

Abstract
Homocysteine is a sulphur‐containing non‐proteinogenic amino acid. Hyperhomocysteinaemia (HHcy), the pathogenic elevation of plasma homocysteine as a result of an imbalance of its metabolism, is an independent risk factor for various vascular diseases, such as atherosclerosis, hypertension, vascular calcification and aneurysm. Treatments aimed at lowering plasma homocysteine via dietary supplementation with folic acids and vitamin B are more effective in preventing vascular disease where the population has a normally low folate consumption than in areas with higher dietary folate. To date, the mechanisms of HHcy‐induced vascular injury are not fully understood. HHcy increases oxidative stress and its downstream signalling pathways, resulting in vascular inflammation. HHcy also causes vascular injury via endoplasmic reticulum stress. Moreover, HHcy up‐regulates pathogenic genes and down‐regulates protective genes via DNA demethylation and methylation respectively. Homocysteinylation of proteins induced by homocysteine also contributes to vascular injury by modulating intracellular redox state and altering protein function. Furthermore, HHcy‐induced vascular injury leads to neuronal damage and disease. Also, an HHcy‐activated sympathetic system and HHcy‐injured adipose tissue also cause vascular injury, thus demonstrating the interactions between the organs injured by HHcy. Here, we have summarized the recent developments in the mechanisms of HHcy‐induced vascular injury, which are further considered as potential therapeutic targets in this condition.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- ADMA
asymmetric dimethylarginine
- Ang II
angiotensin II
- CBS
cystathionine β‐synthase
- CSE
cystathionine γ‐lyase
- DNMT
DNA methyltransferase
- ECs
endothelial cells
- FABP4
fatty acid‐binding protein 4
- GRP78
glucose‐regulated protein 78
- Hcy‐TL
homocysteine thiolactone
- HHcy
hyperhomocysteinaemia
- HO‐1
haem oxygenase‐1
- IGF2
insulin‐like growth factor 2
- MMP
matrix metalloprotease
- MS
methionine synthase
- MT
methyltransferase
- MTHFR
methylene tetrahydrofolate reductase
- NOXs
NADPH oxidases
- SAH
S‐adenosyl‐l‐homocysteine
- SAM
S‐adenosyl‐l‐methionine
- sEH
soluble epoxide hydrolase
- THF
tetrahydrofolate
- VSMCs
vascular smooth muscles cells
Introduction
Homocysteine is a non‐essential, sulphur‐containing, non‐proteinogenic amino acid. It was first isolated from a urinary bladder stone in 1933 by Vincent du Vigneaud, who later received the Nobel Prize in Chemistry, in 1955. Under normal conditions, the concentration of homocysteine ranges from 5 to 10 μM in human plasma but does not exceed 15 μM (Seshadri et al., 2002). Elevation of the plasma levels of homocysteine above 15 μM is defined as hyperhomocysteinaemia (HHcy) (Ji and Kaplowitz, 2004). Plasma homocysteine exists in three different forms defined as free homocysteine, protein‐bound homocysteine and oxidized forms of homocysteine (Jakubowski, 2008). Among these forms, the majority of homocysteine exists in the protein‐bound form and is N‐linked and S‐linked to γ‐globulins or albumins, accounting for 80–90% of the total plasma homocysteine. Moreover, approximately 10–20% of total homocysteine is in the oxidized form, presented as homocysteine‐cysteine (Cys) and homocysteine‐homocysteine dimers in plasma. As a result, less than 1% of total plasma homocysteine exists as the free, reduced amino acid (Jakubowski, 2002).
In 1969, a Harvard pathologist, Kilmer McCully, reported that two children with homocystinuria, a genetic disorder characterized by the existence of homocysteine in the urine as a result of severe HHcy (>100 μM), displayed vascular pathologies (McCully, 1969). Among these children, one 2‐month‐old boy had an advanced stage of arteriosclerosis that closely resembled that observed in elderly individuals with advanced cardiovascular disease. The other child, an 8‐year‐old, died of a stroke related to arteriosclerosis in the carotid artery. These were the first suggestions that elevated plasma homocysteine was a potential cause of premature vascular disease. In 1976, a human population study, which included 25 patients with coronary artery disease and 22 control subjects with normal coronarography, first identified higher homocysteine levels as a result of abnormalities in methionine (Met) metabolism in patients (Wilcken and Wilcken, 1976). To date, a substantial amount of data from case–control cohort studies and meta‐analyses support the association of HHcy with a range of vascular diseases, including hypertension, ischaemic stroke, aortic aneurysm and dissection, coronary heart disease, coronary artery calcification, vascular dementia and cervical artery dissection. as HHcy has also been identified as a risk factor for these diseases (Fallon and Ben‐Shlomo, 2003; Ravaglia et al., 2005; Takagi and Umemoto, 2005; Holmes et al., 2011; Clarke et al., 2012; Liu et al., 2012, 2016; Luo et al., 2014; Kim et al., 2015; Wang et al., 2015b).
Homocysteine metabolism
In this review, homocysteine metabolism includes both the production and the disposal of this amino acid and the normal physiological balance between these two pathways maintains plasma homocysteine at its physiological level (<15 μM). Thus, HHcy is the result of increased production and/or decreased disposal of homocysteine.
Homocysteine is produced in all human tissues, by a single pathway, through the transmethylation of the essential amino acid Met. The process of Met transmethylation involves three steps sequentially catalysed by S‐adenosyl‐l‐methionine (SAM) synthase, methyltransferase (MT) and S‐adenosyl‐l‐homocysteine (SAH) hydrolase. SAM synthetase catalyses the reaction of Met with ATP to form SAM. SAM is converted into SAH via an MT‐catalysed methyl transfer reaction. Finally, SAH is rapidly metabolized by SAH hydrolase to adenosine and homocysteine (Skovierova et al., 2016). Homocysteine production may be enhanced by increased intake of Met‐rich protein, which results in HHcy. Mice fed a high Met diet provide a widely used animal model of HHcy (Yang et al., 2015a) and, clinically, limiting Met intake improved HHcy in patients (Wang et al., 2015b).
Compared with the production of homocysteine, its disposal involves many pathways. First, approximately 50% of homocysteine is re‐methylated to form Met via two distinct mechanisms, folate/vitamin B12‐dependent and folate/vitamin B12‐independent re‐methylation. Folate in the form of N‐5‐methyl tetrahydrofolate (THF), derived from N‐5,10‐methylene tetrahydrofolate reductase (MTHFR)‐catalysed THF modification, donates a methyl group to homocysteine in the re‐methylation catalysed by the vitamin B12‐dependent enzyme methionine synthase (MS) (Castro et al., 2006). In the other mechanism, betaine derived from choline by betaine–homocysteine S‐methyltransferase also behaves as a methyl group donor and contributes to the folate/vitamin B12‐independent re‐methylation of homocysteine to form Met (Skovierova et al., 2016). Second, homocysteine is resynthesized into SAH through the reversal of SAH hydrolase activity. High concentrations of SAH strongly inhibit the transmethylation of Met because SAH is a potent allosteric inhibitor of MT (Kerr, 1972). Third, homocysteine is metabolized to form Cys via trans‐sulphuration, sequentially catalysed by the vitamin B6‐dependent enzymes cystathionine β‐synthase (CBS) and cystathionine γ‐lyase (CSE) (Skovierova et al., 2016). These trans‐sulphuration enzymes CBS and CSE, exhibit other enzymic activities that contribute to the biosynthesis of the gasotransmitter H2S, from homocysteine and/or Cys (Weber et al., 2016). Deficiencies in re‐methylation and trans‐sulphuration are the main causes of the accumulation of homocysteine (Figure 1). A dietary deficiency of folate and vitamin B12 or a dysfunctional mutation in MTHFR also inhibits homocysteine re‐methylation, whereas insufficient intake of vitamin B6 or CBS deficiency impairs the trans‐sulphuration of this amino acid (Chen et al., 2001; Fohr et al., 2002). The MTHFR and CBS genes have been reported to be the genetic determinants in severe HHcy and homocystinuria (Hannibal and Blom, 2017). CBS‐deficient mice provide a model of HHcy that can be used for mechanistic studies (Zhang et al., 2012b). The polymorphisms MTHFR C677T, MTHFR A1298C and MS A2756G, are the critical dominant negative mutations and they jointly add to the risk of folate deficiency, which results in HHcy in humans (Li et al., 2015). The MTHFR C677T polymorphism has been identified as a risk locus in patients with coronary heart disease, coronary artery calcification, cervical artery dissection, aortic aneurysm and dissection (Fallon and Ben‐Shlomo, 2003; Takagi and Umemoto, 2005; Holmes et al., 2011; Clarke et al., 2012; Liu et al., 2012; Luo et al., 2014; Kim et al., 2015; Wang et al., 2015b). To further assess the causality of this single‐nucleotide polymorphism in HHcy‐related vascular injury and disease, Mendelian randomization studies have been carried out by several groups. The results showed that HHcy induced by MTHFR C667T did not have a causal role in high systolic BP, cardiovascular disease mortality, coronary heart disease or ischaemic stroke (Bentley et al., 2010; Clarke et al., 2012; Yang et al., 2012; Borges et al., 2016). However, there are important limitations in these Mendelian randomization analyses (Smulders and Blom, 2011). For instance, it could not be guaranteed that all potential confounders had been excluded; that an unknown indirect association may exist between MTHFR C677T and HHcy and, finally, that genetic heterogeneity, pleiotropy, population stratification and canalization may also be involved in the process of the study. Thus, the outcomes of the Mendelian randomization studies do not provide strong enough evidence to reject the pathogenic effects of homocysteine on vascular injury.
Figure 1.
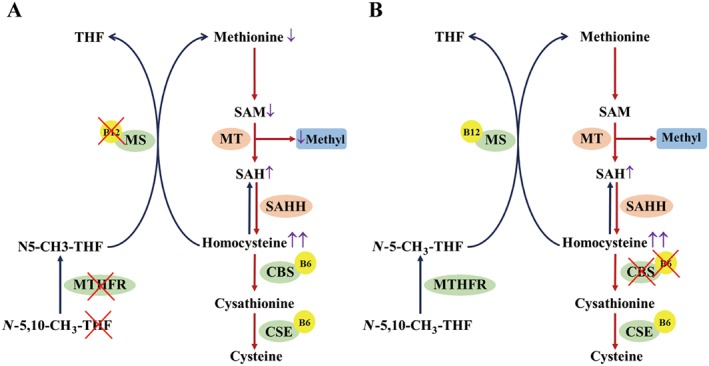
(A) Schematic overview of homocysteine metabolism changes in HHcy induced by re‐methylation (i) or trans‐sulphuration (ii) deficiency. Dietary deficiency of folate (THF) and vitamin B12 or a MTHFR dysfunctional mutation inhibits the re‐methylation of homocysteine into Met and induces homocysteine accumulation. Low levels of Met decrease SAM production, while elevated homocysteine levels contribute to SAH production, catalysed by S‐adenosylhomocysteine hydrolase (SAHH). Thus, the MT‐mediated transformation of SAM into SAH markedly decreases, leading to the lack of methyl donors. (B) Insufficient intake of vitamin B6 or CBS deficiency impairs the trans‐sulphuration of homocysteine and leads to the elevation of this amino acid. Increased levels of homocysteine induce enhanced SAH production, catalysed by SAHH. Because homocysteine re‐methylation is only marginally affected, SAM and methyl donor production is not significantly influenced. B6/B12, vitamin B6/B12.
