependent when the cells were stimulated with 0.5–2.0 mM l‐Hcy; (iv) the d‐type amino acid, being an unnatural amino acid, almost never be metabolized, whereas the half‐life of l‐Hcy is shorter than that of D‐Hcy; (v) in addition, l‐Hcy, but not d‐Hcy, increases IL‐8 expression in human aortic endothelial cells (Poddar et al., 2001); and (vi) SAH is a competitive inhibitor of DNMT, only a high concentration or prolonged action of l‐Hcy can enhance the level of SAH, which then inhibits DNMT.
Among the available inhibitors of cathepsin (such as E64d and Clik148), SID is a potent and specific human cathepsin L/V inhibitor. It has been demonstrated that 30 μM of SID almost abolished cathepsin V activity (Pribis et al., 2015). Consistently, we found that 30 μM SID also significantly attenuated the pro‐inflammatory effect of l‐Hcy. We believe that cathepsin V may become a therapeutic target for the vascular inflammation induced by hyperhomocysteinaemia.
It was recently reported that a first‐generation HIV‐protease inhibitor saquinavir (SQV) significantly depresses the activity of cathepsin V and blocks cathepsin V‐induced TNF‐α production (Pribis et al., 2015). Our next step will, therefore, be to clarify the correlation between cathepsin V, inflammatory cytokines and vascular inflammation in hyperhomocysteinaemic patients and investigate the effects of the antineoplastic drug decitabine (a DNMT inhibitor) and anti‐AIDS drug SQV (a cathepsin V inhibitor) on cathepsin V‐induced vascular inflammation in hyperhomocysteinaemic animal models.
In conclusion, in the present study it was demonstrated that cathepsin V is a potent stimulus for the production of inflammatory cytokines in the presence of high concentrations of homocysteine, and that this effect is partially mediated by the ERK1/2/STAT1 pathway. The cathepsin V inhibitor SID was an effective strategy for reducing inflammatory cytokine levels in this model (Figure 8). These findings may provide a mechanistic basis for therapeutic interventions aimed at reducing l‐Hcy‐induced vascular inflammation in hyperhomocysteinaemia.
Figure 8.
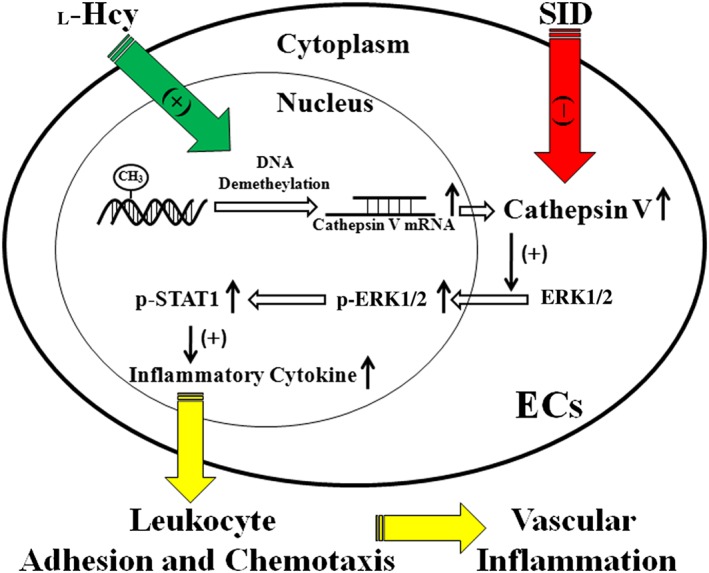
Schema of hypothesis that l‐Hcy stimulates intracellular cathepsin V expression via a DNA demethylation pathway. Increased cathepsin V promotes the phosphorylation and subsequent nuclear translocation of ERK1/2, phosphorylation of STAT1, expression of inflammatory cytokines, adhesion and chemotaxis of leukocytes and vascular inflammation. The cathepsin L/V inhibitor SID suppresses the activity of cathepsin V, and reverses the up‐regulation of inflammatory cytokines and vascular inflammation induced by l‐Hcy.
Author contributions
Y.P.L., Y.S.M., X.G.L., R.F.C., C.F.Q., Y.P.L., X.H.L., P.Y.Z. and Z.Z. performed the research. A.F.C. designed the research study. Y.P.L. and A.F.C. provided the funding.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A) HUVECs were treated with 0.5–2.0 mM l‐Hcy for 24 h, real‐time PCR was used to assess cathepsins mRNA expression, the control group as a reference in each cathepsin expression analysis. (B) The expression of cathepsin B, S, and V induced by l‐Hcy was detected by real‐time PCR. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
Figure S2 In the absence of primary antibody, the nonspecific binding of the second antibody was not observed.
Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program) 2014CB542400, the National Science Foundation of China (NSFC) Projects 81370359, 91339204 and 91639301, the Fundamental Research Funds for the Central Universities of Central South University (2016zzts118) and the Research Foundation of Education Bureau of Hunan Province, China (grant no. 12C0521).
