

Abstract
A 13‐month‐old female Toy Poodle was presented for progressive ataxia and intention tremors of head movement. The diagnosis of Sandhoff's disease (GM2 gangliosidosis) was confirmed by deficient β‐N‐acetylhexosaminidase A and B activity in circulating leukocytes and identification of the homozygous mutation (HEXB: c.283delG). White matter in the cerebrum and cerebellum was hyperintense on T2‐weighted and fluid‐attenuated inversion recovery magnetic resonance images. Over the next 2 years, the white matter lesions expanded, and bilateral lesions appeared in the cerebellum and thalamus, associated with clinical deterioration. Magnetic resonance spectroscopy showed progressive decrease in brain N‐acetylaspartate, and glycine‐myo‐inositol and lactate‐alanine were increased in the terminal clinical stage. The concentrations of myelin basic protein and neuron specific enolase in cerebrospinal fluid were persistently increased. Imaging and spectroscopic appearance correlated with histopathological findings of severe myelin loss in cerebral and cerebellar white matter and destruction of the majority of cerebral and cerebellar neurons.
Keywords: GM2 gangliosidosis, Lysosomal storage diseases, MRI, MRS
Abbreviations
- Cho
choline
- Cr
creatine
- CSF
cerebrospinal fluid
- FLAIR
fluid attenuated inversion recovery
- Gly
glycine
- GM
gray matter
- Ins
myo‐inositol
- Lac‐Ala
lactate‐alanine
- MBP
myelin basic protein
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- NAA
N‐acetylaspartate
- NSE
neuron specific enolase
- SD
Sandhoff's disease
- T1W and T2W
T1 and T2 weighting
- WM
white matter
The GM2 gangliosidoses are a group of lysosomal storage diseases in which GM2 ganglioside and other glycolipids accumulate in lysosomes, especially within neurons.1, 2, 3 These abnormal accumulations lead to delayed myelin formation, and swelling and destruction of neurons, axons, and myelin in the central nervous system, resulting in severe neurologic dysfunction.4 Although GM2 gangliosidoses are thought to affect gray matter primarily, myelin deficiency has been recognized in both the cat and dog.1, 5, 6, 7 As a corollary of these pathological changes, abnormal signal intensity of the cerebral white matter (WM) on magnetic resonance imaging (MRI) has been documented in humans and domestic animals with GM2 gangliosidoses1, 5, 6, 8, 9
Sandhoff's disease (SD) is 1 of 3 main variants of GM2 gangliosidoses‐gangliosidosis variant 0, caused by deleterious mutations of the HEXB gene encoding the β‐subunit that is a common component of the lysosomal acid β‐hexosaminidase A and B.10 In dogs, clinical features and MRI findings in SD have been reported in a Golden Retriever, a family of Toy Poodles, in a mixed‐breed dog and, recently, in Shiba‐Inu dogs.5, 7, 10, 11, 12 However, long‐term serial changes of MRI and magnetic resonance spectroscopy (MRS), and analysis of cerebrospinal fluid (CSF) in dogs with SD are not established. Here, we report changes in MRI and MRS findings and, myelin basic protein (MBP) and neuron‐specific enolase (NSE) concentrations in CSF over a period of 2 years in a dog with SD.
Case Report
A 13‐month‐old 3.8 kg, spayed female red Toy Poodle was presented with a 10‐month history of progressing intention tremor, ataxia, and suspected blindness. The head tremor initially was observed at 3 months of age, and ataxia appeared a few months later. An absent menace response in both eyes was documented at 10 months of age, although the dog was able to chase dropping cotton balls and displayed intact visual placing responses.
The diagnosis of SD was made from a blood sample obtained when the dog was 14 months of age by detection of deficient β‐N‐acetylhexosaminidase A and B activity in leukocytes and by identification of the same homozygous mutation (HEXB: c.283delG) as that previously detected in toy poodles with SD.3, 10 In the absence of effective treatment for SD, euthanasia was proposed but the owner did not consent, instead preferring supportive care with an anti‐inflammatory dosage of prednisone1 (0.5 mg/kg/day) and periodic medical re‐evaluations, including MRI, MRS, and CSF analysis. At each MRI, MRS, and CSF analysis, the dog underwent physical, neurologic, blood, and cardiac examination (ECG and ultrasound) before anesthesia to evaluate safety, and informed consent was obtained. The follow‐up investigation plans for MRI, MRS, and CSF analysis were each ethically evaluated and approved by Nihon University (Animal Medical Center Management and Ethics Committee). Finally, we performed a total of 14 series of MRI and MRS scans, and CSF analysis before the dog died at 37 months of age.
