

Abstract
The endoplasmic reticulum (ER) serves several essential cellular functions including protein synthesis, protein folding, protein translocation, calcium homoeostasis and lipid biosynthesis. Physiological or pathological stimuli, which disrupt ER homoeostasis and disturb its functions, lead to an accumulation of misfolded and unfolded proteins, a condition referred to as ER stress. ER stress triggers the unfolded protein response to restore the homoeostasis of ER, through activating transcriptional and translational pathways. However, prolonged ER stress will lead to cell dysfunction and apoptosis. Recent evidence revealed that ER stress is involved in the development and progression of various heart diseases, such as cardiac hypertrophy, ischaemic heart diseases and heart failure. Therefore, improved understanding of the molecular mechanisms of ER stress in heart disease will help to investigate more potential targets for new therapeutic interventions and drug discovery.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- AAV
Adeno‐associated virus
- ASK1
apoptosis signal‐regulating kinase 1
- ATF4
activating transcription factor 4
- Bip
immunoglobulin‐binding protein
- CaMKII
calcium/calmodulin‐dependent protein kinase II
- CHOP
CCAAT/enhancer‐binding homologous protein
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERAD
ER‐associated degradation
- ERO1
ER oxidoreductase‐1
- ERSE
ER stress response elements
- GK
Ginkgolide K
- GRP78
glucose‐regulated protein 78
- I/R
ischaemia–reperfusion
- IRE1
inositol‐requiring kinase 1
- MI
myocardial infarction
- PDI
protein disulphide isomerase
- PERK
dsRNA‐activated protein kinase‐like ER kinase
- PUMA
p53 up‐regulated modulator of apoptosis
- RIDD
IRE1‐dependent decay of mRNA
- RyR2
ryanodine receptor 2
- SCN5A
sodium channel α‐subunit 5
- SERCA2a
sarco/ER Ca2+ ATPase 2a
- SERCA3f
sarco/ER Ca2+ ATPase isoform 3f
- sXBP1
spliced‐XBP1
- TUDCA
tauroursodeoxycholic acid
- UPR
unfolded protein response
- XBP1
X‐box binding protein 1
Introduction
The endoplasmic reticulum (ER) is a multifunctional intracellular organelle and the primary site of the secretory pathway. It is essential for protein synthesis, protein folding, protein translocation, calcium homoeostasis and lipid biosynthesis. The concentration of proteins and the protein synthesis rate are extremely high within the ER lumen (Stevens and Argon, 1999). The homoeostasis within the ER lumen must be carefully maintained for folding proteins properly. Perturbations of this homoeostasis by physiological or pathological stimuli lead to an accumulation of misfolded and unfolded proteins, a process known as ER stress. ER stress activates complex signalling pathways that deal with the misfolded and unfolded proteins, referred as the unfolded protein response (UPR). UPR activates transcriptional and translational pathways to reduce the rate of general translation and increase the expression of ER resident protein chaperones and protein foldases. ER‐associated degradation (ERAD) is also activated by UPR to clear irreparably misfolded proteins. However, if the UPR fails to reduce ER stress and restore homeostasis, ER stress causes cell dysfunction and apoptosis. Recently, ER stress has received substantial attention and is thought to play an essential role in the development and progression of many human diseases, including cardiovascular diseases, diabetes mellitus, neurodegenerative diseases and liver diseases (Oyadomari et al., 2002; Lindholm et al., 2006; Minamino and Kitakaze, 2010). This review focuses on the molecular mechanisms of ER stress in cardiovascular diseases and the potential therapeutic targets in the process of ER stress.
UPR signalling pathway
In normal conditions, the ER‐resident transmembrane proteins, ATF6, inositol‐requiring kinase 1 (IRE1) and PERK (dsRNA‐activated protein kinase‐like ER kinase), are bound with the ER chaperone, immunoglobulin‐binding protein (Bip)/glucose‐regulated protein 78 (GRP78), to maintain their inactive state (Lee, 2005). When unfolded proteins accumulate in the ER, Bip dissociates from those three sensors to initiate their activity (Bertolotti et al., 2000). The activated UPR then regulates downstream effectors to increase folding and handling efficiency by up‐regulation of ER chaperones, reduce ER workload through attenuation of translation and eliminate unwanted proteins via induction of ERAD.
IRE1α is a transmembrane kinase (Groenendyk et al., 2010), and it is activated by homodimerization and autophosphorylation after release from Bip/GRP78. Activated IRE1α cleaves X‐box binding protein 1 (XBP1) mRNA to initiate translation of transcriptionally active spliced‐XBP1 (sXBP1). Active sXBP1 binds to a variety of UPR‐target gene promoters to up‐regulate a range of ER stress response elements (ERSE) to restore ER homoeostasis and promote cytoprotection.
ATF6α is a 90 kDa ER transmembrane protein under normal conditions but activated ATF6α translocates from ER to the Golgi to be cleaved by site‐1 and site‐2 protease. The cleaved N‐terminal of ATF6α, a 50KDa fragment, migrates from the cytosol into the nucleus to combine with several b‐Zip transcription factors and ERSE for transcriptional induction of several UPR related genes, including CCAAT/enhancer‐binding homologous protein (CHOP), Bip and XBP1 (Haze et al., 1999). In addition, there is an isoform of ATF6 called ATF6β. ATF6β is nonessential for transcriptional induction of ER chaperones (Wu et al., 2007) and may even inhibit ATF6α activity (Thuerauf et al., 2004). Recently, ATF6β has been shown to play a pro‐survival role in chronic ER stress through induction of Wfs1 (Odisho et al., 2015).
PERK is also activated by homodimerization and autophosphorylation. The activated PERK phosphorylates Ser51 on the α‐subunit of the eukaryotic initiation factor 2α (eIF2α) to prevent the formation of translational initiation complexes, which leads to attenuation of cap‐dependent protein translation (Bertolotti et al., 2000). This transient translational arrest helps to recover ER homoeostasis through the reduction of protein synthesis. Meanwhile, eIF2α phosphorylation also induces the translation of the mRNA encoding activating transcription factor 4 (ATF4) to decrease unfolded proteins level in the ER through activation of various UPR genes (Figure 1).
Figure 1.
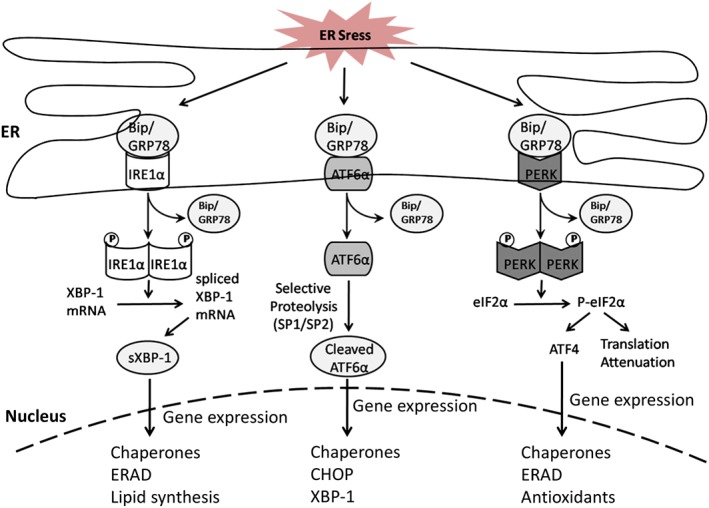
UPR signalling pathway. The three ER‐resident transmembrane proteins, ATF6, IRE1 and PERK, associate with Bip in their inactive state under normal condition. In response to ER stress, these sensors are released and activated. Both IRE1 and PERK are oligomerized and autophosphorylated. Phosphorylated IRE1 catalyses the splicing of XBP1 mRNA leading to the generation of UPR‐target transcription factor. Activated PERK phosphorylates eIF2α leading to general translational attenuation and increased expression of ATF4. ATF6 is cleaved by SP1/SP2, and the cytosolic fragment of ATF6 migrates to the nucleus. The downstream effectors of these three signalling pathways, in combination, induce the expression of proteins, which can help restore the ER protein folding capacity. ERAD is accelerated to remove terminally misfolded proteins.
