

Abstract
Background and Purpose
Metformin, a small molecule, antihyperglycaemic agent, is a well‐known activator of AMP‐activated protein kinase (AMPK) and protects against cardiac fibrosis. However, the underlying mechanisms remain elusive. TGFβ1 is a key cytokine mediating cardiac fibrosis. Here, we investigated the effects of metformin on TGFβ1 production induced by angiotensin II (AngII) and the underlying mechanisms.
Experimental Approach
Wild‐type and AMPKα2−/− C57BL/6 mice were injected s.c. with metformin or saline and infused with AngII (3 mg·kg−1·day−1) for 7 days. Adult mouse cardiac fibroblasts (CFs) were isolated for in vitro experiments.
Key Results
In CFs, metformin inhibited AngII‐induced TGFβ1 expression via AMPK activation. Analysis using bioinformatics predicted a potential hepatocyte nuclear factor 4α (HNF4α)‐binding site in the promoter region of the Tgfb1 gene. Overexpressing HNF4α increased TGFβ1 expression in CFs. HNF4α siRNA attenuated AngII‐induced TGFβ1 production and cardiac fibrosis in vitro and in vivo. Metformin inhibited the AngII‐induced increases in HNF4α protein expression and binding to the Tgfb1 promoter in CFs. In vivo, metformin blocked the AngII‐induced increase in cardiac HNF4α protein levels in wild‐type mice but not in AMPKα2−/− mice. Consequently, metformin inhibited AngII‐induced TGFβ1 production and cardiac fibrosis in wild‐type mice but not in AMPKα2−/− mice.
Conclusions and Implications
HNF4α mediates AngII‐induced TGFβ1 transcription and cardiac fibrosis. Metformin inhibits AngII‐induced HNF4α expression via AMPK activation, thus decreasing TGFβ1 transcription and cardiac fibrosis. These findings reveal a novel antifibrotic mechanism of action of metformin and identify HNF4α as a new potential therapeutic target for cardiac fibrosis.
Linked Articles
This article is part of a themed section on Spotlight on Small Molecules in Cardiovascular Diseases. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.8/issuetoc
Abbreviations
- ACC
acetyl‐CoA carboxylase
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleoside
- AMPK
AMP‐activated protein kinase
- AngII
angiotensin II
- CFs
cardiac fibroblasts
- ChIP
chromatin immunoprecipitation
- EF%
ejection fraction
- eIF5
eukaryotic translation initiation factor 5
- FS%
fractional shortening
- HNF4α
hepatocyte nuclear factor 4α
- MEFs
mouse embryonic fibroblasts
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| AT1 receptor | ACC |
| Nuclear hormone receptors b | AMPK |
| Hepatocyte nuclear factor‐4‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Metformin is a small‐molecule, antihyperglycaemic agent widely used in the treatment of diabetes. It has also been shown to provide cardiovascular protection that cannot be attributed to its glucose‐lowering effects (UK Prospective Diabetes Study (UKPDS) Group, 1998). Our previous study and another group have shown that metformin displays anti‐fibrotic effects, following pressure overload and myocardial infarction in rodents (Xiao et al., 2010; Yin et al., 2011). We also demonstrated that metformin inhibits transverse aortic constriction‐induced myocardial production of TGFβ1 (Xiao et al., 2010). TGFβ1 is a crucial cytokine mediating cardiac fibrosis that plays a causal role in the progression of heart failure (Creemers and Pinto, 2011). However, the molecular mechanisms underlying the inhibitory effect of metformin on TGFβ1 production are unknown. Identifying these mechanisms will reveal new molecular targets for metformin and will identify potential targets for the treatment of cardiac fibrosis.
Using the TRANSFAC database, we predicted several potential transcription factor binding sites, including hepatocyte nuclear factor 4α (HNF4α) binding sites, in the promoter regions of the mouse Tgfb1 gene. HNF4α, a member of the nuclear receptor superfamily, plays a crucial role in regulating metabolic gene expression (Gonzalez, 2008). Despite this knowledge, it is still not known whether HNF4α mediates TGFβ1 transcript expression and cardiac fibrosis.
Previous studies have shown that AMP‐activated protein kinase (AMPK) reduces the ability of HNF4α to bind to DNA and increases the degradation rate of HNF4α (Leclerc et al., 2001; Hong et al., 2003). Metformin is a well‐known AMPK activator. Whether metformin inhibits TGFβ1 production by targeting HNF4α remains to be investigated. Furthermore, the AMPK‐dependence of the inhibitory effect of metformin on the HNF4α ‐TGFβ1 pathway needs to be evaluated.