Magnetic Resonance Imaging Findings
All MRI scans were performed with 1.5‐T superconducting unit2 with the dog under general anesthesia, and T1‐weighted (T1W), T2‐weighted (T2W), fluid‐attenuated inversion recovery (FLAIR), and postcontrast‐T1W sequences were acquired in sagittal, transverse, and dorsal planes at each time point. Concordance between clinical deterioration and MRI changes is summarized in supporting information (Table S1).
On the initial scan at 14 months of age, diffuse T2W and FLAIR hyperintensity of the subcortical and periventricular WM was observed in the frontal and parietal lobes and in parts of the temporal and occipital lobes of the cerebrum (compared with cortical gray matter [GM]), making the border between WM and GM indistinct (Fig 1A–C). These areas exhibited isointensty (subcortical WM) or hyperintensity (periventricular WM) on T1W images, and did not enhance after IV administration of gadoteridol3 at a dosage of 0.3 mL/kg (0.15 mmol/kg). In the cerebellum, the border between GM and WM regions was almost imperceptible on T2W and FLAIR images because the cerebellar WM and GM had become isointense to each other. Diffuse hyperintensity in the brain stem was observed on T2W and FLAIR images (Fig 1A) compared with GM of the cerebrum. Morphologically, the corpus callosum, rostral commissure, and fornix appeared atrophied on mid‐sagittal images (Fig 1A).
Figure 1.
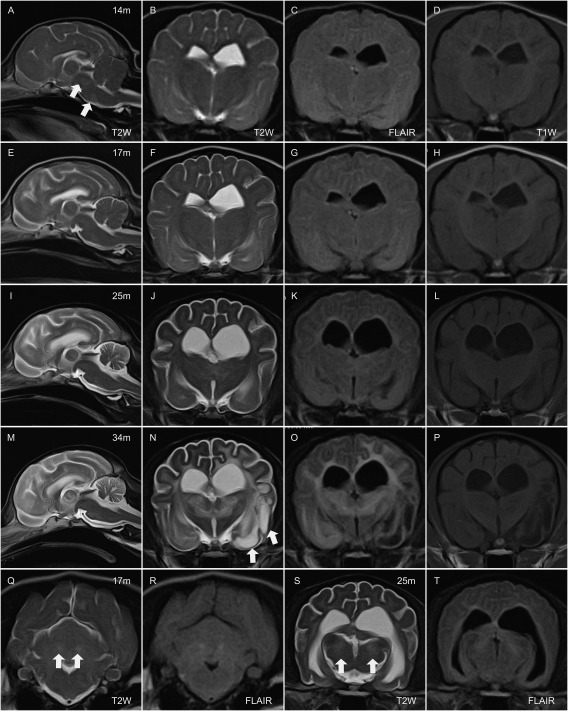
MR images at 14 months (m) (A–D), 17 months (E‐H, Q, and R), 25 months (I–L, S, and T), and 34 months of age (M–P). From left in each row (A–P), mid sagittal T2‐weighted (T2W) image, transverse images of T2W, fluid attenuated inversion recovery (FLAIR), and T1‐weighted at the level of thalamus. In the bottom row, transverse T2W and FLAIR at the level of cerebellum (Q and R) and same sequences at the level of mid brain (S and T). At 14 m, diffused hyperintense area comparing to cerebral cortex are observed in the brain stem on mid‐sagittal T2W (a; arrows) and cerebral white matter (WM) on transverse T2W (B) and FLAIR (C). At 17 m, hyperintense area in the cerebral WM are obvious on T2W and FLAIR images (F and G). Bilateral hyperintense lesion on T2W and FLAIR were observed in the dentate nucleus (Q and R; arrows). Cerebral and cerebellar atrophy are progressed at 25 m (I, J, K, L). Bilateral hyperintense lesion are observed in the medial geniculate nucleus (bodies) on T2W and FLAIR (S and T; arrows). T2W and FLAIR hyperintense lesion in cerebral WM and thalamus are prominent (M, N, O). Some part of cerebral WM in left temporal lobe (piriform lobe) showed hyperintensity on T2W, and hypointensity on T1W and FLAIR (N, O, P; arrows).