ER stress‐induced apoptosis
When the UPR fails to correct the protein‐folding defect for the recovery of ER homoeostasis, the apoptotic signalling pathway is activated. Although all of the UPR sensor proteins are involved in ER stress‐induced apoptosis, it is unclear how the cell decides to commit to death in response to excessive ER stress.
IRE1α mediates apoptosis by interaction with the adaptor molecule TNF‐receptor‐associated factor 2 (TRAF2) and apoptosis signal‐regulating kinase 1 (ASK1), which leads to the activation of JNK and p38 (Urano et al., 2000; Nishitoh et al., 2002). p38 activates CHOP through phosphorylation of its transactivation domain (Wang and Ron, 1996). Both p38 and JNK can phosphorylate the proapoptotic protein Bax to induce its activity (Kim et al., 2006). The association of IRE1α and TRAF2 has also been suggested to induce the activation of caspase‐12 (Nakagawa et al., 2000; Saleh et al., 2006). Caspase‐12 activates caspase‐9 to induce the activation of caspase‐3, which leads to apoptosis. In addition, the IRE1α/TRAF2 complex can also recruit the IκB leading to the activation of NF‐κB (Kaneko et al., 2003), linking ER stress and inflammation. Recently, the regulated IRE1‐dependent decay of mRNA (RIDD) was shown to promote degradation of a number of mRNAs encoding ER‐targeted proteins to reduce a load of incoming proteins during ER stress (Han et al., 2009; Hollien et al., 2009). Under irremediable ER stress, prolonged activation of RIDD degrades mRNAs encoding prosurvival proteins to induce apoptosis (Hollien and Weissman, 2006; Maure et al., 2013). The activation of RIDD requires IRE1 phosphotransfer activity (Son et al., 2014) but the mechanisms involved are poorly understood and require further investigation. Furthermore, IRE1α also acts as an essential factor in calcium homoeostasis disruption induces apoptosis via the inositol‐1,4,5‐trisphosphate (IP3) receptor (Son et al., 2014).
CHOP is a basic leucine zipper‐containing transcription factor, which is regulated by ATF6 and PERK pathways. CHOP can inhibit the expression of the anti‐apoptotic protein Bcl‐2 to induce apoptosis (McCullough et al., 2001). In response to ER stress, CHOP also can directly activate the transcription of Bim leading to the induction of apoptosis (Puthalakath et al., 2007). In addition, CHOP mediates the transcriptional induction of several genes that encode numerous proapoptotic proteins, such as GADD34, death receptor 5, ER oxidoreductase‐1 (ERO1) and carbonic anhydrase VI (Malhotra and Kaufman, 2007). CHOP activates GADD34 to promote dephosphorylation of eIF2α reversing the translational attenuation (Novoa et al., 2009). This accumulates unfolded proteins in the ER and increases the translation of proapoptotic proteins. The activation of ERO1 by CHOP promotes apoptosis by hyperoxidization of the ER and activation of IP3 receptors (Li et al., 2009a,b). Furthermore, recent evidence suggested that CHOP can interact with ATF4 to increase protein synthesis leading to ATP depletion, oxidative stress and cell apoptosis (Han et al., 2013).
Recently, Bcl‐2 family proteins have been suggested to induce apoptosis by calcium signalling during ER stress (Szegezdi et al., 2009). In response to ER stress, proapoptotic Bcl‐2 family proteins, Bak and Bax, undergo a conformational change in the ER membrane to release calcium into the cytoplasm (Scorrano et al., 2003). Released calcium activates the calcium‐dependent protease m‐calpain to cleave procaspase‐12 leading to caspase‐12 activation (Nakagawa and Yuan, 2000). In addition, caspase‐12 can also be activated by the translocation of Bim to the ER membrane in response to ER stress (Morishima et al., 2004). In cardiomyocytes, a Bcl‐2 family protein NIX, which localizes both to the ER and mitochondrial membrane, can induce apoptosis by modulating calcium in the ER in coordination with Bax and Bak (Diwan et al., 2009). Additionally, Bax and Bak can directly associate with IRE1α to induce the activation of the IRE1α signalling pathway (Hetz et al., 2006).
A recent study found that calcium/calmodulin‐dependent protein kinase II (CaMKII) was activated by ER‐released calcium and it acted as a unifying link between ER stress and mitochondrial apoptosis (Ozcan and Tabas, 2010). In response to oxidative stress, CaMKII was activated to mediate ER stress‐induced cardiac dysfunction and apoptosis (Roe and Ren, 2013).
Additionally, ER stress can also trigger necroptosis. Necroptosis is a regulated form of necrosis that involves the activation of receptor‐interacting protein kinase 1 (RIPK1), RIPK3 and mixed lineage kinase domain‐like protein (MLKL) (Vanden Berghe et al., 2015) and has been suggested as another important cell death mediator in cardiomyocytes. Studies using the RIPK1 inhibitor necrostatin‐1 and RIPK3‐deficient mice have shown that inhibition of necroptotic cell death has cardioprotective effects in mouse models of ischaemic injury (Koshinuma et al., 2014; Zhang et al., 2016). Interestingly, induction of ER stress by brefeldin‐A, thapsigargin and tunicamycin triggers RIPK1 kinase‐dependent necroptosis (Saveljeva et al., 2015), and activation of GRP78 and eIF2α has been linked to necroptosis in macrophages/microglia after mouse spinal cord contusion (Fan et al., 2015), further confirming the involvement of ER stress in cardiomyocyte cell death and related pathologies (Figure 2).
Figure 2.
