

Abstract
Enzymes undergo a range of internal motions from local, active site fluctuations to large‐scale, global conformational changes. These motions are often important for enzyme function, including in ligand binding and dissociation and even preparing the active site for chemical catalysis. Protein engineering efforts have been directed towards manipulating enzyme structural dynamics and conformational changes, including targeting specific amino acid interactions and creation of chimeric enzymes with new regulatory functions. Post‐translational covalent modification can provide an additional level of enzyme control. These studies have not only provided insights into the functional role of protein motions, but they offer opportunities to create stimulus‐responsive enzymes. These enzymes can be engineered to respond to a number of external stimuli, including light, pH, and the presence of novel allosteric modulators. Altogether, the ability to engineer and control enzyme structural dynamics can provide new tools for biotechnology and medicine.
Keywords: allostery, enzyme catalysis, external stimulus, networks, protein dynamics, protein engineering, stimulus‐responsive material, structural dynamics
Introduction: The Case for Dynamics
Enzyme catalysis is a very dynamic process: chemical bonds in substrates are broken and formed to yield products, and enzymes often undergo structural changes during these events. In this review, we use the term “structural dynamics” to mean time‐dependent changes in the three‐dimensional protein structure. The amplitudes and timescales for these structural changes can range over many orders of magnitude, from bond vibrations occurring on the femtosecond timescale, to loop motions on the microsecond to millisecond timescales, and to large‐scale domain rearrangements that might occur over seconds or longer;1 all of these events can be important for enzyme function. We recognize that there continues to be controversy about if and how enzyme structural dynamics relates to chemical events and their associated rates.2, 3 Here, we take enzyme catalysis to include all physical and chemical events from initial substrate binding to final product release. Indeed, the overall catalytic rate often depends on events other than chemical catalysis.3 While our understanding of the connections between protein structural dynamics and enzyme function remains poor, even our incomplete understanding has provided a framework to begin engineering protein structural dynamics to change enzyme catalytic activity (e.g., Ref. 4), substrate selectivity (e.g., Ref. 5), the nature of the catalyzed chemical reaction (e.g., Ref. 6), and/or the enzyme's physiochemical properties (e.g., temperature stability7). In this review, we note many of the ways that protein structural dynamics can be altered to modify enzyme function, including site mutations, protein hybrids and other post‐translational chemical modifications. We pay special attention to how these modifications can generate stimulus‐responsive enzymes (e.g., light‐activated), providing unique biotechnology tools. We note that our examples are illustrative, not exhaustive, and that there are other recent reviews related to this area that may highlight alternative cases or approaches.8, 9, 10, 11, 12, 13
Enzyme Structural Dynamics and the Chemical Step
It has been proposed that in some enzymes, there are so‐called rate‐promoting motions that tend to “push” reacting atoms closer together, effectively compressing the reaction coordinate.14, 15, 16, 17, 18 For example, a “network of coupled promoting motions” in Escherichia coli dihydrofolate reductase (DHFR) enzyme has been suggested to sterically push the substrate dihydrofolate (DHF) and the NADPH cofactor closer together, gating the distance between the hydride donor and acceptor atoms19 [Fig. 1(A)]. Amino acid substitutions at these network residues result in changes to the hydride transfer rate and the ability of DHFR to promote hydride quantum tunneling.20, 21, 22, 23, 24 Many of the amino acid changes also lead to changes in DHFR structural dynamics as evidenced by nuclear magnetic resonance (NMR) spectroscopy and molecular dynamics (MD) simulations.25, 26, 27, 28, 29, 30, 31
Figure 1.
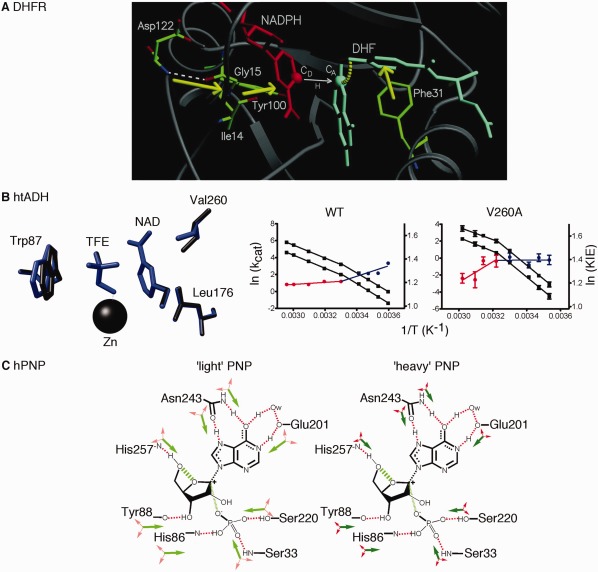
Promoting motions proposed to be important in the compression of the reaction coordinate in enzyme catalyzed reactions. A. Molecular dynamics simulations suggest a network of motions in dihydrofolate reductase that sterically compresses the reaction coordinate and gates the distance between hydride donor (CD) and acceptor (CA) atoms. Reprinted with permission from Ref. 19. B. Formation of small pockets near the NAD cofactor of thermophilic alcohol dehydrogenase through the substitution of Val260 (or Leu176) with Ala leads to changes in the catalytic turnover rate (kcat) and the H/D kinetic isotope effect (KIE) for hydride transfer. The kcat values at different temperatures for hydrogen and deuterium transfer are shown in black (plotted as a function of the left axis), and red and blue indicate the KIE (=kcat,H/kcat,D) highlighting the temperature break in Arrhenius behavior around 30°C (plotted as a function of the right axis). The WT and V260A enzymes have opposing behaviors in the temperature dependence of the KIE. In the V260A variant, hydride tunneling is less important at low temperatures (represented by the blue line) because preorganization is disrupted. At high temperatures (represented by the red line), hydrogen tunneling is more important, but the created cavity increases the need for donor‐acceptor distance sampling (i.e., reorganization is also disrupted). Adapted with permission from Ref. 48. C. Proposed mass‐altered bond vibrational modes in the 1H, 12C, 14N “light” and 2H, 13C, 15N “heavy” human purine nucleoside phosphorylase enzymes. Green arrows tend to promote transition‐state barrier crossing, and red arrows are orthogonal to the reaction coordinate. In the “heavy” enzyme, local motions are reduced, as indicated by the smaller arrows, including those proposed to be important for compressing the distance between the O5’ and O4’ oxygens of the ribose ring, as indicated by the hashed green line. One key motion involves His257; amino acid changes designed to less sterically constrain His257 lead to mass‐independent barrier crossing rates. Adapted with permission from Ref. 56.
Similar findings have been found in other enzymes, especially those in which quantum tunneling may play a role.32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42 Quantum tunneling behavior is often probed using kinetic isotope effects (KIEs) (see reviews43, 44). In many cases, KIEs are temperature‐independent for the wild‐type (WT) enzyme but become more temperature‐dependent when optimal structural dynamics have been perturbed.22, 32, 33, 45 It has been suggested that these perturbations limit the ability of the enzyme to sample tunneling‐ready active site conformations, requiring more motion at higher temperatures to achieve the required optimal donor‐acceptor distance.46 Results from the thermophilic alcohol dehydrogenase from Bacillus stearothermophilus (htADH) provide even more insight. In WT htADH, the KIE is temperature‐dependent below 30°C, but largely temperature‐independent above 30°C (i.e., nearer its temperature optimum).47, 48 The temperature trend in KIEs is reversed in variants in which small cavities have been introduced near the NAD(H) cofactor by substituting residues Leu176 and Val260 for less bulky residues.48, 49 These results have been interpreted in regards to a model in which both large‐scale global conformational sampling, or “preorganization”, and smaller‐scale local adjustments, or “reorganization”, are important for enzyme function.46, 48 The htADH variants are unable to achieve the necessary conformations for hydrogen tunneling at low temperatures (i.e., preorganization is disrupted), so quantum tunneling is a less important rate factor. At higher temperatures, the overall protein structure is more conducive to hydrogen tunneling, but the created cavities increase the need for donor‐acceptor distance sampling (i.e., reorganization is disrupted) [Fig. 1(B)].