By the time of the subsequent scan at 17 months of age, diffuse hyperintensity in cerebral WM extended into the whole of the temporal and occipital lobe on T2W and FLAIR images (Fig 1F,G). Bilateral hyperintense lesions on T2W and FLAIR images were observed in the dentate nucleus of the cerebellum (Fig 1Q,R); these were isointense on T1W and not enhanced after contrast administration. All ventricles, sulci, and the subarachnoid space surrounding the cerebrum and cerebellum were enlarged, suggesting atrophy of brain parenchyma. These abnormalities progressed during the subsequent 4 months, and by 21 months there was, in addition, hyperintensity of the cerebellar WM (compared to GM) and surrounding the interthalamic adhesion on T2W and FLAIR images. These lesions were isointense on T1W and did not enhance. By 24 months of age, the rostral part of the corpus callosum was difficult to recognize and the fornix was poorly delineated. Bilateral hyperintense lesions on T2W and FLAIR images of the medial geniculate nucleus of the thalamus were apparent at 25 months. These were isointense on T1W and did not enhance with gadolinium contrast (Fig 1S,T).
During the terminal clinical stages (34–36 months), most of the diffuse lesions in the cerebral WM and thalamus exhibited greater hyperintensity on T2W and FLAIR images compared to images obtained at earlier stages. In addition, some areas of cerebral WM in the left frontal and piriform lobe showed prominent hyperintensity on T2W, but were hypointense on T1W and FLAIR, and did not enhance after administration of contrast medium (Fig 1N–P). Atrophy of the brain stem and spinal cord also were prominent after 34 months of age (Fig 1M).
Magnetic Resonance Spectroscopy Findings
The protocol and evaluation methods of MRS were similar to those previously described.13 Briefly, a voxel of approximately 3.4 cm3 was placed on the frontal lobe (in the middle of both hemispheres) and spectra were obtained with a point‐resolved spectroscopy sequence using the following conditions: repetition time (TR) 2,000 ms, echo time (TE) 136 ms (Long TE), and single‐voxel. The NAA and Cr concentrations gradually decreased with time (Fig 2 and supporting information [Table S2]). Increases in Gly‐Ins and Lac‐Ala were repeatedly observed in scans after 25 months of age. The NAA/Cr ratio was decreased throughout the whole observation period, and Cho/Cr, Gly‐Ins/Cr, and Lac‐Ala/Cr ratios were increased in the later stages when compared to those of healthy dogs.13
Figure 2.
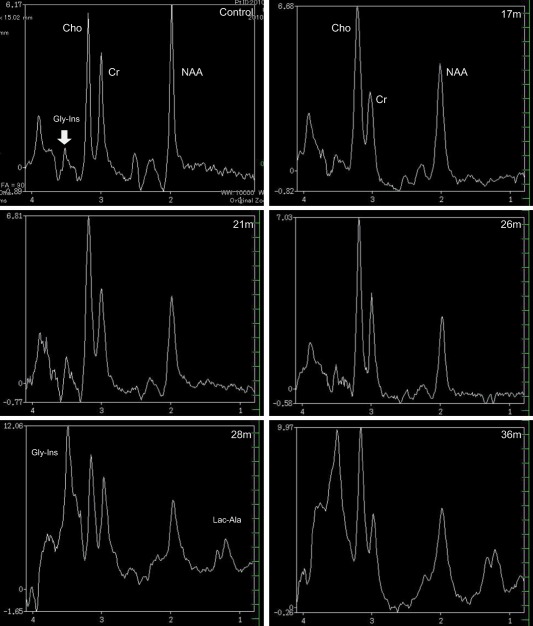
Proton MRS (H1‐MRS) images of the frontal lobe obtained from age‐matched healthy beagle (control) and Sandhoff disease dog using long echo time (TE) sequence. The peak of N‐acetylaspartate (NAA) is lower comparing to control through the periods. In the clinically terminal stage (28 and 34 months of age), high peak of Glycine‐Myo‐inositol (Gly‐Ins) and increased lactate/alanine (Lac‐Ala) were observed.
