

Abstract
Background and Purpose
An appropriate inflammatory response is necessary for cardiac healing after acute myocardial infarction (MI). Resolvin E1 (RvE1) is an anti‐inflammatory and pro‐resolution lipid mediator derived from eicosapentaenoic acid. Here we have investigated the effects of RvE1 on the recovery of cardiac function after MI in mice.
Experimental Approach
Acute MI was induced by surgical ligation of the left anterior descending artery in male C57BL/6 mice. RvE1 (5 ng·g−1·day−1; i.p.) was given to mice at different times following MI. Cardiac function was monitored by transthoracic echocardiography at days 3, 7 and 14 after MI. Effects of RvE1 on the migration of subpopulations of monocytes/macrophages (Mos/Mps, Ly6Chi and Ly6Clow) were examined by flow cytometry and transwell assay.
Key Results
RvE1 administration from days 1 to 7 post‐MI improved cardiac function, whereas treatment from days 7 to 14 markedly inhibited recovery of cardiac function. Early treatment with RvE1 post‐MI suppressed the infiltration of dominant Ly6Chi Mos/Mps and secretion of pro‐inflammatory cytokines in injured hearts, which protected cardiomyocytes against apoptosis in the peri‐infarct zones. Contrastingly, treatment with RvE1 1 week after MI decreased infiltration of Ly6Clow Mos/Mps and expression of pro‐angiogenic factors in cardiac tissue, consequently reducing neovascularization in the peri‐infarct zones. Additionally, RvE1 inhibited Mp migration by activating ChemR23 receptors.
Conclusion and Implications
Treatment with RvE1 during the initial 7 days after MI facilitated cardiac healing by suppressing pro‐inflammatory cytokine secretion, indicating that RvE1 may serve as an early therapeutic agent for acute MI.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- EF
ejection fraction
- MI
myocardial infarction
- Mos/Mps
monocytes/macrophages
- PMNs
polymorphonuclear leukocytes
Introduction
Myocardial infarction (MI) leads to an acute inflammatory response, which is important for debris clearance, tissue repair and regeneration (Aurora et al., 2014; Frantz and Nahrendorf, 2014). As early as 30 min after coronary artery ligation, neutrophils are recruited to the infarcted myocardium in response to chemokines and cytokines produced by apoptotic cardiomyocytes, followed by monocytes/macrophages (Mos/Mps) shortly thereafter. The number of neutrophils peaks 24 h after MI, whereas that of Mos/Mps peaks approximately 3 days after MI and drops to normal after 16 days. In addition, activated Mos/Mps, which participate in phagocytosis and the clearance of necrotic debris, release several pro‐inflammatory cytokines, such as TNFα, IL‐1β and IL‐6 (Maruotti et al., 2013). Following the acute inflammatory response, the infarcted myocardium is subjected to structural remodelling processes, including collagen deposition, angiogenesis and scar formation (Hofmann and Frantz, 2015). Mos/Mps and cardiac fibroblasts secrete large amounts of anti‐inflammatory cytokines, such as IL‐10 and TGF‐β, to promote the resolution of inflammation and angiogenic responses by releasing VEGF (Jeon et al., 2007). Thus, Mos/Mps serve as central cellular protagonists for cardiac repair after ischaemia and may be potential therapeutic targets for acute MI.
Two major subpopulations of Mos/Mps (Ly6Chi and Ly6Clow) have been defined on the basis of their differential expression of Ly6C antigen: Ly6Chi Mos/Mps are classically activated and pro‐inflammatory to clear injured tissue, while Ly6Clow Mos/Mps are alternatively activated to regulate regenerative responses (Nahrendorf et al., 2007). The pathogenesis of various inflammatory diseases, such as atherosclerosis (Brown et al., 2012), mammary cancer (Movahedi et al., 2010), brain ischaemia (Garcia‐Bonilla et al., 2016; Miro‐Mur et al., 2016), liver injury and fibrosis (Tacke and Zimmermann, 2014) and skeletal muscle repair after acute damage (Kharraz et al., 2013), are frequently associated with spatiotemporal dysregulation of Ly6Chi and Ly6Clow Mos/Mps. It has been reported that treatment with bone morphogenetic protein‐7 mitigates atherosclerosis by enhancing Mo/Mp differentiation toward Ly6Clow cells in ApoE−/− mice (Singla et al., 2016). Thus, targeting certain Mo/Mp populations may represent a useful therapeutic strategy for inflammatory diseases.
Anti‐inflammatory and pro‐resolution lipid mediators, such as lipoxins, resolvins (Rvs), protectins and maresins, are produced upon the trafficking of neutrophils‐monocytes and the interactions with neutrophils, platelets and macrophages in inflamed tissues (Serhan, 2017). Unlike most known lipid mediators (such as prostaglandins, leukotrienes and oxygenated eicosanoids), which are strongly pro‐inflammatory, resolvins and protectins showed a remarkable resolving potency when tested in animal models of inflammatory diseases. Their biological functions include limiting neutrophil recruitment, as well as Mp reprogramming, and promoting the resolution of acute inflammation (Serhan, 2014). Rvs are biosynthesized from ω‐3 polyunsaturated fatty acids, including eicosapentaenoic acid (EPA) and docosahexaenoic acid, and are denoted as E‐series (RvE) and D‐series resolvins respectively (Serhan et al., 2002). RvE1 is derived from EPA via aspirin‐acetylated COX‐2 in endothelial cells to produce and release 18R‐hydroxy‐5Z,8Z,11Z,14Z,16E‐EPA (18R‐HEPE) and is subsequently converted by 5‐lipoxygenase (5‐LOX) during the interaction of leukocytes and endothelial cells or through a COX‐independent pathway involving cytochrome P450 and 5‐LOX (Arita et al., 2005). We previously reported that a combination of fish oil and aspirin synergistically inhibited injury‐induced neointimal hyperplasia and reduced inflammatory reactions by increasing RvE1 levels in plasma (Gong et al., 2015). In the present study, we investigated the effect of RvE1 on cardiac healing at different stages after MI in mice.
Methods
Mouse model of MI
All animal care and experimental procedures complied with the guidelines of and were approved by the Animal Care and Use Committee of the Institute for Nutritional Sciences (Chinese Academy of Sciences, Beijing, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
A total of 285 adult C57BL/6 genetic background male mice (aged 8–10 weeks, 23 ± 2 g) were purchased from Shanghai Laboratory Animal Center (Shanghai, China) and were randomized to receive treatment. Mice were housed in a temperature/humidity controlled environment (22 ± 1°C, 60–70% relative humidity) in individual cages (≤5 mice per cage, with wood shaving bedding and nesting material), with a 12:12 h light–dark cycle and were provided with rodent chow (Slacom, Shanghai, China) and tap water ad libitum. Mice were allowed to acclimatize to their housing environment for at least 7 days before experimentation and to the experimental room for 2 h before experiments. The number of mice was not predetermined by a statistical method, and no mice were excluded from statistical analysis. Drug treatment experiments were conducted in a blinded fashion.
All the mice were subjected to permanent left anterior descending artery (LAD) ligation as described previously (Kong et al., 2016). In brief, the mice were lightly anaesthetised with 3.0–4.0% isoflurane gas in an induction chamber and were then fully anaesthetised with 1.0–1.5% isoflurane gas using an intubation tube before being mechanically ventilated with a rodent respirator. Subsequently, the chest cavity was opened to expose the heart and a 6–0 silk suture was used for permanent ligation of the LAD. The suture was passed approximately 2–3 mm below the tip of the left auricle. Complete vessel occlusion was confirmed by the presence of myocardial blanching in the perfusion bed. The thorax was closed with a 5–0 silk suture. Mice that died within 24 h of the surgery were excluded from analysis. Sham‐operated animals underwent the same procedure without coronary artery ligation. At the indicated time points, the mice were killed by inhalation of carbon dioxide, followed by cervical dislocation as a secondary measure, and the hearts were extracted for further analysis.
RvE1
RvE1 (5S,12R,18R–trihydroxy‐6Z,8E,10E,14Z,16E–EPA, purity, 98%, λmax, 272 nm) and d5‐RvE1 (purity, 98%) were synthesized by Wuxi AppTech (Tianjin, China) as previously described (Ogawa and Kobayashi, 2009). Mice were treated with RvE1 (5 ng·g−1·day−1, i.p.) at the indicated time points after LAD ligation.