Rate‐promoting vibrations and active site compression may also be important in other types of enzyme reactions. For example, Bruice and colleagues have suggested that the enzymes chorismate mutase and indole‐3‐glycerol phosphate synthase (InGPS) aid their associated chemical reactions by holding their substrates in constrained, more chemically reactive conformations.50, 51 Active site compaction may also be an important consideration in methyl transfer catalyzed by catechol‐O‐methyltransferase.52, 53 In human purine nucleoside phosphorylase (hPNP), it has been proposed that the movement of His257 helps compress the distance between the O5’ and O4’ oxygens of the ribose ring, which promotes re‐shuttling of the electrons into the purine ring and transition‐state barrier crossing.54 Like in previous examples, mutagenesis studies suggest that there is a coupled network of motions, even including residues on the surface of the enzyme.55 Besides mutagenesis, comparisons between light‐ (i.e., 1H, 12C, 14N) and heavy‐isotope (i.e., 2H, 13C, 15 N) labeled hPNP have been used to gain insight into rate‐promoting vibrations.56, 57, 58 The “heavy” hPNP enzyme had a 9.9% increase in mass compared to the normal “light” enzyme, and the rate constant for the chemical step decreased by 30%.56 Hybrid quantum mechanics/molecular mechanics (QM/MM) calculations indicated that the heavy enzyme had mass‐altered vibrational modes, leading to suboptimal transition‐state barrier crossing58 [Fig. 1(C)]. Consistent with these proposals, the “light” and “heavy” enzymes respond differently to amino acid substitutions designed to affect catalytic site vibrational modes.6 For instance, amino acid substitutions of Glu258 to Asp and Leu261 to Ala were designed to reduce steric contacts with His257 and allow for a greater range of motion, and potential uncoupling of protein and substrate motions. Unlike in the WT enzyme, “light” and “heavy” versions of the E258D/L261A double variant had almost identical rate constants for the chemical step. These results are very analogous to those for the discussed htADH variants discussed above. Studies with other heavy‐isotope labeled enzymes have led to similar ideas.59, 60, 61, 62, 63
Modulation of Loop Dynamics
Loops are among the most structurally dynamic features of proteins.64, 65 They always link more ordered secondary structures (i.e., α‐helices and β‐strands), and can range from well‐defined turns to random‐coil‐like structures. Many of these loops are “invisible” to structure determining methods owing to their conformationally dynamic nature.66 Loops can be important for a range of functions, including interactions with ligands, other parts of the protein, or other macromolecular binding partners.64, 65 Loop motions can also play direct roles in chemical catalysis through positioning catalytically important amino acid residues (e.g., Ref. 67).
While the length, amino acid composition, and the presence of hydrogen‐bonds can all affect loop motions,68 loops often interact with each other in order to gate their motions.69, 70 An illustrative example can be found in E. coli DHFR. There are three major loops in DHFR, those being the Met20, FG, and GH loops71 [Fig. 2(A)]. The Met20 loop folds over the active site and changes conformation between the open, closed, and occluded conformations.71, 72 In the closed conformation, the FG loop contacts the Met20 loop through interactions between the Asp122 side chain and the Gly17 backbone. After the chemical step, the Met20 loop assumes the occluded conformation that blocks the nicotinamide ring of the NADP+ cofactor from entering the active site. In the occluded conformation, the FG‐Met20 loop interactions are broken, and new interactions form between Ser148 of the GH loops and the Asn23 backbone. Amino acid changes to alter the FG‐Met20 or Met20‐GH loop interactions results in changes to the catalytic cycle rate constants.73, 74 For example, the Gly121Val substitution on the FG loop, which perturbs the closed conformation, leads to a 40‐fold decrease in NADPH binding and a 200‐fold decrease in hydride transfer rate.73 The substitution results in changes to the loop motions, and motions elsewhere in the protein, on the picosecond‐nanosecond and microsecond‐millisecond timescales, according to NMR relaxation analyses.25, 27 Amino acid changes to position 148 on the GH loop also result in changes to ligand affinities and ligand off‐rates.74
Figure 2.
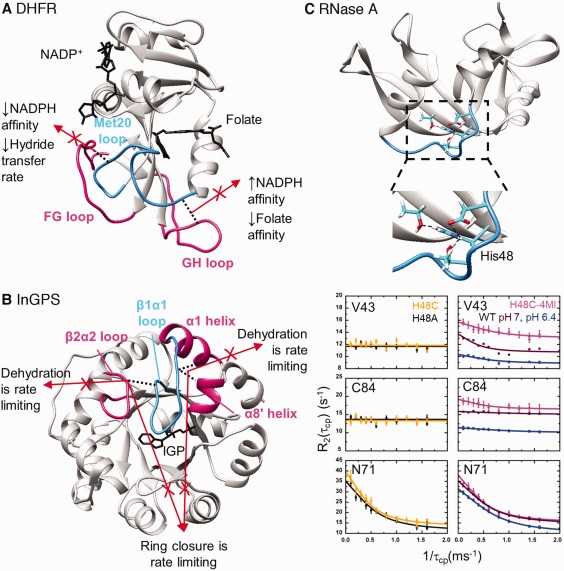
Gated loop motions important in enzyme function. A. In E. coli dihydrofolate reductase, the FG and GH loops compete for hydrogen‐bonding interactions with the active site Met20 loop. When interactions between the FG and Met20 loops are disrupted, the occluded conformation is favored and the hydride transfer rate decreases. When interactions between the GH and Met20 loops are disrupted, the closed conformation is favored and substrate and cofactor affinities are altered. B. In S. solfataricus indole‐3‐glycerol phosphate synthase, there are competing interactions for the active site β1α1 loop. When either set of interactions is lost, the second chemical step (i.e., dehydration) is rate limiting, but if both sets of interactions are disrupted, the first chemical step (i.e., ring closure) is rate limiting similar to the WT enzyme. Results suggest that the competing interactions keep the β1α1 loop appropriately dynamic. C. In ribonuclease A, the protonation state of His48 helps to control μs‐ms timescale motions. His48 makes interactions with loop 1 and β‐strands housing functionally important residues. The μs‐ms timescale motions can be evaluated using NMR relaxation dispersion methods (see182). When the time between successive 180° refocusing pulses (τcp) is increased in the NMR relaxation dispersion experiments, conformational exchange can contribute to a higher apparent R2 transverse relaxation rate. Contributions to the R2 from conformational exchange are suppressed when τcp is decreased. Some motions are repressed when His48 is substituted with Cys (yellow curves, left) or Ala (black curves, left), but these motions are essentially restored by re‐introducing the imidazole moiety through reaction of the H48C variant with 4‐chloromethyl imidazole (pink curves, right). Some motions in the WT enzyme are also pH dependent (pH 7, brown curves; pH 6.4, blue curves). The motions of Asn71 are not perturbed by His48 substitution or by a change in pH. These studies indicate that important motions can be pH modulated and changed through covalent modification. Reprinted from Ref. 90 with permission from Biophysical Society.
The loop interactions in DHFR are also likely important for catalytic flux, which may depend on the cellular environment and NADP+/NADPH concentrations.75, 76 For example, the occluded conformation is disfavored by a polyproline sequence at the C‐terminal end of the Met20 loop in human DHFR. When this sequence was introduced in the E. coli homolog (i.e., N23PP), the µs‐ms timescale dynamics were ablated, although the global protein structure did not change substantially.77 The hydride transfer step was also severely impaired, which was put forth as evidence that the structural dynamics of these loops was essential for chemical catalysis in this enzyme,77 although there exists skepticism regarding the direct connection between protein motions and hydride transfer.78 The loop dynamics may also be important for biological function, considering that human DHFR is unable to rescue DHFR‐deficient E. coli cells.76
Similar loop‐loop interactions are important in other enzymes,69, 70 including for InGPS. As with other (β/α)8 barrel enzymes, the residues directly involved in chemical catalysis lie on the β‐strands and on loops connecting the β‐strands to the proceeding α‐helices.79 InGPS catalyzes the ring closure and dehydration of 1‐(o‐carboxyphenylamino)‐1‐deoxyribulose‐5‐phosphate (CdRP) to form indole‐3‐glycerol phosphate (IGP) in two discrete steps. In the Sulfolobus solfataricus enzyme (ssInGPS), the rate‐limiting step changes based on the temperature, suggesting a connection between enzyme structural dynamics and the kinetic mechanism.80 It has been suggested that substrate binding, product release and intermediate reorientation are coordinated by the β1α1 loop dynamics.51, 81, 82, 83 The β1α1 loop interacts with the β2α2 loop through a hydrogen bonding interaction between Arg54 and Asn90 [Fig. 2(B)]. The N90A substitution (on the β2α2 loop) induces small changes to the InGPS kinetic parameters, changes the identity of the rate‐limiting step to the dehydration step, and leads to a surprising decrease in β1α1 loop flexibility.84 The β1α1 loop also interacts with residues on the adjacent α1 and α8’ helices. Removal of these interactions through the R64A/D65A substitutions also results in the dehydration step becoming rate‐limiting and a decrease in the β1α1 loop flexibility.85 However, severing the hydrogen bonds on both the N‐ and C‐terminal sides of the loop as in the R64A/D65A/N90A variant restores some flexibility to the β1α1 loop and the ring closure step becomes rate‐limiting again.85 We have suggested that this “tug‐of‐war” between hydrogen‐bonding partners on either side of the β1α1 loop facilitates the motions needed to properly catalyze the ssInGPS reaction.85 This type of “frustration” (i.e., the inability to fulfill all potential interactions simultaneously) may in general be important for controlling protein structural dynamics.86
Changing loop motions offers a means to control enzyme structural dynamics in a stimulus‐dependent manner. For example, hydrogen bonding interactions from the side chain of His48 helps to control loop dynamics and function in ribonuclease A (RNase A).87, 88, 89, 90 As also observed for E. coli DHFR,91 there are both pH‐sensitive and pH‐insensitive motions in RNase.90 Interestingly, the pH‐dependence of the motions closely resembles the pH‐dependence of the RNase A catalytic activity,90 which might both be dependent on the protonation state of His48. The H48C substitution abrogates the pH‐dependent motions in loop 1 and connected regions, but covalent attachment of 4‐methylimidazole at Cys48 restores these motions90 [Fig. 2(C)]. These studies elegantly demonstrate that functionally important motions can be pH‐modulated, and these motions can be activated (or potentially deactivated) through covalent modification at critical residues. Chemical rescue of perturbed noncovalent interactions may be a general way to allosterically control enzyme catalytic activity as also shown in indole rescue of Trp to Gly variants of β‐glycosidase and β‐glucuronidase enzymes.92
Enzymes as Amino Acid Interaction Networks
Globular proteins can be viewed as three‐dimensional interaction networks.9, 12, 93, 94 Many structure‐ and sequence‐based methods have been developed to identify amino acid interaction networks in proteins.95 For example, networks have been proposed for E. coli DHFR based on amino acid covariation in multiple sequence alignments,96, 97 molecular dynamics simulations19 and X‐ray crystallography analysis.98, 99 The CONTACT (Contact Networks Through Alternate Conformation Transitions) algorithm uses high resolution X‐ray data to identify alternative side chain conformations that may propagate motions through steric interactions98, 100 [Fig. 3(A)]. This method provided atomic‐level insight into how distal amino acid substitutions, like G121V and S148A, can propagate their effects throughout the enzyme. Similar types of network analyses have contributed to our understanding in many other enzyme systems.101, 102, 103, 104, 105, 106, 107
Figure 3.