Cerebrospinal Fluid Analysis
Cerebrospinal fluid was obtained from the cisterna magna at each time point. Cell population, pH, and concentrations of protein and glucose were within reference intervals at all time points. However, up until 26 months of age, occasional mononuclear cells contained eosinophilic granular cytoplasmic inclusions (Fig 3); these were not apparent after this time. Possible CSF biomarker concentrations including NSE and MBP, were measured by ELISA, and results are summarized in Fig 3. The concentrations of NSE and MBP varied: NSE, 154.3 ± 94.70 μg/L (mean ± SD; range, 5.4–286 μg/L) and MBP 121.88 ± 40.74 pg/mL, but were higher than those in CSF obtained from healthy dogs (healthy adult dog [aged 1–2 years], n = 4; NSE, 12.32 ± 2.01 μg/L; MBP, 0.17 ± 0.10 pg/mL).
Figure 3.
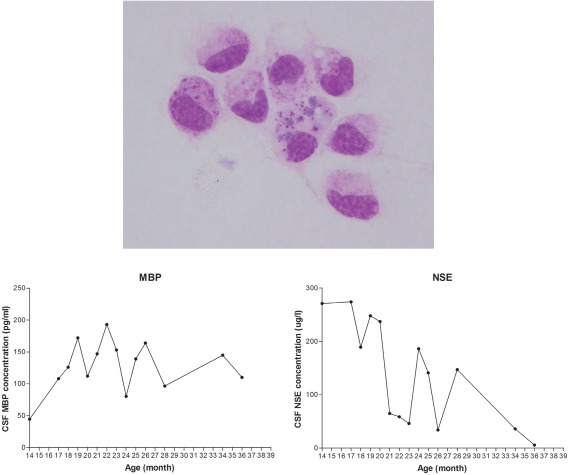
Analysis of cerebrospinal fluid (CSF). Some of mononuclear cells in CSF contained eosinophilic granules within the cytoplasm. These cells could be seen in CSF until 26 months of age. Follow‐up measurement of neuron specific enolase and myelin basic protein revealed increased concentration with time associated with neurons and myelin destruction.
Histopathological Findings
At 37 months of age, the dog died after aspiration of vomitus. Tissue samples of brain, spinal cord, liver, and spleen were fixed in 10% formalin for histopathological evaluation. Paraffin‐embedded sections were prepared, and stained with hematoxylin and eosin (HE), Luxol fast blue (LFB), periodic acid‐Schiff (PAS), and Sudan black B (SBB; Fig 4). Most neurons in the cerebral GM had been destroyed and the few remaining neurons were swollen with LFB‐, PAS‐, and SBB‐positive granules that corresponded with the eosinophilic granules visible on HE‐stained material (Fig 4). Many glial cells contained similar granules. Almost all myelin was lost in the cerebral WM and many enlarged granule‐containing astrocytes also were recognized. The left pyriform lobe of the cerebrum was necrotic and formed a cystic lesion but, although the hippocampus contained many severely degenerate neurons, the right piriform lobe structure was preserved. In the thalamus, most neurons were absent and there were many degenerate astrocytes. The neurons in the brain stem were preserved but swollen with inclusions. The molecular layer in the cerebellum was thin and there was severe loss of myelin and neurons including Purkinje cells; many degenerate glial cells were observed. The neurons in the dentate nucleus were relatively well preserved but grossly swollen with inclusions. Although neurons in the spinal cord were severely swollen with inclusions, myelin in the WM was relatively well preserved. Cells in the liver and spleen also contained LFB‐ and SBB‐positive granules.
Figure 4.