In the present study, we identified HNF4α as a novel transcriptional factor targeting Tgfb1 gene expression and as a crucial regulator of cardiac fibrosis induced by angiotensin II (AngII). We demonstrated that metformin inhibits TGFβ1 production by targeting HNF4α via an AMPK‐dependent pathway.
Methods
Isolation of adult mouse cardiac fibroblasts (CFs)
CFs were isolated from 8‐ to 10‐week‐old male C57BL/6 mice as previously described (Du et al., 2005). Ventricles were minced and digested with 0.1% (g·mL−1) collagenase II (Worthington, Columbia, NJ, USA) at 37°C. Cells were collected and plated in a culture dish at 37°C. Two hours later, the dish was washed with PBS to remove cell debris and non‐adherent cells. CFs were cultured in DMEM containing 10% FBS (Ausbian, Australia) at 37°C in 5% CO2. Cells in the second passage were used and were randomly divided into multiple groups for the further experiments.
Western blot
Cell and heart tissue (left ventricular) lysates were separated on SDS‐PAGE gels and transferred to nitrocellulose membranes (Pall, Port Washington, NY, USA). The membranes were incubated with primary antibodies overnight at 4°C. Antibodies against TGFβ1 and eukaryotic translation initiation factor 5 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against HNF4α and AMPKα2 were purchased from Abcam (Cambridge, MA, USA). Antibodies against phospho‐AMPKα Thr172 (p‐AMPK), AMPK, acetyl‐CoA carboxylase (ACC) and phospho‐ACC Ser79 were purchased from Cell Signalling Technology (Danvers, MA, USA). After incubating the membranes with corresponding HRP‐conjugated secondary antibodies, protein bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Corporation, USA). The protein level was quantified by calculating the grey value of each protein band using Image J software.
Binding site prediction
Putative HNF4α binding sites on the mouse Tgfb1 gene were predicted using the position weight matrix algorithm in TRANSFAC to scan the promoter regions of the gene (Wingender et al., 1997). The promoter regions were defined as −3000 ~ 500 nucleotides from the transcriptional start site of the gene.
HNF4α overexpression and knock‐down
Adenovirus expressing HNF4α (Ad‐HNF4α) was purchased from HanBio Co., Ltd. (Shanghai, China). CFs were infected with Ad‐HNF4α followed by RNA or protein extraction. The adenoviral vector Ad‐GFP was used as a control. HNF4α was knocked down in CFs using HNF4α siRNA‐expressing lentivirus, which was purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China).
Dual luciferase reporter gene assay
Mouse embryonic fibroblasts (MEFs) were used in this assay. A total of 105 MEFs were seeded in 24‐well plates in complete medium without antibiotics and were randomly divided into four groups for the further treatment. After 24 h, the MEFs were transfected with the wild type (containing the TGFβ1 promoter region with an intact −1259 ~ −1255 fragment) or mutant luciferase reporter plasmid (in which the −1259 ~ −1255 fragment was deleted). The Renilla luciferase plasmid was transfected as a reference. After treatment with AngII for 24 h, transfected cells were lysed and analysed using a Dual‐luciferase Reporter Assay System (Promega). The results are expressed as the luminescence ratio of firefly (luc)/Renilla luciferase.
Chromatin immunoprecipitation (ChIP) assay
Formaldehyde was used to crosslink proteins and their interacting DNA in live CFs. The lysates were then collected and sonicated to shear DNA into fragments of 500–1000 bp in length. An antibody against HNF4α (Abcam, ab41898, ChIP‐grade) and mouse IgG were used for immunoprecipitation. The primers used for detection of the binding of HNF4α to the HNF4α binding sites in the mouse TGFβ1 promoter are listed in Supporting Information Table S1.
Animal model
The investigations conformed to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 2011). All animal experiments were approved by the Animal Ethics Committee of Peking University (LA 2010–048) and were conducted in accordance with the Guidelines for Animal Experiments of the Peking University Health Science Centre. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). AMPKα2−/− mice were kindly provided by Professor Benoit Viollet (Institute National de la Santé et de la Recherche Médicale U567, Paris, France). AMPKα2−/− mice and their wild type littermates AMPKα2+/+ mice (C57BL/6 background, male, 10 weeks old, approximately 25 g body weight) were housed in a specific pathogen‐free environment under a 12/12 h light/dark cycle.