Transthoracic echocardiography
Cardiac function was evaluated using a Vevo2100 Ultrasound system (VisualSonics, Toronto, Canada) at different time points after the surgery. The investigator was blinded to group assignment. Mice were first anaesthetised via 1% isoflurane and moved to a warming plate that maintained core body temperature. Mice were then evaluated under light anaesthesia (0.5% isoflurane). Two‐dimensional parasternal long axis views of the left ventricle were obtained. The left ventricle internal diameters at end‐diastole and end‐systole, as well as the interventricular septal wall and posterior wall thicknesses, were measured by M‐mode tracing.
Measurement of myocardial infarct size
Infarct size was determined as previously described (Yan et al., 2013). Briefly, 0.2 mL of 2% Evans blue dye was injected into the tail vein in mice, and then the hearts of the mice were collected, frozen at −30°C, cut transversely into 1‐mm‐thick slices using a Mouse Heart Slicer Matrix (Leica Biosystems, Wetzlar, Germany) and stained with 1.5% triphenyltetrazolium chloride in PBS (pH 7.4) for 15 min in a 37°C water bath. Each slice was weighed and photographed after fixation in 4% neutral‐buffered formaldehyde for 4–6 h. Infarct size was measured and calculated using ImageJ software (National Institutes of Health, USA).
Immunofluorescence staining and TUNEL assay
Immunostaining was performed according to a reported protocol (Troidl et al., 2009). Frozen tissue sections were fixed in cold acetone, washed with PBS and incubated in a mixture of Tris‐buffered saline and Tween® 20 containing 3% BSA for 30 min at room temperature to prevent non‐specific binding of antibodies. Tissue slides were then incubated with the following primary antibodies: anti‐CD68 (1:200; AbD Serotec, Kidlington, UK), anti‐CD11b (1:200; BD Biosciences, San Jose, CA, USA) or anti‐CD31 (1:200; BD Biosciences, San Diego, CA, USA) antibody at 4°C overnight. The slides were then washed with PBS three times before incubation with Alexa Fluor®‐conjugated secondary antibodies (1:1000; Invitrogen, Carlsbad, CA, USA) for 2 h at room temperature. ProLong® Gold antifade reagent with 4′,6‐diamidino‐2‐phenylindole (Invitrogen) was applied to counterstain the slides. The slides were then mounted, and the stained sections were visualized under a laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) and analysed using Image‐Pro Plus software (version 6.0; Media Cybernetics, Rockville, MD, USA). At least three different heart sections were obtained from each mouse, and more than six random images of at‐risk areas were taken of each section. For the TUNEL assay, cardiac tissues from mice that were administered PBS or RvE1 were embedded in optimal cutting temperature (OCT) compound, frozen and cut into 8‐μm‐thick sections. The frozen sections were stained using a TUNEL fluorescence In Situ Apoptosis Detection kit (Yeasen, Shanghai, China), according to the manufacturer's instructions.
RT‐PCR
Total RNA was extracted from the heart tissues using TRIzol reagent (Invitrogen) and an RNA isolation kit (RNeasy Mini kit; QIAGEN, Valencia, CA, USA), according to the manufacturers' instructions, followed by quantitation with a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA (1.0 μg) was subsequently reverse transcribed into cDNA using Reverse Transcription Reagent kits (Takara, Otsu, Shiga, Japan). RT‐PCR was performed using a CFX96™ Real‐Time System (Bio‐Rad, Hercules, CA, USA) and iQ™ SYBR® Green Supermix (Applied Biosystems, Foster City, CA, USA) according to the manufacturers' instructions. Raw data were normalized to the internal control, GAPDH, and analysed using the ΔΔCT method. Each sample was analysed in triplicate and normalized to a reference RNA. PCR products were confirmed by a single band of the expected size on 2% agarose gels. All of the primers used for RT‐PCR are shown in Supporting Information Table S1.
Isolation of peritoneal macrophages
Peritoneal macrophages were induced by an i.p. injection of 3% Brewer's thioglycollate 3 days before collection, as described previously (Yu et al., 2006). Next, 10 mL of cold, sterile PBS was injected into the peritoneal cavity. The macrophages were then collected and seeded into 60 mm dishes. The cells (1–2.5 × 107) were allowed to adhere for 2 h in RPMI‐1640 medium supplemented with 10% FBS (HyClone, Logan, UT, USA) and 1% penicillin–streptomycin solution (Sigma‐Aldrich, St. Louis, MO, USA) at 37°C in a humidified atmosphere containing 5% CO2. The dishes were washed with fresh PBS before use to remove non‐adherent cells. The adherent macrophages were used in various assays described below.
Isolation of neonatal mouse cardiac fibroblasts
Neonatal mouse cardiac fibroblasts were prepared as previously described (Wang et al., 2015). Briefly, neonatal mouse ventricles (from 20 to 30 hearts) were harvested into a 60 mm dish with ice‐cold PBS and trimmed to remove fat tissue. The hearts were cut into small pieces and transferred into a 50 mL conical tube with 0.05% trypsin–EDTA and incubated at 37°C for 15 min. Type II collagenase buffer (0.5 mg·mL−1 in HBSS, Sigma‐Aldrich) was then added and the tube was incubated for 10–15 min. The tissue suspension was collected and passed through a 40 μm cell strainer. After centrifugation, the cells were resuspended in DMEM supplemented with 10% FBS (HyClone) and 1% penicillin–streptomycin solution (Sigma‐Aldrich). Thy1.2+ fibroblasts were isolated by magnetic‐activated cell sorting (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany).
RNA interference
Cultured peritoneal macrophages were transfected with siRNA for mouse leukotriene B4 BLT1receptor, for the chemerin receptor 23 (ChemR23) or scrambled siRNA (100 nM, GenePharma, Shanghai, China) using RNAiFect Transfection Reagent (QIAGEN, Crawley, UK) according to the manufacturer's instructions. The siRNAs were designed to target 21 nucleotide sequences. Control siRNAs were scrambled derivatives of the BLT1 and ChemR23 siRNA sequences. One microgram of siRNA and RNAiFect transfection reagent were mixed and incubated for 20 min at room temperature to allow the transfection complexes to form before delivery to the cells. Knockdown efficiency was assessed by RT‐PCR 48 h later.
Flow cytometric analysis
Flow cytometric analysis was performed as previously described (Kong et al., 2016). The infarcted hearts were triturated and digested at 37°C for 1.5 h in PBS (2 mL), containing type II collagenase (1.5 mg·mL−1, Worthington Biochemical Corporation, Lakewood, NJ, USA), elastase (0.25 mg·mL−1, Worthington Biochemical Corporation) and DNase I (0.5 mg·mL−1, Worthington Biochemical Corporation) to isolate the macrophages. After digestion, the tissues were passed through a 70 μm cell strainer, and leukocyte‐enriched fractions were isolated with 37–70% Percoll® (Biosharp, Hefei, China) by density gradient centrifugation. Next, the cells were collected from the interface and washed with RPMI‐1640 medium. The cells were then incubated with the following antibodies: CD45‐PE (BD Biosciences, San Diego), CD11b‐FITC (Miltenyi Biotec, Bergisch Gladbach, Germany), F4/80‐BV421 (Brilliant Violet 421; BioLegend, San Diego, CA, USA), Ly‐6G‐APC (BD Biosciences, San Diego) and Ly6C‐PE‐cy7 (BD Biosciences) at 4°C for 30 min. Flow cytometric analysis and sorting were performed using a FACSAria™ flow cytometer (BD Biosciences, San Jose, USA), and the data obtained were analysed with FlowJo 7.6.1 software (Tree Star Inc., Ashland, OR, USA).
Adoptive transfer of monocytes
The adoptive transfer assay was performed as previously described (Hilgendorf et al., 2014). Bone marrow cells from CD45.1+ WT mice were harvested, counted and then incubated at 4°C for 30 min in the dark with the following fluorophore‐conjugated antibodies: CD11b‐PE, Ly6G–APC‐cy7 and Ly6C‐PE‐cy7 (BD Pharmingen™, San Diego, CA, USA). The cells were then purified by high‐speed cell sorting with the flow cytometer. Next, CD11b+Ly6G−Ly6Chi Mos (1.0 × 106) were injected through the tail vein into congenic CD45.2+ mice treated with PBS or RvE1 at 7 days after the induction of MI. The mice were killed 3 days after the adoptive transfer was initiated. Mos/Mps were isolated from the infarcted hearts and analysed by flow cytometry using CD45.1‐FITC, CD45.2‐APC, F4/80‐BV421 and Ly6C‐PE‐cy7 antibodies (BD Pharmingen™), which were allowed to bind at 4°C for 30 min. Flow cytometric analysis was performed as described in the preceding section.