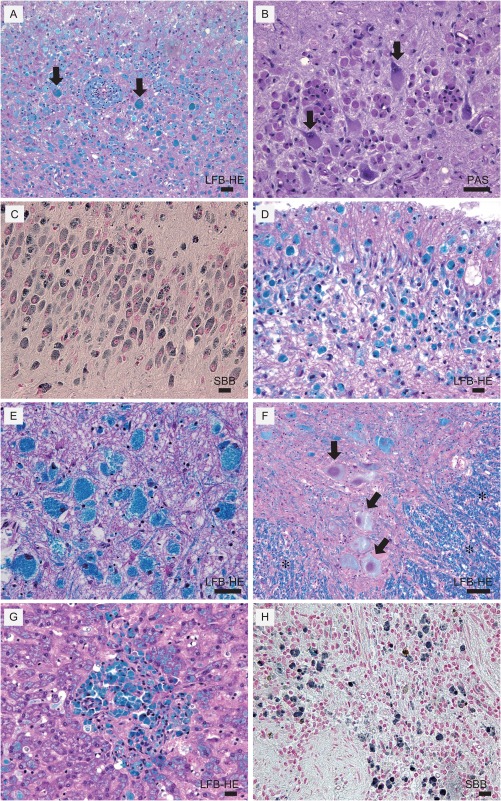
Histopathology of dog with SD. (A) Cerebral cortex stained with Luxol fast blue and, hematoxylin and eosin (LFB‐HE). There is severe neuron loss and remaining neurons are swollen with LFB positive granular inclusions (arrows). Microglia also contained LFB‐positive materials. (B) Cerebral cortex stained with periodic acid‐Schiff (PAS). Inclusions within cytoplasm were PAS‐positive (arrows). (C) Hippocampus stained with Sudan black B (SBB) revealed inter‐cellar inclusions are SBB‐positive. (D) Cerebellum, LFB‐HE. (E) Brain stem, LFB‐HE. Though neurons are preserved they are severely swollen with LFB positive granular inclusions. (F) Spinal cord: LFB‐HE. Well‐preserved myelin in white matter (*) and severely swollen neurons with LFB positive inclusions were observed (arrows). (G) Liver, LFB‐HE. LFB‐positive cells in the liver. (H) Spleen, SBB‐positive cells. Scale Bar represents 100 μm for all panels.
Discussion
Although unusual in a clinical case, we were able to follow changes in MRI, MRS, and CSF of a dog with SD over 2 years. The major changes in MRI appearance included enlarging hyperintense lesions in cerebral and cerebellar WM, thalamus and brain stem on T2W and FLAIR, appearance of bilateral T2 hyperintense lesions in the nucleus, and progressive brain atrophy. There was a clear relationship between clinical deterioration and changes in the MRI findings. Previously, T2W hyperintense lesions in cerebral WM have been inconsistently documented in dogs with SD.5, 7, 14 Progression of the disease apparently leads to the abnormal signal changes on T2W because, in our present case, hyperintense T2W lesions were not apparent at the earliest stages but became more obvious and extensive with time, similar to observations in human SD patients during a 2‐year follow‐up period.15
In humans and small animals, signal changes in cerebral WM are thought to represent a combination of abnormal myelination and myelin loss. Indeed, histopathological evaluation of cats and dogs with SD has confirmed these changes.1, 5, 7 In a mixed breed, dog with SD that did not show cerebral WM abnormality on MRI, there was little observed myelin loss.7 In contrast, our present case showed a T2W hyperintense lesion in the WM and severe myelin loss in addition to axon loss.
In 2 previously reported SD dogs, bilateral lesions were observed in the caudate nucleus,5, 14 but our case had bilateral lesions in the cerebellum and thalamus. In humans with SD, bilateral lesions often are described in the thalamus and basal nuclei including the caudate nucleus, globus pallidus, and putamen, and few reports mentioned lesions in the dentate nucleus of the cerebellum.16, 17, 18 The signal intensity of these lesions however contrasted between humans and dogs: lesions in humans generally show T2 hypointensity and T1 hyperintensity and in dogs lesions show T2 hyperintensity and T1 hypointensity. Although the cause of bilateral T1W hyperintensity in the lesions of affected humans is still unknown, loss of axons and myelin, gliosis and intralysosomal storage in addition to calcium accumulation are thought to be likely,17 whereas the imaging characteristics in dogs are attributed to loss of axons and myelin, gliosis, and intralysosomal storage.
In our case, brain atrophy progressed with age, differing from human patients in which brain atrophy was not observed during a 2‐year follow‐up of serial MRI.15
In the cerebrum and cerebellum, abnormal signal changes were seen in early stages and progressed on MRI, corresponding to widespread destruction of neurons on histological examination. Therefore, we conclude that T2‐hyperintense WM lesions represent the severity of lesions in the affected myelin and axons, gliosis and intralysosomal storage, and progression of destruction and degeneration would lead to brain atrophy. Recently, the morphology of the corpus callosum, rostral commissure, and fornix on nonserial MRI of dogs with GM 1 and GM2 gangliosidosis was evaluated.19 In this study, it was difficult to recognize the corpus callosum and rostral commissure on midsagittal images of dogs with SD.19 Our case showed similar findings, and in the later stages it was increasingly difficult to recognize those structures, corresponding to progressive atrophy.