Both wild type and AMPKα2−/− mice were randomly divided into four groups (vehicle, metformin, AngII, and AngII + metformin). The mice were infused with AngII (3 mg·kg−1·day−1) through an implanted osmotic mini‐pump (Alzet MODEL 1007D, DURECT, Cupertino, CA) for 7 days. The surgical procedures were performed after the righting reflex disappeared under anaesthesia with 1–2% isoflurane. Metformin (200 mg·kg−1·day−1, Sigma, Saint Louis, USA) or saline was injected s.c. daily beginning 3 days prior to the AngII infusion. After the 7 day AngII infusion period, fasting blood glucose levels were measured using ACCU‐CHEK Active Glucose Test Strips (Roche, Germany), and blood pressure was measured using a tail‐cuff system (BP‐98A, Softron, Japan). Mice were anaesthetized with 2–3% isoflurane in oxygen, and pedal pinch reflexes were completely inhibited prior to them being killed. This model has been in use for several years (Yang et al., 2012).
In vivo gene silencing
Chemically modified siRNA specific for HNF4α and a non‐specific control siRNA were synthesized by Ribobio Co., Ltd. (Guangzhou, China). C57BL/6 mice (male, 10 weeks old, approximately 25 g body weight) were randomly divided into four groups [negative control (NC) siRNA, HNF4α siRNA, NC siRNA + AngII and HNF4α siRNA + AngII]. The mice were treated with 10 nmol siRNA (diluted in 0.12 mL of saline) via a tail vein injection for three consecutive days (Soutschek et al., 2004; Yuan et al., 2008; Li et al., 2013). The mice were then infused with AngII for 7 days. siRNA was injected once every 2 days after initiation of the AngII infusion. The sequences of the siRNAs used are listed in supplemental methods.
Echocardiography
After 7 days of AngII infusion, mice were anaesthetized with 1–2% isoflurane and underwent echocardiography using a high‐resolution Vevo 2100 system (Visualsonics Inc., Toronto, Canada). The peak flow velocities during early diastole (E wave) as well as the early diastolic peak velocity (E’ wave) of the mitral valve ring were measured. Then, E/E’, which reflects left ventricular diastolic function, was calculated. The left ventricular ejection fraction (EF%) and the left ventricular fractional shortening (FS%) were calculated.
elisa
Heart tissues were harvested, immediately frozen in liquid nitrogen and then homogenized in lysis buffer. The content of TGFβ1 was measured via elisa (R&D Systems, Inc., Minneapolis, MN, USA). All absorbance values were in the linear range of the standard curve and were normalized to the total protein concentration.
Histological analysis
Heart tissues were fixed in 4% paraformaldehyde for 6–8 h. After fixation, the tissues were embedded in paraffin and transversely sectioned at a thickness of 5 μm. Sirius red staining was used to evaluate fibrosis in paraffin‐embedded heart sections. Images of the sections were captured using a Leica Q550 IW imaging workstation (Leica Microsystems Imaging Solutions Ltd., Cambridge, UK) and were analysed using Image Pro Plus 6.0 software (Media Cybernetics, LP, USA).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) and IBM SPSS Statistics 21.0 (IBM Corporation, Armonk, NY, USA). Values are presented as means ± SEM, and data analyses were carried out without knowledge of treatments (blinded assessment). To control for unwanted sources of variation, some values are expressed as ‘fold of control mean’. For parametric data with a normal distribution based on the K–S test, Student's t‐test or ANOVA combined with the Bonferroni post hoc test was used to analyse the differences between groups. Bonferroni post hoc tests were run when F achieved P < 0.05 and there was no significant inhomogeneity. For data with unequal variances, Welch's t‐test or Welch's ANOVA with post hoc Games–Howell test was used. For non‐parametric data, Kruskal–Wallis ANOVA with post hoc Dunn's multiple comparison test was used. A P value <0.05 was considered statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
Metformin and AngII were purchased from Sigma (Saint Louis, USA), 5‐aminoimidazole‐4‐carboxamide ribonucleoside (AICAR) was purchased from Toronto Research Chemicals (Toronto, Canada) and Compound C was purchased from EMD Millipore Corp., (Billerica, MA, USA).
Results
Metformin inhibition of AngII‐induced TGFβ1 production depends on AMPK activity
CFs are the major cellular source of TGFβ1 production in the heart (Eghbali, 1989). We initially investigated whether metformin attenuates AngII‐induced TGFβ1 expression in cultured CFs and determined the role of AMPK. In wild type CFs, metformin (1 mM) significantly reduced AngII‐induced TGFβ1 mRNA and protein expression, and these effects were blocked by pretreatment with the AMPK inhibitor Compound C (1 μM) (Figure 1A–B). Consistently, metformin did not reduce AngII‐induced TGFβ1 expression at the mRNA or protein level in AMPKα2−/− CFs (Figure 1C–D). These results suggest that the inhibitory effects of metformin on AngII‐induced TGFβ1 production in the cultured CFs depend on AMPK activity.