The mechanisms of vascular injury induced by HHcy
HHcy displays a close relationship with many vascular diseases, even though the underlying mechanisms are not fully understood. Vascular remodelling, the architectural alteration of the vascular wall, contributes substantially to the pathogenesis of vascular injury. The vascular wall is composed of the intima, media and adventitia, with endothelial cells (ECs) constituting the main cellular fraction of the vascular intima. These cells regulate the vascular tone and maintain the inflammatory balance. Endothelial dysfunction, defined as a disorder of endothelial‐dependent dilation, as well as endothelial activation, which refers to the overexpression of adhesion molecules, are two major pathological changes taking place in ECs in vascular diseases. Vascular smooth muscles cells (VSMCs) located in the media exhibit an extensive plasticity in which fully differentiated VSMCs may de‐differentiate from a contractile phenotype into other phenotypes in physiological processes, including vascular remodelling and angiogenesis, and in pathological processes, such as hypertension, atherosclerosis and vascular calcification. The de‐differentiation of VSMCs, defined as the alteration in contractile genes and functions, contributes to vascular remodelling. The vascular adventitia mainly consists of adventitial fibroblasts. Over the last 10 years, an outside‐in vascular injury model has emerged, which suggests an initiating role for the adventitial layer in vascular injury (Stenmark et al., 2013; Majesky, 2015). Most vascular injuries involve vascular inflammation through the interaction between vascular resident cells and activated inflammatory cells. Circulating leukocytes, including monocytes/macrophages, lymphocytes and neutrophils, are all involved in the pathogenesis of vascular injuries. Several in vitro and in vivo studies, using homocysteine at pathogenic concentrations and HHcy animal models, respectively, have demonstrated that homocysteine induces pathogenic effects on both vascular resident cells and circulating leukocytes.
Oxidative stress in HHCy
Oxidative stress as a result of ROS accumulation is the major mechanism that mediates homocysteine‐induced vascular injury (Tyagi et al., 2005). In addition to the interaction of homocysteine with Cys to form a disulphide bond, homocysteine also directly inhibits the activity of antioxidants, thereby disrupting SOD, activating NADPH oxidases (NOXs) and subsequently producing superoxide anion, causing an accumulation of ROS. The generated ROS further activate the transcriptional activity of NF‐κB, which results in the expression of pro‐inflammatory genes and vascular inflammation (Skovierova et al., 2016) (Figure 2).
Figure 2.
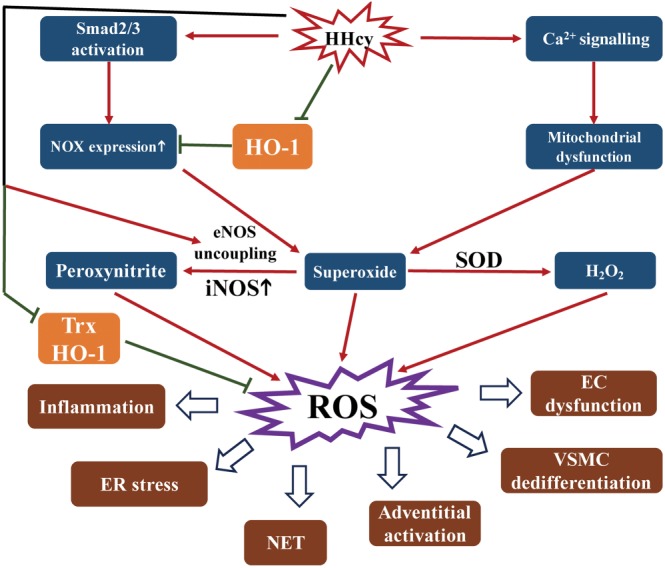
Schematic overview of HHcy‐induced oxidative stress. HHcy up‐regulates NOX expression by Smad2/3 activation, while HHcy induces mitochondrial dysfunction via activating calcium signalling. Both these effects of HHcy result in superoxide production. Superoxide is converted to H2O2 catalysed by SOD. Moreover, HHcy up‐regulates iNOS and enhances eNOS uncoupling, which further induces peroxynitrite formed by NO and superoxide. Superoxide, H2O2 and peroxynitrite all are ROS. HHcy also inhibits the activity of antioxidants, such as thioredoxin (Trx) and HO‐1, to up‐regulate NOX expression and attenuate antioxidant‐mediated elimination of ROS. NET, neutrophil extracellular trap.
ROS production
Homocysteine at a pathogenic concentration leads to significant ROS production in both vascular resident cells and circulating leukocytes (Zhang et al., 2001; Zhang et al., 2002). The mechanisms underlying the generation of ROS by homocysteine includes the up‐regulation of NOX, mitochondrial dysfunction, abnormal NO generation and inhibition of antioxidants, as discussed below in more detail.
NADPH oxidase
NOXs are enzymes that mediate ROS production. Homocysteine increases NOX2 and NOX4 expression in ECs (Lai and Kan, 2015) and, in adventitial fibroblasts, up‐regulates the expression of NOX4 via Smad2/3 activation, in vitro (Liu et al., 2012). Furthermore, ROS production induced by homocysteine is blocked by NOX inhibitors in VSMCs, monocytes/macrophages, neutrophils and lymphocytes, which indicates that NOX also contributes to homocysteine‐induced ROS production in these cells (Zeng et al., 2003; Dai et al., 2008; Zanin et al., 2015).
Mitochondrial dysfunction
Mitochondria are an important source of ROS within most mammalian cells. Superoxide, the proximal mitochondrial ROS, further forms peroxynitrite by interacting with nitrate and is the substrate for SOD to form H2O2. In general, superoxide accumulates in mitochondria following the mitochondrial disorders that exhibit dysfunctions of oxidative phosphorylation and the respiratory chain (Murphy, 2009). In ECs, homocysteine at a pathological concentration leads to mitochondrial toxicity. First, homocysteine induces an elevated Ca2+ level that impairs mitochondrial function. Second, homocysteine enhances the mitochondrial dehydrogenase activity, which suggests increased oxidation in mitochondria, whereas the down‐regulation by homocysteine of nitrate and SOD2 leads to a decreased consumption of superoxide. Third, homocysteine decreases mitochondria membrane potentials and ATP production, which suggests the dysfunction of the respiratory chain and accumulation of superoxide (Kamat et al., 2015).
Abnormal NO generation
In general, endothelial NOS (eNOS) plays a vasoprotective role by producing NO in ECs. homocysteine inhibits eNOS‐mediated NO production, as well as inducing eNOS uncoupling, both of which synergistically generate peroxynitrite production derived from superoxide and NO. Moreover, homocysteine up‐regulates iNOS in ECs and VSMCs, which further increases peroxynitrite production (Welch et al., 1998; Wang et al., 2000).
Antioxidant levels
Inhibition or down‐regulation of antioxidants is involved in the process of HHcy‐related vascular injury and remodelling. Homocysteine markedly down‐regulates the expression of the antioxidant proteins, thioredoxin and haem oxygenase‐1 (HO‐1), which also contribute to oxidative stress (Sawle et al., 2001; Dong et al., 2005). Moreover, the antioxidant compound N‐acetyl cysteine (NAC) and thioredoxin markedly inhibited the generation of ROS induced by homocysteine in monocytes/macrophages (Zeng et al., 2003; Dai et al., 2008; Zanin et al., 2015). Piceatannol, a resveratrol analogue, protected ECs against homocysteine‐induced apoptosis, oxidative stress and endoplasmic reticulum (ER) stress. Moreover, piceatannol markedly increases HO‐1 expression via the activation of the transcription factor Nrf2. The inhibitory effects of piceatannol on apoptosis, ROS generation and ER stress were blocked by silencing HO‐1 and were mimicked by treating the ECs with the HO‐1 inducer, haemin. These results suggest the antioxidant enzyme HO‐1 may protect ECs against apoptosis, oxidative stress and ER stress induced by homocysteine (Sawle et al., 2001).
Effects of ROS generated by homocysteine
The major effect of ROS‐induced oxidative stress is to induce inflammatory responses, mediated particularly by NF‐κB, downstream of ROS. In addition to inflammation, homocysteine‐induced oxidative stress contributes to dysfunction of EC, differentiation of VSMCs, adventitial activation and formation of the neutrophil extracellular trap.
Inflammation
Inflammation is the major pathogenic process involved in several vascular injuries and diseases, and homocysteine is a potent inducer of inflammatory interactions between vascular resident cells and leukocytes. NF‐κB, ERK1/2 and redox factor 1, as downstream targets of ROS, as well as PKC and calmodulin as signalling molecules upstream of ROS, are involved in the secretion of inflammatory cytokines, induced by homocysteine, from monocytes/macrophages in atherosclerosis (Zeng et al., 2003; Dai et al., 2006; Zanin et al., 2015). In ECs, homocysteine up‐regulated VCAM‐1 and matrix metalloprotease (MMP)‐9 via NF‐κB signalling, both in vitro and in vivo (Au‐Yeung et al., 2004). Moreover, homocysteine induced production and secretion of the chemokine CCL2, IL‐8 and IL‐1β from monocytes/macrophages which was blocked by inhibitors of NOX or antioxidants, such as NAC and thioredoxin, indicating that oxidative stress was involved in this effect of homocysteine (Zeng et al., 2003; Dai et al., 2008; Zanin et al., 2015). Furthermore, homocysteine‐induced oxidative stress potentiated the proliferation and inflammatory activation of concanavalin A‐elicited splenic T‐lymphocytes in vitro, which implied overactivation of T‐lymphocytes (Zhang et al., 2002).
In addition to these in vitro studies, several in vivo studies have also demonstrated that HHcy activates inflammation partly via increased oxidative stress. CBS‐deficient mice exhibit HHcy as a result of a disorder of homocysteine trans‐sulphuration. In vivo studies have shown that CBS deficiency‐induced HHcy increases inflammatory monocyte (ly6Chigh) differentiation, a process dependent on superoxide anion production, and HHcy‐activated monocytes produce TNF‐α, IL‐6 and CCL2, which exacerbates atherosclerosis in mice (Zhang et al., 2009, 2012b). Interestingly, CBS‐mediated trans‐sulphuration of homocysteine to Cys also plays a critical role in the macrophage‐involved host defence against invading pathogens. Inhibition of the trans‐sulphuration pathway by propargylglycine impairs pathogen clearance by macrophages (Garg et al., 2006). Recently, pyruvate kinase 2‐dependent metabolic reprogramming has been demonstrated to mediate proliferation and IgG secretion of B cells induced by homocysteine and HHcy‐exacerbated atherosclerosis (Deng et al., 2017). In apolipoprotein E‐deficient (ApoE−/−) mice with HHcy, treatment with CTLA4‐IgG attenuated the atherosclerotic lesions, by competing with CD28 to inhibit T‐lymphocyte overactivation (Ma et al., 2013). Moreover, regulatory T‐cells ameliorated HHcy‐exacerbated atherosclerosis in ApoE−/− mice by inhibiting the inflammatory activation of T‐lymphocytes (Feng et al., 2009). In contrast, homocysteine, in vitro, inhibited proliferation of concanavalin A‐elicited blood T‐lymphocytes via induction of DNA fragmentation, which is suggestive of immunosenescence (Picerno et al., 2007). Homocysteine also promoted LPS‐induced B‐lymphocyte proliferation and IgG secretion dependent on the PKC–ROS–p38MAPK–NF‐κB pathways. Treatment with the liver X receptor agonist T0901317 decreased formation of ROS, activation of NF‐κB and secretion of IgG, induced by homocysteine, in B‐lymphocytes in vitro (Zhang et al., 2001; Chang et al., 2007).
EC dysfunction
Dysfunction of ECs is usually assessed as impaired NO production by eNOS. High levels of ROS inhibit the enzymic activity of dimethylarginine dimethylaminohydrolase, which results in an accumulation of asymmetric dimethylarginine (ADMA). As a competitor of the normal eNOS substrate l‐arginine, ADMA can bind to eNOS and inhibit its activity (Dayal and Lentz, 2005). Homocysteine‐induced ROS also decrease the activity of the l‐arginine transporter CAT‐1 in ECs (Jin et al., 2007). A lack of l‐arginine causes eNOS uncoupling (Forstermann and Munzel, 2006), which further impairs NO production and enhances ROS production.
VSMC de‐differentiation
VSMC de‐differentiation is mainly regulated by homocysteine‐generated oxidative stress. ROS production, induced by this amino acid, up‐regulated CCL2 and MMP‐2 via the NF‐κB pathway in VSMCs, and led to VSMC de‐differentiation into an inflammatory and secretory phenotype with a high proliferation rate (Wang et al., 2000; Ke et al., 2010). In addition to oxidative stress, homocysteine increased VSMC proliferation via a ROS‐independent pathway (Taha et al., 1999). Moreover, this amino acid increased caspase‐3‐mediated VSMC apoptosis induced by other pathogenic stimulators, such as beta amyloid (Mok et al., 2002).