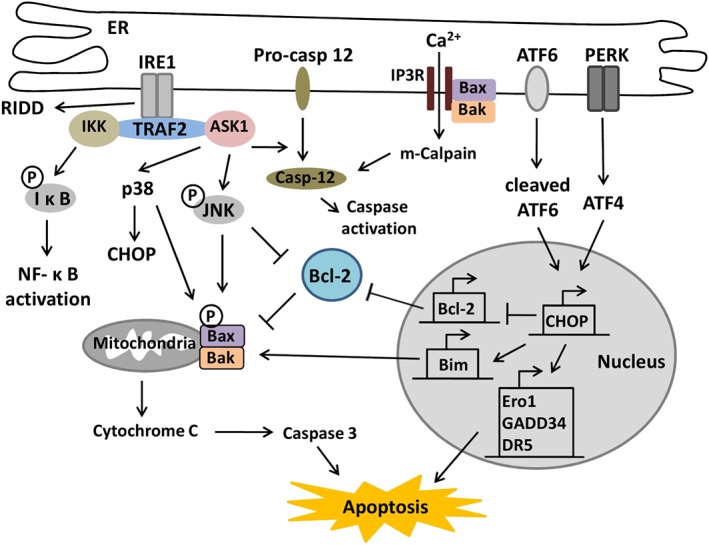
ER stress‐induced apoptosis. Activated IRE1 recruits TRAF2 and ASK1 leading to the activation of JNK and p38 and also the release of procaspase‐12 from the ER. IRE1/TRAF2 also recruits IκB for the activation of NFκB. In addition, IRE1 induces RIDD leading to apoptosis. During ER stress, Bax and Bak in the ER membrane undergo conformational change to release calcium into the cytoplasm, which activates m‐calpain and caspase 12 leading to the activation of the caspase cascade. ATF6 and PERK activate CHOP to inhibit the expression of Bcl‐2 and activate Bim leading to apoptosis. Activated CHOP increases the expression of GADD34, DR5 and Ero1 to induce apoptosis.
ER stress in cardiac hypertrophy and heart failure
In failing hearts, the ER is overloaded and ER stress can be induced by enhanced protein synthesis, oxidative stress and hypoxia (Maron et al., 1975). In 2004, GRP78 was first found to be increased in hearts from patients with heart failure, suggesting that UPR activation is induced in this condition (Okada et al., 2004). In addition, extensive splicing of XBP1 was found in these patients and XBP1 has been shown to be a regulator of brain natriuretic peptide in cardiomyocytes (Sawada et al., 2010). CHOP knockout mice developed less cardiac hypertrophy, fibrosis and cardiac dysfunction compared with wild‐type mice after TAC, suggesting CHOP may also contribute to the transition from cardiac hypertrophy to heart failure (Fu et al., 2010). The inhibition of GADD34 was lost after CHOP deletion, which leads to the increased phosphorylation of eIF2α and decreased global translation (Harding et al., 2000). This could explain the mechanism of how CHOP deletion contributes to preventing the development of cardiac hypertrophy.
Heart failure can also be induced by protein accumulation. The Lys‐Asp‐Glu‐Leu (KDEL) receptor is a retrieval receptor for ER chaperones in the early secretory pathway. Transgenic mice expressing a mutant KDEL receptor, which disturbs the recycling and protein quality control in the ER, showed aggregation of misfolded proteins, increased expression of CHOP and apoptosis in mutant hearts (Hamada et al., 2004). Furthermore, rabbits immunized against β‐adrenoceptors exhibited left ventricular dilation, systolic dysfunction and cardiomyocyte apoptosis in association with enhanced expression of GRP78 and CHOP (Mao et al., 2007). These findings suggest that ER stress plays a critical role in dilated and autoimmune cardiomyopathy. In cultured rat ventricular myocytes, stimulation of β‐adrenoceptors activated ER stress to induce apoptosis (Dala et al., 2012). Antagonists of β‐adrenoceptors can ameliorate ER stress to attenuate cardiac hypertrophy and heart failure (Ni et al., 2011).
ASK1 plays an essential role in ER stress‐induced apoptosis. ASK1 knockout mice exhibited less cardiac dysfunction and reduced cardiomyocytes apoptosis after 4 weeks TAC (Yamaguchi et al., 2003). Meanwhile, neonatal cardiomyocytes with ASK1 deletion showed resistance to H2O2‐induced apoptosis (Yamaguchi et al., 2003). These results suggest that ASK1 could be involved in the development of heart failure. Mice with cardiac‐specific and inducible overexpression of ASK1 showed greater TUNEL but no increase in myocyte or whole organ hypertrophy after 8 weeks TAC (Liu et al., 2009). Thus, ASK1 is a key regulator of cardiomyocyte apoptosis but not hypertrophy. Additionally, a small molecule inhibitor of ASK1 can reduce cardiomyocyte apoptosis and myocardial infarct size in a rat ischaemia/reperfusion model (Gerczauk et al., 2012).
Prostatic androgen‐repressed message‐1 (PARM‐1) is a transmembrane protein, which specifically expressed in hearts and skeletal muscles and predominantly localized in ER. Stimulation of PARM‐1 and enhanced expression of GRP78 and CHOP were observed in Dahl salt‐sensitive rats with cardiac hypertrophy and heart failure (Isodono et al., 2010). In response to ER stress stimuli, cardiomyocytes with silencing PARM‐1 showed enhanced apoptosis, repressed expression of ATF6 and PERK and increased induction of CHOP (Isodono et al., 2010). These results indicate that PARM‐1 act as an antiapoptotic protein to protect the heart from heart failure through regulating the expression of ATF6, PERK and CHOP.
Recently, PERK has been demonstrated to play a cardioprotective role in heart failure. The inducible cardiac‐specific PERK knockout mice showed enhanced cardiac dysfunction, fibrosis and apoptosis compared with wild‐type mice after TAC (Liu et al., 2014a,b). In PERK knockout heart, the expression of CHOP was increased in response to TAC, which indicates that CHOP‐induced apoptosis may contribute to heart failure. The ATPase SERCA2a, a key regulator of calcium homoeostasis in cardiomyocytes, was significantly reduced in PERK knockout mice after TAC (Mekahli et al., 2011). Previous studies demonstrated that reduction of SERCA2a activity can induce ER calcium depletion leading to ER stress (Mekahli et al., 2011) and disruption of the SERCA2 gene in cardiomyocytes causes ER stress and promotes heart failure (Liu et al., 2011). Therefore, maintaining the expression of SERCA2a by PERK is probably an important mechanism to protect the heart from heart failure. In addition, the isoform SERCA3f was also up‐regulated in human failing hearts (Dally et al., 2009). Remarkably, overexpression of SERCA3f induces the increased expression of GRP78 and XBP1 splicing in cardiomyocytes (Dally et al., 2009). Thus, SERCA3f may be involved in the mechanism(s) of ER stress leading to heart failure.
Chronic alcohol consumption is a risk factor of cardiac hypertrophy (Piano, 2002), and recent evidence has demonstrated that it can lead to ER stress. Friend virus‐B‐type mice chronically fed with alcohol showed the development of cardiac hypertrophy in association with increased expression of GRP78, CHOP and IRE1α (Li and Ren, 2008). In addition, mice with cardiac‐specific overexpression of alcohol dehydrogenase exhibited enhanced expression of GRP78, IRE1α and CHOP compared with wild‐type mice in response to chronic alcohol treatment (Li and Ren, 2008). Meanwhile, mice overexpressing aldehyde dehydrogenase‐2, an enzyme which metabolizes acetaldehyde, showed less cardiac hypertrophy and significantly reduced expression of GRP78, IRE1α and CHOP (Li et al., 2009a,b). Taken together, these results suggest that acetaldehyde may induce ER stress and cardiac hypertrophy in response to chronic alcohol consumption.
In 2015, XBP1 was first shown to be a critical angiogenic factor for maintaining normal cardiac function in the early stage of cardiac hypertrophy, and its activity was inhibited by miR‐214 and miR‐30* (recently designated as the miR‐30‐3p family) in hypertrophic and failing hearts (Duan et al., 2015). Further, XBP1 regulated the expression of VEGFA in cardiomyocytes, which is consistent with the binding of XBP1s to the promoter of VEGFA, in response to ER stress (Ghosh et al., 2010). This finding suggests that XBP1 plays a cardioprotective role through promoting VEGF mediated‐cardiac angiogenesis. Thus, miR‐214 and miR‐30* inhibit the expression of XBP1 and VEGFA in the progression from adaptive hypertrophy to heart failure.