Figure 1.
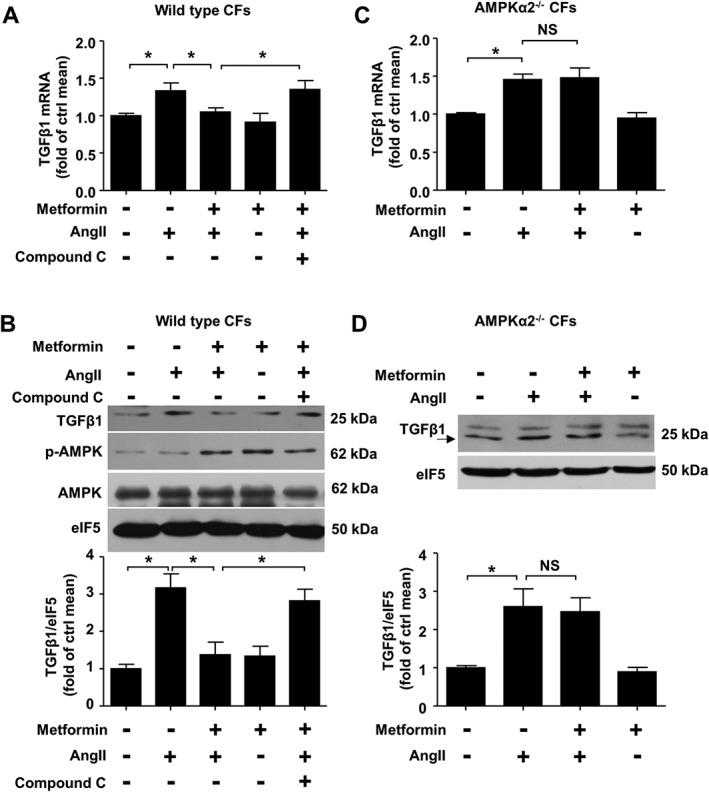
Metformin inhibits AngII‐induced TGFβ1 production in cardiac fibroblasts (CFs) via AMPK activation. CFs were pretreated with Compound C (1 μM) for 0.5 h, and metformin (1 mM) was applied for an additional 2 h. Next, AngII (1 μM) was applied for 24 h before harvesting. (A) TGFβ1 mRNA expression was determined via real‐time PCR analysis. (B) TGFβ1 protein contents were determined via western blot analysis. (C) Real‐time PCR analysis of TGFβ1 mRNA expression in AMPKα2−/− CFs treated with AngII and/or metformin. (D) Western blot analysis of TGFβ1 expression in AMPKα2−/− CFs treated with AngII and/or metformin. Data are expressed as means ± SEM from five independent experiments. *P < 0.05, NS = not significant. One‐way ANOVA (A and B) or two‐way ANOVA (C) with the Bonferroni post hoc test was used. Welch's ANOVA with post hoc Games–Howell test was used in (D).
HNF4α mediates AngII‐induced TGFβ1 expression and cardiac fibrosis
To identify the target of metformin on TGFβ1, bioinformatics’ analysis was used to predict potential transcriptional factors targeting the mouse Tgfb1 gene. A putative binding site of HNF4α, which was down‐regulated by AMPK, was found to be located in the mouse Tgfb1 gene promoter region. We first investigated whether HNF4α is modulated by AngII. As shown in Supporting Information Fig. S1a, AngII significantly promoted HNF4α protein expression in the heart. Similarly, AngII induced HNF4α protein expression in CFs, which are the main cells participating in cardiac fibrosis (Supporting Information Fig. S1b). Furthermore, pretreatment with the AT1 receptor antagonist losartan blocked AngII‐induced HNF4α protein up‐regulation (Supporting Information Fig. S1c), suggesting that AngII promotes HNF4α protein expression via AT1 receptor activity.
Moreover, the organ distribution of HNF4α expression was examined. Although HNF4α is highly expressed in the heart, it is most abundantly expressed in the liver and lungs (Supporting Information Fig. S2a). HNF4α protein variants arise from mRNAs transcribed from two promoters, the proximal promoter P1 and the distal promoter P2 (Babeu and Boudreau, 2014). Of note, P1‐derived HNF4α transcripts are more abundant in the liver, but P2‐derived HNF4α transcripts are more abundant in the heart (Supporting Information Fig. S2b).