Adventitial activation
The adventitia plays a deleterious role via the production of NOX‐derived ROS and recruitment of inflammatory cells involved in vascular injury and disease (Meijles and Pagano, 2016). HHcy induced by drinking water supplemented with homocysteine, enhanced aortic adventitial inflammation in ApoE−/− mice, caused by infused angiotensin II (Ang II), as shown by increased macrophage infiltration, IL‐6 and CCL2 production, and MMP proteolysis activity, preferentially in the tunica adventitia. Correspondingly, the incidence of Ang II‐induced abdominal aortic aneurysm (AAA) and aortic dissection in ApoE−/− mice was increased by such supplementation with homocysteine. Mechanistically, administration of a NOX4 inhibitor or siRNA abolished the homocysteine‐induced adventitial activation (Liu et al., 2012). Moreover, NOX4 deficiency inhibited AAA formation‐induced by Ang II in mice with hyperphenylalaninaemia and uncoupled eNOS (Siu et al., 2016). Thus, NOX4 mediates the adverse effects of homocysteine in adventitial inflammation and AAA formation (Liu et al., 2012). Furthermore, HHcy in rats, induced by a high Met diet, potentiated balloon injury‐induced adventitial hyperplasia and collagen I deposition. Mechanistically, homocysteine inhibits MMP‐2 activity, which thus decreases collagen I degradation, and favours its deposition in adventitial fibroblasts in vitro (Guo et al., 2008). Furthermore, HHcy‐aggravated adventitial hyperplasia and collagen I deposition were reversed by the angiotensin AT1 receptor antagonist, valsartan, in vivo, implying the involvement of these receptors in the pathogenic effects of homocysteine on adventitial remodelling (Yao and Sun, 2014).
Neutrophil extracellular trap
Neutrophil recruitment in the vascular wall is considered to be the early stage of inflammation related to atherosclerosis and aneurysm. Recently, a human study demonstrated that elevated homocysteine levels in type 2 diabetes induce the formation of neutrophil extracellular traps, a network of fibres composed of neutrophil DNA and proteins which bind and kill pathogens, without phagocytosis. Such traps are considered to represent an inflammatory activation of neutrophils. Mechanistically, homocysteine induces superoxide anion generated by NOX, which subsequently leads to the activation of ERK1/2 and Akt signalling related to the formation of neutrophil extracellular traps (Joshi et al., 2016). Furthermore, homocysteine promotes the chemotaxis and migration of human peripheral neutrophils via NOX‐derived ROS production and subsequent activation of ERK1/2 (Alvarez‐Maqueda et al., 2004).
Potential cell surface receptors for homocysteine
Both groups of glutamate receptors, AMPA as well as NMDA receptors, have been identified as potential cell surface receptors for homocysteine. NMDA receptors are expressed in ECs, VSMCs, monocytes/macrophages, lymphocytes (Boldyrev et al., 2012) and neutrophils (Bryushkova et al., 2011; Boldyrev et al., 2012) (Figure 3). An NMDA receptor antagonist blocked oxidative stress induced by homocysteine in various types of cells (Boldyrev et al., 2013). Inhibition of NMDA signalling by a receptor antagonist or the relevant siRNA blocked homocysteine‐induced proliferation and mitochondrial toxicity in ECs (Chen et al., 2005; Kamat et al., 2015). Moreover, homocysteine decreases claudin‐5 expression in brain microvascular ECs via nuclear translocation of β‐catenin, which further results in the disruption of the cell junction in the EC layer in vitro and the increased permeability of the blood–brain barrier in CBS‐deficient mice in vivo. This homocysteine‐induced EC injury was rescued by an NMDA receptor antagonist (Qureshi et al., 2005). In murine macrophages, homocysteine enhanced COX‐2 expression by ROS generated via NMDA receptor‐mediated calcium signalling pathways, as shown by the inhibition of these homocysteine‐induced effects by the NMDA receptor inhibitor MK‐801 or NMDA receptor siRNA (Lee et al., 2013). In lymphocytes, homocysteine‐induced ROS production and inflammatory cytokine release in vitro were attenuated by MK‐801, implying that NMDA receptors may also mediate the effect of homocysteine in these cells (Boldyrev, 2005; Vladychenskaya et al., 2011). However, most of these experiments have been performed in vitro and in vivo studies are required for confirmation. The possibility that other cell surface receptors are involved in the effects of homocysteine has still to be investigated. As homocysteine is not readily radiolabelled, because of the lack of a phenolic hydroxyl group and its ability to be easily oxidized (Tsen et al., 2003; Yang et al., 2015b), no direct evidence of the interaction of homocysteine with possible receptors, for example, ligand–receptor binding assays, is available to date.
Figure 3.
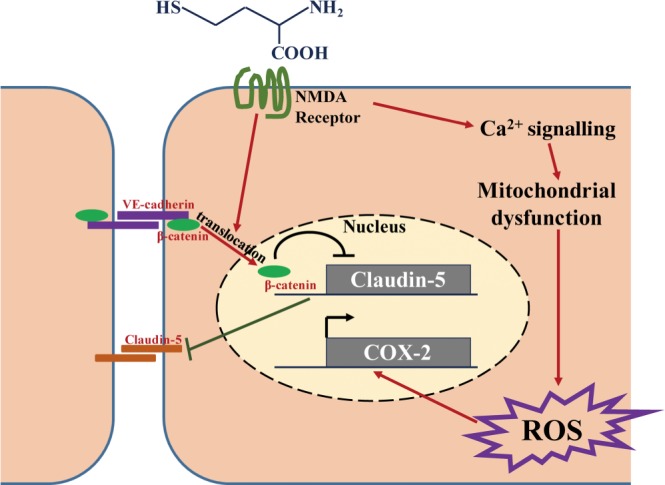
NMDA receptors may act as the cell surface receptor for homocysteine. NMDA receptor mediates homocysteine‐induced ROS production by activating calcium signalling and impairing mitochondrial function. ROS accumulation further up‐regulates COX‐2 gene transcription. Homocysteine also leads to the nuclear translocation of β‐catenin through NMDA receptors. The nuclear translocation of β‐catenin results in disruption of vascular endothelial (VE)‐cadherin–β‐catenin interaction and inhibition of claudin‐5 transcription. Thus, the tight junction of cells is damaged as the homophilic interactions of VE‐cadherin and claudin‐5 are reduced.
ER stress in HHcy
The ER is the site in which newly synthesized proteins, undergoing post‐translational modifications, fold into the correct conformation. Only correctly folded proteins are exported to the Golgi apparatus, whereas misfolded proteins are retained in the ER for refolding or ER‐associated degradation. When unfolded and aggregated proteins accumulate in the ER they cause “ER stress” (Binet and Sapieha, 2015) and ER stress is known to be involved in the pathogenesis of vascular diseases. Homocysteine is known to elicit ER stress (Figure 4), as demonstrated by its up‐regulation of the expression of ER stress response genes, including those for glucose‐regulated protein 78 (GRP78), activating transcription factor‐6 (ATF6), protein kinase RNA‐like endoplasmic reticulum kinase (PERK), ATF4, IRE1α and XBP1 spliced form (Outinen et al., 1999; Hossain et al., 2003). ER stress elicited by homocysteine mediates EC apoptosis and death and further accelerates the development of atherosclerosis in mice (Hossain et al., 2003; Kim et al., 2007). Furthermore, ER stress elevates both intracellular and plasma membrane cholesterol, which is mediated by the inhibition of neutral sphingomyelinase 2 activity in ECs. Vasorelaxation is mediated by the KCa3.1 (IKCa) and SKCa ion channels, and NO production derived from ECs. Homocysteine‐induced ER stress impairs NO production, IKCa and SKCa channels, as well as NOX‐generated ROS (Chaube et al., 2012; Wang et al., 2015a). Another target of homocysteine is soluble epoxide hydrolase (sEH), an enzyme that hydrolyses epoxyeicosatrienoic acids and attenuates their vasorelaxing and protective effects in ECs. Treatment of cultured human ECs with homocysteine dose‐ and time‐dependently up‐regulated sEH mRNA and protein, which was associated with up‐regulation of adhesion molecules and activation of ATF6. Mechanistically, ATF6 binds to the promoter of sEH and up‐regulates its expression. Consequently, siRNA knockdown of ATF6α blocks and ATF6 overexpression mimicks the effect of homocysteine on sEH up‐regulation, indicating that ATF6 activation is involved in homocysteine‐induced sEH expression and endothelial activation (Zhang et al., 2012c). Similarly, in VSMCs, homocysteine causes ER stress, as indicated by an up‐regulation of GRP78 and PERK activation, and further enhances the secretion of the inflammatory cytokines CRP and CCL2 (Wang et al. 2000; Pang et al., 2014). In lymphocytes, homocysteine enhanced ER stress, as indicated by PERK activation and IRE1α‐spliced XBP1, and inhibition of ER stress with 4‐phenylbutyric acid blocked T‐cell activation induced by homocysteine. Mechanistically, homocysteine increased ER–mitochondria coupling, and uncoupling ER from mitochondria with the microtubule inhibitor nocodazole attenuated homocysteine‐stimulated mitochondrial reprogramming, IFN‐γ secretion and proliferation in T‐cells. These results suggest that juxtaposition of the ER and mitochondria is required for homocysteine‐promoted mitochondrial function and T‐cell activation in vitro (Feng et al., 2016). Furthermore, as previously described, the inhibition of oxidative stress or ROS production relieves ER stress, which suggests homocysteine‐induced ROS production mediates ER stress (Kil et al., 2017). Interestingly, homocysteine thiolactone (Hcy‐TL) directly up‐regulates GRP78 expression and induces ER stress independent of N‐homocysteinylation and subsequently leads to oxidative stress and endothelial inflammation (Wu et al., 2015). Overall, homocysteine‐induced ER stress and oxidative stress constitute a mutually reinforcing system.
Figure 4.
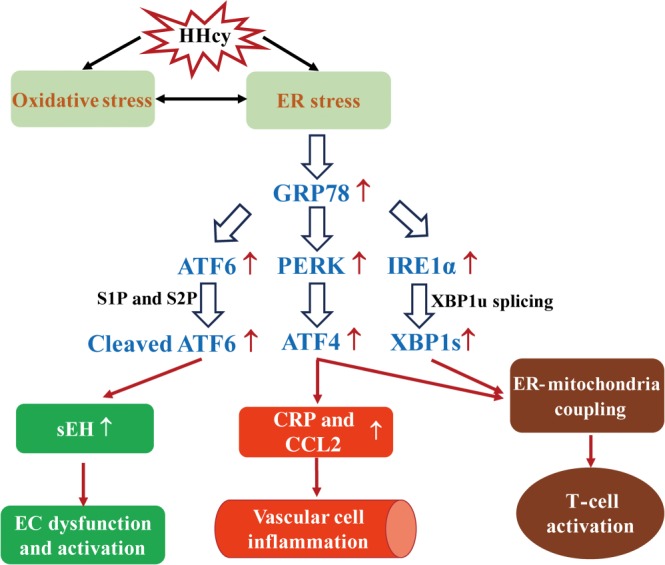
Schematic overview of HHcy‐induced ER stress. HHcy‐induced ER stress is mediated by oxidative stress. Conversely, HHcy can directly contribute to ER stress, which further leads to oxidative stress. Cleaved ATF6, ATF4 and X‐box binding protein 1, spliced form (XBP1s) transcriptionally regulates their downstream genes separately. S1P, site 1 protease; S2P, site 2 protease; XBP1u, X‐box binding protein 1, unspliced form.
Epigenetic modification in HHcy
Homocysteine also exerts its pathogenic effects via increasing methylation (Kamat et al., 2016). SAM and SAH derived from the homocysteine metabolic pathway regulate DNA methylation. SAM serves as a methyl donor via transmethylation into SAH. HHcy‐related MS or MTHFR deficiency inhibits the transformation of homocysteine into Met and further decreases SAM production, whereas the accumulation of homocysteine leads to the formation of SAH from homocysteine, thus substantially elevating the level of SAH, which further inhibits the transmethylation of SAM (Kamat et al., 2016). Taken together, the SAM/SAH ratio, reduced by the elevation of SAH and decrease of SAM, corresponds with the levels of global hypomethylation in cells. In apparent contradiction, HHcy was associated with a reduced SAM/SAH ratio but not global hypomethylation in CBS‐deficient mice (Lee et al., 2017). One possible explanation is that the level of SAM did not decrease but rather increased as a result of the functional MS and MTHFR. Moreover, homocysteine also directly regulates the MT ,e.g. DNA methyltransferases (DNMTs), activity, which causes DNA, RNA and histone methylation (Castro et al., 2005; Yu et al., 2009).