Compared with our previous report of MRS in healthy dogs,13 there was a pattern of decreased NAA/Cr and increased Cho/Cr, Gly‐Ins/Cr, and Lac‐Ala/Cr ratios (Gly‐Ins/Cr and Lac‐Ala/Cr ratio increased at later stage). N‐acetylaspartate is a marker for intact neuroaxonal tissue20 and decreased NAA/Cr implies neuroaxonal damage in humans with SD18, 21, 22 and decreased NAA and NAA/Cr have been described in a dog with GM2 B variant.6 Increased Cho and Cho/Cr were thought to be associated with demyelination in a human SD patient.21 In our MRS protocol using a long TE (136 ms), it was difficult to differentiate metabolites that had a peak at approximately 3.5–3.6 ppm because the spectra of Gly and Ins overlap. Generally, it is thought that the peak seen at approximately 3.5–3.6 ppm obtained with long TE (136 ms) represents Gly, and those with short TE (35 ms) represent Ins.23 However, the spectrum of Ins is also detectable using a long TE (135 and 270 ms) although the peak tends to decrease with increasing TE.24 We believe the peak at approximately 3.5–3.6 ppm in our case represented increased Ins rather than Gly because Ins is an activated glial marker predominantly seen in astrocytes,25, 26 and prominently increased Ins has been detected in GM2 gangliosidoses, including SD, as a generalized sign of gliosis in humans.18, 21, 22 Whereas Gly is an inhibitory neurotransmitter and co‐agonist at excitatory N‐methyl‐d‐aspartate receptors,27, 28 and increased Gly was detected in nonketotic hyperglycemia and malignant brain tumors,29, 30 it is not reported in GM1 and GM2 gangliosidoses. The increased Lac‐Ala/Cr in later stages may correspond to severe neural cell destruction, anaerobic metabolism, or both similar to that occurring in humans with SD.21
The eosinophilic inclusions in CSF monocytes were thought to be debris of degenerated neurons or glia, or accumulated storage materials. Vacuolated neurons and glia containing foamy eosinophilic materials were apparent in HE sections and we considered that, when these cells were destroyed, the debris was phagocytosed by macrophages. Similar pathological changes have been reported in humans, dogs, and cats with SD.1, 7, 31 Although cells containing inclusions in CSF have not previously been described, they may represent a corollary of lysosomal storage diseases. Because of destruction of myelin and neural cells, the concentrations of MBP and NSE in CSF were higher than those of control dogs, similar to dogs with GM1 gangliosidoses.32 Similar to previous cases with GM1, the efficacy of glucocorticoids (ie, prednisone) was doubtful in light of the progressive clinical signs and concentration of MBP and NSE in CSF before and after treatment, although there is evidence of an inflammatory reaction in the central nervous system associated with GM1 and GM2 gangliosidoses.2, 33, 34, 35
The MRI changes we report might explain the variation in reported MRI findings in previously reported SD dogs.5, 7, 14 Furthermore, the data here suggest that MRS and CSF might be useful for diagnosing lysosomal storage diseases, and MRS also is useful to evaluate progress of the disease.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article:
Table S1. Changes of MRI findings and clinical deteriorations.
Table S2. Results of area under the MRS peak (arbitrary unit) of five metabolites and metabolite ratio in the frontal lobe of a dog with Sandhoff's disease.
Acknowledgments
Grant support
The work was not supported by any grant. The authors gratefully acknowledge the contributions of owners and veterinary nurses (Ami Tsuzuki, Chisato Komori, Mariko Kawai, and Yukino Yabe) for their care of the dog.
Conflict of Interest Declaration
Authors declare no conflict of interest.
Off‐Label Antimicrobial Declaration
Authors declare no off‐label use of antimicrobials.
Institutional Animal Care and Use Committee (IACUC) or Other Approval Declaration
Nihon University Animal Medical Center Management and Ethics Committee (individual case approval number: NU‐15874).