To determine whether HNF4α mediates AngII‐induced TGFβ1 expression, we first investigated whether HNF4α overexpression increases TGFβ1 expression. As expected, TGFβ1 mRNA and protein levels were increased in HNF4α adenovirus‐infected CFs (Figure 2A–B). Furthermore, HNF4α siRNA inhibited the increase in TGFβ1 mRNA and protein expression induced by AngII (Figure 2C–D). These observations suggest that HNF4α mediated the expression of TGFβ1 upon stimulation with AngII.
Figure 2.
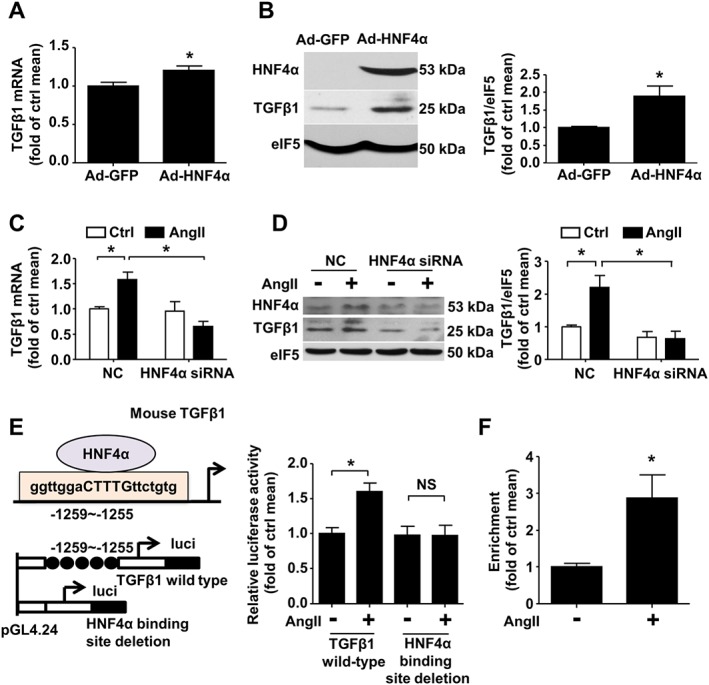
AngII increases the HNF4α protein levels and HNF4α binding activity. (A) Quantitative real‐time PCR analysis of TGFβ1 mRNA expression in CFs treated with Ad‐GFP or Ad‐HNF4α at a multiplicity of infection (MOI) of 15. (B) Western blot analysis of TGFβ1 and HNF4α expression in CFs infected with Ad‐GFP or Ad‐HNF4α. (C) Real‐time PCR analysis of TGFβ1 mRNA expression and (D) western blot analysis of TGFβ1 protein expression in CFs transfected with negative control (NC) or HNF4α siRNA and then exposed to AngII for 24 h. (E) Left panel: Schematic of the interaction of HNF4α with the Tgfb1 promoter region and the structures of the reporter plasmids carrying the full Tgfb1 promoter region (wild type) or carrying the −1259/−1255 bp site deletion fragment (mutant). Right panel: MEFs were transfected with the wild type or mutant plasmid and then treated with AngII or untreated. A dual luciferase reporter assay was performed. (F) ChIP analysis using antibodies against HNF4α or IgG, soluble chromatin (~500 bp in length) from CFs treated with AngII, and primers targeting the region spanning the HNF4α binding sites in the Tgfb1 promoter, n = 6. For (A–E), data are expressed as means ± SEM from five independent experiments. *P < 0.05, NS = not significant. Student's unpaired two‐tailed t‐test (A) or Welch's t‐test (B and F) was used. Two‐way ANOVA with the Bonferroni post hoc test was used in (C, D and E).
To further examine whether HNF4α mediates TGFβ1 transcription, a dual luciferase reporter assay was used. A putative binding site of HNF4α was found to be located at −1259/−1255 bp in the mouse Tgfb1 gene promoter region (Figure 2E). The wild type or mutant HNF4α luciferase reporter plasmid, harbouring the wild type Tgfb1 promoter and the HNF4α binding site‐deleted Tgfb1 promoter, respectively, was transfected into MEFs. AngII treatment for 24 h significantly increased luciferase activity in cells transfected with the reporter plasmid carrying the wild type Tgfb1 promoter but had no effect on cells transfected with the reporter plasmid carrying the mutant Tgfb1 promoter (Figure 2E). A ChIP assay further demonstrated an increase in the binding of HNF4α to the Tgfb1 promoter region upon AngII exposure (Figure 2F). Taken together, these results suggest that HNF4α mediates AngII‐induced TGFβ1 transcription in fibroblasts.