SAM/SAH ratio
The decrease of the SAM/SAH ratio is related to reduced DNA methylation under conditions of HHcy. HHcy‐induced SAH elevation and SAM reduction contribute to the genome‐wide hypomethylation in homocysteine‐treated VSMCs (Yideng et al., 2007) and in rats with HHcy induced by a high Met diet, there was DNA hypomethylation in aortic tissue (Jiang et al., 2007a), as demonstrated by a high‐throughput quantitative methylation assay. Furthermore, an HHcy‐attenuated SAM/SAH ratio leads to demethylation of DNA for human telomerase reverse transcriptase and p21 and subsequent up‐regulation of the expression of these two genes, which contributes to the shortening of the telomere length and cell senescence in ECs both in vitro and in vivo (Wang et al., 1997; Zhang et al., 2015). In VSMCs, expression of PDGF, an inducer of VSMC proliferation, is up‐regulated as a result of DNA demethylation induced by the HHcy‐reduced SAM/SAH ratio (Han et al., 2014). Moreover, SAM and SAH exhibit directly opposite effects on VSMC proliferation and migration and neointimal formation in vivo. SAM protects against neointimal formation via the inhibition of ER stress and inflammation of VSMCs in obese diabetic rats (Lim et al., 2011), whereas SAH induces the proliferation and migration of VSMCs and enhances atherosclerotic lesions through an oxidative stress‐ERK1/2 pathway in ApoE−/− mice (Luo et al., 2012).
Regulation of DNA methylation
DNA methylation is one of several epigenetic mechanisms that cells employ to control gene expression. DNA methylation occurs at the cytosine bases of eukaryotic DNA, which are converted to 5‐methylcytosine by DNMT enzymes. The altered cytosine residues are typically adjacent to a guanine nucleotide as CpG islands, which results in two methylated cytosine residues placed diagonally to each other on opposing DNA strands. In the human genome, 75% of all promoters are within CpG islands. Thus, the methylated cytosine typically leads to the decrease of interaction between promoters and transcriptional regulators (including transcriptional factors, suppressors and repressors). This decreased interaction results in the repression of transcription, whereas the blockage of suppressors and repressors that interacts with methylated promoter elements alternatively up‐regulates gene expression (Robertson, 2005; Edwards et al., 2017). In addition, DNA methylation brings about the deacetylation of histone H4 and methylation of Lys9 of histone H3 (H3‐Lys9) and prevents the methylation of Lys4 of histone H3 (H3‐Lys4), thus generating a chromatin structure identical to that of methylated sequences in the genome (Hashimshony et al., 2003). HHcy has been reported to contribute to vascular injury and remodelling by directly modulating DNMT activity and the corresponding gene expression.
The amino acid, homocysteine, down‐regulates DNMT1 activity and further induces DNA demethylation. Thus, down‐regulation of DNMT1 by homocysteine enhanced PDGF and sEH expression by increasing the binding of ATF6 and SP‐1 to the demethylated promoter of these genes in ECs. The increased expression of PDGF and sEH subsequently results in the degradation of EC‐derived vasodilator epoxyeicosatrienoic acids and VSMC proliferation (Zhang et al., 2012c,b). Both in vitro and in vivo studies have further demonstrated that DNMT1‐mediated demethylation of the gene for fatty acid‐binding protein 4 (FABP4), induced by homocysteine at a pathogenic concentration, or by HHcy, induced by a high Met diet, up‐regulate FABP4 expression, which leads to intracellular cholesterol accumulation in THP‐1 monocyte‐derived macrophages (Jiang et al., 2016) and accelerates atherosclerosis in ApoE−/− mice (Yang et al., 2015a) respectively. However, the inhibition of DNMT1 by homocysteine attenuates the expression of cyclin A as a result of the enhanced transcriptional suppressors bound to the demethylated promoter element and subsequently mediates homocysteine‐inhibited EC growth (Jamaluddin et al., 2007).
In contrast, homocysteine has been reported to increase DNMT activity and induce DNA methylation. Interestingly, this amino acid decreases the expression of the protective genes DDAH2 and p66shc through DNA methylation dependent on DNMT1 or DNMT3b activity. The down‐regulation of DDAH2 and p66shc mediates homocysteine‐induced ROS production, dysfunctional NO production and apoptosis in ECs (Zhang et al., 2007; Kim et al., 2011; Jia et al., 2013). Moreover, homocysteine down‐regulates the expression of FGF2 via DNA methylation and further inhibits EC growth and survival via the Gi protein pathway (Chang et al., 2008). Similarly, as observed in homocysteine‐treated VSMCs, the up‐regulated DNMT3a (Zhang et al., 2016) and DNMT3b increase p53 DNA methylation and decrease its gene expression, which further contributes to VSMC proliferation (Cao et al., 2016). Furthermore, in an insulin‐like growth factor 2 (IGF2)/H19 imprinting system of VSMCs (Sasaki et al., 2000), homocysteine exhibits an inductive effect of methylation on the H19 enhancer element, which results in the down‐regulation of H19 and up‐regulation of IGF2 involved in VSMC proliferation (Li et al., 2009). In monocyte/macrophages, in vitro studies have indicated that homocysteine up‐regulates DNMT expression and modulates gene expression. First, the DNA methylation of the atheroprotective genes for PPARα, PPARγ and ApoE is significantly induced by homocysteine, which leads to the down‐regulation of the expression of these genes and the promotion of atherogenesis (Yi‐Deng et al., 2007; Yideng et al., 2008). Second, the expression of iNOS is down‐regulated by homocysteine via DNA methylation, which is blocked by PPARα/γ ligands (Jiang et al., 2007b). As the cholesterol efflux pathway protects against monocyte/macrophage‐derived foam cell formation, homocysteine also enhances the DNA methylation of ABCA1 transporter and acetyl‐CoA acetyltransferase 1, two key proteins mediating cholesterol efflux and further markedly down‐regulates their expression in THP‐1 monocytes, which accelerates foam cell formation (Liang et al., 2013).
Regulation of RNA methyltransferase
RNA methylation, usually present at the cytosine and adenine bases of eukaryotic DNA (5‐methylcytosine and 6‐methyladenine, respectively), has been observed in numerous types of RNA molecules, including mRNAs, tRNAs and non‐coding RNAs; however, the function of RNA methylation remains unclear. A tRNA MT NSun2 activated by homocysteine increases ICAM‐1 expression via mRNA methylation in ECs, and HHcy‐induced ICAM‐1 expression in ECs is abolished by NSun2 deficiency in vivo (Luo et al., 2016). This study indicates that mRNA methylation is also involved in the pathogenic effect of homocysteine in EC injuries.
Regulation of histone methyltransferase
Histone methylation mainly occurs on the side chains of lysines or arginines. These residues are able to be methylated multiple times and repress or activate gene transcription. The histone lysine MTs, including G9a and Suv39h1, catalyse H3‐Lys9 methylation in mammalian cells. A recent study demonstrated that unstable plaque formation and the number of apoptotic cells in the lesion are significantly increased in ApoE−/− mice with HHcy, induced by a high‐Met diet, accompanied by a decreased expression of H3‐Lys9 dimethylation. HHcy increases the apoptosis of macrophages and inhibits H3‐Lys9 dimethylation, as well as the expression of histone MT G9a in vitro. The inhibition of histone methylation by BIX01294 enhances macrophage apoptosis and foam cell formation in vitro. In conclusion, HHcy‐induced macrophage apoptosis mediates the progression of atherosclerosis, and histone methylation attributed to HHcy may be involved in this process (Cong et al., 2017).
Collectively, homocysteine has been reported to regulate gene expression via DNA, RNA and histone methylation in ECs, VSMCs and monocytes/macrophages (Figure 5). In general, a decreased reduced SAM/SAH ratio in HHcy leads to genome‐wide hypomethylation in vivo. In contrast, the intake of a folate‐deficient or vitamin B6‐deficient high‐fat diet markedly elevated plasma homocysteine levels and aggravated atherosclerotic plaques in ApoE−/− mice compared with a regular high‐fat diet, with no effect on 5‐methyldeoxycytidine levels in vascular tissues (McNeil et al., 2011). Moreover, there are other discrepancies associated with DNMTs regulated by homocysteine at pathological concentration, between different in vitro studies. An explanation of the differing effects of HHcy on regulation of DNMTs expression is still to be provided. Because the conditions in cell cultures in vitro are not usually identical to those in vivo, additional studies in HHcy animal models in vivo are required. Furthermore, in view of the complexity of epigenetic modulation, other hidden mechanisms, particularly the mechanism by which homocysteine regulates DNMTs, should be investigated in the future.
Figure 5.
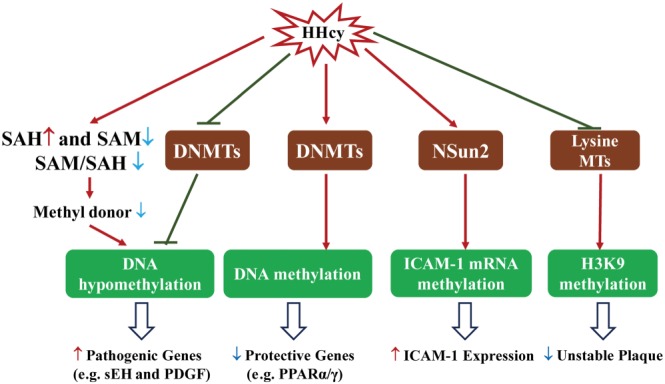
Schematic overview of HHcy‐induced epigenetic methylation. HHcy decreases SAM/SAH ratio and inhibits expression of DNMTs, thus leading to DNA hypomethylation and up‐regulation of pathogenic gene expression. In contrast, HHcy enhances DNMT expression and DNA methylation to down‐regulate expression of protective genes. Furthermore, HHcy up‐regulates tRNA MT NSun2 and promotes ICAM‐1 mRNA methylation, which further increases its gene expression. Histone H3‐Lys9 (H3K9) methylation protects against unstable atherosclerosis plaque. HHcy inhibits lysine MTs and H3K9 methylation, further inducing unstable plaque.
Homocysteinylation in HHcy
Homocysteinylation mainly targets proteins and is classified as S‐homocysteinylation and N‐homocysteinylation (Figure 6). S‐Homocysteinylation occurs when homocysteine binds a protein through its free thiol group to another free thiol group of a Cys residue in the protein molecule to form a disulphide bond. N‐Homocysteinylation is mainly performed by the corresponding thiolactone, Hcy‐TL, which is generated by an error‐editing function of aminoacyl‐tRNA synthetases with homocysteine and ATP. N‐Homocysteinylation comprises the interaction of amino groups from Hcy‐TL and a Lys of a target protein, which further produces a new free thiol group derived from Hcy‐TL. Thus, homocysteinylation may not only affect protein function via amino acid modification but also affects the thiol‐dependent redox status (Jakubowski, 1999). Protein homocysteinylation is enhanced in diabetes and atherosclerosis patients with HHcy (Jakubowski et al., 2000; Jakubowski, 2001). Homocysteinylated LDL isolated from circulating blood exerts cytotoxic effects on ECs in vitro (Ferretti et al., 2004, 2006; Nanetti et al., 2012), whereas fibronectin homocysteinylated by homocysteine attenuates its interaction with fibrin, which indicates a potential delay in haemostasis (Majors et al., 2002). Metallothionein homocysteinylated by homocysteine results in an antioxidative activity disorder and favours ROS accumulation in ECs (Barbato et al., 2007), whereas homocysteinylated eNOS reduces the activity of NOS (Zhang et al., 2000). In contrast, homocysteinylated ACE exhibits an increased ability to produce Ang II and consequently activate the Ang II–NOX–ROS pathway in ECs (Zhang et al., 2007). However, the function of a substantial number of proteins homocysteinylated by homocysteine or its thiolactone in the vascular system has not been fully investigated.
Figure 6.