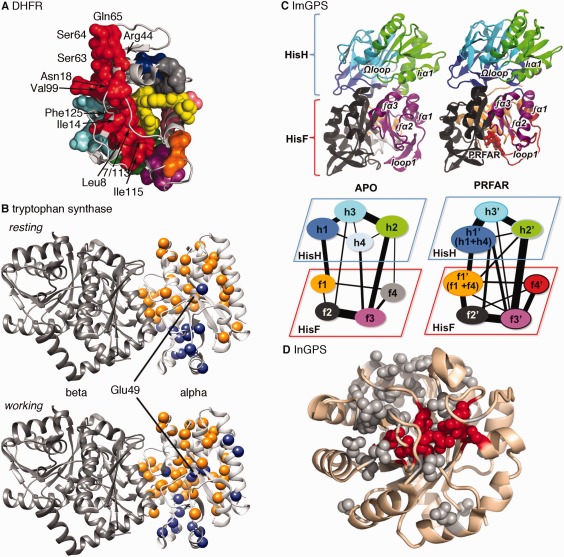
Dynamic amino acid interaction networks in the function and allosteric regulation of enzymes. A. The CONTACT approach, based on fitting alternative side‐chain conformations in X‐ray diffraction maps, was used to identify amino acid interaction networks in E. coli dihydrofolate reductase. This approach provides experimental insight into the connections between distant parts of the enzyme, including those regions identified as being part of a network of coupled promoting motions [see Fig. 1(A)]. Reprinted by permission from Macmillan Publishers Ltd.98 B. The CHESCA approach,111 based on analyzing NMR chemical shift changes induced by protein perturbations, was used to identify amino acid interactions networks in the alpha subunit of tryptophan synthase. The orange and blue spheres indicate two different networks. The catalytic residue Glu49 has a different network association in the apo resting state and in the catalytically active working state. Network residues also include those at the alpha/beta interface. C. Amino acid interaction network communities (represented by different colors) in imidazole glycerol phosphate synthase from the histidine biosynthetic pathway, which includes both HisH and HisF subunits. Ammonia is passed from the HisH to the HisF subunits. Binding of PRFAR (N’‐[(5’‐phosphoribulosyl)formimino]‐5‐aminoimidazole‐4‐carboxamide ribonucleotide) changes community interactions. Reprinted with permission from Ref. 116. D. Statistical coupling analysis, based on amino acid covariation in a multiple sequence alignment,166 was used to identify amino acid interaction networks (or sectors) in indole‐3‐glycerol phosphate synthase. Residues highlighted in red are those at the active site. Amino acid substitutions at many of these positions lead to changes in competitive effectiveness of yeast strains carrying these variants, according to high throughput mutational analyses. Reprinted with permission from Ref. 132.
These long‐range networks may also offer means through which enzymes communicate in multi‐enzyme complexes.97 For example, we used NMR chemical shift covariance analysis108, 109among a series of site‐directed protein variants to identify amino acid interaction networks in the alpha subunit of E. coli tryptophan synthase (αTS).110, 111 Our analysis revealed that there are two networks of residues [colored orange and blue in Fig. 3(B)], and the residues involved in these networks change depending on what is bound to the protein.110, 111, 112 Intriguingly, Glu49, a residue directly involved in catalysis, belongs to different networks when comparing the apo resting state and when the enzyme is actively turning over substrate/products (i.e., the working state).111 Molecular dynamics (MD) simulations also indicated that the motions of Glu49 are correlated to different sets of residues in these states.111, 113, 114 Amino acid substitutions at surface‐exposed network positions led to modest changes in αTS kinetic parameters.111 These networks may also be important for how αTS communicates and coordinates channeling of its product indole into the active site of the beta subunit of tryptophan synthase (βTS). An amino acid substitution known to disrupt direct channeling of indole from the α‐ to β‐active sites also disrupts networks that might transmit structural dynamic information from the α‐active site to the α/β binding interface.112
The functional coordination of different active sites through amino acid interaction networks is exemplified by the histidine biosynthetic enzyme imidazole glycerol phosphate synthase (ImGPS).115, 116, 117, 118 ImGPS is a heterodimeric enzyme consisting of the HisH subunit, which catalyzes the hydrolysis of glutamine to produce ammonia, and the HisF subunit, which receives the ammonia and catalyzes its reaction with PRFAR (N’‐[(5'phosphoribusyl}formimino]‐5]aminoimidazle‐4‐carboxamide ribonucleotide).119 The glutaminase activity of the HisH subunit is dependent on binding of PRFAR to the HisH subunit.120 The Batista and Loria groups have used MD simulations to identify correlated motions in ImGPS and community analysis to identify allosteric pathways116 [Fig. 3(C)]. Amino acid substitutions at network positions in the HisH subunit led to changes in the microsecond‐millisecond timescale dynamics according to NMR relaxation dispersion spectroscopy, and decreased PRFAR‐enhancement of the HisF catalytic activity.117 Intriguingly, these groups have also found a small molecule inhibitor that binds at the HisH‐HisF interface also disrupts ImGPS motions coordinated by the proposed network.121 These results and many others (e.g., Refs. 10, 122, 123, 124, 125, 126, 127) have suggested that surface‐exposed network residues may serve as druggable interfaces for allosteric modulators.
High throughput mutational analyses128, 129, 130, 131 may allow for more comprehensive interrogation and validation of these amino acid interaction networks. For example, the Matthews groups conducted a comprehensive mutational analysis of three InGPS enzymes (i.e., >5000 mutations) expressed in yeast cells in which the endogenous IGPS gene was deleted,132 following the deep mutational scan approach developed by the Bolon group.133 One of their most intriguing findings was the discovery of a long‐range allosteric pathway connecting the outer α‐helical shell to the active site, as elucidated by the analysis of beneficial mutations in their high throughput analysis, and consistent with amino acid covariation in the naturally occurring sequences of InGPS enzymes132 [Fig. 3(D)]. The connection between surface‐exposed sites and the active site is very reminiscent of the NMR‐derived networks we elucidated for the structurally and functionally related αTS enzyme.111
Controlling Enzyme Structural Dynamics through Covalent Constraints
In most of the above discussion, the goal was to modify noncovalent interactions to affect enzyme function and structural dynamics. Motions might also be altered through the introduction of covalent constraints, or “molecular stapling.” Cysteine mutagenesis has been commonly done to introduce disulfide bonds to measure their impact on protein structure and stability,134, 135, 136 including classic enzyme examples.137, 138, 139, 140 For example, Ma et al. introduced disulfide bonds to target the fluctuations of mobile elements in T7 RNA polymerase139 [Fig. 4(A)]. Through these studies, they were able to identify conformations and their associated transitions most important for different stages of RNA replication, including initiation and elongation. There is also the potential to control the catalytic function of enzymes with introduced disulfide bonds through their redox states,138 or even potentially through microwave irradiation.141 Other types of crosslinking technologies are also being developed,142, 143, 144 including those that can be genetically encoded.145, 146
Figure 4.