To confirm the role of HNF4α in AngII‐induced TGFβ1 expression and cardiac fibrosis in vivo, mice were injected with HNF4α siRNA via the tail vein. HNF4α expression in the heart was successfully knocked down via siRNA injection (Figure 3A). Consistent with the results of the in vitro experiments, the AngII‐induced increase in the TGFβ1 mRNA level was significantly inhibited by injection with HNF4α siRNA (Figure 3B). Concerning cardiac fibrosis, silencing HNF4α significantly reduced the area of collagen deposition upon AngII exposure (Figure 3C–D). Similarly, HNF4α knockdown decreased AngII‐induced collagen I and III mRNA expression (Figure 3E–F). Echocardiography demonstrated that E/E’ was decreased, indicating improved diastolic function, in the HNF4α siRNA treatment group compared with the non‐specific control siRNA treatment group (Figure 3G–H). These results suggest that HNF4α mediates cardiac TGFβ1 production and fibrosis upon stimulation with AngII.
Figure 3.
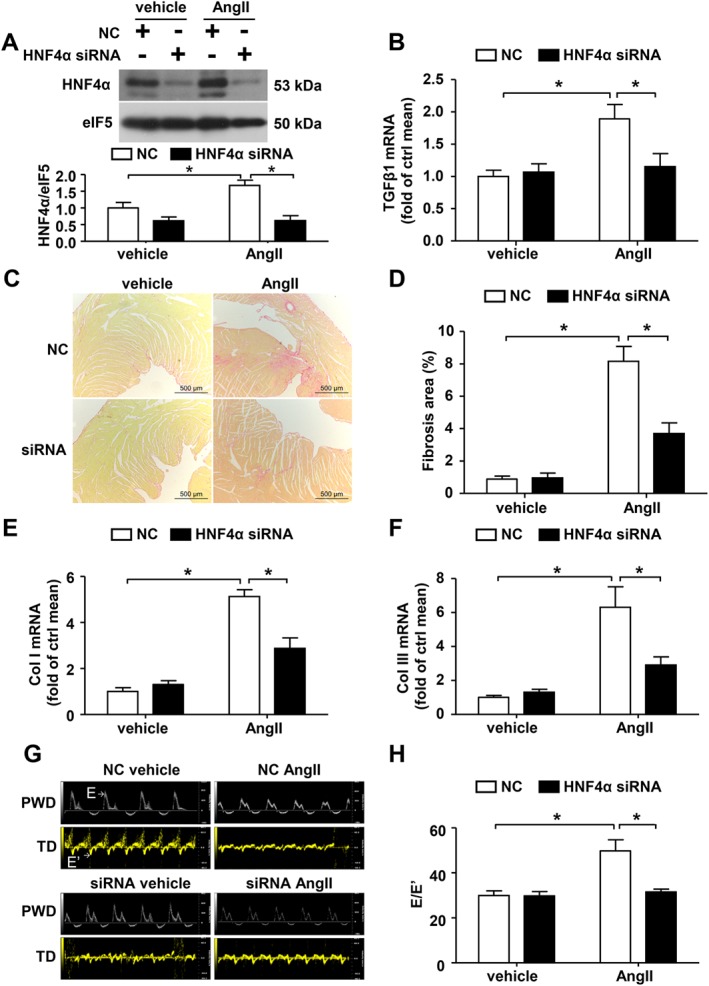
HNF4α mediates AngII‐induced TGFβ1 expression and cardiac fibrosis in vivo. The mice were treated with negative control (NC) or HNF4α siRNA followed by AngII infusion. (A) Western blot analysis of HNF4α expression in the hearts. (B) Quantitative real‐time PCR analysis of TGFβ1 mRNA expression in heart lysates. (C) Representative micrographs of Sirius red‐stained heart sections; the red area represents collagen. Bars = 500 μm. (D) Quantification of the fibrotic area is expressed as a percentage of the total cardiac area. Collagen I (E) and collagen III (F) mRNA expression were measured via real‐time PCR analysis. (G) Representative pulsed wave Doppler (PWD) images across the mitral flow and tissue Doppler (TD) images of the mitral valve ring on the seventh day of AngII infusion in wild type mice. (H) E/E’: Bars represent means ± SEM of six mice per group. *P < 0.05. Two‐way ANOVA with the Bonferroni post hoc test was used (A, B and E). Welch's ANOVA with post hoc Games–Howell test was used in (D, F and H).