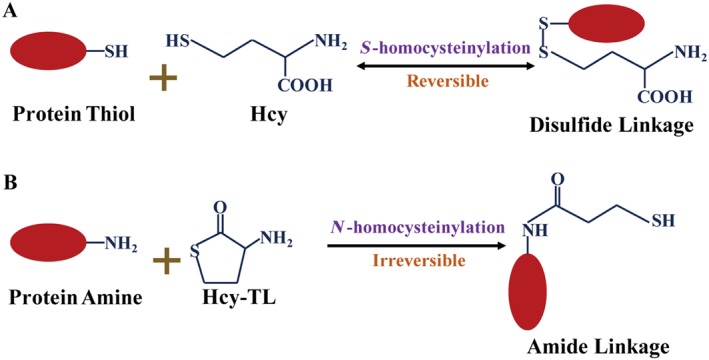
Chemical reactions of S‐homocysteinylation (A) and N‐homocysteinylation (B).
The interaction between vascular injury and injury to other systems, induced by HHcy
As the vascular system is distributed in all organs throughout the entire body, HHcy‐induced vascular injury may influence other organs. HHcy increases the permeability of the blood–brain barrier by disrupting the cell junction of the brain microvascular ECs (Qureshi et al., 2005). A substantial amount of homocysteine subsequently accumulates in the CSF. Homocysteine may act as an agonist of the NMDA receptor, which further causes neural damage via calcium ion efflux or free radial generation (Williams and Schalinske, 2010). HHcy has been associated with pathogenic alterations in mental health, such as cognitive impairment, dementia, depression, Alzheimer's and Parkinson's diseases (Williams and Schalinske, 2010). Moreover, homocysteine‐induced vascular injury contributes to renal vascular dysfunction and results in chronic renal disease (Skovierova et al., 2016).
HHcy‐induced injury of other systems secondarily contributes to vascular injury. Thus, homocysteine is excitatory on hypothalamic neurons, controlling cardiac functions via the sympathetic system. Consequently, homocysteine can cause hypertension via the neuronal system in addition to its effects on NO production (Ganguly and Alam, 2015). Resistin, an adipokine produced by perivascular adipose tissue, contributes to intimal hyperplasia and atherosclerosis (Nosalski and Guzik, 2017). Resistin induces the proliferation and migration of VSMCs via the ERK1/2 or PI3K‐Akt (Calabro et al., 2004) and integrin α5β1‐FAK/paxillin‐Rac1 (Jiang et al., 2009) or PKC (Raghuraman et al., 2016) signalling pathways respectively. Moreover, resistin leads to endothelial dysfunction as shown by the up‐regulation of VCAM‐1 and ICAM‐1 expression, as well as the increased secretion of endothelin‐1 and CCL2 by ECs (Verma et al., 2003; Jamaluddin et al., 2012). Homocysteine promotes the production and secretion of resistin by adipocytes via the ROS–PKC–NF‐κB pathways, and the expression of resistin is enhanced in epididymal adipose tissue from C57BL mice with HHcy, compared with normal mice (Li et al., 2008). Taken together, these data indicate that homocysteine may indirectly induce EC and VSMC injury via the paracrine release of adipokines from perivascular adipose tissue.
Drug targets for HHcy‐induced vascular injury
The development of drugs for the treatment of homocysteine‐induced vascular injury should mainly target on the pathways of homocysteine metabolism and the underlying mechanisms of pathogenic effects of HHcy. Treatments aimed at lowering homocysteine levels have been clinically applied, while many natural chemicals and already available drugs have been shown to exert therapeutic effects in HHcy‐induced vascular injury by inhibiting oxidative or ER stress (Figure 7).
Figure 7.
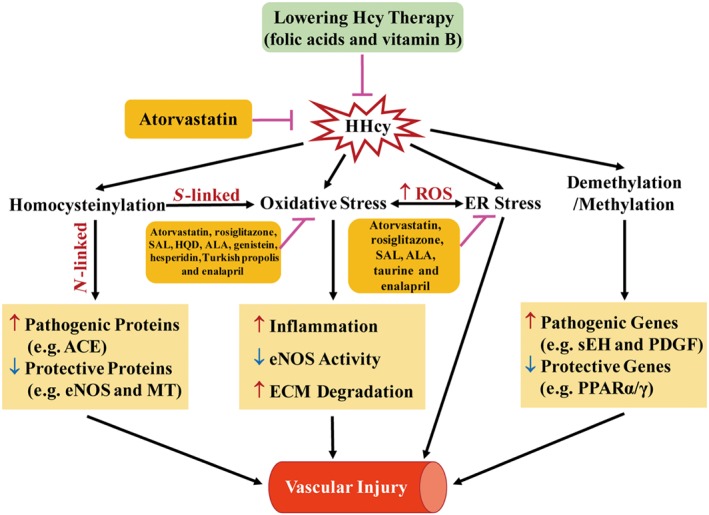
Mechanisms and drug targets of HHcy‐induced vascular injury. HHcy induces vascular injury by promoting oxidative stress, ER stress and protein homocysteinylation as well as regulating methylation. Among them, oxidative stress and ER stress aggravate mutually via ROS as a mediator. For homocysteinylation, the S‐linked reaction induces oxidative stress, whereas the N‐linked reaction regulates the property and activity of target proteins. Lowering homocysteine levels and inhibition of oxidative/ER stress are the potential therapeutic targets for drugs or natural chemicals. ALA, α‐lipoic acid; ECM, extracellular matrix; HQD, Huang Qi decoction; MT, metallothionein; SAL, salidroside.
Homocysteine‐lowering therapy
As low levels of folic acids and vitamin B in the human body cause HHcy, dietary supplementation with folic acids and vitamin B may favour the lowering of homocysteine in the plasma and improve HHcy‐related vascular diseases. Folic acid supplementation has been associated with a reduction in the progression of carotid atherosclerosis (Vermeulen et al., 2000). Further clinical trials have also shown that lowering homocysteine with folic acid and vitamin B reduces the risk of stroke (Saposnik et al., 2009; Liu et al., 2015). However, several clinical trials did not show beneficial effects of lowering plasma homocysteine through folate and vitamin B supplementation on the risk of major cardiovascular events in patients with vascular disease (Lonn et al., 2006; Ebbing et al., 2008) and kidney disease (Bostom et al., 2011; Jardine et al., 2012). These trials with negative results were performed in a population with normally high folate consumption (the USA, Australia and New Zealand). High folate consumption may induce a state of HHcy with a higher basal level of plasma folate. As a result, the further administration of folic acids and vitamin B results in a less effective lowering of plasma homocysteine. Strikingly, it has been suggested that clinical trials of homocysteine‐lowering interventions through folic acids and vitamin B supplementation in vascular disease prevention should be conducted in regions with low folate consumption rather than in areas where foods are more commonly fortified with folate (Holmes et al., 2011). Besides folic acids and vitamin B supplementation, a recent clinical trial has indicated an association between atorvastatin and lowering homocysteine in renal transplantation patients (Monfared et al., 2016), implying that atorvastatin may be used as a homocysteine‐lowering therapy, especially in the areas with dietary folate fortification.
Oxidative stress
Because oxidative stress is the major mechanism mediating homocysteine‐induced vascular injury, several natural chemicals or known drugs are protective against homocysteine‐induced injury, through their antioxidant activity. Atorvastatin is widely used to lower serum cholesterol levels and has been found to protect against HHcy‐induced oxidative stress in ECs both in vitro and in vivo (Bao et al., 2010; Jia et al., 2012, 2016). The PPAR agonist rosiglitazone, developed for type II diabetes, also ameliorates EC dysfunction via suppressing HHcy‐induced oxidative stress in rats (Yang et al., 2015c). Salidroside, an active component of Rhodiola rosea, attenuated homocysteine‐induced EC dysfunction by reducing oxidative stress (Leung et al., 2013). Genistein, a soy isoflavone, blocks homocysteine‐induced pathogenic alterations in human ECs via down‐regulating antioxidative pathways (Han et al., 2015). Hesperidin, a citrus flavonoid, protects against HHcy induced by a high Met diet, by blocking oxidative stress, EC dysfunction and neurotoxicity in Wistar rats (Hemanth Kumar et al., 2017). Huang Qi decoction (HQD) is a traditional Chinese medical formula, and its components exert antioxidant effects. HQD alleviates EC dysfunction initiated by HHcy through antioxidant mechanisms (Chu et al., 2016). α‐Lipoic acid, a disulphide‐containing compound, can scavenge ROS, inhibit the formation of free radicals and chelate metal ions, to maintain cellular homeostasis. Thus, α‐lipoic acid suppressed homocysteine‐induced oxidation and reduced EC apoptosis and inflammation (Hu et al., 2016). Turkish propolis is a natural product, made by honey bees from various plant oils, pollens, resins and wax materials. Turkish propolis protects human ECs in vitro from HHcy‐induced apoptosis by decreasing HHcy‐induced ROS overproduction and lipid peroxidation levels (Darendelioglu et al., 2016). Enalapril, a widely used ACE inhibitor, effectively inhibits aortic ROS production induced by a high Met diet in rats (Zhou et al., 2015).
ER stress
As ER stress also plays a critical role in HHcy‐induced vascular injury, it could be another therapeutic target for HHcy‐related vascular diseases. Atorvastatin attenuates atherosclerotic plaque destabilization by inhibiting endothelial ER stress in HHcy mice (Jia et al., 2016) and the activation of AMP‐activated protein kinase may be involved in this protective effect (Jia et al., 2012). Taurine prevents the decrease in expression and secretion of extracellular SOD induced by homocysteine in VSMCs by decreasing ER stress (Nonaka et al., 2001). Salidroside protects against HHcy‐induced injury in HUVEC via the regulation of ER stress (Zhu et al., 2017). Moreover, α‐lipoic acid and enalapril protect ECs and improve hypertension through inhibiting ER stress as well (Zhou et al., 2015; Hu et al., 2016).
Conclusion and perspective
HHcy exerts its pathogenic effects on vascular injury via various pathways, including oxidative stress, ER stress, epigenetic modulation and protein homocysteinylation. To date, the therapy for HHcy mainly focuses on lowering plasma homocysteine via dietary supplementation with folic acids and vitamin B. However, many in vitro and in vivo studies have demonstrated that many natural chemicals or established drugs are protective against homocysteine‐induced vascular injury. Further animal studies and clinical trials will be required to confirm these potential therapeutic applications.
Nevertheless, several problems and controversies remain unsolved. First, many mechanisms have only been demonstrated in vitro, such as the agonist‐like effect of homocysteine on NMDA receptors. Clearly, the role of NMDA receptors in HHcy‐induced vascular injury should be further explored in vivo. Moreover, the concentration of homocysteine applied in cell culture experiments varies considerably between different studies, even up to millimolar concentrations, which are rarely observed in HHcy patients. Some effects such as the regulation of DNMTs by homocysteine, appear to provide opposing results, related to the different times and concentrations used. Thus, many of the findings from in vitro experiments should be further validated in in vivo studies. Second, many new studies have demonstrated that the effects of HHcy derive from the targeting of multiple systems, rather than a single system. For example, HHcy‐induced vascular injury further induces the injury of other organs, whereas vascular injury could also be secondarily caused by HHcy‐induced injury of other systems. This observation indicates that HHcy is more likely to be a disease that involves the dysfunction of several organs. Advanced studies should be broadened to focus on the interaction of multiple organs or systems in the entire body. Third, homocysteine‐lowering therapy by the intake of folic acids and vitamin B shows little or no curative effect in patients who are normally exposed to diets with folate fortification. Thus, the application of this therapy may be more preferable for patients with low daily folate intake (as in Asia), which would indicate a region‐dependent use. For patients accustomed exposed to dietary folate fortification, the development of another therapy to lower homocysteine levels may be beneficial for their HHcy‐induced vascular injury.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015,a,b,c,d,e,f,g).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This research was supported by funding from the National Natural Science Foundation of the P. R. China (91539203, 81220108004, 81670436), the National Key R&D Program of P. R. China (2016YFC09030000), and the 111 Project of Chinese Ministry of Education (no. B07001).