A recent study showed that 4‐phenylbutyric acid (4‐PBA) could prevent TAC‐induced cardiac hypertrophy through attenuation of ER stress (Luo et al., 2015). After mechanical unloading with a left ventricular assist device, heart failure patients showed reduced expression of ER stress markers and improved expression of Ca2+ cycling proteins (Castillero et al., 2015). These findings indicate that mechanical unloading contributes to reverse remodelling of the failing heart in association with the restoration of ER homoeostasis.
ER stress in ischaemic heart disease
Ischaemia can lead to the impairment of protein folding in the ER, resulting in the activation of the UPR (Glembotski, 2008). Activation of the UPR has been observed in ischaemic heart disease, and ER stress appears to mediate the progression of ischaemic cardiomyopathy in a range of models from mice to humans (Azfer et al., 2006; Szegezdi et al., 2006). The increase in UPR activation by ischaemia is shown by early expression of XBP1, eIF2α and ATF6 with the consequent activation of ATF4 and GRP78 in cardiomyocytes to restore ER homoeostasis and efficient protein folding (Azfer et al., 2006). However, under prolonged ischaemia and ischaemia–reperfusion (I/R) injury, persistent UPR activation leads to apoptosis by promoting the activation of JNK, cleaved caspase‐3, caspase‐12, p53 up‐regulated modulator of apoptosis (PUMA) and CHOP contributing to the onset of heart failure (Thuerauf et al., 2006).
The IRE1 branch of the UPR appears to have a protective role in ischaemia. In vitro and in vivo experimental models of ischaemia show a robust induction of XBP1 and GRP78 (Thuerauf et al., 2006; Qi et al., 2007). Importantly, increased expression of both XBP1 and GRP78 is also found in the ischaemic human heart (Sawada et al., 2010). In addition, in mice subjected to I/R injury, cardiomyocyte‐specific deletion of XBP1 shows an increase in myocardial infarct size, impairment in cardiac function and hypertrophic remodelling. Conversely, transgenic mice overexpressing of XBP1s, show reduced infarct size and significant improvement of cardiac function after I/R injury, further highlighting the protective role of XBP1 in cardiomyocytes (Wang et al., 2014). Similarly, overexpression of GRP94 (HSP90B1), a downstream target of XBP1, reduces cardiomyocyte cell death induced by calcium overload and simulated ischaemia (Vitadello et al., 2003).
Activation of the PERK branch of the UPR is also observed under ischaemic conditions (Szegezdi et al., 2006). Phosphorylation of eIF2α is an early event observed in cardiomyocytes after ischaemia in vitro and after I/R in vivo (Szegezdi et al., 2006; Miyazaki et al., 2011). PERK overexpression promotes cell survival under hypoxic conditions, and PERK down‐regulation or the expression of a dominant‐negative mutant leads to decrease in cell viability (Lu et al., 2004). However, when the activation of PERK is prolonged, cardiomyocyte cell death is triggered. In heart cells, persistent ER stress induced by ischaemia promotes the activation of the PERK/ATF4/CHOP axis (Mughal and Kirshenbaum, 2011). Furthermore, CHOP deficiency has been shown to reduce reperfusion injury in a mouse model of myocardial infarction (MI) (Terai et al., 2005). Silencing of both CHOP and caspase‐12 shows cardioprotective effects following exposure to hypoxia (Terai et al., 2005). CHOP expression and cleavage of caspase‐12 can both be inhibited by the activation of AMP‐activated protein kinase (Nickson et al., 2007), highlighting the importance of ER stress‐induced apoptosis in hypoxic conditions. In addition, the pro‐apoptotic member of the Bcl‐2 family PUMA, also a downstream effector of PERK, is an important regulator of the ischaemia/reperfusion (I/R)‐induced cell death under ER stress in cardiomyocytes. Overexpression of PUMA induces cell apoptosis in cardiomyocytes under ER stress, and PUMA deletion is protective after I/R injury in vitro and in vivo (Nickson et al., 2007). Furthermore, I/R injury‐associated apoptosis and myocardial dysfunction can be also alleviated by 4‐PBA by attenuating the onset of ER stress through the inhibition of GRP78 expression and PERK phosphorylation (Jian et al., 2016). Another downstream effector of PERK, Tribbles3, is elevated following myocardial infarction (MI) and cardiomyocyte‐specific overexpression of Tribbles3 in mice shows increased pathological cardiac remodelling and apoptosis after MI (Avery et al., 2010). Thus, sustained activation of PERK and, consequently, triggering of the ER stress‐initiated apoptotic signalling mediates cell death in I/R myocardium, playing an overall negative role in the ischaemic heart.
The activation of the ATF6 branch of the UPR appears to have a cardioprotective role. Primary cardiomyocytes show ATF6 activation under hypoxia and nutrient deprivation conditions (Doroudgar et al., 2009), and ATF6‐inducible genes show cardioprotection under stress, including I/R. Consistent with this, cardiac‐specific expression of active ATF6 in mice shows increased expression of ER stress‐inducible mRNAs and proteins including GRP78 and GRP94 and protection against tissue damage, necrosis and apoptosis after I/R injury (Martindale et al., 2006). Moreover, expression of a dominant‐negative ATF6 or using ATF6‐targeted miRNA significantly decreases the induction of GRP78 and increases cardiomyocyte death upon simulated reperfusion (Doroudgar et al., 2009). Additionally, in mice after MI, inhibition of ATF6 activation impairs cardiac function and increased mortality, further demonstrating the protective role of ATF6 (Toko et al., 2010). Also, ATF6 is protective in heart tissue, by induction of ERAD, promoting the degradation of terminally misfolded protein in the ER (Nakatsukasa et al., 2008). The ability to clear misfolded proteins from the ER appears to be especially critical during ischaemic stress. One of the early ERAD components, Derlin‐3, a retro translocation channel, is induced by ATF6 in the heart (Belmont et al., 2010). Overexpression of Derlin‐3 enhances the clearance of misfolded proteins and attenuates ER stress activation and caspase activity, protecting cardiomyocytes from ischaemia‐induced apoptosis. Conversely, genetic deletion or knockdown of Derlin‐3 impairs the clearance of misfolded proteins and shows an increase in cell death after simulated I/R (Belmont et al., 2010).
The cardioprotective effects of the UPR can be attributed to the induction of ER chaperones and the consequent enhancement of protein folding (Glembotski, 2008). Hypoxia impairs disulphide bond formation resulting in oxidative protein misfolding in the ER in which protein disulphide isomerase (PDI), a marker of the UPR, plays a key role (Glembotski, 2008). In human hearts, PDI acts as a cardiomyocyte survival factor in ischaemic cardiomyopathy (Glembotski, 2008), and an increase of PDI is observed in the viable peri‐infarcted myocardium. Adenoviral‐mediated expression of PDI results in a significant decrease in infarct size and decrease in pathological remodelling with improvements in contractility and shows reduced cardiomyocyte apoptosis in the peri‐infarct region in an in vivo mouse model of MI (Severino et al., 2007).