Metformin suppresses AngII‐induced HNF4α and TGFβ1 expression both in vitro and in vivo
The effects of metformin on HNF4α were further investigated in CFs; metformin (1 mM) reduced the increase in HNF4α protein expression induced by AngII (Figure 4A). Metformin decreased luciferase activity induced by AngII in cells transfected with the reporter plasmid carrying the wild type Tgfb1 promoter (Figure 4B). A ChIP assay demonstrated that metformin blocked the increase in the binding of HNF4α to the Tgfb1 promoter region upon AngII exposure (Figure 4C).
Figure 4.
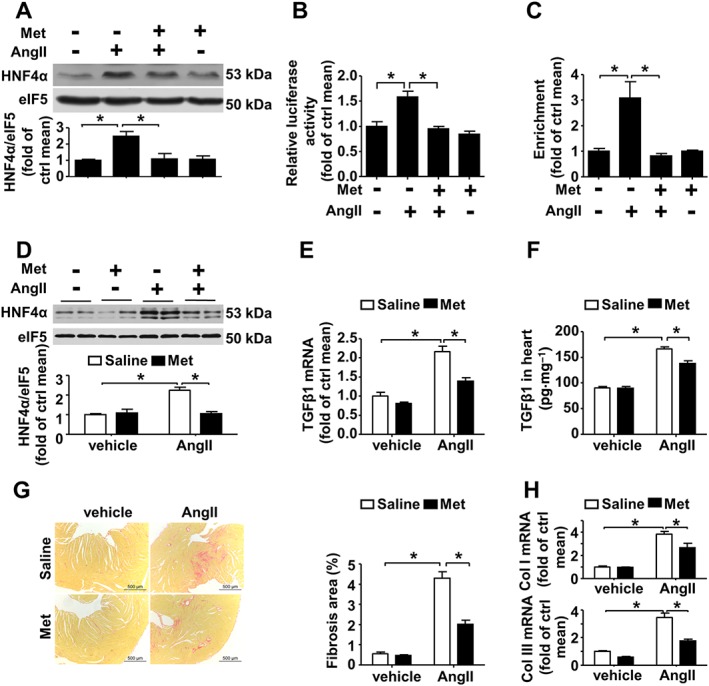
Metformin inhibits AngII‐induced HNF4α and TGFβ1 expression. (A) Western blot analysis of HNF4α expression in CFs. Met: metformin, n = 6. (B) MEFs were transfected with the reporter plasmid carrying the full Tgfb1 promoter region and then treated with AngII or metformin. A dual luciferase reporter assay was performed, n = 6. (C) ChIP analysis using antibodies against HNF4α or IgG, soluble chromatin (~500 bp in length) from CFs treated with AngII and/or metformin, and primers targeting the region spanning the HNF4α binding sites in the Tgfb1 promoter, n = 5. (D) Western blot analysis of HNF4α expression in wild type mice treated with AngII and/or metformin. (E) Quantitative real‐time PCR analysis of TGFβ1 mRNA expression in heart tissue lysates. (F) The TGFβ1 protein level in the heart as determined via elisa. (G) Left panel: representative micrographs of Sirius red‐stained heart sections; the red areas represent collagen. Bars = 500 μm. Right panel: the fibrotic area was quantified as the percentage of the total cardiac area. (H) Collagen I (upper panel) and collagen III (lower panel) mRNA expression was measured via real‐time PCR analysis. Data are presented as means ± SEM of eight mice per group. *P < 0.05. Welch's ANOVA with post hoc Games–Howell test was used (C, G and H). Two‐way ANOVA with the Bonferroni post hoc test was used for the other panels.
In vivo, metformin inhibited HNF4α protein expression induced by AngII (Figure 4D) and subsequently inhibited TGFβ1 mRNA and protein up‐regulation induced by AngII (Figure 4E–F). As a consequence, collagen deposition and increased collagen I and III mRNA expression induced by AngII infusion were alleviated by metformin (Figure 4G–H). Echocardiography demonstrated that E/E’ was significantly lower in the metformin treatment group than in the non‐metformin‐treated group (Supporting Information Fig. S3a). This decrease in E/E’ suggest heart function is improved in metformin‐treated mice upon AngII exposure. EF% and FS% were comparable between all groups (Supporting Information Fig. S3b–d), suggesting systolic function was preserved upon AngII exposure and metformin had no effect on systolic function in the present study. These results indicate that metformin suppresses the AngII‐induced increase in HNF4α expression and subsequently decreases cardiac TGFβ1 expression and cardiac fibrosis.