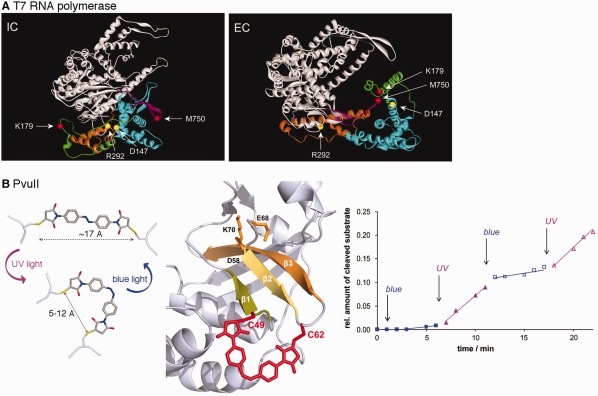
Stimulus‐responsive covalent constraints can control enzyme conformational changes and function. A. Disulfide bridges were engineered to capture the initiation (IC; C147‐C292) and elongation (EC; C179‐C750) complexes of T7 RNA polymerase. Reprinted with permission from Ref. 139. B. Light‐dependent enzyme activity through engineering azobenzene‐crosslinked PvuII restriction endonuclease. Photoconversion of the cross‐linked azobenzene moiety to the cis form results in greater restriction endonuclease activity. Reprinted with permission from Ref. 152.
Some of the most interesting cross‐linkers are those sensitive to environmental conditions. For example, azobenzene and its derivatives have proven to be useful photoswitches, as they will undergo a cis/trans isomerization which can lead to structural changes to their attached biomolecule.147, 148, 149 An azobenzene photoswitch was covalently linked across a binding groove of a PDZ domain, resulting in conformational changes that mimic those involved in ligand binding.150 MD simulations also supported the notion that the photoswitch mimicked the transitions between ligand free and bound forms of the protein.151 In another example, multiple azobenzene‐crosslinked derivatives of the restriction enzyme PvuII were generated,152 guided by the sGAL program that helps to find suitable surface‐exposed sites for Cys‐mediated crosslinks153 [Fig. 4(B)]. Those variants with double crosslinks, especially those that included crosslinks closer to the active site, gave the greatest enzyme catalytic activity difference between the cis and trans configuration of the azobenzene photoswitches.152 It should be noted that in some cases the azobenzene does not need to act as part of a crosslink to affect an enzyme's catalytic activity. Azobenzene‐maleimide probes were attached through a single Cys substitution on an important loop in bacterial histone deacetylase amidohydrolase (HDAH) to create a light‐switchable enzyme.154 The modified HDAH enzyme was more active when the azobenzene photoswitch was in the trans‐ compared to the cis‐configuration, potentially because in the cis‐configuration the moiety interacts with the enzyme surface.155
Surface‐exposed Cys residues may in general serve as attachment points for novel, allosteric modulators. Recently, Wu and coworkers have created the “cysteinome”—a comprehensive database of proteins with targetable cysteines along with any known covalent inhibitors.156 Indeed, Backus and coworkers performed a quantitative analysis of small molecule fragments reactive towards cysteine.157 Covalent allosteric drugs were previously known for several high priority drug targets, including GTPases, kinases, GPCRs, heat‐shock proteins and ribonucleases.158, 159 Natural post‐translation modifications may likewise affect protein function through allosteric mechanisms.160 All of these effects may depend in part on how covalent modification alters protein structural dynamics.
Grafting Proteins Domains to Control Enzyme Function
Larger global changes to protein structural dynamics can be accomplished by creating hybrid or fusion proteins, in which existing regulatory domains are engineered onto another protein.161, 162, 163, 164, 165 The hybrid enzymes can then respond to the presence of different ligands or even light, likely through induced conformational changes.162 For example, the Ranganathan and Benkovic groups created a light‐dependent DHFR enzyme by inserting the light‐sensitive LOV2 domain into the FG loop of DHFR based on network sites derived from their amino acid covariation analysis known as statistical coupling analysis96, 166, 167, 168 [Fig. 5(A)]. Other network methods169 and knowledge of the structural dynamics of the regulatory and catalytic domains can inform on the appropriate attachment points. Hahn and colleagues created a number of ligand‐ and light‐dependent hybrid enzymes by inserting regulatory domains into loops that were determined by MD simulations to be mechanically coupled to the active site170, 171 [Fig. 5(B)]. These studies also exemplify the vast practical utility of these biotechnology tools to explore biological interactions and function.170, 172
Figure 5.
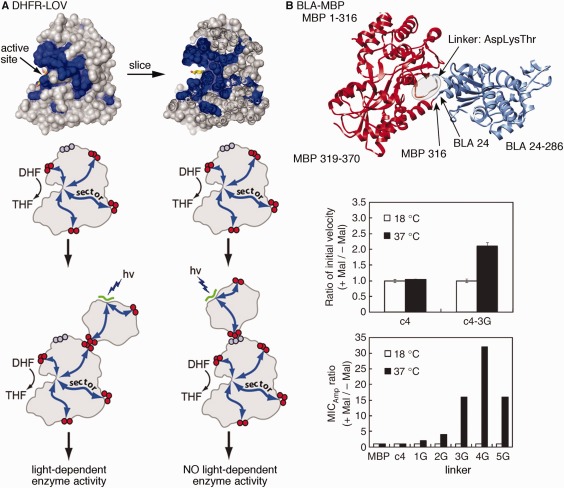
Chimeric enzymes to control structural dynamics and catalytic function according to external stimuli. A. Statistical coupling analysis was used to identify surface‐exposed network residues in E. coli dihydrofolate reductase and the light‐responsive LOV domain. Engineering of chimeric enzymes to join these positions results in light‐dependent catalytic conversion of dihydrofolate (DHF) to tetrahydrofolate (THF); use of other non‐network, surface‐exposed sites does not lead to light‐dependent enzyme activity. Reprinted from Ref. 97 with permission from Cell Press. B. Engineering of chimeric β‐lactamase‐maltose binding proteins. The c4 fusion protein as shown in the ribbon diagram is not responsive to the presence of maltose, but the introduction of additional glycine residues into the linker, which presumably increases molecular flexibility, led to maltose‐ and temperature‐dependent catalytic activity and biological response (according to ampicillin minimum inhibitory concentration, MIC). Reprinted with permission from Ref. 163.
Future Outlook
There have been tremendous advancements over the past decade in our understanding of protein structure and design173 and enzyme engineering,174 along with a continued acknowledgement of the importance of protein structural dynamics to enzyme function and regulation. We may be approaching a time when not only de novo protein design yields desired structures, but fluctuations between two or more protein conformations on desired timescales can be more aptly engineered.175, 176, 177 For example, the Goto and Chica labs used multistate design to induce Streptococcal protein G domain β1 protein to fluctuate between two designed conformations.177 Similar methods might be used to more rationally engineer enzyme structural dynamics. Studies presented in this review and elsewhere have shown that enzyme structural dynamics are naturally, or can be engineered to be, sensitive to external stimuli, including changes in pH, light, pressure and covalent modification. This ability to control enzyme structural dynamics may provide many powerful biotechnology tools. It should also be pointed out that issues pertaining to molecular structural dynamics extend to non‐biological catalysts (e.g., Refs. 178, 179, 180). As these molecular machines181 become larger and more complex, there will be an increased need to control the magnitude, trajectories, and timescales of their moving parts. Lessons learned from enzymes and other proteins will likely be applicable for resolving these issues.
Acknowledgments
We thank Jingjing Shi for helping with the initial conception of this review. The work in the Boehr laboratory on enzyme structural dynamics and amino acid interaction networks is supported by NSF grant MCB1615032 and NIH grant R01AI104878.