ER stress in arrhythmias
Over the last decades, new evidence has emerged for the involvement of ER stress in arrhythmias. Two cardiac cation channels, Nav1.5 and Kv4.3 were inhibited by activation of PERK (Gao et al., 2013). In human heart failure, the gene for the α‐subunit of cardiac Nav1.5 channels (SCN5A), was abnormally spliced and resulted in truncated mRNA variants. The truncated mRNA variants translated into nonfunctional channel proteins and were trapped in the ER inducing the activation of UPR to down‐regulate the protein expression of the full‐length Na+ channel (Gao et al., 2013). In addition, inhibition of PERK prevented the degradation of full‐length SCN5A mRNA and the reduction in Na+ currents, suggesting that PERK activation could induce Na+ current reduction through destabilization of full‐length SCN5A mRNA to contribute to arrhythmic risk (Gao et al., 2013). The effect of PERK activation was not specific to cardiac Nav1.5 channels. Blocking PERK also prevented the down‐regulation of the gene for the α‐subunit of cardiac Kv4.3 channels (Liu and Dudley, 2015). Cardiac Kv4.3 channels contribute to the cardiac transient outward potassium current (Ito), which is the main contributor to the repolarizing phase 1 of the cardiac action potential. Therefore, PERK activation could reduce Ito, resulting in shortening of the cardiac action potential duration and phase 2 reentry (Liu and Dudley, 2015). Thus, inhibition of PERK may reverse the down‐regulation of arrhythmogenic ion channels to prevent arrhythmias.
In rats with diabetic cardiomyopathy, inhibition of PERK in association with reduced activity of calcineurin decreased arrhythmias (Liu et al., 2014a,b). An in vitro study also demonstrated that PERK activation was required in the activation of calcineurin and the dissociation of the FK506 binding protein 1B, 12.6 kDa from the ryanodine receptor 2 (RyR2) (Liu et al., 2014a,b). Thus, PERK activated calcineurin to facilitate degradation of FKBP‐RyR2 complex leading to intracellular calcium accumulation, which might be a mechanism inducing arrhythmias. ER‐dependent ion channel glycosylation could be another mechanism contributing to cardiac arrhythmias. A recent study indicated that only the fully glycosylated form of Nav1.5 channel protein was trafficked normally (Mercier et al., 2015). Hence, altered glycosylation during ER stress might be involved in alterations of ion channels and induction of cardiac arrhythmias.
UPR as a therapeutic target in cardiac diseases
Pharmacological agents that directly modulate the UPR are emerging as promising tools towards effective treatment of cardiovascular diseases. It has been shown that salubrinal, an eIF2α phosphatase inhibitor, significantly increases GRP78 expression and appears to be protective against ER stress‐induced cardiomyocyte apoptosis in a rat MI model (Li et al., 2015). SIRT1‐activating compounds such as resveratrol appear to have protective roles in cardiovascular disease (Hubbard and Sinclair, 2014). Interestingly, activation of SIRT1 prevents cardiomyocytes ER stress‐induced apoptosis through eIF2α deacetylation (Prola et al., 2017), supporting the therapeutic potential of SIRT1 activators for the treatment of cardiac pathologies associated with ER stress. However, it is important to consider that, as discussed above, the UPR response participates in both protective and proapoptotic responses and that very little is known about the mechanistic aspects of the switch from pro‐survival to pro‐apoptosis. In this context, the kinase inhibitor sunitinib can directly activate IRE1 with the consequent activation of XBP1 and reduction of ER stress. However, in patients with previous history of hypertension and heart disease, sunitinib appears to increase the risk for cardiovascular disease (Chu et al., 2007). Recently, Ginkgolide K (1,10‐dihydroxy‐3,14‐didehydroginkgolide, GK), a diterpene lactone isolated from the leaves of Ginkgo biloba, has been demonstrated to protect cardiomyocytes from ER stress‐induced apoptosis both in vitro and in vivo (Wang et al., 2016). In response to ER stress, GK can selectively activate the IRE1α/XBP1 pathway and inhibit the activation of RIDD and JNK (Wang et al., 2016). Therefore, GK has a potential for treating cardiovascular diseases.
Pharmacological alleviation of ER stress can also be achieved by stabilizing and rescuing protein misfolding using chemical chaperones that mimic ER chaperones (Perlmutter, 2002). Two such compounds, 4‐PBA and tauroursodeoxycholic acid (TUDCA), have been approved by the FDA for clinical use, and demonstrate the opportunity for pharmacological treatment. Oral administration of PBA in mice reduced ER stress and apoptosis and reduced pressure‐overload cardiac hypertrophy, after TAC (Park et al., 2012). Interestingly, PBA prevented doxorubicin‐induced cardiac injury and isoprenaline‐induced cardiac fibrosis, further highlighting its potential as a cardioprotective drug (Ayala et al., 2012). Additionally, TUDCA appears to restore the reduced contractile function in mouse cardiomyocytes under oxidative stress (Guo et al., 2009). Thus, by relief of ER stress, chemical chaperones can play a protective role against cardiac hypertrophy and, potentially, heart failure. Similarly, ROS‐induced ER stress can be prevented by curcumin and masoprocol through GRP94 induction, reduced caspase‐12 activation and preservation of PDI integrity (Pal et al., 2010). However, it is important to note that PBA treatment unexpectedly led to a higher mortality, promoted cardiac hypertrophy and dysfunction in mice after TAC. This is in part explained by actions of PBA outside its role as a chemical chaperone (Ma et al., 2016). Thus whereas alleviation of ER stress shows protection in the heart, ‘off target’ effects of the compounds used as chaperones have to be taken into consideration.
The increased expression of CHOP found in pressure‐overloaded hearts also appears as an attractive target for inhibition of ER stress‐induced apoptosis. As there are no direct pharmacological agents targeting CHOP, the indirect modulation of its activity appears as a promising therapeutic strategy. The statin atorvastatin, used for prevention of cardiovascular disease, was shown to decrease the expression of caspase‐12 and CHOP and decrease cardiomyocyte apoptosis in a post‐MI‐induced heart failure model (Song et al., 2011). The decrease in CHOP expression and phosphorylation was also observed after treatment with SP600125, a JNK inhibitor, which prevents CHOP up‐regulation under cyclic stretching in cardiomyocytes (Cheng et al., 2009). Similarly, angiotensin AT1 receptor antagonists reduce apoptosis and cardiac hypertrophy by attenuation of ER stress‐mediated apoptosis. Telmisartan prevented the increase in GRP78, CHOP, caspase‐12 and p‐JNK in rats after abdominal aortic constriction, and olmesartan decreased the expression of GRP78, caspase‐12 and p‐JNK in cardiomyocytes from rats with heart failure (Sukumaran et al., 2011). Additionally, calcitriol and paricalcitol, agonists of Vitamin D receptors, were protective in MI/R injury by inhibition of caspase‐12 and CHOP expression in mice (Yao et al., 2015). This information supports the potential use of inhibitors of ER stress‐induced apoptosis, in pressure‐overloaded hearts, as a novel therapeutic approach.