Fu, Y. , Wang, X. , and Kong, W. (2018) Hyperhomocysteinaemia and vascular injury: advances in mechanisms and drug targets. British Journal of Pharmacology, 175: 1173–1189. doi: 10.1111/bph.13988.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015f). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015g). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Maqueda M, El Bekay R, Monteseirin J, Alba G, Chacon P, Vega A et al (2004). Homocysteine enhances superoxide anion release and NADPH oxidase assembly by human neutrophils. Effects on MAPK activation and neutrophil migration. Atherosclerosis 172: 229–238. [DOI] [PubMed] [Google Scholar]
- Au‐Yeung KK, Woo CW, Sung FL, Yip JC, Siow YL, O K (2004). Hyperhomocysteinemia activates nuclear factor‐kappaB in endothelial cells via oxidative stress. Circ Res 94: 28–36. [DOI] [PubMed] [Google Scholar]
- Bao XM, Wu CF, Lu GP (2010). Atorvastatin inhibits homocysteine‐induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis 210: 114–121. [DOI] [PubMed] [Google Scholar]
- Barbato JC, Catanescu O, Murray K, DiBello PM, Jacobsen DW (2007). Targeting of metallothionein by l‐homocysteine: a novel mechanism for disruption of zinc and redox homeostasis. Arterioscler Thromb Vasc Biol 27: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley P, Peck G, Smeeth L, Whittaker J, Sharma P (2010). Causal relationship of susceptibility genes to ischemic stroke: comparison to ischemic heart disease and biochemical determinants. PLoS One 5: e9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet F, Sapieha P (2015). ER stress and angiogenesis. Cell Metab 22: 560–575. [DOI] [PubMed] [Google Scholar]
- Boldyrev A, Bryushkova E, Mashkina A, Vladychenskaya E (2013). Why is homocysteine toxic for the nervous and immune systems? Curr Aging Sci 6: 29–36. [DOI] [PubMed] [Google Scholar]
- Boldyrev AA (2005). Homocysteinic acid causes oxidative stress in lymphocytes by potentiating toxic effect of NMDA. Bull Exp Biol Med 140: 33–37. [DOI] [PubMed] [Google Scholar]
- Boldyrev AA, Bryushkova EA, Vladychenskaya EA (2012). NMDA receptors in immune competent cells. Biochemistry (Mosc) 77: 128–134. [DOI] [PubMed] [Google Scholar]
- Borges MC, Hartwig FP, Oliveira IO, Horta BL (2016). Is there a causal role for homocysteine concentration in blood pressure? A Mendelian randomization study. Am J Clin Nutr 103: 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostom AG, Carpenter MA, Kusek JW, Levey AS, Hunsicker L, Pfeffer MA et al (2011). Homocysteine‐lowering and cardiovascular disease outcomes in kidney transplant recipients: primary results from the folic acid for vascular outcome reduction in transplantation trial. Circulation 123: 1763–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryushkova EA, Vladychenskaya EA, Stepanova MS, Boldyrev AA (2011). Effect of homocysteine on properties of neutrophils activated in vivo. Biochemistry (Mosc) 76: 467–472. [DOI] [PubMed] [Google Scholar]
- Calabro P, Samudio I, Willerson JT, Yeh ET (2004). Resistin promotes smooth muscle cell proliferation through activation of extracellular signal‐regulated kinase 1/2 and phosphatidylinositol 3‐kinase pathways. Circulation 110: 3335–3340. [DOI] [PubMed] [Google Scholar]
- Cao C, Zhang H, Zhao L, Zhou L, Zhang M, Xu H et al (2016). miR‐125b targets DNMT3b and mediates p53 DNA methylation involving in the vascular smooth muscle cells proliferation induced by homocysteine. Exp Cell Res 347: 95–104. [DOI] [PubMed] [Google Scholar]
- Castro R, Rivera I, Blom HJ, Jakobs C, Tavares de Almeida I (2006). Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: an overview. J Inherit Metab Dis 29: 3–20. [DOI] [PubMed] [Google Scholar]
- Castro R, Rivera I, Martins C, Struys EA, Jansen EE, Clode N et al (2005). Intracellular S‐adenosylhomocysteine increased levels are associated with DNA hypomethylation in HUVEC. J Mol Med (Berl) 83: 831–836. [DOI] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Li W, Dai J, Guan Y, Wang X (2007). Liver‐X‐receptor activator prevents homocysteine‐induced production of IgG antibodies from murine B lymphocytes via the ROS‐NF‐kappaB pathway. Biochem Biophys Res Commun 357: 772–778. [DOI] [PubMed] [Google Scholar]
- Chang PY, Lu SC, Lee CM, Chen YJ, Dugan TA, Huang WH et al (2008). Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor‐2 involving G protein and DNA methylation. Circ Res 102: 933–941. [DOI] [PubMed] [Google Scholar]
- Chaube R, Kallakunta VM, Espey MG, McLarty R, Faccenda A, Ananvoranich S et al (2012). Endoplasmic reticulum stress‐mediated inhibition of NSMase2 elevates plasma membrane cholesterol and attenuates NO production in endothelial cells. Biochim Biophys Acta 1821: 313–323. [DOI] [PubMed] [Google Scholar]
- Chen H, Fitzgerald R, Brown AT, Qureshi I, Breckenridge J, Kazi R et al (2005). Identification of a homocysteine receptor in the peripheral endothelium and its role in proliferation. J Vasc Surg 41: 853–860. [DOI] [PubMed] [Google Scholar]
- Chen Z, Karaplis AC, Ackerman SL, Pogribny IP, Melnyk S, Lussier‐Cacan S et al (2001). Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet 10: 433–443. [DOI] [PubMed] [Google Scholar]
- Chu S, Mao XD, Wang L, Peng W (2016). Effects of Huang Qi decoction on endothelial dysfunction induced by homocysteine. Evid Based Complement Alternat Med 2016 7272694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Bennett DA, Parish S, Verhoef P, Dotsch‐Klerk M, Lathrop M et al (2012). Homocysteine and coronary heart disease: meta‐analysis of MTHFR case‐control studies, avoiding publication bias. PLoS Med 9: e1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong G, Yan R, Huang H, Wang K, Yan N, Jin P et al (2017). Involvement of histone methylation in macrophage apoptosis and unstable plaque formation in methionine‐induced hyperhomocysteinemic ApoE−/− mice. Life Sci 173: 135–144. [DOI] [PubMed] [Google Scholar]
- Dai J, Li W, Chang L, Zhang Z, Tang C, Wang N et al (2006). Role of redox factor‐1 in hyperhomocysteinemia‐accelerated atherosclerosis. Free Radic Biol Med 41: 1566–1577. [DOI] [PubMed] [Google Scholar]
- Dai J, Wang X, Feng J, Kong W, Xu Q, Shen X et al (2008). Regulatory role of thioredoxin in homocysteine‐induced monocyte chemoattractant protein‐1 secretion in monocytes/macrophages. FEBS Lett 582: 3893–3898. [DOI] [PubMed] [Google Scholar]
- Darendelioglu E, Aykutoglu G, Tartik M, Baydas G (2016). Turkish propolis protects human endothelial cells in vitro from homocysteine‐induced apoptosis. Acta Histochem 118: 369–376. [DOI] [PubMed] [Google Scholar]
- Dayal S, Lentz SR (2005). ADMA and hyperhomocysteinemia. Vasc Med (London, England) 10 (Suppl 1): S27–S33. [DOI] [PubMed] [Google Scholar]
- Deng J, Lu S, Liu H, Liu B, Jiang C, Xu Q et al (2017). Homocysteine activates B cells via regulating PKM2‐dependent metabolic reprogramming. J Immunol 198: 170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Zhang X, Li SY, Zhang Z, Ren Q, Culver B et al (2005). Possible involvement of NADPH oxidase and JNK in homocysteine‐induced oxidative stress and apoptosis in human umbilical vein endothelial cells. Cardiovasc Toxicol 5: 9–20. [DOI] [PubMed] [Google Scholar]
- Ebbing M, Bleie O, Ueland PM, Nordrehaug JE, Nilsen DW, Vollset SE et al (2008). Mortality and cardiovascular events in patients treated with homocysteine‐lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA 300: 795–804. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Yarychkivska O, Boulard M, Bestor TH (2017). DNA methylation and DNA methyltransferases. Epigenetics Chromatin 10: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon UB, Ben‐Shlomo Y (2003). Homocysteine, MTHFR 677C→T polymorphism, and risk of ischemic stroke: results of a meta‐analysis. Neurology 60: 526–527 author reply 526‐527. [DOI] [PubMed] [Google Scholar]
- Feng J, Lu S, Ding Y, Zheng M, Wang X (2016). Homocysteine activates T cells by enhancing endoplasmic reticulum‐mitochondria coupling and increasing mitochondrial respiration. Protein Cell 7: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhang Z, Kong W, Liu B, Xu Q, Wang X (2009). Regulatory T cells ameliorate hyperhomocysteinaemia‐accelerated atherosclerosis in apoE−/− mice. Cardiovasc Res 84: 155–163. [DOI] [PubMed] [Google Scholar]
- Ferretti G, Bacchetti T, Moroni C, Vignini A, Nanetti L, Curatola G (2004). Effect of homocysteinylation of low density lipoproteins on lipid peroxidation of human endothelial cells. J Cell Biochem 92: 351–360. [DOI] [PubMed] [Google Scholar]
- Ferretti G, Bacchetti T, Rabini RA, Vignini A, Nanetti L, Moroni C et al (2006). Homocysteinylation of low‐density lipoproteins (LDL) from subjects with type 1 diabetes: effect on oxidative damage of human endothelial cells. Diabet Med 23: 808–813. [DOI] [PubMed] [Google Scholar]
- Fohr IP, Prinz‐Langenohl R, Bronstrup A, Bohlmann AM, Nau H, Berthold HK et al (2002). 5,10‐Methylenetetrahydrofolate reductase genotype determines the plasma homocysteine‐lowering effect of supplementation with 5‐methyltetrahydrofolate or folic acid in healthy young women. Am J Clin Nutr 75: 275–282. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Munzel T (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Ganguly P, Alam SF (2015). Role of homocysteine in the development of cardiovascular disease. Nutr J 14: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Vitvitsky V, Gendelman HE, Banerjee R (2006). Monocyte differentiation, activation, and mycobacterial killing are linked to transsulfuration‐dependent redox metabolism. J Biol Chem 281: 38712–38720. [DOI] [PubMed] [Google Scholar]
- Guo YH, Chen FY, Wang GS, Chen L, Gao W (2008). Diet‐induced hyperhomocysteinemia exacerbates vascular reverse remodeling of balloon‐injured arteries in rat. Chin Med J (Engl) 121: 2265–2271. [PubMed] [Google Scholar]
- Han S, Wu H, Li W, Gao P (2015). Protective effects of genistein in homocysteine‐induced endothelial cell inflammatory injury. Mol Cell Biochem 403: 43–49. [DOI] [PubMed] [Google Scholar]
- Han XB, Zhang HP, Cao CJ, Wang YH, Tian J, Yang XL et al (2014). Aberrant DNA methylation of the PDGF gene in homocysteinemediated VSMC proliferation and its underlying mechanism. Mol Med Rep 10: 947–954. [DOI] [PubMed] [Google Scholar]
- Hannibal L, Blom HJ (2017). Homocysteine and disease: causal associations or epiphenomenons? Mol Aspects Med 53: 36–42. [DOI] [PubMed] [Google Scholar]
- Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H (2003). The role of DNA methylation in setting up chromatin structure during development. Nat Genet 34: 187–192. [DOI] [PubMed] [Google Scholar]
- Hemanth Kumar B, Dinesh Kumar B, Diwan PV (2017). Hesperidin, a citrus flavonoid, protects against l‐methionine‐induced hyperhomocysteinemia by abrogation of oxidative stress, endothelial dysfunction and neurotoxicity in Wistar rats. Pharm Biol 55: 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Newcombe P, Hubacek JA, Sofat R, Ricketts SL, Cooper J et al (2011). Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: a meta‐analysis of genetic studies and randomised trials. Lancet (London, England) 378: 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain GS, van Thienen JV, Werstuck GH, Zhou J, Sood SK, Dickhout JG et al (2003). TDAG51 is induced by homocysteine, promotes detachment‐mediated programmed cell death, and contributes to the cevelopment of atherosclerosis in hyperhomocysteinemia. J Biol Chem 278: 30317–30327. [DOI] [PubMed] [Google Scholar]
- Hu H, Wang C, Jin Y, Meng Q, Liu Q, Liu K et al (2016). Alpha‐lipoic acid defends homocysteine‐induced endoplasmic reticulum and oxidative stress in HAECs. Biomed Pharmacother 80: 63–72. [DOI] [PubMed] [Google Scholar]