Transwell assay
The transwell assay was performed as previously described (Ancuta et al., 2003; Lee et al., 2015). In brief, HUVECs (Cell Resource Center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences) were cultured on filters (6.5 mm diameter and 8 μm pore size; Corning Costar, Corning, NY, USA) at 2 × 105 cells per well in RPMI‐1640 medium (Invitrogen) supplemented with 10% FBS (HyClone) and maintained at 37°C in a humidified atmosphere containing 5% CO2 for 24 h. Peritoneal macrophage polarization was subsequently induced with LPS (1 μg·mL−1; Sigma‐Aldrich, St. Louis, MO, USA) or IL‐4 (20 ng·mL−1; PeproTech, Rocky Hill, NJ, USA) for 12 h. Next, Mps (1 × 106), pretreated with methanol or RvE1 (100 nM), were seeded into the top chamber. Culture medium containing the chemokine CCL2 (20 ng·mL−1, PeproTech) or the chemokine CXCL3 (20 ng·mL−1, PeproTech) was added to the lower chamber. For Mp and cardiac fibroblast co‐culture experiments, Mps (1 × 106) were seeded onto the upper wells without serum, and cardiac fibroblasts (5.0 × 105), pretreated with RvE1 (100 nM) or methanol, were seeded into the lower wells. After incubation for 4 or 12 h, non‐migrating Mps were removed from the upper surfaces of the filters, and the cells on the underside were counted using a microscope at 100× magnification. The migration rate was calculated as the ratio of the number of migrated cells to the total number of cells. Each experiment was performed in triplicate.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data were expressed as means ± SEM. Data were analysed using GraphPad Prism 5.0 software (GraphPad Software, Inc., San Diego, CA, USA), using either Student's unpaired t‐tests, Mann–Whitney U‐tests or one‐way ANOVA, followed by Bonferroni post hoc tests where appropriate. Differences were considered statistically significant when P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Early treatment (initial 7 days) with RvE1 accelerates cardiac recovery, while late treatment (after 7 days) with RvE1 delays cardiac healing after MI
To fully evaluate the effect of RvE1 on cardiac healing post‐MI, RvE1 (5 ng·g−1·day−1) was injected i.p. in mice over different periods after LAD ligation. Cardiac function (ejection fraction, EF) was markedly improved at days 3 and 7 after MI in the mice that received RvE1 compared to that in the control mice. However, EF declined at day 14 after MI (Figure 1A; Supporting Information Table S2), indicating that RvE1 has a cardioprotective effect early after MI, but not at later stages. Indeed, RvE1 administered during the first 3 days [RvE1 (1–3)] and during the first week [RvE1 (1–7)] (Figure 1B, C; Supporting Information Table S2) after the MI significantly enhanced cardiac recovery in mice. Moreover, administration of RvE1 at day 3 to day 14 [RvE1 (3–14)] after the MI induction increased cardiac output at day 7 but reduced cardiac function at day 14 (Figure 1D; Supporting Information Table S2). Furthermore, treatment with RvE1 that was started at day 7 [RvE1 (7–14)] after MI dramatically delayed cardiac recovery compared with that of PBS‐treated mice (Figure 1E; Supporting Information Table S2). Taken together, the results show that early treatment with RvE1 facilitates cardiac recovery after MI, whereas late treatment impedes cardiac healing.
Figure 1.
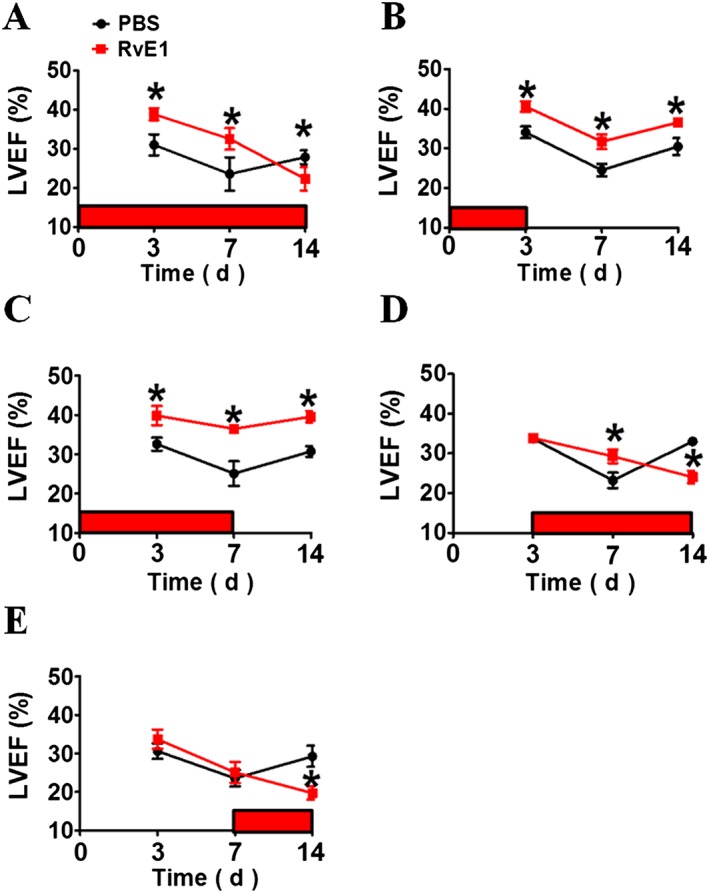
Effect of RvE1 treatment during different periods on cardiac function in mice after acute MI. Acute MI was induced by surgical ligation of the LAD coronary artery in mice. RvE1 (5 ng·g−1·day−1) was injected i.p. from days 1 to 14 (A), days 1 to 3 (B), days 1 to 7 (C), days 3 to 14 (D) and days 7 to 14 (E) after LAD ligation (indicated with horizontal red bars on each plot). Heart function was evaluated by echocardiography as indicated. LVEF, left ventricular ejection fraction. Results are from experiments on two separate occasions. *P < 0.05, significantly different from corresponding PBS value. (A) PBS, n = 9; RvE1, n = 14. (B) PBS, n = 8; RvE1, n = 8. (C) PBS, n = 8; RvE1, n = 10. (D) PBS, n = 8; RvE1, n = 10. (E) PBS, n = 9; RvE1, n = 8.
Early treatment with RvE1 reduces infarct size through the inhibition of inflammatory cell recruitment and cardiomyocyte apoptosis
We found that infarct size after MI significantly decreased in the RvE1 (1–7) treatment group, compared with that in the PBS treatment group (Figure 2A, B). A compensatory myocardial hypertrophy always occurs post‐MI, which can be assessed by an increase in heart weight to body weight (HW/BW) ratio (Vlasov and Volkov, 2004). The HW/BW ratio after treatment with RvE1 within the first 7 days following MI [RvE1 (1–7)] was much lower than that in the mice treated with PBS at 14 days post‐MI (Figure 2C). These findings suggest that treatment with RvE1 at an early stage (1–7 days) after MI exerts a protective effect against heart failure. The neutrophils (CD11b+) and Mps (CD68+) at day 3 after MI, but not at day 14, were markedly reduced in number in the infarcted hearts (Figure 2D, E; Supporting Information Figure S1A, B), indicating that the acute inflammatory response to ischaemia was suppressed by RvE1. Furthermore, a decrease in TUNEL‐positive cardiomyocytes in the RvE1 (1–7) group was observed (Figure 2F, G), suggesting that RvE1 protects the myocardium from apoptotic injury after MI in mice. However, the number of CD31+ endothelial cells in the peri‐infarct zones, which is an indicator of angiogenesis (Zhong et al., 2014), was not significantly altered in response to the RvE1 treatment from days 1 to 7 after MI (Figure 2H, I).
Figure 2.