Recently, cardiac‐specific gene transfer has appeared as a novel therapeutic approach in the treatment of heart disease (Zacchigna et al., 2014). The use of Adeno‐associated virus (AAV) has become one of the most promising gene transfer tools for gene therapy, and understanding the molecular basis of myocardial dysfunction has allowed the development of AAV‐mediated cardiac gene transfer strategies in animal models. Importantly, the use of these delivery vectors has proven to be safe and effective, as clinical applications of AAV‐mediated gene therapy have been tested in an increasing number of Phase I–III clinical trials, with promising results (Collins and Thrasher, 2015). However, this therapy approach is still in its infancy. Administration of an AAV1 vector containing SERCA2 initially resulted in improvement of patients with advanced heart failure (Zsebo et al., 2014). However, no improvement was observed in patients with heart failure in the CUPID‐2b trial for the AAV1/SERCA2a vector (Greenberg et al., 2016). Thus, strategies to further improve the efficiency and effectiveness of these delivery systems, along with the development of vectors with higher cardiac tropism, have to be considered. Among the different AAV serotypes, AAV9 appears to be the most efficient in gene transfer studies in rodents, making it the preferred AAV serotype for potential clinical approaches in cardiac disease (Inagaki et al., 2006). In line with this, Hrd1, a key component of ERAD, has been shown to positively act in the adaptive ER stress response in mammalian cardiac myocytes (Doroudgar et al., 2015). AAV9‐mediated Hrd1 expression directed to ventricular myocytes contributed to preserving heart function and reduced cardiac hypertrophy in mice with pressure overload‐induced cardiac pathology (Doroudgar et al., 2015). Similarly, AAV9‐mediated ATF6 cardiac overexpression reversed the damage and decreased function observed in ATF6 knockout mice under I/R by ER stress and oxidative stress alleviation in heart (Jin et al., 2016), further highlighting the protective role of the UPR and the potential use of AAV‐mediated gene transfer as a therapeutic strategy in ER stress‐related cardiac pathology (Figure 3).
Figure 3.
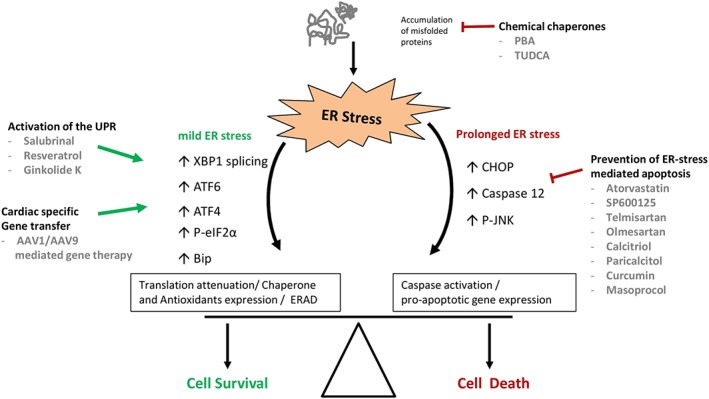
Restoring ER homeostasis as a therapeutic target in cardiac diseases. Different therapeutic approaches aim to restore the balance between the pro‐survival and pro‐apoptotic ER responses, towards a protective outcome in cardiomyocytes under stress. The main approaches are as follows: (i) the direct alleviation of ER stress by lessening protein misfolding using chemical chaperones; (ii) The use of a compound that is able to directly enhance the protective responses of the UPR; and (iii) the use of a compound that prevents the trigger of ER stress‐mediated apoptosis by suppression of CHOP, caspase 12 and JNK activity and expression. In addition, gene therapy based on cardiac‐specific expression of protective genes may contribute to the maintenance of cardiomyocyte homeostasis under stress conditions.
In conclusion, ER stress is involved in many pathological processes of cardiovascular disease. The UPR is a defensive mechanism, which can protect cardiomyocytes by maintaining ER homoeostasis. However, prolonged ER stress will cause dysfunction and apoptosis of cardiomyocytes, leading to cardiovascular diseases. Over the recent years, understanding of the pathophysiological role of ER stress in cardiovascular disease has progressed significantly and several potential therapeutic agents have been investigated. However, there are still many unresolved questions that need to be answered. An improved understanding of the molecular mechanisms underlying ER stress in heart diseases will help to identify novel potential targets for new therapeutic interventions and drug discovery.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d,e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by the British Heart Foundation (PG/14/71/31063, PG/14/70/31039 and FS/15/16/31477).
Wang, S. , Binder, P. , Fang, Q. , Wang, Z. , Xiao, W. , Liu, W. , and Wang, X. (2018) Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. British Journal of Pharmacology, 175: 1293–1304. doi: 10.1111/bph.13888.
Contributor Information
Wei Xiao, Email: xiaowei1959@yahoo.com.
Wei Liu, Email: wei.liu@manchester.ac.uk.
Xin Wang, Email: xin.wang@manchester.ac.uk.
References
- Alexander SPH, Kelly E, Marrion N, Peters J, Benson H, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: –5744, 5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery J, Etzion S, DeBosch BJ, Jin X, Lupu TS, Beitinjaneh B et al. (2010). TRB3 function in cardiac endoplasmic reticulum stress. Circ Res 106: 1516–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala P, Montenegro J, Vivar R, Letelier A, Urroz PA, Copaja M et al. (2012). Attenuation of endoplasmic reticulum stress using the chemical chaperone 4‐phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol 92: 97–104. [DOI] [PubMed] [Google Scholar]
- Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE (2006). Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol 291: H1411–H1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont PJ, Chen WJ, San Pedro MN, Thuerauf DJ, Lowe NG, Gude N et al. (2010). Roles for ER‐associated degradation (ERAD) and the novel ER stress response gene, derlin‐3, in the ischemic heart. Circ Res 106: 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000). Dynamic interaction of Bip and ER stress transducers in the unfolded‐protein response. Nat Cell Biol 2: 326–332. [DOI] [PubMed] [Google Scholar]
- Castillero E, Akashi H, Pendrak K, Yerebakan H, Najjar M, Wang C et al. (2015). Attenuation of the unfolded protein response and endoplasmic reticulum stress after mechanical unloading in dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 309: H459–H470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WP, Wang BW, Shyu KG (2009). Regulation of GADD153 induced by mechanical stress in cardiomyocytes. Eur J Clin Invest 39: 960–971. [DOI] [PubMed] [Google Scholar]
- Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L et al. (2007). Cardiotoxicity associated with the tyrosine kinase inhibitor Sunitinib. Lancet 370: 2011–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Thrasher A (2015). Gene therapy: progress and predictions. Proc Biol Sci 282 20143003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dala S, Foster CR, Das BC, Singh M, Singh K (2012). B‐adrenergic receptor stimulation induces endoplasmic reticulum stress in adult cardiac myocytes: role in apoptosis. Mol Cell Biochem 364: 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dally S, Monceau V, Corvazier E, Bredoux R, Raies A, Bobe R et al. (2009). Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non‐failing and failing human heart. Cell Calcium 45: 144–156. [DOI] [PubMed] [Google Scholar]
- Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH et al. (2009). Endoplasmic reticulum‐mitochondria crosstalk in NIX‐mediated murine cell death. J Clin Invest 119: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC (2009). Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem 284: 29735–29745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Völkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL et al. (2015). Hrd1 and ER‐associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res 117: 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q, Chen C, Yang L, Li N, Gong W, Li S et al. (2015). microRNA regulation of unfolded protein response transcription factor XBP1 in the progression of cardiac hypertrophy and heart failure in vivo. J Transl Med 13: –363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Tang HB, Kang J, Shan L, Song H, Zhu K et al. (2015). Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience 311: 362–373. [DOI] [PubMed] [Google Scholar]
- Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M et al. (2010). Ablation of C/EBP homologous protein attenuates ER‐mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122: 361–369. [DOI] [PubMed] [Google Scholar]