- Jakubowski H (1999). Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J 13: 2277–2283. [PubMed] [Google Scholar]
- Jakubowski H (2001). Protein N‐homocysteinylation: implications for atherosclerosis. Biomed Pharmacother 55: 443–447. [DOI] [PubMed] [Google Scholar]
- Jakubowski H (2002). Homocysteine is a protein amino acid in humans. Implications for homocysteine‐linked disease. J Biol Chem 277: 30425–30428. [DOI] [PubMed] [Google Scholar]
- Jakubowski H (2008). The pathophysiological hypothesis of homocysteine thiolactone‐mediated vascular disease. J Physiol Pharmacol 59 (Suppl 9): 155–167. [PubMed] [Google Scholar]
- Jakubowski H, Zhang L, Bardeguez A, Aviv A (2000). Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res 87: 45–51. [DOI] [PubMed] [Google Scholar]
- Jamaluddin MD, Chen I, Yang F, Jiang X, Jan M, Liu X et al (2007). Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood 110: 3648–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamaluddin MS, Weakley SM, Yao Q, Chen C (2012). Resistin: functional roles and therapeutic considerations for cardiovascular disease. Br J Pharmacol 165: 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine MJ, Kang A, Zoungas S, Navaneethan SD, Ninomiya T, Nigwekar SU et al (2012). The effect of folic acid based homocysteine lowering on cardiovascular events in people with kidney disease: systematic review and meta‐analysis. BMJ 344: e3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N (2004). Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J Gastroenterol 10: 1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia F, Wu C, Chen Z, Lu G (2012). Atorvastatin inhibits homocysteine‐induced endoplasmic reticulum stress through activation of AMP‐activated protein kinase. Cardiovasc Ther 30: 317–325. [DOI] [PubMed] [Google Scholar]
- Jia F, Wu C, Chen Z, Lu G, Sun J (2016). Atorvastatin attenuates atherosclerotic plaque destabilization by inhibiting endoplasmic reticulum stress in hyperhomocysteinemic mice. Mol Med Rep 13: 3574–3580. [DOI] [PubMed] [Google Scholar]
- Jia SJ, Lai YQ, Zhao M, Gong T, Zhang BK (2013). Homocysteine‐induced hypermethylation of DDAH2 promoter contributes to apoptosis of endothelial cells. Pharmazie 68: 282–286. [PubMed] [Google Scholar]
- Jiang C, Zhang H, Zhang W, Kong W, Zhu Y, Zhang H et al (2009). Homocysteine promotes vascular smooth muscle cell migration by induction of the adipokine resistin. Am J Physiol Cell Physiol 297: C1466–C1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Ma S, Zhang H, Yang X, Lu GJ, Zhang H et al (2016). FABP4‐mediated homocysteine‐induced cholesterol accumulation in THP‐1 monocyte‐derived macrophages and the potential epigenetic mechanism. Mol Med Rep 14: 969–976. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Sun T, Xiong J, Cao J, Li G, Wang S (2007a). Hyperhomocysteinemia‐mediated DNA hypomethylation and its potential epigenetic role in rats. Acta Biochim Biophys Sin (Shanghai) 39: 657–667. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zhang J, Xiong J, Cao J, Li G, Wang S (2007b). Ligands of peroxisome proliferator‐activated receptor inhibit homocysteine‐induced DNA methylation of inducible nitric oxide synthase gene. Acta Biochim Biophys Sin (Shanghai) 39: 366–376. [DOI] [PubMed] [Google Scholar]
- Jin L, Caldwell RB, Li‐Masters T, Caldwell RW (2007). Homocysteine induces endothelial dysfunction via inhibition of arginine transport. J Physiol Pharmacol 58: 191–206. [PubMed] [Google Scholar]
- Joshi MB, Baipadithaya G, Balakrishnan A, Hegde M, Vohra M, Ahamed R et al (2016). Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci Rep 6: 36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Tyagi SC, Tyagi N (2015). Hydrogen sulfide epigenetically attenuates homocysteine‐induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J Cell Physiol 230: 378–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Mallonee CJ, George AK, Tyagi SC, Tyagi N (2016). Homocysteine, alcoholism, and its potential epigenetic mechanism. Alcohol Clin Exp Res 40: 2474–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke XD, Foucault‐Bertaud A, Genovesio C, Dignat‐George F, Lamy E, Charpiot P (2010). Homocysteine modulates the proteolytic potential of human arterial smooth muscle cells through a reactive oxygen species dependant mechanism. Mol Cell Biochem 335: 203–210. [DOI] [PubMed] [Google Scholar]
- Kerr SJ (1972). Competing methyltransferase systems. J Biol Chem 247: 4248–4252. [PubMed] [Google Scholar]
- Kil JS, Jeong SO, Chung HT, Pae HO (2017). Piceatannol attenuates homocysteine‐induced endoplasmic reticulum stress and endothelial cell damage via heme oxygenase‐1 expression. Amino Acids 49: 735–745. [DOI] [PubMed] [Google Scholar]
- Kim BJ, Kim BS, Kang JH (2015). Plasma homocysteine and coronary artery calcification in Korean men. Eur J Prev Cardiol 22: 478–485. [DOI] [PubMed] [Google Scholar]
- Kim CS, Kim YR, Naqvi A, Kumar S, Hoffman TA, Jung SB et al (2011). Homocysteine promotes human endothelial cell dysfunction via site‐specific epigenetic regulation of p66shc. Cardiovasc Res 92: 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Pae HO, Zheng M, Park R, Kim YM, Chung HT (2007). Carbon monoxide induces heme oxygenase‐1 via activation of protein kinase R‐like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res 101: 919–927. [DOI] [PubMed] [Google Scholar]
- Lai WK, Kan MY (2015). Homocysteine‐induced endothelial dysfunction. Ann Nutr Metab 67: 1–12. [DOI] [PubMed] [Google Scholar]
- Lee HO, Wang L, Kuo YM, Gupta S, Slifker MJ, Li YS et al (2017). Lack of global epigenetic methylation defects in CBS deficient mice. J Inherit Metab Dis 40: 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Lee SJ, Seo KW, Bae JU, Park SY, Kim CD (2013). Homocysteine induces COX‐2 expression in macrophages through ROS generated by NMDA receptor‐calcium signaling pathways. Free Radic Res 47: 422–431. [DOI] [PubMed] [Google Scholar]
- Leung SB, Zhang H, Lau CW, Huang Y, Lin Z (2013). Salidroside improves homocysteine‐induced endothelial dysfunction by reducing oxidative stress. Evid Based Complement Alternat Med 2013: 679635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Xie J, Zhang M, Wang S (2009). Homocysteine harasses the imprinting expression of IGF2 and H19 by demethylation of differentially methylated region between IGF2/H19 genes. Acta Biochim Biophys Sin (Shanghai) 41: 464–471. [DOI] [PubMed] [Google Scholar]
- Li WX, Dai SX, Zheng JJ, Liu JQ, Huang JF (2015). Homocysteine metabolism gene polymorphisms (MTHFR C677T, MTHFR A1298C, MTR A2756G and MTRR A66G) jointly elevate the risk of folate deficiency. Forum Nutr 7: 6670–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jiang C, Xu G, Wang N, Zhu Y, Tang C et al (2008). Homocysteine upregulates resistin production from adipocytes in vivo and in vitro. Diabetes 57: 817–827. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yang X, Ma L, Cai X, Wang L, Yang C et al (2013). Homocysteine‐m IKCa ediated cholesterol efflux via ABCA1 and ACAT1 DNA methylation in THP‐1 monocyte‐derived foam cells. Acta Biochim Biophys Sin (Shanghai) 45: 220–228. [DOI] [PubMed] [Google Scholar]
- Lim S, Moon MK, Shin H, Kim TH, Cho BJ, Kim M et al (2011). Effect of S‐adenosylmethionine on neointimal formation after balloon injury in obese diabetic rats. Cardiovasc Res 90: 383–393. [DOI] [PubMed] [Google Scholar]
- Liu J, Zuo SW, Li Y, Jia X, Jia SH, Zhang T et al (2016). Hyperhomocysteinaemia is an independent risk factor of abdominal aortic aneurysm in a Chinese Han population. Sci Rep 6: 17966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shi M, Xia F, Han J, Liu Z, Wang B et al (2015). The China Stroke Secondary Prevention Trial (CSSPT) protocol: a double‐blinded, randomized, controlled trial of combined folic acid and B vitamins for secondary prevention of stroke. Int J Stroke 10: 264–268. [DOI] [PubMed] [Google Scholar]
- Liu Z, Luo H, Zhang L, Huang Y, Liu B, Ma K et al (2012). Hyperhomocysteinemia exaggerates adventitial inflammation and angiotensin II‐induced abdominal aortic aneurysm in mice. Circ Res 111: 1261–1273. [DOI] [PubMed] [Google Scholar]
- Lonn E, Yusuf S, Arnold MJ, Sheridan P, Pogue J, Micks M et al (2006). Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med 354: 1567–1577. [DOI] [PubMed] [Google Scholar]
- Luo H, Liu B, Hu J, Wang X, Zhan S, Kong W (2014). Hyperhomocysteinemia and methylenetetrahydrofolate reductase polymorphism in cervical artery dissection: a meta‐analysis. Cerebrovasc Dis 37: 313–322. [DOI] [PubMed] [Google Scholar]
- Luo X, Xiao Y, Song F, Yang Y, Xia M, Ling W (2012). Increased plasma S‐adenosyl‐homocysteine levels induce the proliferation and migration of VSMCs through an oxidative stress‐ERK1/2 pathway in apoE(−/−) mice. Cardiovasc Res 95: 241–250. [DOI] [PubMed] [Google Scholar]
- Luo Y, Feng J, Xu Q, Wang W, Wang X (2016). NSun2 deficiency protects endothelium from inflammation via mRNA methylation of ICAM‐1. Circ Res 118: 944–956. [DOI] [PubMed] [Google Scholar]
- Ma K, Lv S, Liu B, Liu Z, Luo Y, Kong W et al (2013). CTLA4‐IgG ameliorates homocysteine‐accelerated atherosclerosis by inhibiting T‐cell overactivation in apoE(−/−) mice. Cardiovasc Res 97: 349–359. [DOI] [PubMed] [Google Scholar]
- Majesky MW (2015). Adventitia and perivascular cells. Arterioscler Thromb Vasc Biol 35: e31–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majors AK, Sengupta S, Willard B, Kinter MT, Pyeritz RE, Jacobsen DW (2002). Homocysteine binds to human plasma fibronectin and inhibits its interaction with fibrin. Arterioscler Thromb Vasc Biol 22: 1354–1359. [DOI] [PubMed] [Google Scholar]
- McCully KS (1969). Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 56: 111–128. [PMC free article] [PubMed] [Google Scholar]
- McNeil CJ, Beattie JH, Gordon MJ, Pirie LP, Duthie SJ (2011). Differential effects of nutritional folic acid deficiency and moderate hyperhomocysteinemia on aortic plaque formation and genome‐wide DNA methylation in vascular tissue from ApoE−/− mice. Clin Epigenetics 2: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijles DN, Pagano PJ (2016). Nox and inflammation in the vascular adventitia. Hypertension 67: 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok SS, Turner BJ, Beyreuther K, Masters CL, Barrow CJ, Small DH (2002). Toxicity of substrate‐bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur J Biochem 269: 3014–3022. [DOI] [PubMed] [Google Scholar]
- Monfared A, Azimi SZ, Kazemnezhad E (2016). The association between atorvastatin administration and plasma total homocysteine levels in renal transplant recipients. J Nephropathol 5: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanetti L, Raffaelli F, Ferretti G, Bacchetti T, Rabini RA, Mazzanti L et al (2012). Homocysteinylation of low‐density lipoproteins (LDL) from subjects with type 1 diabetes and human aortic endothelial cells: an in vitro study. Nutr Metab Cardiovasc Dis 22: e9–e10. [DOI] [PubMed] [Google Scholar]
- Nonaka H, Tsujino T, Watari Y, Emoto N, Yokoyama M (2001). Taurine prevents the decrease in expression and secretion of extracellular superoxide dismutase induced by homocysteine: amelioration of homocysteine‐induced endoplasmic reticulum stress by taurine. Circulation 104: 1165–1170. [DOI] [PubMed] [Google Scholar]
- Nosalski R, Guzik TJ (2017). Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol 174: 3496–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outinen PA, Sood SK, Pfeifer SI, Pamidi S, Podor TJ, Li J et al (1999). Homocysteine‐induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 94: 959–967. [PubMed] [Google Scholar]