Leng, Y.‐P. , Ma, Y.‐S. , Li, X.‐G. , Chen, R.‐F. , Zeng, P.‐Y. , Li, X.‐H. , Qiu, C. , Li, Y.‐P. , Zhang, Z. , and Chen, A. F. (2018) l‐Homocysteine‐induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. British Journal of Pharmacology, 175: 1157–1172. doi: 10.1111/bph.13920.
References
- Aavik E, Lumivuori H, Leppänen O, Wirth T, Häkkinen SK, Bräsen JH et al. (2015). Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur Heart J 36: 993–1000. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Imazeki F, Sakai Y, Mikata R, Tada M, Seki N et al. (2008). Analysis of the methylation status of genes up‐regulated by the demethylating agent, 5‐aza‐2′‐deoxycytidine, in esophageal squamous cell carcinoma. Oncol Rep 20: 405–412. [PubMed] [Google Scholar]
- Chen H, Wang J, Xiang MX, Lin Y, He A, Jin CN et al. (2013). Cathepsin S‐mediated fibroblast trans‐differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovasc Res 100: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y, Lin HC, Chen KC, Chang CC, Lee WS, Juan SH (2013). Molecular mechanisms underlying the anti‐proliferative and anti‐migratory effects of folate on homocysteine‐challenged rat aortic smooth muscle cells. Br J Pharmacol 169: 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Morgan RG, Walker AE, Lesniewski LA (2015). Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol 89: 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Chen NL, Wong A, Craik CS, Brömme D (2013). Elastin degradation by cathepsin V requires two exosites. J Biol Chem 288: 34871–34881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elordieta G, Calleja N (2005). Microvariation in accentual alignment in Basque Spanish. Lang Speech 48: 397–439. [DOI] [PubMed] [Google Scholar]
- Familtseva A, Chaturvedi P, Kalani A, Jeremic N, Metreveli N, Kunkel GH et al. (2016). Toll‐like receptor 4 mutation suppresses hyperhomocysteinemia‐induced hypertension. Am J Physiol Cell Physiol 311: C596–C606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frau M, Feo F, Pascale RM (2013). Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J Hepatol 59: 830–841. [DOI] [PubMed] [Google Scholar]
- Gulati A (2016). Vascular endothelium and hypovolemic shock. Curr Vasc Pharmacol 14: 187–195. [DOI] [PubMed] [Google Scholar]
- Han S, Wu H, Li W, Gao P (2015). Protective effects of genistein in homocysteine‐induced endothelial cell inflammatory injury. Mol Cell Biochem 403: 43–49. [DOI] [PubMed] [Google Scholar]
- Han XB, Zhang HP, Cao CJ, Wang YH, Tian J, Yang XL et al. (2014). Aberrant DNA methylation of the PDGF gene in homocysteine‐mediated VSMC proliferation and its underlying mechanism. Mol Med Rep 10: 947–954. [DOI] [PubMed] [Google Scholar]
- Hu L, Cheng XW, Song H, Inoue A, Jiang H, Li X et al. (2014). Cathepsin K activity controls injury‐related vascular repair in mice. Hypertension 63: 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Li J, Qin X, Huang Y, Wang X, Gottesman RF et al. (2015). Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: the CSPPT randomized clinical trial. JAMA 313: 1325–1335. [DOI] [PubMed] [Google Scholar]
- Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y et al. (2014). Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun 5: 3838. [DOI] [PubMed] [Google Scholar]
- Jung CH, Lee MJ, Kang YM, Lee YL, Yoon HK, Kang SW et al. (2014). Vaspin inhibits cytokine‐induced nuclear factor‐kappa B activation and adhesion molecule expression via AMP‐activated protein kinase activation in vascular endothelial cells. Cardiovasc Diabetol 13: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, Tyagi N (2013). Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral‐administered homocysteine in mice. Neuroscience 252: 302–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan PM, Wilder CL, Platt MO (2012). Tumor necrosis factor alpha stimulates cathepsin K and V activity via juxtacrine monocyte‐endothelial cell signaling and JNK activation. Mol Cell Biochem 367: 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Luo M, Xie N, Wang J, Chen L (2016). Curcumin protects endothelial cells against homocysteine induced injury through inhibiting inflammation. Am J Transl Res 8: 4598–4604. [PMC free article] [PubMed] [Google Scholar]
- Li TW, Yang H, Peng H, Xia M, Mato JM, Lu SC (2012a). Effects of S‐adenosylmethionine and methylthioadenosine on inflammation‐induced colon cancer in mice. Carcinogenesis 33: 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cheng XW, Hu L, Wu H, Guo‐Ping, Hao CN et al. (2015). Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol 183: 198–208. [DOI] [PubMed] [Google Scholar]