- Gao G, Xie A, Zhang J, Herman A, Jeong E, Gu L et al. (2013). Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol 6: 1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerczauk PZ, Breckenridge DG, Liles JT, Budas GR, Shryock JC, Belardinelli L et al. (2012). An apoptosis signal‐regulating kinase 1 inhibitor reduces cardiomyocyte apoptosis and infarct size in a rat ischemia‐reperfusion model. J Cardiovasc Pharmacol 60: 276–282. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D et al. (2010). Transcriptional regulation of VEGF‐A by the unfolded protein response pathway. PLoS One 5: e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glembotski CC (2008). The role of the unfolded protein response in the heart. J Mol Cell Cardiol 44: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS et al. (2016). Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double‐blind, placebo‐controlled, phase 2b trial. Lancet 387: 1178–1186. [DOI] [PubMed] [Google Scholar]
- Groenendyk J, Sreenivasaiah PK, Kim DOH, Agellon LB, Michalak M (2010). Biology of endoplasmic reticulum stress in the heart. Circ Res 107: 1185–1197. [DOI] [PubMed] [Google Scholar]
- Guo R, Ma H, Gao F, Zhong L, Ren J (2009). Metallothionein alleviates oxidative stress‐induced endoplasmic reticulum stress and myocardial dysfunction. J Mol Cell Cardiol 47: 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y et al. (2004). Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol 24: 8007–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M et al. (2000). Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6: 1099–1108. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Walle LV, Upton JP, Xu WH, Hagen A et al. (2009). IRE1 alpha kinase activation modes control alternative endoribonuclease outputs to determine divergent cell fates. Cell 138: 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J et al. (2013). ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B et al. (2006). Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312: 572–576. [DOI] [PubMed] [Google Scholar]
- Hollien J, Lin JH, Stevens N, Walter P, Weissman JS (2009). Regulated Ire‐1 dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS (2006). Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science 313: 104–107. [DOI] [PubMed] [Google Scholar]
- Hubbard BP, Sinclair DA (2014). Small molecule SIRT1 activators for the treatment of aging and age‐related diseases. Trends Pharmacol Sci 35: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA et al. (2006). Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 14: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isodono K, Takahashi T, Imoto H, Nakanishi N, Ogata T, Asada S et al. (2010). PARM‐1is an endoplasmic reticulum molecule involved in endoplasmic reticulum stress‐induced apoptosis in rat cardiacmyocytes. PLoS One 5: e9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian L, Lu Y, Lu S, Lu C (2016). Chemical chaperone 4‐phenylbutyric acid reduces cardiac ischemia/reperfusion injury by alleviating endoplasmic reticulum stress and oxidative stress. Med Sci Monit 22: 5218–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JK, Blackwood EA, Azizi KM, Thuerauf DJ, Fahem AG, Hofmann C et al. (2016). ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res 116: 310266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshinuma S, Miyamae M, Kaneda K, Kotani J, Figueredo VM (2014). Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia‐reperfusion injury. J Anesth 28: 235–241. [DOI] [PubMed] [Google Scholar]
- Lee AS (2005). The ER chaperone and signaling regulator GRP78/bip as a monitor of endoplasmic reticulum stress. Methods 35: 378–381. [DOI] [PubMed] [Google Scholar]
- Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AT et al. (2009b). Roles of ERO1‐alpha‐mediated stimulation of inositol 1,4,5‐triphosphate receptor activity in endoplasmic reticulum stress‐induced apoptosis. J Cell Biol 186: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RJ, He KL, Li X, Wang LL, Liu CL, He YY (2015). Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2α. Mol Med Rep 12: 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SY, Ren J (2008). Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion‐induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. J Mol Cell Cardiol 44: 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li SY, Gilbert SA, Li Q, Ren J (2009a). Aldehyde dehydrogenase‐2 (ALDH2) ameliorates chronic alcohol ingestion‐induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol 47: 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L (2006). ER stress and neurodegenerative diseases. Cell Death Differ 13: 385–392. [DOI] [PubMed] [Google Scholar]
- Liu M, Dudley S (2015). Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci 17: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sargent M, York A, Molkentin J (2009). Ask1 regulates cardiomyocyte death but not hypertrophy in transgenic mice. Circ Res 105: 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H et al. (2014b). Endoplasmic reticulum stress sensor protein kinase R‐like endoplasmic reticulum kinase (PERK) protects against pressure overload‐induced heart failure and lung remodeling. Hypertension 64: 738–744L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Zhang ZY, Andersson KB, Husberg C, Enger UH, Ræder MG et al. (2011). Cardiomyocyte‐specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium 49: 201–207. [DOI] [PubMed] [Google Scholar]
- Liu Z, Cai H, Zhu H, Toque H, Zhao N, Qiu C et al. (2014a). Protein kinase RNA‐like endoplasmic reticulum kinase (PERK)/calcineurin signaling is a novel pathway regulating intracellular calcium accumulation which might be involved in ventricular arrhythmias in diabetic cardiomyopathy. Cell Signal 26: 2591–2600. [DOI] [PubMed] [Google Scholar]
- Luo T, Chen B, Wang X (2015). 4‐PBA prevents pressure overload‐induced myocardial hypertrophy and interstitial fibrosis by attenuating endoplasmic reticulum stress. Chem Biol Interact 242: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D et al. (2004). Cytoprotection by pre‐emptive conditional phosphorylation of translation initiation factor 2. EMBO J 23: 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Niinuma Y, Nomura Y (2003). Activation signal of nuclear factor‐kappa B in response to endoplasmic reticulum stress is transduced via IRE and tumor necrosis factor receptor‐associated factor 2. Biol Pharm Bull 26: 931–935. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Ryu SW, Song BJ (2006). JNK‐ and p38 kinase‐mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 281: 21256–21265. [DOI] [PubMed] [Google Scholar]
- Ma J, Luo T, Zeng Z, Fu H, Asano Y, Liao Y et al. (2016). Histone deacetylase inhibitor phenylbutyrate exaggerates heart failure in pressure overloaded mice independently of HDAC inhibition. Sci Rep 6: 34036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ (2007). Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double‐edged sword? Antioxid Redox Signal 9: 2277–2293. [DOI] [PubMed] [Google Scholar]
- Mao W, Fukuoka S, Iwai C, Liu J, Sharma VK, Sheu SS et al. (2007). Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol 293: H1636–H1645. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Ferrans VJ, Roberts WC (1975). Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol 79: 387–434. [PMC free article] [PubMed] [Google Scholar]
- Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA et al. (2006). Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen‐regulated form of ATF6. Circ Res 98: 1186–1193. [DOI] [PubMed] [Google Scholar]
- Maure M, Degeans N, Taouji S, Chevet E, Grosset CF (2013). MicroRNA‐1291‐mediated silencing of IREalpha enhances glypican‐3 expression. RNA 19: 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier A, Clément R, Harnois T, Bourmeyster N, Bois P, Chatelier A (2015). Nav1.5 channels can reach the plasma membrane through distinct N‐glycosylation states. BBA‐Genl Subj 1850: 1215–1223. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down‐regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21: 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L (2011). Endoplasmic reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3: a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Kitakaze M (2010). ER stress in cardiovascular disease. J Mol Cell Cardiol 48: 1105–1110. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K et al. (2011). C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol 31: 1124–1132. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E (2004). Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase‐12 during ER stress‐induced apoptosis. J Biol Chem 279: 50375–50381. [DOI] [PubMed] [Google Scholar]