The genetic analysis was performed at Kagoshima University, and histopathological examination was done in the University of Tokyo. Other work including serial MRI, MRS, and CSF analysis were performed in Nihon University. Interpretation of the results and writing were done in Nihon University and Texas A&M University.
Footnotes
PREDONINE, Shionogi, Tokyo, Japan
EXELART Vantage, Toshiba, Tokyo, Japan
ProHance, Bracco‐Eisai Co, Ltd, Tokyo, Japan
References
- 1. Kroll RA, Pagel MA, Roman‐Goldstein S, et al. White matter changes associated with feline GM2 gangliosidosis (Sandhoff disease): Correlation of MR findings with pathologic and ultrastructural abnormalities. AJNR Am J Neuroradiol 1995;16:1219–1226. [PMC free article] [PubMed] [Google Scholar]
- 2. Myerowitz R, Lawson D, Mizukami H, et al. Molecular pathophysiology in Tay‐Sachs and Sandhoff diseases as revealed by gene expression profiling. Hum Mol Genet 2002;11:1343–1350. [DOI] [PubMed] [Google Scholar]
- 3. Yamato O, Satoh H, Matsuki N, et al. Laboratory diagnosis of canine GM2‐gangliosidosis using blood and cerebrospinal fluid. J Vet Diagn Invest 2004;16:39–44. [DOI] [PubMed] [Google Scholar]
- 4. Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 2004;5:554–565. [DOI] [PubMed] [Google Scholar]
- 5. Tamura S, Tamura Y, Uchida K, et al. GM2 gangliosidosis variant 0 (Sandhoff‐like disease) in a family of toy poodles. J Vet Intern Med 2010;24:1013–1019. [DOI] [PubMed] [Google Scholar]
- 6. Freeman AC, Platt SR, Vandenberg M, et al. GM2 gangliosidosis (B variant) in two Japanese Chins: Clinical, magnetic resonance imaging and pathological characteristics. J Vet Intern Med 2013;27:771–776. [DOI] [PubMed] [Google Scholar]
- 7. Kohyama M, Yabuki A, Kawasaki Y, et al. GM2 gangliosidosis variant 0 (Sandhoff disease) in a mixed‐breed dog. J Am Anim Hosp Assoc 2015;51:396–400. [DOI] [PubMed] [Google Scholar]
- 8. Van der Knaap MS, Valk J. GM2 gangliosidosis In: Vam der Knaap MS, Valk J, eds. Magnetic Resonance of Myelin, Myelination, and Myelin Disorders, Vol. 10, 2nd ed Berlin: Springer Verlag; 1995:81–89 [Google Scholar]
- 9. Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rahman MM, Chang HS, Mizukami K, et al. A frameshift mutation in the canine HEXB gene in toy poodles with GM2 gangliosidosis variant 0 (Sandhoff disease). Vet J 2012;194:412–416. [DOI] [PubMed] [Google Scholar]
- 11. Yamato O, Matsuki N, Satoh H, et al. Sandhoff disease in a golden retriever dog. J Inherit Metab Dis 2002;25:319–320. [DOI] [PubMed] [Google Scholar]
- 12. Kolicheski A, Johnson GS, Villani NA, et al. GM2 gangliosidosis in Shiba Inu dogs with an in‐frame deletion in HEXB. J Vet Intern Med 2017;31:1520–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ono K, Kitagawa M, Ito D, et al. Regional variations and age‐related changes detected with magnetic resonance spectroscopy in the brain of healthy dogs. Am J Vet Res 2014;75:179–186. [DOI] [PubMed] [Google Scholar]
- 14. Matsuki N, Yamato O, Kusuda M, et al. Magnetic resonance imaging of GM2‐gangliosidosis in a golden retriever. Can Vet J 2005;46:275–278. [PMC free article] [PubMed] [Google Scholar]
- 15. Koelfen W, Freund M, Jaschke W, et al. GM‐2 gangliosidosis (Sandhoff's disease): Two year follow‐up by MRI. Neuroradiology 1994;36:152–154. [DOI] [PubMed] [Google Scholar]
- 16. Hittmair K, Wimberger D, Bernert G, et al. MRI in a case of Sandhoff's disease. Neuroradiology 1996;38:178–180. [DOI] [PubMed] [Google Scholar]
- 17. Yüksel A, Yalçinkaya C, Işlak C, et al. Neuroimaging findings of four patients with Sandhoff disease. Pediatr Neurol 1999;21:562–565. [DOI] [PubMed] [Google Scholar]