References
- 1. Henzler‐Wildman K, Kern D (2007) Dynamic personalities of proteins. Nature 450:964–972. [DOI] [PubMed] [Google Scholar]
- 2. Warshel A, Bora RP (2016) Perspective: defining and quantifying the role of dynamics in enzyme catalysis. J Chem Phys 144:180901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kohen A (2015) Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc Chem Res 48:466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suliman M, Santosh V, Seegar TCM, Dalton AC, Schultz KM, Klug CS, Barton WA (2017) Directed evolution provides insight into conformational substrate sampling by SrtA. PLoS One 12:e0184271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vogeli B, Bibow S, Chi CN (2016) Enzyme selectivity fine‐tuned through dynamic control of a loop. Angew Chem Int Ed Engl 55:3096–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zoi I, Suarez J, Antoniou D, Cameron SA, Schramm VL, Schwartz SD (2016) Modulating enzyme catalysis through mutations designed to alter rapid protein dynamics. J Am Chem Soc 138:3403–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Childers MC, Daggett V (2017) Insights from molecular dynamics simulations for computational protein design. Mol Syst Des Eng 2:9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Narayanan C, Bernard DN, Doucet N (2016) Role of conformational motions in enzyme function: selected methodologies and case studies. Catalysts 6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhabha G, Biel JT, Fraser JS (2015) Keep on moving: discovering and perturbing the conformational dynamics of enzymes. Acc Chem Res 48:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dokholyan NV (2016) Controlling allosteric networks in proteins. Chem Rev 116:6463–6487. [DOI] [PubMed] [Google Scholar]
- 11. McAuley M, Timson DJ (2017) Modulating mobility: a paradigm for protein engineering?. Appl Biochem Biotechnol 181:83–90. [DOI] [PubMed] [Google Scholar]
- 12. Lisi GP, Loria JP (2017) Allostery in enzyme catalysis. Curr Opin Struct Biol 47:123–130. [DOI] [PubMed] [Google Scholar]
- 13. Motlagh HN, Wrabl JO, Li J, Hilser VJ (2014) The ensemble nature of allostery. Nature 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nashine VC, Hammes‐Schiffer S, Benkovic SJ (2010) Coupled motions in enzyme catalysis. Curr Opin Chem Biol 14:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz SD (2013) Protein dynamics and the enzymatic reaction coordinate. Top Curr Chem 337:189–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agarwal PK (2006) Enzymes: an integrated view of structure, dynamics and function. Microb Cell Fact 5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klinman JP (2015) Dynamically achieved active site precision in enzyme catalysis. Acc Chem Res 48:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hay S, Pudney CR, Scrutton NS (2009) Structural and mechanistic aspects of flavoproteins: probes of hydrogen tunnelling. FEBS J 276:3930–3941. [DOI] [PubMed] [Google Scholar]
- 19. Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes‐Schiffer S (2002) Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA 99:2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajagopalan PT, Lutz S, Benkovic SJ (2002) Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry 41:12618–12628. [DOI] [PubMed] [Google Scholar]
- 21. Wang L, Goodey NM, Benkovic SJ, Kohen A (2006) The role of enzyme dynamics and tunnelling in catalysing hydride transfer: studies of distal mutants of dihydrofolate reductase. Philos Trans R Soc Lond B Biol Sci 361:1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang L, Goodey NM, Benkovic SJ, Kohen A (2006) Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci USA 103:15753–15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stojkovic V, Perissinotti LL, Lee J, Benkovic SJ, Kohen A (2010) The effect of active‐site isoleucine to alanine mutation on the DHFR catalyzed hydride‐transfer. Chem Commun 46:8974–8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh P, Sen A, Francis K, Kohen A (2014) Extension and limits of the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. J Am Chem Soc 136:2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mauldin RV, Sapienza PJ, Petit CM, Lee AL (2012) Structure and dynamics of the g121v dihydrofolate reductase mutant: lessons from a transition‐state inhibitor complex. PLoS One 7:e33252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mauldin RV, Lee AL (2010) Nuclear magnetic resonance study of the role of m42 in the solution dynamics of Escherichia coli dihydrofolate reductase. Biochemistry 49:1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boehr DD, Schnell JR, McElheny D, Bae SH, Duggan BM, Benkovic SJ, Dyson HJ, Wright PE (2013) A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry 52:4605–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watney JB, Agarwal PK, Hammes‐Schiffer S (2003) Effect of mutation on enzyme motion in dihydrofolate reductase. J Am Chem Soc 125:3745–3750. [DOI] [PubMed] [Google Scholar]
- 29. Wong KF, Selzer T, Benkovic SJ, Hammes‐Schiffer S (2005) Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc Natl Acad Sci USA 102:6807–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doron D, Stojkovic V, Gakhar L, Vardi‐Kilshtain A, Kohen A, Major DT (2015) Free energy simulations of active‐site mutants of dihydrofolate reductase. J Phys Chem B 119:906–916. [DOI] [PubMed] [Google Scholar]
- 31. Roston D, Kohen A, Doron D, Major DT (2014) Simulations of remote mutants of dihydrofolate reductase reveal the nature of a network of residues coupled to hydride transfer. J Comput Chem 35:1411–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hong B, Maley F, Kohen A (2007) Role of y94 in proton and hydride transfers catalyzed by thymidylate synthase. Biochemistry 46:14188–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meyer MP, Tomchick DR, Klinman JP (2008) Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (i553) in soybean lipoxygenase‐1. Proc Natl Acad Sci USA 105:1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilde TC, Blotny G, Pollack RM (2008) Experimental evidence for enzyme‐enhanced coupled motion/quantum mechanical hydrogen tunneling by ketosteroid isomerase. J Am Chem Soc 130:6577–6585. [DOI] [PubMed] [Google Scholar]
- 35. Wang Z, Abeysinghe T, Finer‐Moore JS, Stroud RM, Kohen A (2012) A remote mutation affects the hydride transfer by disrupting concerted protein motions in thymidylate synthase. J Am Chem Soc 134:17722–17730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hay S, Pudney CR, McGrory TA, Pang J, Sutcliffe MJ, Scrutton NS (2009) Barrier compression enhances an enzymatic hydrogen‐transfer reaction. Angew Chem Int Ed Engl 48:1452–1454. [DOI] [PubMed] [Google Scholar]
- 37. Pudney CR, Johannissen LO, Sutcliffe MJ, Hay S, Scrutton NS (2010) Direct analysis of donor‐acceptor distance and relationship to isotope effects and the force constant for barrier compression in enzymatic h‐tunneling reactions. J Am Chem Soc 132:11329–11335. [DOI] [PubMed] [Google Scholar]
- 38. Pudney CR, Khara B, Johannissen LO, Scrutton NS (2011) Coupled motions direct electrons along human microsomal p450 chains. PLoS Biol 9:e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davarifar A, Antoniou D, Schwartz SD (2011) The promoting vibration in human heart lactate dehydrogenase is a preferred vibrational channel. J Phys Chem B 115:15439–15444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Russell HJ, Jones AR, Hay S, Greetham GM, Towrie M, Scrutton NS (2012) Protein motions are coupled to the reaction chemistry in coenzyme b12‐dependent ethanolamine ammonia lyase. Angew Chem Int Ed Engl 51:9306–9310. [DOI] [PubMed] [Google Scholar]
- 41. Hardman SJ, Pudney CR, Hay S, Scrutton NS (2013) Excited state dynamics can be used to probe donor‐acceptor distances for h‐tunneling reactions catalyzed by flavoproteins. Biophys J 105:2549–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoeven R, Hardman SJ, Heyes DJ, Scrutton NS (2016) Cross‐species analysis of protein dynamics associated with hydride and proton transfer in the catalytic cycle of the light‐driven enzyme protochlorophyllide oxidoreductase. Biochemistry 55:903–913. [DOI] [PubMed] [Google Scholar]
- 43. Klinman JP, Kohen A (2013) Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu Rev Biochem 82:471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roston D, Islam Z, Kohen A (2014) Kinetic isotope effects as a probe of hydrogen transfers to and from common enzymatic cofactors. Arch Biochem Biophys 544:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stojkovic V, Perissinotti LL, Willmer D, Benkovic SJ, Kohen A (2012) Effects of the donor‐acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction. J Am Chem Soc 134:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nagel ZD, Klinman JP (2009) A 21st century revisionist's view at a turning point in enzymology. Nat Chem Biol 5:543–550. [DOI] [PubMed] [Google Scholar]
- 47. Klinman JP, Kohen A, Cannio R, Bartolucci S, Klinman JP (1999) Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 399:496–499. [DOI] [PubMed] [Google Scholar]