- Mughal W, Kirshenbaum LA (2011). Cell death signalling mechanisms in heart failure. Exp Clin Cardiol 16: 102–108. [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J (2000). Cross‐talk between two cysteine protease families. Activation of caspase‐12 by calpain in apoptosis. J Cell Biol 150: 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA et al. (2000). Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐beta. Nature 403: 98–103. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL (2008). Dissecting the ER‐associated degradation of a misfolded polytopic membrane protein. Cell 132: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickson P, Toth A, Erhardt P (2007). PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc Res 73: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K et al. (2002). ASK1 is essential for endoplasmic reticulum stress‐induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L, Zhou C, Duan Q, Lv J, Fu X, Xia Y et al. (2011). beta‐AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PLoS One 6: e27294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Hardding HP, Ron D (2009). Feedback inhibition of the unfolded protein response by GAD344‐mediated dephosphorylation of eIF2alpha. J Cell Biol 186: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odisho T, Zhang L, Volchuk A (2015). ATF6β reulates the Wfs1 gene and has a cell survival role in the ER stress response in pancreatic β‐cell. Exp Cell Res 330: 111–122. [DOI] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S et al. (2004). Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic. Circulation 110: 705–712. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Akira E et al. (2002). Targeted disruption of the Chop gene delays endoplasmic reticulum stress‐mediated diabetes. J Clin Invest 109: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, Tabas I (2010). Pivotal role of calcium/calmodulin‐dependent protein kinase II in ER stress‐induced apoptosis. Cell Cycle 9: 223–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal R, Cristan EA, Schnittker K, Narayan M (2010). Rescue of ER oxidoreductase function through polyphenolic phytochemical intervention: implications for subcellular traffic and neurodegenerative disorders. Biochem Biophys Res Commun 392: 567–571. [DOI] [PubMed] [Google Scholar]
- Park CS, Cha H, Kwon EJ, Sreenivasaiah PK, do Kim H (2012). The chemical chaperone 4‐phenylbutyric acid attenuates pressure‐overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun 421: 578–584. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH (2002). Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res 52: 832–836. [DOI] [PubMed] [Google Scholar]
- Piano MR (2002). Alcoholic cardiomyopathy: incidence, clinical characteristics, and pathophysiology. Chest 121: 1638–1650. [DOI] [PubMed] [Google Scholar]
- Prola A, Pires Da Silva J, Guilbert A, Lecru L, Piquereau J, Ribeiro M et al. (2017). SIRT1 protects the heart from ER stress‐induced cell death through eIF2α deacetylation. Cell Death Differ 24: 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Cunn P, Lee L, Kelly PN, Huntington ND et al. (2007). ER stress triggers apoptosis by activating BH3‐only protein Bim. Cell 129: 1337–1349. [DOI] [PubMed] [Google Scholar]
- Qi X, Vallentin A, Churchill E, Mochly‐Rosen D (2007). δPKC participates in the endoplasmic reticulum stress‐induced response in cultured cardiac myocytes and ischemic heart. J Mol Cell Cardiol 43: 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe N, Ren J (2013). Oxidative activation of Ca2+/calmodulin‐activated kinase II mediates ER stress‐induced cardiac dysfunction and apoptosis. Am J Physiol Heart Circ Physiol 304: H828–H839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N et al. (2006). Enhanced bacterial clearance and sepsis resistance in caspase‐12‐deficient mice. Nature 440: 1064–1068. [DOI] [PubMed] [Google Scholar]
- Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJM (2015). Endoplasmic reticulum stress induces ligand‐independent TNFR1‐mediated necroptosis in L929 cells. Cell Death Dis 6: e1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T et al. (2010). X‐box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE‐like element in cardiomyocytes. J Mol Cell Cardiol 48: 1280–1289. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139. [DOI] [PubMed] [Google Scholar]
- Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U et al. (2007). Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol 50: 1029–1037. [DOI] [PubMed] [Google Scholar]
- Son SM, Byun J, Roh SE, Kim SJ, Mook‐Jung I (2014). Reduced IRE1α mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis 5: e1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY (2011). Atorvastatin inhibits myocardial cell apoptosis in a rat model with post‐myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci 8: 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens FJ, Argon Y (1999). Protein folding in the ER. Semin Cell Dev Biol 10: 443–454. [DOI] [PubMed] [Google Scholar]
- Sukumaran V, Watanabe K, Veeraveedu PT, Gurusamy N, Ma M, Thandavarayan RA et al. (2011). Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int J Biol Sci 7: 154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Duffy A, O'Mahoney ME, Logue SE, Mylotte L, O'Brien T et al. (2006). ER stress contributes to ischemia‐induced cardiomyocyte apoptosis. Biochem Biophys Res Commun 349: 1406–1411. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A (2009). Bcl‐2 family on guard at the ER. Am J Physiol 296: C941–C953. [DOI] [PubMed] [Google Scholar]
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M et al. (2005). AMP‐activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 25: 9554–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuerauf DJ, Morrison L, Glembotski CC (2004). Opposing roles for ATF6alpha and ATF6beta in endoplasmic reticulum stress response gene induction. J Biol Chem 279: 21078–21084. [DOI] [PubMed] [Google Scholar]
- Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC (2006). Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 99: 275–282. [DOI] [PubMed] [Google Scholar]
- Toko H, Takahashi H, Kayama Y, Okada S, Minamino T, Terasaki F et al. (2010). ATF6 is important under both pathological and physiological states in the heart. J Mol Cell Cardiol 49: 113–120. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P (2015). Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2: e975093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitadello M, Penzo D, Petronilli V, Michieli G, Gomirato S, Menabo R et al. (2003). Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. FASEB J 17: 923–925. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang Z, Fan Q, Guo J, Galli G, Du G et al. (2016). Ginkgolide K protects the heart against endoplasmic reticulum stress injury by activating the inositol‐requiring enzyme 1α/X box‐binding protein‐1 pathway. Br J Pharmacol 173: 2402–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ron D (1996). Stress‐induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272: 1347–1349. [DOI] [PubMed] [Google Scholar]
- Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR et al. (2014). Spliced X‐box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156: 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J et al. (2007). ATF6 alpha optimizes long‐term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 13: 351–364. [DOI] [PubMed] [Google Scholar]
- Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S et al. (2003). Targeted deletion of apoptosis signal‐regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci U S A 100: 15883–15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong H et al. (2015). Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid Redox Signal 22: 633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchigna S, Zentilin L, Giacca M (2014). Adeno‐associated virus vectors as therapeutic and investigational tools in the cardiovascular system. Circ Res 114: 1827–1846. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F et al. (2016). CaMKII is a RIP3 substrate mediating ischemia‐ and oxidative stress‐induced myocardial necroptosis. Nat Med 22: 175–182. [DOI] [PubMed] [Google Scholar]
- Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M et al. (2014). Long‐term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 114: 101–108. [DOI] [PubMed] [Google Scholar]