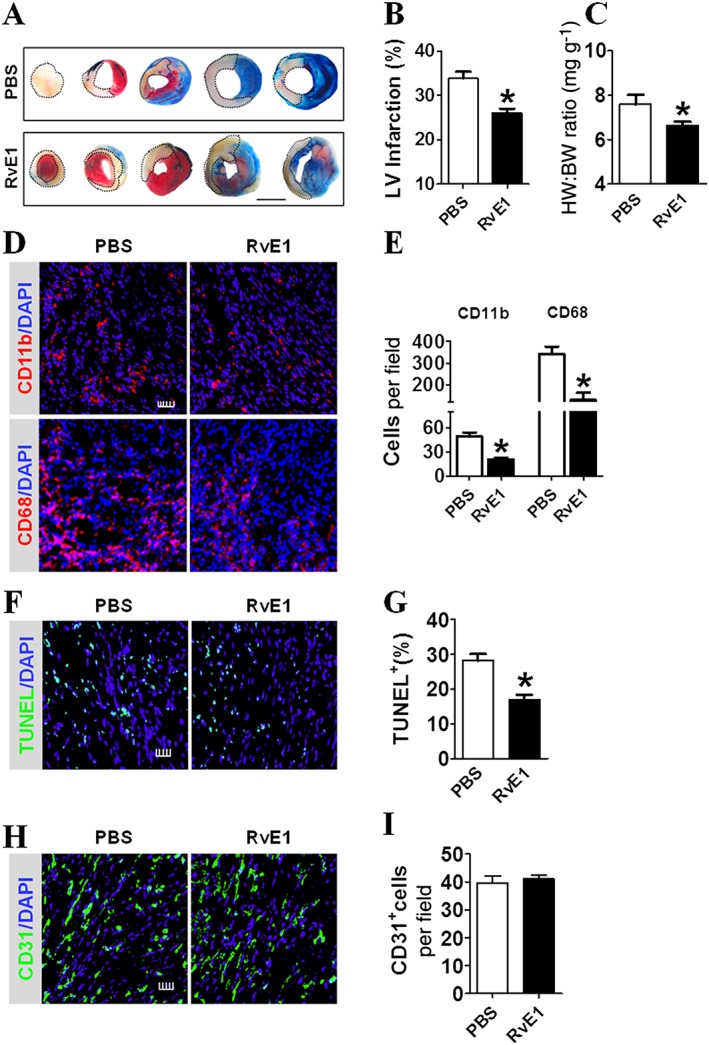
RvE1 treatment from days 1 to 7 improves cardiac recovery after MI in mice. (A) Representative images of tetrazolium chloride (TTC) stained heart sections (2 weeks post‐MI) from RvE1‐treated (days 1–7) mice. The dotted line denotes the infarct zone. Scale bar, 10 mm. (B) Infarct sizes were quantified and plotted. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 7; RvE1, n = 7). (C) Ratio of heart weight to body weight (HW/BW) of mice was measured 2 weeks after MI. *P < 0.05. significantly different from corresponding PBS value; PBS, n = 9; RvE1, n = 14). (D) Representative CD11b+ and CD68+ inflammatory cell staining images of infarcted hearts from PBS‐ and RvE1‐treated (days 1–7) mice, at 3 days post‐MI. Scale bar, 50 μm. (E) Quantification of CD11b+ and CD68+ inflammatory cells from D. *P < 0.05, significantly different from corresponding PBS value; CD11b: PBS, n = 9; RvE1, n = 8; CD68: PBS, n = 9; RvE1, n = 10). (F) Representative TUNEL staining of cardiomyocytes from PBS‐ and RvE1‐treated (days 1–7) mice at 3 days post‐MI. Scale bar, 100 μm. (G) Quantification of TUNEL+ cardiomyocytes from F. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 6; RvE1, n = 8). (H) Representative CD31 staining in hearts from PBS‐ and RvE1‐treated (days 1–7) mice at 14 days post‐MI. Scale bar, 100 μm. (I) Quantification of CD31+ cells from H (PBS, n = 6; RvE1, n = 6).
RvE1 suppresses the expression of pro‐inflammatory cytokines by inhibiting Ly6Chi Mos/Mps recruitment
We examined the mRNA levels of IL‐4, IL‐6, TNF‐α and IFN‐γ in the areas at risk in the heart tissues. The expression levels of the four pro‐inflammatory cytokines were significantly lower in the RvE1 treatment group [RvE1 (1–7)] compared to the levels in the PBS treatment group at day 3 after MI (Figure 3A). Moreover, the mRNA levels of IL‐6 and TNF‐α were reduced by RvE1 treatment on day 7 after MI (Figure 3B). However, the expression of factors involved in repair and healing, such as VEGFs and TGF‐β, was not markedly changed in the infarcted hearts by early treatment with RvE1 (Figure 3C). Infiltrating Ly6Chi and Ly6Clow Mos/Mps play an important role in cardiac repair after ischaemia. Therefore, we examined the influence of RvE1 on Ly6Chi and Ly6Clow Mo/Mp recruitment in hearts after the LAD ligation (Figure 3D). The total number of infiltrating Mos/Mps gradually increased in the first 3 days after MI, which was markedly suppressed by RvE1 treatment (Figure 3E). Indeed, both Ly6Chi (Figure 3F) and Ly6Clow Mos/Mps (Figure 3G) recruited into hearts were suppressed in the RvE1 treatment group during the acute phase after MI (days 1–3), despite Ly6Chi Mos/Mps dominating in the first 3 days. These results indicate that RvE1 inhibits the acute inflammatory responses after MI, mainly by reducing Mo/Mp infiltration and pro‐inflammatory cytokine secretion.
Figure 3.
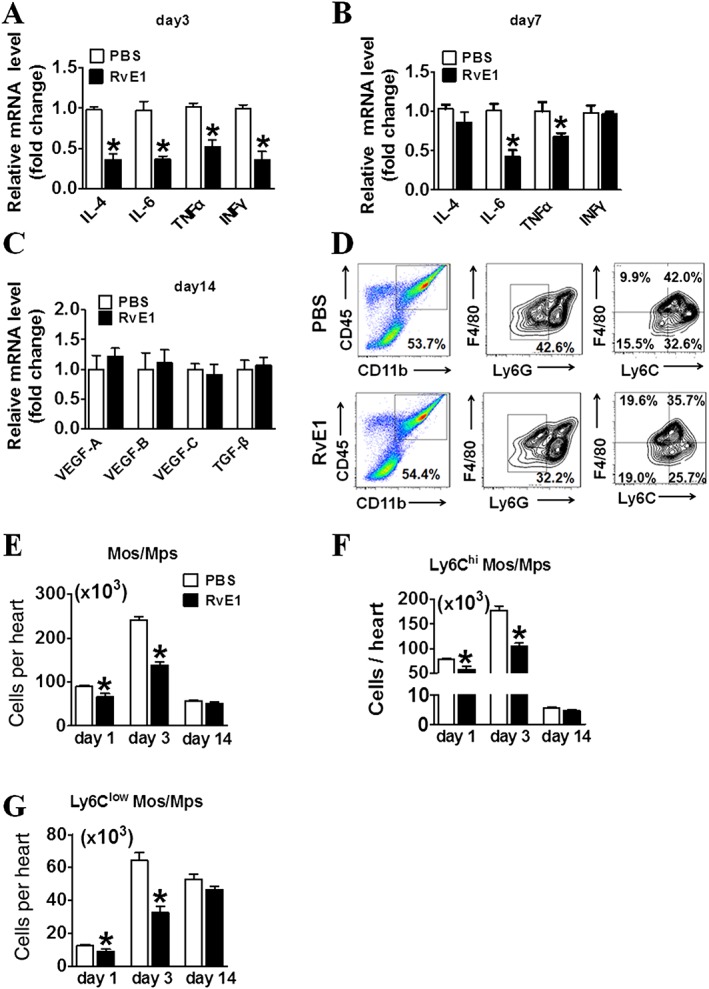
Early treatment with RvE1 suppresses pro‐inflammatory Ly6Chi monocyte/macrophage (Mo/Mp) recruitment in hearts of mice after MI. (A–B) Effect of RvE1 treatment (days 1–7) on expression of pro‐inflammatory markers in heart tissues in mice at day 3 (A) or day 7 (B) after MI. (C) Effect of RvE1 treatment (days 1–7) on expression of pro‐angiogenic cytokines in hearts of mice at day 14 after MI. (D) Gating strategy for CD11b+CD45+Ly6G−F4/80+Ly6Chi and CD11b+CD45+Ly6G−F4/80+Ly6Clow Mos/Mps in hearts from RvE1‐treated mice after MI. (E–G) Effect of RvE1 treatment on the recruitment of total Mos/Mps (E), Ly6Chi (F) and Ly6Clow (G) Mos/Mps in injured hearts from RvE1‐treated mice on day 1, day 3 and day 14 after MI. All data were from experiments on two separate occasions, *P < 0.05, significantly different from corresponding PBS value. (A) PBS, n = 5; RvE1, n = 5. (B) PBS, n = 8; RvE1, n = 5. (C) PBS, n = 5; RvE1, n = 5. (E–G) day 1: PBS, n = 6; RvE1, n = 6; day 3: PBS, n = 6; RvE1, n = 7; day 14: PBS, n = 5; RvE1, n = 5.