- 48. Nagel ZD, Meadows CW, Dong M, Bahnson BJ, Klinman JP (2012) Active site hydrophobic residues impact hydrogen tunneling differently in a thermophilic alcohol dehydrogenase at optimal versus nonoptimal temperatures. Biochemistry 51:4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagel ZD, Dong M, Bahnson BJ, Klinman JP (2011) Impaired protein conformational landscapes as revealed in anomalous Arrhenius prefactors. Proc Natl Acad Sci USA 108:10520–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang X, Bruice TC (2005) The proficiency of a thermophilic chorismate mutase enzyme is solely through an entropic advantage in the enzyme reaction. Proc Natl Acad Sci USA 102:18356–18360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazumder‐Shivakumar D, Bruice TC (2004) Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole‐3‐glycerol phosphate synthase. Proc Natl Acad Sci USA 101:14379–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J, Kulik HJ, Martinez TJ, Klinman JP (2015) Mediation of donor‐acceptor distance in an enzymatic methyl transfer reaction. Proc Natl Acad Sci USA 112:7954–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J, Klinman JP (2011) Enzymatic methyl transfer: role of an active site residue in generating active site compaction that correlates with catalytic efficiency. J Am Chem Soc 133:17134–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nunez S, Antoniou D, Schramm VL, Schwartz SD (2004) Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J Am Chem Soc 126:15720–15729. [DOI] [PubMed] [Google Scholar]
- 55. Saen‐Oon S, Ghanem M, Schramm VL, Schwartz SD (2008) Remote mutations and active site dynamics correlate with catalytic properties of purine nucleoside phosphorylase. Biophys J 94:4078–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Silva RG, Murkin AS, Schramm VL (2011) Femtosecond dynamics coupled to chemical barrier crossing in a born‐oppenheimer enzyme. Proc Natl Acad Sci USA 108:18661–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suarez J, Schramm VL (2015) Isotope‐specific and amino acid‐specific heavy atom substitutions alter barrier crossing in human purine nucleoside phosphorylase. Proc Natl Acad Sci USA 112:11247–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Antoniou D, Ge X, Schramm VL, Schwartz SD (2012) Mass modulation of protein dynamics associated with barrier crossing in purine nucleoside phosphorylase. J Phys Chem Lett 3:3538–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Swiderek K, Ruiz‐Pernía JJ, Moliner V, Tuñón I (2014) Heavy enzymes–experimental and computational insights in enzyme dynamics. Curr Opin Chem Biol 21:11–18. [DOI] [PubMed] [Google Scholar]
- 60. Wang Z, Singh P, Czekster CM, Kohen A, Schramm VL (2014) Protein mass‐modulated effects in the catalytic mechanism of dihydrofolate reductase: beyond promoting vibrations. J Am Chem Soc 136:8333–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kipp DR, Silva RG, Schramm VL (2011) Mass‐dependent bond vibrational dynamics influence catalysis by hiv‐1 protease. J Am Chem Soc 133:19358–19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Z, Chang EP, Schramm VL (2016) Triple isotope effects support concerted hydride and proton transfer and promoting vibrations in human heart lactate dehydrogenase. J Am Chem Soc 138:15004–15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Longbotham JE, Hardman SJ, Gorlich S, Scrutton NS, Hay S (2016) Untangling heavy protein and cofactor isotope effects on enzyme‐catalyzed hydride transfer. J Am Chem Soc 138:13693–13699. [DOI] [PubMed] [Google Scholar]
- 64. Nestl BM, Hauer B (2014) Engineering of flexible loops in enzymes. ACS Catal 4:3201–3211. [Google Scholar]
- 65. Papaleo E, Saladino G, Lambrughi M, Lindorff‐Larsen K, Gervasio FL, Nussinov R (2016) The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev 116:6391–6423. [DOI] [PubMed] [Google Scholar]
- 66. Levantino M, Yorke BA, Monteiro DC, Cammarata M, Pearson AR (2015) Using synchrotrons and XFELs for time‐resolved X‐ray crystallography and solution scattering experiments on biomolecules. Curr Opin Struct Biol 35:41–48. [DOI] [PubMed] [Google Scholar]
- 67. Whittier SK, Hengge AC, Loria JP (2013) Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 341:899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gu Y, Li DW, Bruschweiler R (2015) Decoding the mobility and time scales of protein loops. J Chem Theory Comput 11:1308–1314. [DOI] [PubMed] [Google Scholar]
- 69. Gunasekaran K, Ma B, Nussinov R (2003) Triggering loops and enzyme function: identification of loops that trigger and modulate movements. J Mol Biol 332:143–159. [DOI] [PubMed] [Google Scholar]
- 70. Gunasekaran K, Nussinov R (2004) Modulating functional loop movements: the role of highly conserved residues in the correlated loop motions. Chembiochem 5:224–230. [DOI] [PubMed] [Google Scholar]
- 71. Schnell JR, Dyson HJ, Wright PE (2004) Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct 33:119–140. [DOI] [PubMed] [Google Scholar]
- 72. Sawaya MR, Kraut J (1997) Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 36:586–603. [DOI] [PubMed] [Google Scholar]
- 73. Cameron CE, Benkovic SJ (1997) Evidence for a functional role of the dynamics of glycine‐121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site‐directed mutant. Biochemistry 36:15792–15800. [DOI] [PubMed] [Google Scholar]
- 74. Miller GP, Benkovic SJ (1998) Strength of an interloop hydrogen bond determines the kinetic pathway in catalysis by Escherichia coli dihydrofolate reductase. Biochemistry 37:6336–6342. [DOI] [PubMed] [Google Scholar]
- 75. Weikl TR, Boehr DD (2012) Conformational selection and induced changes along the catalytic cycle of Escherichia coli dihydrofolate reductase. Proteins 80:2369–2383. [DOI] [PubMed] [Google Scholar]
- 76. Bhabha G, Ekiert DC, Jennewein M, Zmasek CM, Tuttle LM, Kroon G, Dyson HJ, Godzik A, Wilson IA, Wright PE (2013) Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat Struct Mol Biol 20:1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE (2011) A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 332:234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Loveridge EJ, Behiry EM, Guo J, Allemann RK (2012) Evidence that a 'dynamic knockout' in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem 4:292–297. [DOI] [PubMed] [Google Scholar]
- 79. Sterner R, Hocker B (2005) Catalytic versatility, stability, and evolution of the (betaalpha)8‐barrel enzyme fold. Chem Rev 105:4038–4055. [DOI] [PubMed] [Google Scholar]
- 80. Zaccardi MJ, Mannweiler O, Boehr DD (2012) Differences in the catalytic mechanisms of mesophilic and thermophilic indole‐3‐glycerol phosphate synthase enzymes at their adaptive temperatures. Biochem Biophys Res Commun 418:324–329. [DOI] [PubMed] [Google Scholar]
- 81. Zaccardi MJ, Yezdimer EM, Boehr DD (2013) Functional identification of the general acid and base in the dehydration step of indole‐3‐glycerol phosphate synthase catalysis. J Biol Chem 288:26350–26356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schlee S, Dietrich S, Kurcon T, Delaney P, Goodey NM, Sterner R (2013) Kinetic mechanism of indole‐3‐glycerol phosphate synthase. Biochemistry 52:132–142. [DOI] [PubMed] [Google Scholar]
- 83. Mazumder‐Shivakumar D, Kahn K, Bruice TC (2004) Computational study of the ground state of thermophilic indole glycerol phosphate synthase: structural alterations at the active site with temperature. J Am Chem Soc 126:5936–5937. [DOI] [PubMed] [Google Scholar]
- 84. Zaccardi MJ, O'Rourke KF, Yezdimer EM, Loggia LJ, Woldt S, Boehr DD (2014) Loop‐loop interactions govern multiple steps in indole‐3‐glycerol phosphate synthase catalysis. Protein Sci 23:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. O'Rourke KF, Jelowicki AM, Boehr DD (2016) Controlling active site loop dynamics in the (beta/alpha)(8) barrel enzyme indole‐3‐glycerol phosphate synthase. Catalysts 6:129. [Google Scholar]
- 86. Ferreiro DU, Komives EA, Wolynes PG (2014) Frustration in biomolecules. Q Rev Biophys 47:285–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Watt ED, Shimada H, Kovrigin EL, Loria JP (2007) The mechanism of rate‐limiting motions in enzyme function. Proc Natl Acad Sci USA 104:11981–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Doucet N, Watt ED, Loria JP (2009) The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry 48:7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Doucet N, Khirich G, Kovrigin EL, Loria JP (2011) Alteration of hydrogen bonding in the vicinity of histidine 48 disrupts millisecond motions in RNAse A. Biochemistry 50:1723–1730. [DOI] [PubMed] [Google Scholar]
- 90. Watt ED, Rivalta I, Whittier SK, Batista VS, Loria JP (2011) Reengineering rate‐limiting, millisecond enzyme motions by introduction of an unnatural amino acid. Biophys J 101:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Boehr DD, Dyson HJ, Wright PE (2008) Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry 47:9227–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Deckert K, Budiardjo SJ, Brunner LC, Lovell S, Karanicolas J (2012) Designing allosteric control into enzymes by chemical rescue of structure. J Am Chem Soc 134:10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boulton S, Melacini G (2016) Advances in NMR methods to map allosteric sites: from models to translation. Chem Rev 116:6267–6304. [DOI] [PubMed] [Google Scholar]