- Li X, Liu Z, Cheng Z, Cheng X (2012b). Cysteinyl cathepsins: multifunctional enzymes in cardiovascular disease. Chonnam Med J 48: 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P (2012). Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin N, Qin S, Luo S, Cui S, Huang G, Zhang X (2014). Homocysteine induces cytotoxicity and proliferation inhibition in neural stem cells via DNA methylation in vitro. FEBS J 281: 2088–2096. [DOI] [PubMed] [Google Scholar]
- Luo Y, Feng J, Xu Q, Wang W, Wang X (2016). NSun2 deficiency protects endothelium from inflammation via mRNA methylation of ICAM‐1. Circ Res 118: 944–956. [DOI] [PubMed] [Google Scholar]
- Lv BJ, Lindholt JS, Wang J, Cheng X, Shi GP (2013). Plasma levels of cathepsins L, K, and V and risks of abdominal aortic aneurysms: a randomized population‐based study. Atherosclerosis 230: 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumnik W, Ossolińska M, Płońska I, Chyczewska E, Nikliński J (2015). Endostatin and cathepsin‐V in bronchoalveolar lavage fluid of patients with pulmonary sarcoidosis. Adv Exp Med Biol 833: 55–61. [DOI] [PubMed] [Google Scholar]
- Nienaber‐Rousseau C, Ellis SM, Moss SJ, Melse‐Boonstra A, Towers GW (2013). Gene–environment and gene–gene interactions of specific MTHFR, MTR and CBS gene variants in relation to homocysteine in black South Africans. Gene 530: 113–118. [DOI] [PubMed] [Google Scholar]
- Owens DM, Keyse SM (2007). Differential regulation of MAP kinase signalling by dual‐specificity protein phosphatases. Oncogene 26: 3203–3213. [DOI] [PubMed] [Google Scholar]
- Platt MO, Shockey WA (2016). Endothelial cells and cathepsins: biochemical and biomechanical regulation. Biochimie 122: 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW (2001). Homocysteine induces expression and secretion of monocyte chemoattractant protein‐1 and interleukin‐8 in human aortic endothelial cells: implications for vascular disease. Circulation 103: 2717–2723. [DOI] [PubMed] [Google Scholar]
- Pribis JP, Al‐Abed Y, Yang H, Gero D, Xu H, Montenegro MF et al. (2015). The HIV protease inhibitor saquinavir inhibits HMGB1 driven inflammation by targeting the interaction of cathepsin V with TLR4/MyD88. Mol Med . https://doi.org/10.2119/molmed.2015.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach G, Starzinski‐Powitz A, Sloane BF, Doll M, Kippenberger S, Bernd A et al. (2013). PPARα agonist Wy14643 suppresses cathepsin B in human endothelial cells via transcriptional, post‐transcriptional and post‐translational mechanisms. Angiogenesis 16: 223–233. [DOI] [PubMed] [Google Scholar]
- Robert K, Pagès C, Ledru A, Delabar J, Caboche J, Janel N (2005). Regulation of extracellular signal‐regulated kinase by homocysteine in hippocampus. Neuroscience 133: 925–935. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanowicz‐Hajduk J, Adamska A, Bartoszewski R, Ochocka JR (2016). Reuse of E‐plate cell sensor arrays in the xCELLigence Real‐Time Cell Analyzer. Biotechniques 61: 117–122. [DOI] [PubMed] [Google Scholar]
- Tan H, Jiang X, Yang F, Li Z, Liao D, Trial J et al. (2006). Hyperhomocysteinemia inhibits post‐injury reendothelialization in mice. Cardiovasc Res 69: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnády GE, Simon I, Váradi A, Arányi T (2005). BiSearch: primer‐design and search tool for PCR on bisulfite‐treated genomes. Nucleic Acids Res 33: e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C, Heeschen C, Aicher A, Sasaki K, Bruhl T, Farhadi MR et al. (2005). Cathepsin L is required for endothelial progenitor cell‐induced neovascularization. Nat Med 11: 206–213. [DOI] [PubMed] [Google Scholar]
- Xu S, Ren J, Chen HB, Wang Y, Liu Q, Zhang R et al. (2014). Cytostatic and apoptotic effects of DNMT and HDAC inhibitors in endometrial cancer cells. Curr Pharm Des 20: 1881–1887. [DOI] [PubMed] [Google Scholar]
- Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Brömme D (2004). Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J Biol Chem 279: 36761–36770. [DOI] [PubMed] [Google Scholar]
- Zanin RF, Bergamin LS, Morrone FB, Coutinho‐Silva R, de Souza Wyse AT, Battastini AM (2015). Pathological concentrations of homocysteine increases IL‐1β production in macrophages in a P2X7, NF‐κB, and erk‐dependent manner. Purinergic Signal 11: 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhang YY, Li QY, Cai ZH (2015). Evolutionary history of cathepsin L (L‐like) family genes in vertebrates. Int J Biol Sci 11: 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) HUVECs were treated with 0.5–2.0 mM l‐Hcy for 24 h, real‐time PCR was used to assess cathepsins mRNA expression, the control group as a reference in each cathepsin expression analysis. (B) The expression of cathepsin B, S, and V induced by l‐Hcy was detected by real‐time PCR. The data shown are the mean ± SD of five independent experiments. * P < 0.05.
Figure S2 In the absence of primary antibody, the nonspecific binding of the second antibody was not observed.