Late treatment with RvE1 impairs post‐MI recovery by reducing infiltration of Ly6Clow Mo/Mp involved in repair
Treatment with RvE1 from days 7 to 14 after MI [RvE1(7–14)] led to the formation of a larger infarct than that without treatment (Figure 4A, B), suggesting that late‐stage RvE1 treatment post‐MI resulted in adverse left ventricular remodelling. The HW/BW ratio was also higher in the RvE1 (7–14) group than in the control group (Figure 4C). These findings suggest that the treatment with RvE1 from days 7 to 14 after LAD ligation impaired heart function in the mice. The immunostaining study showed that Mos/Mps infiltration at day 14 post‐MI was also decreased in the RvE1 (7–14) group (Figure 4D, E). Interestingly, the number of CD31+ endothelial cells in the peri‐infarct zones was significantly reduced in response to RvE1 treatment from days 7 to 14 after MI (Figure 4F, G). Taken together, the results show that administration of RvE1 at the late stage delayed cardiac repair and healing, possibly via suppression of neovascularization in the peri‐infarct zones of the injured hearts.
Figure 4.
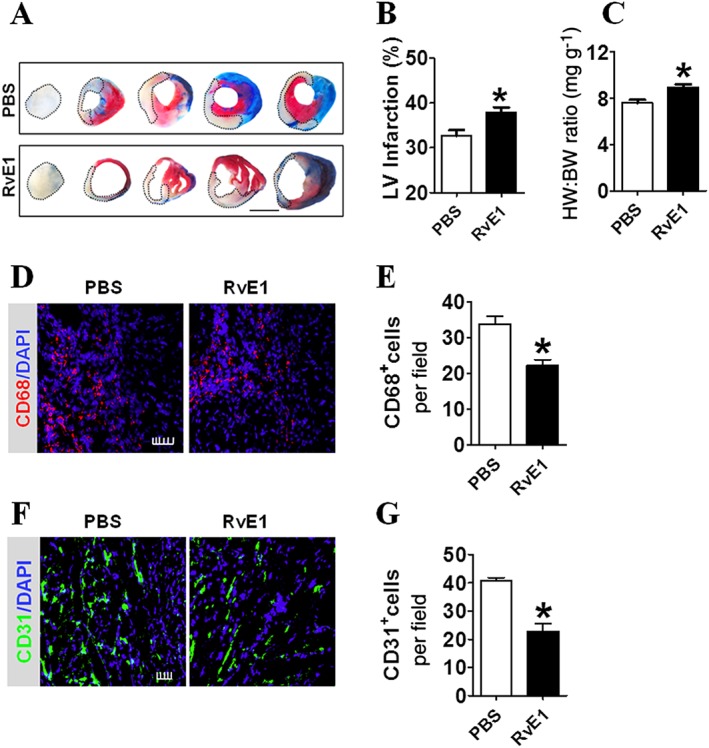
RvE1 infusion from days 7 to 14 impairs cardiac recovery after MI in mice. (A) Representative images of tetrazolium chloride (TTC)‐stained heart sections 2 weeks post‐MI, from RvE1‐treated (days 7–14) mice. The dotted line denotes the infarct zone. Scale bar, 10 mm. (B) Infarct sizes from A were quantitated. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 8; RvE1, n = 8. (C) HW/BW of mice was measured 2 weeks after MI. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 9; RvE1, n = 9. (D) Representative CD68+ inflammatory cell staining images of infarcted hearts from PBS‐ and RvE1‐treated (days 7–14) mice at day 14 after MI. Scale bar, 50 μm. (E) Quantification of CD68+ inflammatory cells from D. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 8; RvE1, n = 10. (F) Representative CD31 staining in hearts from PBS‐ and RvE1‐treated (days 7–14) mice at day 14 after MI. Scale bar, 100 μm. (G) Quantification of CD31+ cells from F. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 8; RvE1, n = 9.
Next, we examined the expression of proangiogenic factors in the injured hearts. RvE1(given from days 7–14) reduced the expression of proangiogenic factors, such as VEGF‐A, VEGF‐B, VEGF‐C and TGF‐β in the peri‐infarct zones of the hearts (Figure 5A). During the recovery stage after MI, infiltrating Ly6Clow Mos/Mps are a major source of secreted pro‐angiogenic factors (Nahrendorf et al., 2007). Indeed, the RvE1 treatment that was started at day 7 reduced total Mo/Mp infiltration in the injured hearts (Figure 5B). Notably, the dominant (>70%) Mos/Mps (Ly6Clow), which had infiltrated the hearts at days 8 and 10 after MI, were markedly suppressed by the RvE1 treatment starting from day 7 post‐MI (Figure 5C, D). Moreover, down‐regulation of mRNA levels of VEGF, TGFβ, FGF and HGF was observed in sorted Mos/Mps from injured hearts in the RvE1‐treated mice (Figure 5E).
Figure 5.
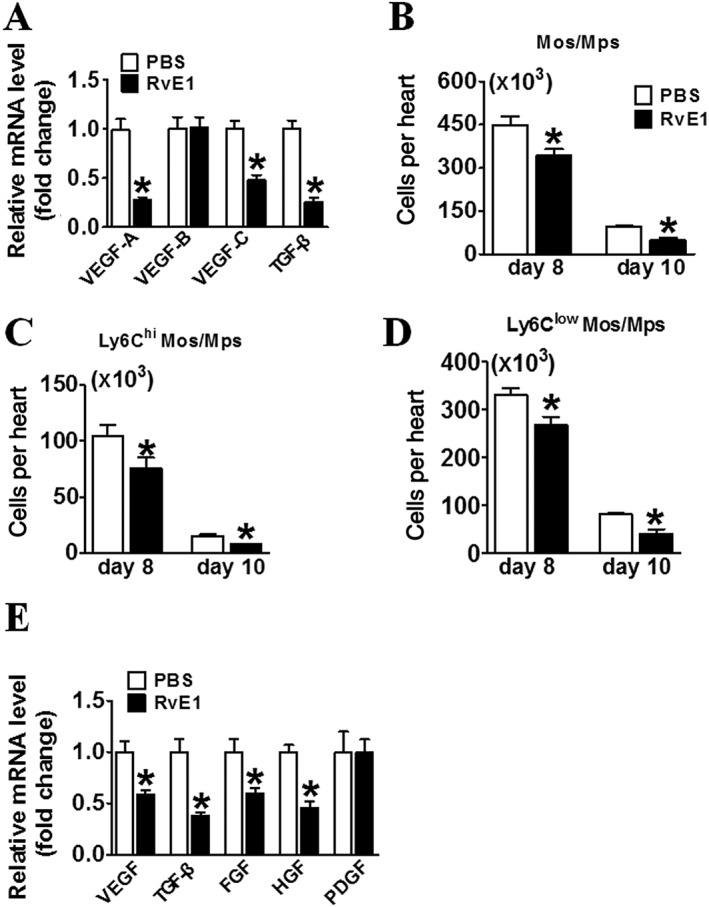
RvE1 suppresses recruitment of Ly6Clow Mos/Mps in the infarcted hearts of mice. (A) Effect of RvE1 treatment (days 7–14) on expression of pro‐angiogenic cytokines in hearts in mice at day 14 after MI. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 7; RvE1, n = 7). (B–D) Effect of RvE1 treatment starting from day 7 on the recruitment of total Mos/Mps (B), Ly6Chi (C), and Ly6Clow (D) Mos/Mps in injured hearts from mice at days 8 and 10 after MI were quantified and plotted. (E) Effect of RvE1 treatment starting from day 7 on VEGF, TGF‐β, FGF, hepatocyte growth factor and PDGF mRNA expression in Mos/Mps sorted from hearts of mice at day 10 after MI. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 5; RvE1, n = 5). Data are from at least two separate occasions (B–D). *P < 0.05, significantly different from corresponding PBS value; day 8: PBS, n = 6; RvE1, n = 6; day 10: PBS, n = 5; RvE1, n = 5.