- 94. Ideker T, Nussinov R (2017) Network approaches and applications in biology. PLoS Comput Biol 13:e1005771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. O'Rourke KF, Gorman SD, Boehr DD (2016) Biophysical and computational methods to analyze amino acid interaction networks in proteins. Comput Struct Biotechnol J 14:245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee J, Natarajan M, Nashine VC, Socolich M, Vo T, Russ WP, Benkovic SJ, Ranganathan R (2008) Surface sites for engineering allosteric control in proteins. Science 322:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Reynolds KA, McLaughlin RN, Ranganathan R (2011) Hot spots for allosteric regulation on protein surfaces. Cell 147:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS (2013) Automated identification of functional dynamic contact networks from x‐ray crystallography. Nat Methods 10:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fenwick RB, van den Bedem H, Fraser JS, Wright PE (2014) Integrated description of protein dynamics from room‐temperature x‐ray crystallography and NMR. Proc Natl Acad Sci USA 111:E445–E454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van den Bedem H, Dhanik A, Latombe J‐C, Deacon AM (2009) Modeling discrete heterogeneity in X‐ray diffraction data by fitting multi‐conformers. Acta Cryst 65:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Narayanan C, Gagne D, Reynolds KA, Doucet N (2017) Conserved amino acid networks modulate discrete functional properties in an enzyme superfamily. Sci Rep 7:3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kim J, Ahuja LG, Chao FA, Xia Y, McClendon CL, Kornev AP, Taylor SS, Veglia G (2017) A dynamic hydrophobic core orchestrates allostery in protein kinases. Sci Adv 3:e1600663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xia B, Liu Y, Guevara J, Li J, Jilich C, Yang Y, Wang L, Dominy BN, Cao W (2017) Correlated mutation in the evolution of catalysis in uracil DNA glycosylase superfamily. Sci Rep 7:45978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Simakov N, Leonard DA, Smith JC, Wymore T, Szarecka A (2017) A distal disulfide bridge in oxa‐1 beta‐lactamase stabilizes the catalytic center and alters the dynamics of the specificity determining omega loop. J Phys Chem B 121:3285–3296. [DOI] [PubMed] [Google Scholar]
- 105. Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D (2016) Dynamical network of residue‐residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci USA 113:4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aoto PC, Martin BT, Wright PE (2016) NMR characterization of information flow and allosteric communities in the map kinase p38gamma. Sci Rep 6:28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Nagel ZD, Cun S, Klinman JP (2013) Identification of a long‐range protein network that modulates active site dynamics in extremophilic alcohol dehydrogenases. J Biol Chem 288:14087–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Boulton S, Akimoto M, Selvaratnam R, Bashiri A, Melacini G (2014) A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci Rep 4:7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G (2011) Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA 108:6133–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Axe JM, Boehr DD (2013) Long‐range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling. J Mol Biol 425:1527–1545. [DOI] [PubMed] [Google Scholar]
- 111. Axe JM, Yezdimer EM, O'Rourke KF, Kerstetter NE, You W, Chang C‐e. A, Boehr DD (2014) Amino acid networks in a (beta/alpha)(8) barrel enzyme change during catalytic turnover. J Am Chem Soc 136:6818–6821. [DOI] [PubMed] [Google Scholar]
- 112. Axe JM, O'Rourke KF, Kerstetter NE, Yezdimer EM, Chan YM, Chasin A, Boehr DD (2015) Severing of a hydrogen bond disrupts amino acid networks in the catalytically active state of the alpha subunit of tryptophan synthase. Protein Sci 24:484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fatmi MQ, Chang CE (2010) The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase. PLoS Comput Biol 6:e1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ai R, Fatmi MQ, Chang CEA (2010) T‐analyst: a program for efficient analysis of protein conformational changes by torsion angles. J Comput Aid Mol Des 24:819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lipchock JM, Loria JP (2010) Nanometer propagation of millisecond motions in v‐type allostery. Structure 18:1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rivalta I, Sultan MM, Lee NS, Manley GA, Loria JP, Batista VS (2012) Allosteric pathways in imidazole glycerol phosphate synthase. Proc Natl Acad Sci USA 109:E1428–E1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lisi GP, East KW, Batista VS, Loria JP (2017) Altering the allosteric pathway in igps suppresses millisecond motions and catalytic activity. Proc Natl Acad Sci USA 114:E3414–E3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lisi GP, Manley GA, Hendrickson H, Rivalta I, Batista VS, Loria JP (2016) Dissecting dynamic allosteric pathways using chemically related small‐molecule activators. Structure 24:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Beismann‐Driemeyer S, Sterner R (2001) Imidazole glycerol phosphate synthase from Thermotoga maritima. Quaternary structure, steady‐state kinetics, and reaction mechanism of the bienzyme complex. J Biol Chem 276:20387–20396. [DOI] [PubMed] [Google Scholar]
- 120. Myers RS, Jensen JR, Deras IL, Smith JL, Davisson VJ (2003) Substrate‐induced changes in the ammonia channel for imidazole glycerol phosphate synthase. Biochemistry 42:7013–7022. [DOI] [PubMed] [Google Scholar]
- 121. Rivalta I, Lisi GP, Snoeberger NS, Manley G, Loria JP, Batista VS (2016) Allosteric communication disrupted by a small molecule binding to the imidazole glycerol phosphate synthase protein‐protein interface. Biochemistry 55:6484–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Taguchi J, Kitao A (2016) Dynamic profile analysis to characterize dynamics‐driven allosteric sites in enzymes. Biophys Physicobiol 13:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Qi Y, Wang Q, Tang B, Lai L (2012) Identifying allosteric binding sites in proteins with a two‐state go model for novel allosteric effector discovery. J Chem Theory Comput 8:2962–2971. [DOI] [PubMed] [Google Scholar]
- 124. Wang Q, Qi Y, Yin N, Lai L (2014) Discovery of novel allosteric effectors based on the predicted allosteric sites for Escherichia coli d‐3‐phosphoglycerate dehydrogenase. PLoS One 9:e94829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. DeLaBarre B, Hurov J, Cianchetta G, Murray S, Dang L (2014) Action at a distance: allostery and the development of drugs to target cancer cell metabolism. Chem Biol 21:1143–1161. [DOI] [PubMed] [Google Scholar]
- 126. Nussinov R, Tsai CJ (2014) Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol Sci 35:256–264. [DOI] [PubMed] [Google Scholar]
- 127. Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA, Amaro RE (2016) Emerging computational methods for the rational discovery of allosteric drugs. Chem Rev 116:6370–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Boucher JI, Bolon DN, Tawfik DS (2016) Quantifying and understanding the fitness effects of protein mutations: laboratory versus nature. Protein Sci 25:1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cobb RE, Sun N, Zhao H (2013) Directed evolution as a powerful synthetic biology tool. Methods 60:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gray VE, Hause RJ, Fowler DM (2017) Analysis of large‐scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics 207:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wrenbeck EE, Faber MS, Whitehead TA (2017) Deep sequencing methods for protein engineering and design. Curr Opin Struct Biol 45:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chan YH, Venev SV, Zeldovich KB, Matthews CR (2017) Correlation of fitness landscapes from three orthologous TIM barrels originates from sequence and structure constraints. Nat Commun 8:14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hietpas R, Roscoe B, Jiang L, Bolon DN (2012) Fitness analyses of all possible point mutations for regions of genes in yeast. Nat Protoc 7:1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hagihara Y, Saerens D (2014) Engineering disulfide bonds within an antibody. Biochim Biophys Acta 1844:2016–2023. [DOI] [PubMed] [Google Scholar]
- 135. Trivedi MV, Laurence JS, Siahaan TJ (2009) The role of thiols and disulfides on protein stability. Curr Protein Pept Sci 10:614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wedemeyer WJ, Welker E, Narayan M, Scheraga HA (2000) Disulfide bonds and protein folding. Biochemistry 39:7032. [DOI] [PubMed] [Google Scholar]