- Pang X, Liu J, Zhao J, Mao J, Zhang X, Feng L et al (2014). Homocysteine induces the expression of C‐reactive protein via NMDAr‐ROS‐MAPK‐NF‐kappaB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 236: 73–81. [DOI] [PubMed] [Google Scholar]
- Picerno I, Chirico C, Condello S, Visalli G, Ferlazzo N, Gorgone G et al (2007). Homocysteine induces DNA damage and alterations in proliferative capacity of T‐lymphocytes: a model for immunosenescence? Biogerontology 8: 111–119. [DOI] [PubMed] [Google Scholar]
- Qureshi I, Chen H, Brown AT, Fitzgerald R, Zhang X, Breckenridge J et al (2005). Homocysteine‐induced vascular dysregulation is mediated by the NMDA receptor. Vasc Med (London, England) 10: 215–223. [DOI] [PubMed] [Google Scholar]
- Raghuraman G, Zuniga MC, Yuan H, Zhou W (2016). PKCepsilon mediates resistin‐induced NADPH oxidase activation and inflammation leading to smooth muscle cell dysfunction and intimal hyperplasia. Atherosclerosis 253: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N et al (2005). Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr 82: 636–643. [DOI] [PubMed] [Google Scholar]
- Robertson KD (2005). DNA methylation and human disease. Nat Rev Genet 6: 597–610. [DOI] [PubMed] [Google Scholar]
- Saposnik G, Ray JG, Sheridan P, McQueen M, Lonn E, Heart Outcomes Prevention Evaluation I (2009). Homocysteine‐lowering therapy and stroke risk, severity, and disability: additional findings from the HOPE 2 trial. Stroke 40: 1365–1372. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Ishihara K, Kato R (2000). Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long‐distance gene regulation. J Biochem 127: 711–715. [DOI] [PubMed] [Google Scholar]
- Sawle P, Foresti R, Green CJ, Motterlini R (2001). Homocysteine attenuates endothelial haem oxygenase‐1 induction by nitric oxide (NO) and hypoxia. FEBS Lett 508: 403–406. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB et al (2002). Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med 346: 476–483. [DOI] [PubMed] [Google Scholar]
- Siu KL, Li Q, Zhang Y, Guo J, Youn JY, Du J et al (2016). NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol 11: 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovierova H, Vidomanova E, Mahmood S, Sopkova J, Drgova A, Cervenova T et al (2016). The molecular and cellular effect of homocysteine metabolism imbalance on human health. Int J Mol Sci 17: 1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulders YM, Blom HJ (2011). The homocysteine controversy. J Inherit Metab Dis 34: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark KR, Yeager ME, El Kasmi KC, Nozik‐Grayck E, Gerasimovskaya EV, Li M et al (2013). The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 75: 23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha S, Azzi A, Ozer NK (1999). Homocysteine induces DNA synthesis and proliferation of vascular smooth muscle cells by a hydrogen peroxide‐independent mechanism. Antioxid Redox Signal 1: 365–369. [DOI] [PubMed] [Google Scholar]
- Takagi H, Umemoto T (2005). Homocysteinemia is a risk factor for aortic dissection. Med Hypotheses 64: 1007–1010. [DOI] [PubMed] [Google Scholar]
- Tsen CM, Hsieh CC, Yen CH, Lau YT (2003). Homocysteine altered ROS generation and NO accumulation in endothelial cells. Chin J Physiol 46: 129–136. [PubMed] [Google Scholar]
- Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC (2005). Mechanisms of homocysteine‐induced oxidative stress. Am J Physiol Heart Circ Physiol 289: H2649–H2656. [DOI] [PubMed] [Google Scholar]
- Verma S, Li SH, Wang CH, Fedak PW, Li RK, Weisel RD et al (2003). Resistin promotes endothelial cell activation: further evidence of adipokine‐endothelial interaction. Circulation 108: 736–740. [DOI] [PubMed] [Google Scholar]
- Vermeulen EG, Stehouwer CD, Twisk JW, van den Berg M, de Jong SC, Mackaay AJ et al (2000). Effect of homocysteine‐lowering treatment with folic acid plus vitamin B6 on progression of subclinical atherosclerosis: a randomised, placebo‐controlled trial. Lancet (London, England) 355: 517–522. [DOI] [PubMed] [Google Scholar]
- Vladychenskaya E, Tyulina O, Urano S, Boldyrev A (2011). Rat lymphocytes express NMDA receptors that take part in regulation of cytokine production. Cell Biochem Funct 29: 527–533. [DOI] [PubMed] [Google Scholar]
- Wang G, Siow YL, O K (2000). Homocysteine stimulates nuclear factor kappaB activity and monocyte chemoattractant protein‐1 expression in vascular smooth‐muscle cells: a possible role for protein kinase C. Biochem J 352 (Pt 3): 817–826. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yoshizumi M, Lai K, Tsai JC, Perrella MA, Haber E et al (1997). Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem 272: 25380–25385. [DOI] [PubMed] [Google Scholar]
- Wang XC, Sun WT, Yu CM, Pun SH, Underwood MJ, He GW et al (2015a). ER stress mediates homocysteine‐induced endothelial dysfunction: modulation of IKCa and SKCa channels. Atherosclerosis 242: 191–198. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xu X, Huo Y, Liu D, Cui Y, Liu Z et al (2015b). Predicting hyperhomocysteinemia by methylenetetrahydrofolate reductase C677T polymorphism in Chinese patients with hypertension. Clin Appl Thromb Hemost 21: 661–666. [DOI] [PubMed] [Google Scholar]
- Weber GJ, Pushpakumar S, Tyagi SC, Sen U (2016). Homocysteine and hydrogen sulfide in epigenetic, metabolic and microbiota related renovascular hypertension. Pharmacol Res 113: 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch GN, Upchurch GR Jr, Farivar RS, Pigazzi A, Vu K, Brecher P et al (1998). Homocysteine‐induced nitric oxide production in vascular smooth‐muscle cells by NF‐kappa B‐dependent transcriptional activation of Nos2. Proc Assoc Am Physicians 110: 22–31. [PubMed] [Google Scholar]
- Wilcken DE, Wilcken B (1976). The pathogenesis of coronary artery disease. A possible role for methionine metabolism. J Clin Invest 57: 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KT, Schalinske KL (2010). Homocysteine metabolism and its relation to health and disease. Biofactors 36: 19–24. [DOI] [PubMed] [Google Scholar]
- Wu S, Gao X, Yang S, Meng M, Yang X, Ge B (2015). The role of endoplasmic reticulum stress in endothelial dysfunction induced by homocysteine thiolactone. Fundam Clin Pharmacol 29: 252–259. [DOI] [PubMed] [Google Scholar]
- Yang AN, Zhang HP, Sun Y, Yang XL, Wang N, Zhu G et al (2015a). High‐methionine diets accelerate atherosclerosis by HHcy‐mediated FABP4 gene demethylation pathway via DNMT1 in ApoE(−/−) mice. FEBS Lett 589: 3998–4009. [DOI] [PubMed] [Google Scholar]
- Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD et al (2015b). Phospho‐selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F‐NMR. Nat Commun 6: 8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Bailey L, Clarke R, Flanders WD, Liu T, Yesupriya A et al (2012). Prospective study of methylenetetrahydrofolate reductase (MTHFR) variant C677T and risk of all‐cause and cardiovascular disease mortality among 6000 US adults. Am J Clin Nutr 95: 1245–1253. [DOI] [PubMed] [Google Scholar]
- Yang XH, Li P, Yin YL, Tu JH, Dai W, Liu LY et al (2015c). Rosiglitazone via PPARgamma‐dependent suppression of oxidative stress attenuates endothelial dysfunction in rats fed homocysteine thiolactone. J Cell Mol Med 19: 826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Sun NL (2014). Hyperhomocysteinemia accelerates collagen accumulation in the adventitia of balloon‐injured rat carotid arteries via angiotensin II type 1 receptor. Int J Mol Sci 15: 19487–19498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yideng J, Jianzhong Z, Ying H, Juan S, Jinge Z, Shenglan W et al (2007). Homocysteine‐mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol 26: 603–611. [DOI] [PubMed] [Google Scholar]
- Yi‐Deng J, Tao S, Hui‐Ping Z, Jian‐Tuan X, Jun C, Gui‐Zhong L et al (2007). Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol 26: 737–744. [DOI] [PubMed] [Google Scholar]
- Yideng J, Zhihong L, Jiantuan X, Jun C, Guizhong L, Shuren W (2008). Homocysteine‐mediated PPARalpha,gamma DNA methylation and its potential pathogenic mechanism in monocytes. DNA Cell Biol 27: 143–150. [DOI] [PubMed] [Google Scholar]
- Yu X, Ling W, Mi M (2009). Relationship of impairment induced by intracellular S‐adenosylhomocysteine accumulation with DNA methylation in human umbilical vein endothelial cells treated with 3‐deazaadenosine. Int J Exp Pathol 90: 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanin RF, Bergamin LS, Morrone FB, Coutinho‐Silva R, de Souza Wyse AT, Battastini AM (2015). Pathological concentrations of homocysteine increases IL‐1beta production in macrophages in a P2X7, NF‐kB, and erk‐dependent manner. Purinergic Signal 11: 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Dai J, Remick DG, Wang X (2003). Homocysteine mediated expression and secretion of monocyte chemoattractant protein‐1 and interleukin‐8 in human monocytes. Circ Res 93: 311–320. [DOI] [PubMed] [Google Scholar]
- Zhang D, Chen Y, Xie X, Liu J, Wang Q, Kong W et al (2012a). Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet‐derived growth factor in endothelial cells. J Mol Cell Cardiol 53: 487–496. [DOI] [PubMed] [Google Scholar]
- Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD et al (2012b). Severe hyperhomocysteinemia promotes bone marrow‐derived and resident inflammatory monocyte differentiation and atherosclerosis in LDLr/CBS‐deficient mice. Circ Res 111: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Jiang X, Fang P, Yan Y, Song J, Gupta S et al (2009). Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta‐synthase‐deficient mice. Circulation 120: 1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Sun X, Liu J, Xie X, Cui W, Zhu Y (2015). Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler Thromb Vasc Biol 35: 71–78. [DOI] [PubMed] [Google Scholar]
- Zhang D, Xie X, Chen Y, Hammock BD, Kong W, Zhu Y (2012c). Homocysteine upregulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Circ Res 110: 808–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HP, Wang YH, Cao CJ, Yang XM, Ma SC, Han XB et al (2016). A regulatory circuit involving miR‐143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol Med Rep 13: 483–490. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Liu JX, Li ZH, Wang LZ, Jiang YD, Wang SR (2007). Dysfunction of endothelial NO system originated from homocysteine‐induced aberrant methylation pattern in promoter region of DDAH2 gene. Chin Med J (Engl) 120: 2132–2137. [PubMed] [Google Scholar]
- Zhang Q, Zeng X, Guo J, Wang X (2001). Effects of homocysteine on murine splenic B lymphocyte proliferation and its signal transduction mechanism. Cardiovasc Res 52: 328–336. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zeng X, Guo J, Wang X (2002). Oxidant stress mechanism of homocysteine potentiating Con A‐induced proliferation in murine splenic T lymphocytes. Cardiovasc Res 53: 1035–1042. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li H, Jin H, Ebin Z, Brodsky S, Goligorsky MS (2000). Effects of homocysteine on endothelial nitric oxide production. Am J Physiol Renal Physiol 279: F671–F678. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhao L, Zhang Z, Lu X (2015). Protective effect of enalapril against methionine‐enriched diet‐induced hypertension: role of endoplasmic reticulum and oxidative stress. Biomed Res Int 2015: 724876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jia F, Wei J, Yu Y, Yu T, Wang Y et al (2017). Salidroside protects against homocysteine‐induced injury in human umbilical vein endothelial cells via the regulation of endoplasmic reticulum stress. Cardiovasc Ther 35: 33–39. [DOI] [PubMed] [Google Scholar]