Ly6Clow Mos/Mps recruited into infarcted hearts may originate from either circulating Mo subsets or local Ly6Chi Mo differentiation (Hilgendorf et al., 2014). Therefore, we examined the effect of the RvE1 treatment on Ly6Chi Mo transformation in the infarcted hearts, using an adoptive transfer assay. Ly6Chi Mos were sorted from CD45.1+ mice and injected into congenic CD45.2+ mice after LAD ligation (Figure 6A). CD45.1+ Mos/Mps were sorted from the infarcted hearts 3 days after the adoptive transfer for differentiation analysis (Figure 6B). Indeed, the total infiltrated CD45.1+ Mos were significantly reduced by the RvE1 treatment (Figure 6C), whereas the Ly6Clow Mos differentiated from CD45.1+ Ly6Chi Mos were not notably affected (Figure 6D). Collectively, RvE1 treatment reduced proangiogenic factors secretion by inhibiting recruitment of Ly6Clow Mo/Mp which involved in repair, to the infarcted hearts.
Figure 6.
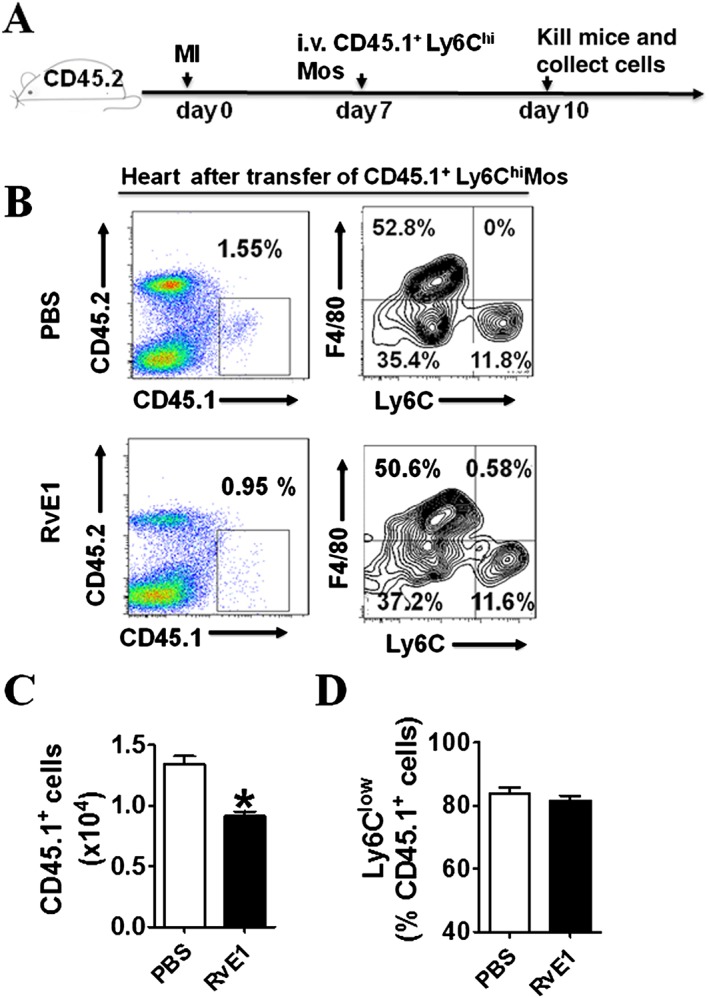
Effect of RvE1 treatment on trans‐differentiation of Ly6Chi Mos to Ly6Clow Mos. (A) Schematic of protocol for adoptive transfer and administration of RvE1 to mice. (B) Representative flow cytometric analysis for CD11b+Ly6G−Ly6Chi Mos 3 days after adoptive transfer. (C) Quantification of infiltrated CD45.1+ Mos/Mps in hearts after MI. (D) Ratio of Ly6Clowcells transdifferentiated from CD45.1+ Ly6Chi cells. All data are from experiments on two separate occasions (C–D). *P < 0.05, significantly different from corresponding PBS value; PBS, n = 7; RvE1, n = 7.
RvE1 inhibits the migration of Mps through ChemR23 receptors
Functionally, Ly6Chiand Ly6Clow Mps are similar to M1‐ and M2‐type Mps in the context of inflammation (Nahrendorf and Swirski, 2013). Mps can be differentiated in vitro toward M1 and M2 polarization by LPS and IL‐4 respectively. We monitored migration capacities of RvE1‐treated M1‐ and M2‐type Mps in response to the chemotactic factors CCL2 and CXCL3. Consistent with the observations in the infarcted hearts, we noted that RvE1 inhibited the migrations of both M1‐ and M2‐type Mps (Figure 7A). RvE1 can bind to two different receptors: BLT1 and the chemerin receptor ChemR23. As previously described (Arita et al., 2007), ChemR23, but not BLT1 receptors, were abundantly expressed in Mps (Figure 7B), and ChemR23 receptor expression was down‐regulated in Mps by LPS stimulation (Supporting Information Figure S2A, B). Accordingly, knockdown of ChemR23, but not of BLT1 receptors (Figure 7C), abolished the suppressed migration of RvE1‐treated Mps in response to both MCP‐1 and CXCL3 (Figure 7D). Cardiac fibroblasts play an important role in cardiac inflammation and repair after MI (Shinde and Frangogiannis, 2014). Expression of both ChemR23 and BLT1 receptors was relatively low in cardiac fibroblasts (Supporting Information Figure S3A). Silencing of either ChemR23 or BLT1 receptors in cardiac fibroblasts did not affect Mp migration in the co‐culture system (Supporting Information Figure S3B, C). In addition, RvE1 treatment had no significant influence on matrix protein and cytokine expression in cardiac fibroblasts (Supporting Information Figure S3D). Taken together, our data suggest that RvE1 facilitated cardiac recovery in the early stages by suppressing the infiltration of dominant Ly6Chi Mos/Mps in the heart after MI, while late treatment impaired healing by inhibiting recruitment of Ly6Clow Mos/Mps which are involved in repair (Figure 7E).
Figure 7.
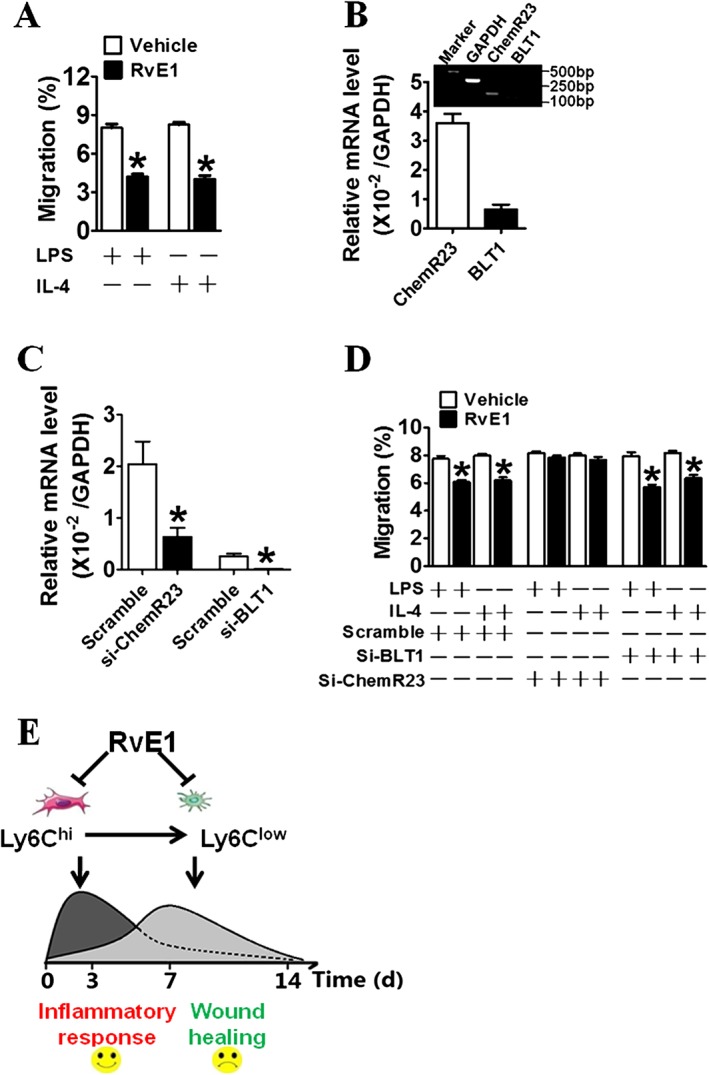
RvE1 inhibits transendothelial migration of Mps in vitro. (A) The effect of RvE1 on migration of polarized Mps through a confluent HUVEC monolayer in a transwell assay. *P < 0.05, significantly different from corresponding PBS value; PBS, n = 7; RvE1, n = 7. (B) Relative mRNA levels for ChemR23 and BLT1 receptors in murine peritoneal Mps (n = 5). (C) Knockdown effects of siRNAs for BLT1 receptors (right side) and ChemR23 receptors (left side) in primary peritoneal Mps. Data are normalized to GAPDH levels. *P < 0.05, significantly different from corresponding scrambled value; Scramble, n = 7; Si‐ChemR23, n = 7; Si‐BLT1, n = 7). (D) Effect of silencing ChemR23 or BLT1 receptors on RvE1‐induced suppression of Mp trans‐endothelial migration in vitro. *P < 0.05, significantly different from vehicle; vehicle, n = 7; RvE1, n = 7. (E) Schematic diagram of RvE1‐mediated suppression on Ly6Chi and Ly6Clow Mos/Mps recruited in hearts after MI.