- 137. Matsumura M, Becktel WJ, Levitt M, Matthews BW (1989) Stabilization of phage t4 lysozyme by engineered disulfide bonds. Proc Natl Acad Sci USA 86:6562–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pan P, Jakob CA, Sandmeier E, Christen P, Gehring H (1994) Modulation of the activity of mitochondrial aspartate aminotransferase h352c by the redox state of the engineered interdomain disulfide bond. J Biol Chem 269:25432–25436. [PubMed] [Google Scholar]
- 139. Ma K, Temiakov D, Anikin M, McAllister WT (2005) Probing conformational changes in t7 RNA polymerase during initiation and termination by using engineered disulfide linkages. Proc Natl Acad Sci USA 102:17612–17617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ivens A, Mayans O, Szadkowski H, Jurgens C, Wilmanns M, Kirschner K (2002) Stabilization of a (betaalpha)8‐barrel protein by an engineered disulfide bridge. Eur J Biochem 269:1145–1153. [DOI] [PubMed] [Google Scholar]
- 141. Sengupta D, Roy B, Basu B (2017) Microwave‐assisted formation of organic disulfides of biochemical significance. Curr Med Chem 24:4627–4637. [DOI] [PubMed] [Google Scholar]
- 142. Lau YH, de Andrade P, Wu Y, Spring DR (2015) Peptide stapling techniques based on different macrocyclisation chemistries. Chem Soc Rev 44:91–102. [DOI] [PubMed] [Google Scholar]
- 143. Abdeljabbar DM, Piscotta FJ, Zhang S, James Link A (2014) Protein stapling via azide‐alkyne ligation. Chem Commun 50:14900–14903. [DOI] [PubMed] [Google Scholar]
- 144. Brown SP, Smith AB 3rd (2015) Peptide/protein stapling and unstapling: introduction of s‐tetrazine, photochemical release, and regeneration of the peptide/protein. J Am Chem Soc 137:4034–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chen XH, Xiang Z, Hu YS, Lacey VK, Cang H, Wang L (2014) Genetically encoding an electrophilic amino acid for protein stapling and covalent binding to native receptors. ACS Chem Biol 9:1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Rabuka D (2010) Chemoenzymatic methods for site‐specific protein modification. Curr Opin Chem Biol 14:790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Beharry AA, Woolley GA (2011) Azobenzene photoswitches for biomolecules. Chem Soc Rev 40:4422–4437. [DOI] [PubMed] [Google Scholar]
- 148. Hoersch D (2017) Let there be light: how to use photoswitchable cross‐linker to reprogram proteins. Biochem Soc Trans 45:831–837. [DOI] [PubMed] [Google Scholar]
- 149. Fehrentz T, Schonberger M, Trauner D (2011) Optochemical genetics. Angew Chem Int Ed Engl 50:12156–12182. [DOI] [PubMed] [Google Scholar]
- 150. Buchli B, Waldauer SA, Walser R, Donten ML, Pfister R, Blochliger N, Steiner S, Caflisch A, Zerbe O, Hamm P (2013) Kinetic response of a photoperturbed allosteric protein. Proc Natl Acad Sci USA 110:11725–11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Buchenberg S, Knecht V, Walser R, Hamm P, Stock G (2014) Long‐range conformational transition of a photoswitchable allosteric protein: molecular dynamics simulation study. J Phys Chem B 118:13468–13476. [DOI] [PubMed] [Google Scholar]
- 152. Schierling B, Noel AJ, Wende W, Hien le T, Volkov E, Kubareva E, Oretskaya T, Kokkinidis M, Rompp A, Spengler B, Pingoud A (2010) Controlling the enzymatic activity of a restriction enzyme by light. Proc Natl Acad Sci USA 107:1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Woolley GA, Lee ES, Zhang F (2006) Sgal: a computational method for finding surface exposed sites in proteins suitable for cys‐mediated cross‐linking. Bioinformatics 22:3101–3102. [DOI] [PubMed] [Google Scholar]
- 154. Horstmann B, Korbus M, Friedmann T, Wolff C, Thiele CM, Meyer‐Almes FJ (2014) Synthesis of azobenzenealkylmaleimide probes to photocontrol the enzyme activity of a bacterial histone deacetylase‐like amidohydrolase. Bioorg Chem 57:155–161. [DOI] [PubMed] [Google Scholar]
- 155. Korbus M, Backe S, Meyer‐Almes FJ (2015) The cis‐state of an azobenzene photoswitch is stabilized through specific interactions with a protein surface. J Mol Recognit 28:201–209. [DOI] [PubMed] [Google Scholar]
- 156. Wu S, Luo Howard H, Wang H, Zhao W, Hu Q, Yang Y (2016) Cysteinome: the first comprehensive database for proteins with targetable cysteine and their covalent inhibitors. Biochem Biophys Res Commun 478:1268–1273. [DOI] [PubMed] [Google Scholar]
- 157. Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez‐Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF (2016) Proteome‐wide covalent ligand discovery in native biological systems. Nature 534:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nussinov R, Tsai CJ (2015) The design of covalent allosteric drugs. Annu Rev Pharmacol Toxicol 55:249–267. [DOI] [PubMed] [Google Scholar]
- 159. Lu S, Zhang J (2017) Designed covalent allosteric modulators: an emerging paradigm in drug discovery. Drug Discov Today 22:447–453. [DOI] [PubMed] [Google Scholar]
- 160. Nussinov R, Tsai CJ, Xin F, Radivojac P (2012) Allosteric post‐translational modification codes. Trends Biochem Sci 37:447–455. [DOI] [PubMed] [Google Scholar]
- 161. Fastrez J (2009) Engineering allosteric regulation into biological catalysts. Chembiochem 10:2824–2835. [DOI] [PubMed] [Google Scholar]
- 162. Ostermeier M (2009) Designing switchable enzymes. Curr Opin Struct Biol 19:442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Choi JH, Laurent AH, Hilser VJ, Ostermeier M (2015) Design of protein switches based on an ensemble model of allostery. Nat Commun 6:6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Guntas G, Ostermeier M (2004) Creation of an allosteric enzyme by domain insertion. J Mol Biol 336:263–273. [DOI] [PubMed] [Google Scholar]
- 165. Cross PJ, Allison TM, Dobson RC, Jameson GB, Parker EJ (2013) Engineering allosteric control to an unregulated enzyme by transfer of a regulatory domain. Proc Natl Acad Sci USA 110:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Halabi N, Rivoire O, Leibler S, Ranganathan R (2009) Protein sectors: evolutionary units of three‐dimensional structure. Cell 138:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Lockless SW, Ranganathan R (1999) Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286:295–299. [DOI] [PubMed] [Google Scholar]
- 168. Suel GM, Lockless SW, Wall MA, Ranganathan R (2003) Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol 10:59–69. [DOI] [PubMed] [Google Scholar]
- 169. Dagliyan O, Karginov AV, Yagishita S, Gale ME, Wang H, DerMardirossian C, Wells CM, Dokholyan NV, Kasai H, Hahn KM (2017) Engineering pak1 allosteric switches. ACS Synth Biol 6:1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Dagliyan O, Tarnawski M, Chu PH, Shirvanyants D, Schlichting I, Dokholyan NV, Hahn KM (2016) Engineering extrinsic disorder to control protein activity in living cells. Science 354:1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Dagliyan O, Shirvanyants D, Karginov AV, Ding F, Fee L, Chandrasekaran SN, Freisinger CM, Smolen GA, Huttenlocher A, Hahn KM, Dokholyan NV (2013) Rational design of a ligand‐controlled protein conformational switch. Proc Natl Acad Sci USA 110:6800–6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM (2010) Engineered allosteric activation of kinases in living cells. Nat Biotechnol 28:743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Huang PS, Boyken SE, Baker D (2016) The coming of age of de novo protein design. Nature 537:320–327. [DOI] [PubMed] [Google Scholar]
- 174. Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Engineering the third wave of biocatalysis. Nature 485:185–194. [DOI] [PubMed] [Google Scholar]
- 175. Davey JA, Chica RA (2012) Multistate approaches in computational protein design. Protein Sci 21:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Lassila JK (2010) Conformational diversity and computational enzyme design. Curr Opin Chem Biol 14:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Davey JA, Damry AM, Goto NK, Chica RA (2017) Rational design of proteins that exchange on functional timescales. Nat Chem Biol 13:1280–1285. [DOI] [PubMed] [Google Scholar]
- 178. Nojiri A, Kumagai N, Shibasaki M (2009) Linking structural dynamics and functional diversity in asymmetric catalysis. J Am Chem Soc 131:3779–3784. [DOI] [PubMed] [Google Scholar]
- 179. Stoll RS, Peters MV, Kuhn A, Heiles S, Goddard R, Buhl M, Thiele CM, Hecht S (2009) Photoswitchable catalysts: correlating structure and conformational dynamics with reactivity by a combined experimental and computational approach. J Am Chem Soc 131:357–367. [DOI] [PubMed] [Google Scholar]
- 180. Cardenas AJ, Ginovska B, Kumar N, Hou J, Raugei S, Helm ML, Appel AM, Bullock RM, O'Hagan M (2016) Controlling proton delivery through catalyst structural dynamics. Angew Chem Int Ed Engl 55:13509–13513. [DOI] [PubMed] [Google Scholar]
- 181. Stoddart JF (2017) Mechanically interlocked molecules (mims)‐molecular shuttles, switches, and machines (nobel lecture). Angew Chem Int Ed Engl 56:11094–11125. [DOI] [PubMed] [Google Scholar]
- 182. Loria JP, Berlow RB, Watt ED (2008) Characterization of enzyme motions by solution NMR relaxation dispersion. Acc Chem Res 41:214–221. [DOI] [PubMed] [Google Scholar]