- 18. Wilken B, Dechent P, Hanefeld F, et al. Proton MRS of a child with Sandhoff disease reveals elevated brain hexosamine. Eur J Paediatr Neurol 2008;12:56–60. [DOI] [PubMed] [Google Scholar]
- 19. Hasegawa D, Tamura S, Nakamoto Y, et al. Magnetic resonance findings of the corpus callosum in canine and feline lysosomal storage diseases. PLoS One 2013;8:e83455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bjartmar C, Battistuta J, Terada N, et al. N‐acetylaspartate is an axon‐specific marker of mature white matter in vivo: A biochemical and immunohistochemical study on the rat optic nerve. Ann Neurol 2002;51:51–58. [DOI] [PubMed] [Google Scholar]
- 21. Alkan A, Kutlu R, Yakinci C, et al. Infantile Sandhoff's disease: Multivoxel magnetic resonance spectrosecopy findings. J Child Neurol 2003;18:425–428. [DOI] [PubMed] [Google Scholar]
- 22. Assadi M, Baseman S, Janson C, et al. Serial 1H‐MRS in GM2 gangliosidoses. Eur J Pediatr 2008;167:347–352. [DOI] [PubMed] [Google Scholar]
- 23. Choi C, Bhardwaj PP, Seres P, et al. Measurement of glycine in human brain by triple refocusing 1H‐MRS in vivo at 3.0T. Magn Reson Med 2008;59:59–64. [DOI] [PubMed] [Google Scholar]
- 24. Kreis R, Ernst T, Ross BD. Development of the human brain: In vivo quantification of metabolite and water content with proton magnetic resonance spectroscopy. Magn Reson Med 1993;30:424–437. [DOI] [PubMed] [Google Scholar]
- 25. Brand A, Richter‐Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci 1993;15:289–298. [DOI] [PubMed] [Google Scholar]
- 26. Michaelis T, Helms G, Merboldt KD, et al. Identification of Scyllo‐inositol in proton NMR spectra of human brain in vivo. NMR Biomed 1993;6:105–109. [DOI] [PubMed] [Google Scholar]
- 27. Eulenburg V, Armsen W, Betz H, et al. Glycine transporters: Essential regulators of neurotransmission. Trends Biochem Sci 2005;30:325–333. [DOI] [PubMed] [Google Scholar]
- 28. Legendre P. The glycinergic inhibitory synapse. Cell Mol Life Sci 2001;58:760–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heindel W, Kugel H, Roth B. Noninvasive detection of increased glycine content by proton MR spectroscopy in the brains of two infants with nonketotic hyperglycinemia. AJNR Am J Neuroradiol 1993;14:629–635. [PMC free article] [PubMed] [Google Scholar]
- 30. Ganji SK, Maher EA, Choi C. In vivo (1)H MRSI of glycine in brain tumors at 3T. Magn Reson Med 2016;75:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nassogne MC, Commare MC, Lellouch‐Tubiana A, et al. Unusual presentation of GM2 gangliosidosis mimicking a brain stem tumor in a 3‐year‐old girl. AJNR Am J Neuroradiol 2003;24:840–842. [PMC free article] [PubMed] [Google Scholar]
- 32. Satoh H, Yamato O, Asano T, et al. Cerebrospinal fluid biomarkers showing neurodegeneration in dogs with GM1 gangliosidosis: Possible use for assessment of a therapeutic regimen. Brain Res 2007;1133:200–208. [DOI] [PubMed] [Google Scholar]
- 33. Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA 2000;97:10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeyakumar M, Thomas R, Elliot‐Smith E, et al. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003;126:974–987. [DOI] [PubMed] [Google Scholar]
- 35. Yamaguchi A, Katsuyama K, Nagahama K, et al. Possible role of autoantibodies in the pathophysiology of GM2 gangliosidoses. J Clin Invest 2004;113:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article:
Table S1. Changes of MRI findings and clinical deteriorations.
Table S2. Results of area under the MRS peak (arbitrary unit) of five metabolites and metabolite ratio in the frontal lobe of a dog with Sandhoff's disease.