Discussion
The lipid mediator RvE1 exerts clearly beneficial effects by attenuating inflammation in a variety of diseases, including asthma, colitis, rheumatoid arthritis, atherosclerosis, dry eye and retinopathy (Lim et al., 2015; Hesselink et al., 2016). In the present study, we found that RvE1 conferred early cardioprotection after MI by inhibiting recruitment of Ly6Chi Mo/Mp and secretion of pro‐inflammatory cytokines, thereby suppressing cardiomyocyte apoptosis during the acute phase (days 1–7) after MI. Paradoxically, the Ly6Clow Mos/Mps which have repair and healing functions and also infiltrate the infarcted hearts, were suppressed by RvE1 during resolution of inflammation (days 7–14 after MI), which resulted in a delayed recovery from MI. Thus, our findings indicate that RvE1 may act as an early beneficial agent for acute MI.
MI triggers an acute inflammatory response that is critical for cardiac repair but also participates in cardiac remodelling after ischaemia, which can contribute to heart failure. Pro‐inflammatory Ly6Chi and healing Ly6Clow Mos are recruited into the infarcted myocardium sequentially (Nahrendorf et al., 2007). The Ly6Chi Mo subset accumulates, via CCL2, in the injured heart on the first day post‐MI and peaks at day 5 (Dewald et al., 2005). Along with the resolution of inflammation in the myocardium, the Ly6Clow Mo subset is recruited later, via CX3CR1, and reaches its peak at day 7 post‐MI (Nahrendorf et al., 2007). Both subsets exhibit a similar phagocytic capacity (Nahrendorf et al., 2007). Ly6Chi Mos have highly inflammatory functions, as they secrete pro‐inflammatory cytokines, such as IL‐1β, IL‐6 and TNF‐α, whereas Ly6Clow Mos show attenuated inflammatory and pro‐angiogenic properties by secreting pro‐resolution cytokines, such as VEGF and TGF (Nahrendorf and Swirski, 2013). Thus, at days 1–4 after MI, the milieu in the heart is highly inflammatory, while during the recovery stage, the inflammatory reaction resolves and pro‐angiogenic factors and fibrotic proteins are released instead. We found that RvE1 inhibited the migration of both Ly6Chi and Ly6Clow subsets into the infarcted hearts. This facilitated cardiac repair by suppressing pro‐inflammatory cytokine expression at the early stages after MI, but delayed cardiac healing by attenuating of pro‐resolution factor secretion at later stages.
Ly6Chi Mos are able to convert to Ly6Clow Mos in the circulation (Yona et al., 2013) and in infarcted hearts (Hilgendorf et al., 2014). The turnover of Mos/Mps in the heart occurs very rapidly; the infiltrated Mos reside in injured hearts for approximately 20 h (Leuschner et al., 2012). However, the dynamic alterations of Mos in infarcts have also been observed in circulation after MI in mice (Nahrendorf et al., 2007) and in patients with acute MI (Tsujioka et al., 2009). Treatment with RvE1 does not influence the trans‐differentiation of Ly6Chi Mos to Ly6Clow Mos in infarcted hearts. Thus, monitoring the Ly6Chi/Ly6Clow ratio in the circulation may be helpful for guiding RvE1 therapy for acute MI.
RvE1 is a potent anti‐inflammatory and pro‐resolving lipid mediator biosynthesized from ω‐3 fatty acids. It promotes the resolution of acute inflammation by inhibiting transendothelial migration of polymorphonuclear leukocytes (PMNs), stimulating Mp phagocytosis and reducing pro‐inflammatory cytokine levels (Serhan, 2014). Two different receptors (ChemR23 and BLT1) have been identified to specifically bind RvE1 (Serhan, 2014). Moreover, RvE1 attenuates LTB4‐induced NF‐kB activation in PMNs and reduces experimental peritonitis in a BLT1 receptor‐dependent manner (Arita et al., 2007). We and others (Serhan, 2014) found that ChemR23, but not BLT1 receptors, are highly expressed in Mos/Mps and that RvE1 initiates ChemR23 receptor activation, signals phosphorylation and facilitates phagocytosis in human Mps (Ohira et al., 2010). Interestingly, ChemR23 receptors are highly expressed in human M1 Mps, but not in M2 Mps (Herova et al., 2015). In the present study, we observed that RvE1 suppressed Mo/Mp migration in the infarcted hearts and in vitro via ChemR23 receptors, thereby reducing the secretion of both pro‐inflammatory and pro‐resolving cytokines in the hearts of the mice at the early and late recovery stages post‐MI respectively. In ChemR23 receptor‐deficient mice, Mp recruitment was sharply reduced in zymosan‐induced peritonitis (Cash et al., 2008). Similarly, RvE1 also inhibits dendritic cell migration in the skin and attenuates inflammatory responses (Sawada et al., 2015), as ChemR23 receptors are abundantly expressed in dendritic cells (Arita et al., 2005).
In summary, RvE1 confers cardioprotection after acute MI at early stages through decreased recruitment of pro‐inflammatory Ly6Chi Mo/Mp to the infarcted areas but impairs cardiac recovery at late stages through the suppression of recruitment of Ly6Clow Mo/Mp which mediate repair and healing. These observations suggest that RvE1 could be used as an adjuvant therapeutic agent only during the early stages after acute MI.
Author contributions
G.L., C.D.F., H.Y. and Y.Y. designed the research associated with the project. G.L., Q.L. and Y.S. performed experiments. D.K., Y.G., B.T., G.C., S.G., J.L., S.Z., Y.Y., L.Z., B.Z. and C.D.F. provided important technical support. G.L., J.Z. and Y.Y. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primers used for RT‐PCR analysis.
Table S2 Effect of RvE1 treatment during different periods on cardiac healing after MI in mice.
Figure S1 Effect of early treatment of RvE1 (day 1–7) on CD68+ cell recruitment in hearts on day 14 after MI. (A) Representative CD68+ inflammatory cell immunofluorescence images of infarcted hearts from RvE1‐treated (day 1–7) mice on day 14 after MI. Scale bar, 50 μm. (B) Quantification of CD68+ inflammatory cells from A (mean ± SEM; PBS, n = 6; RvE1, n = 6).
Figure S2 ChemR23 expression levels in LPS‐treated (A) and IL‐4‐treated (B) murine Mps. ChemR23 expression were analysed by RT‐PCR (mean ± SEM, n = 6; *, P < 0.05).
Figure S3 Effect of knock down of ChemR23 or BLT1 receptors in cardiac fibroblasts on Mo/Mp migration. (A) Relative mRNA levels of ChemR23 and BLT1 receptors in cardiac fibroblasts (ChemR23, n = 6; BLT1, n = 6). (B) Knockdown efficiency of ChemR23 and BLT1 receptors in cardiac fibroblasts by siRNAs. Data are normalized to GAPDH levels (*P < 0.05 vs. Scramble siRNA; Scramble, n = 5; si‐ChemR23, n = 5; si‐BLT1, n = 5). (C) Mp transmembrane migration in co‐culture with cardiac fibroblasts pretreated with si‐ChemR23 or si‐BLT1 (n = 6). (D) Effect of RvE1 treatment on expression of matrix protein and cytokine mRNA in cardiac fibroblasts (n = 6). All plotted values are means ± SEM.
Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (81525004, 91439204, 31200860, 81400239 and 91639302), and the Shanghai Committee of Science and Technology (14JC1407400 and 15140902000). Y.Y. is a fellow at the Jiangsu Collaborative Innovation Center for Cardiovascular Disease Translational Medicine. C.D.F. holds a Canada Research Chair position and is supported by CIHR (MOP142476).
Liu, G. , Liu, Q. , Shen, Y. , Kong, D. , Gong, Y. , Tao, B. , Chen, G. , Guo, S. , Li, J. , Zuo, S. , Yu, Y. , Yin, H. , Zhang, L. , Zhou, B. , Funk, C. D. , Zhang, J. , and Yu, Y. (2018) Early treatment with Resolvin E1 facilitates myocardial recovery from ischaemia in mice. British Journal of Pharmacology, 175: 1205–1216. doi: 10.1111/bph.14041.
Contributor Information
Jian Zhang, Email: zhangjian_tina@163.com.
Ying Yu, Email: yuying@sibs.ac.cn, Email: yuying@tmu.edu.cn.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16:Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW et al (2003). Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 197: 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S et al (2005). Stereochemical assignment, antiinflammatory properties, and receptor for the omega‐3 lipid mediator resolvin E1. J Exp Med 201: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol 178: 3912–3917. [DOI] [PubMed] [Google Scholar]
- Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel‐Duby R et al (2014). Macrophages are required for neonatal heart regeneration. J Clin Invest 124: 1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Zhu X, Rong S, Shewale S, Seo J, Boudyguina E et al (2012). Omega‐3 fatty acids ameliorate atherosclerosis by favorably altering monocyte subsets and limiting monocyte recruitment to aortic lesions. Arterioscler Thromb Vasc Biol 32: 2122–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash JL, Hart R, Russ A, Dixon JP, Colledge WH, Doran J et al (2008). Synthetic chemerin‐derived peptides suppress inflammation through ChemR23. J Exp Med 205: 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou‐Khamis T et al (2005). CCL2/Monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 96: 881–889. [DOI] [PubMed] [Google Scholar]
- Frantz S, Nahrendorf M (2014). Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res 102: 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J et al (2016). Spatio‐temporal profile, phenotypic diversity, and fate of recruited monocytes into the post‐ischemic brain. J Neuroinflammation 13: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Lin M, Piao L, Li X, Yang F, Zhang J et al (2015). Aspirin enhances protective effect of fish oil against thrombosis and injury‐induced vascular remodelling. Br J Pharmacol 172: 5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herova M, Schmid M, Gemperle C, Hersberger M (2015). ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J Immunol 194: 2330–2337. [DOI] [PubMed] [Google Scholar]
- Hesselink JM, Chiosi F, Costagliola C (2016). Resolvins and aliamides: lipid autacoids in ophthalmology – what promise do they hold? Drug Des Devel Ther 10: 3133–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG et al (2014). Ly‐6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 114: 1611–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann U, Frantz S (2015). Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res 116: 354–367. [DOI] [PubMed] [Google Scholar]
- Jeon SH, Chae BC, Kim HA, Seo GY, Seo DW, Chun GT et al (2007). Mechanisms underlying TGF‐beta1‐induced expression of VEGF and Flk‐1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol 81: 557–566. [DOI] [PubMed] [Google Scholar]
- Kharraz Y, Guerra J, Mann CJ, Serrano AL, Munoz‐Canoves P (2013). Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm 2013: 491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Shen Y, Liu G, Zuo S, Ji Y, Lu A et al (2016). PKA regulatory IIalpha subunit is essential for PGD2‐mediated resolution of inflammation. J Exp Med 213: 2209–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kang JH, Boo HJ, Hwang SJ, Hong S, Lee SC et al (2015). STAT3‐mediated IGF‐2 secretion in the tumour microenvironment elicits innate resistance to anti‐IGF‐1R antibody. Nat Commun 6: 8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW et al (2012). Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209: 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JY, Park CK, Hwang SW (2015). Biological roles of resolvins and related substances in the resolution of pain. Biomed Res Int 2015: 830930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruotti N, Annese T, Cantatore FP, Ribatti D (2013). Macrophages and angiogenesis in rheumatic diseases. Vascular cell 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miro‐Mur F, Perez‐de‐Puig I, Ferrer‐Ferrer M, Urra X, Justicia C, Chamorro A et al (2016). Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun 53: 18–33. [DOI] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J et al (2010). Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70: 5728–5739. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK (2013). Monocyte and macrophage heterogeneity in the heart. Circ Res 112: 1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL et al (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204: 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa N, Kobayashi Y (2009). Total synthesis of resolvin E1. Tetrahedron Lett 50: 6079–6082. [Google Scholar]
- Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE, Serhan CN (2010). Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem 285: 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Honda T, Hanakawa S, Nakamizo S, Murata T, Ueharaguchi‐Tanada Y et al (2015). Resolvin E1 inhibits dendritic cell migration in the skin and attenuates contact hypersensitivity responses. J Exp Med 212: 1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro‐resolving lipid mediators are leads for resolution physiology. Nature 510: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2017). Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J 31: 1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G et al (2002). Resolvins. J Exp Med 196: 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde AV, Frangogiannis NG (2014). Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 70: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla DK, Singla R, Wang J (2016). BMP‐7 treatment increases M2 macrophage differentiation and reduces inflammation and plaque formation in Apo E−/− mice. PLoS One 11: e0147897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Zimmermann HW (2014). Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 60: 1090–1096. [DOI] [PubMed] [Google Scholar]
- Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S et al (2009). Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med 13: 3485–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T et al (2009). Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol 54: 130–138. [DOI] [PubMed] [Google Scholar]
- Vlasov IA, Volkov AM (2004). Dependence of heart weight on body weight in patients with cardiovascular diseases. Fiziol Cheloveka 30: 62–68. [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Zhou Y, Liu J, Qian L (2015). Improved generation of induced cardiomyocytes using a polycistronic construct expressing optimal ratio of Gata4, Mef2c and Tbx5. J Vis Exp. https://doi.org/10.3791/53426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J et al (2013). Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62: 24–35. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M et al (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Fan J, Chen XS, Wang D, Klein‐Szanto AJ, Campbell RL et al (2006). Genetic model of selective COX2 inhibition reveals novel heterodimer signaling. Nat Med 12: 699–704. [DOI] [PubMed] [Google Scholar]
- Zhong GQ, Tu RH, Zeng ZY, Li QJ, He Y, Li S et al (2014). Novel functional role of heat shock protein 90 in protein kinase C‐mediated ischemic postconditioning. J Surg Res 189: 198–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used for RT‐PCR analysis.
Table S2 Effect of RvE1 treatment during different periods on cardiac healing after MI in mice.
Figure S1 Effect of early treatment of RvE1 (day 1–7) on CD68+ cell recruitment in hearts on day 14 after MI. (A) Representative CD68+ inflammatory cell immunofluorescence images of infarcted hearts from RvE1‐treated (day 1–7) mice on day 14 after MI. Scale bar, 50 μm. (B) Quantification of CD68+ inflammatory cells from A (mean ± SEM; PBS, n = 6; RvE1, n = 6).
Figure S2 ChemR23 expression levels in LPS‐treated (A) and IL‐4‐treated (B) murine Mps. ChemR23 expression were analysed by RT‐PCR (mean ± SEM, n = 6; *, P < 0.05).
Figure S3 Effect of knock down of ChemR23 or BLT1 receptors in cardiac fibroblasts on Mo/Mp migration. (A) Relative mRNA levels of ChemR23 and BLT1 receptors in cardiac fibroblasts (ChemR23, n = 6; BLT1, n = 6). (B) Knockdown efficiency of ChemR23 and BLT1 receptors in cardiac fibroblasts by siRNAs. Data are normalized to GAPDH levels (*P < 0.05 vs. Scramble siRNA; Scramble, n = 5; si‐ChemR23, n = 5; si‐BLT1, n = 5). (C) Mp transmembrane migration in co‐culture with cardiac fibroblasts pretreated with si‐ChemR23 or si‐BLT1 (n = 6). (D) Effect of RvE1 treatment on expression of matrix protein and cytokine mRNA in cardiac fibroblasts (n = 6). All plotted values are means ± SEM.