Neither blood pressure nor heart rate was affected by metformin treatment (Supporting Information Fig. S4). This result indicates that the anti‐fibrosis effects of metformin are not attributable to a reduction in haemodynamic overload induced by AngII. The levels of fasting blood glucose in mice were comparable between all groups (Supporting Information Fig. S5), and this finding suggests that the antihyperglycaemic effect of metformin is not related to its activities in this model.
Metformin inhibits AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis in an AMPK‐dependent manner
Furthermore, we validated the involvement of AMPK in the inhibitory effects of metformin on AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis. Metformin did not reduce the increase in HNF4α protein expression induced by AngII in CFs treated with the AMPK inhibitor Compound C (Figure 5A). Similarly, AICAR, another well‐known AMPK activator, also inhibited AngII‐induced HNF4α protein expression in CFs (Figure 5B). As AMPKα2 is the major AMPKα subunit in the heart (Kim and Tian, 2011), AMPKα2−/− mice (Supporting Information Fig. S6) were used to elucidate the role of AMPKs in the inhibitory effects of metformin on TGFβ1 production and cardiac fibrosis. Although AMPKα1 was detectable, the AMPK activity, as indicated by phosphor‐AMPK(Thr172), was indeed decreased in AMPKα2−/− mice(Supporting Information Fig. S6). This AMPKα2 deficiency amplified AngII‐induced HNF4α protein expression and TGFβ1 production (Supporting Information Fig. S7a–c). As a consequence, AngII‐induced cardiac fibrosis was exacerbated (Supporting Information Fig. S7d), and collagen I and collagen III expression were further up‐regulated (Supporting Information Fig. S7e). Diastolic dysfunction was significantly decreased in AMPKα2−/− mice compared with wild type mice (Supporting Information Fig. S7f).
Figure 5.
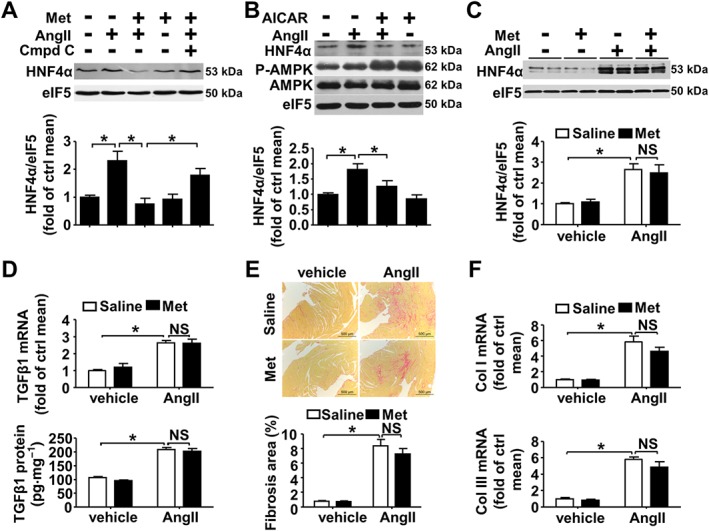
Metformin inhibits AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis in an AMPK‐dependent manner. (A–B) Western blot analysis of HNF4α expression in CFs. (A) CFs were pretreated with Compound C (1 μM) for 0.5 h and then treated with metformin (1 mM) for 2 h. Afterwards, AngII (1 μM) was added for 24 h before harvesting. n = 5. Met: metformin; Cmpd C: Compound C. (B) CFs were pretreated with AICAR (0.1 mM) for 1 h and then treated with AngII (1 μM) for 24 h before harvesting, n = 6. (C) Western blot analysis of HNF4α expression in AMPKα2−/− mice treated with AngII and/or metformin. (D) Upper panel: quantitative real‐time PCR analysis of TGFβ1 mRNA expression. Lower panel: the TGFβ1 protein level was determined via elisa. (E) Upper panel: representative micrographs of Sirius red‐stained heart sections; the red areas represent collagen. Bars = 500 μm. Lower panel: the fibrotic area was quantified as the percentage of the total cardiac area. (F) Collagen I (upper panel) and collagen III (lower panel) mRNA expression was measured via real‐time PCR analysis. Bars represent means ± SEM of eight mice per group. *P < 0.05, NS = not significant. One‐way (A) or two‐way ANOVA with the Bonferroni post hoc test was used (B and D). Welch's ANOVA with post hoc Games–Howell test was used in (C, E and F).
In vivo, metformin had no effect on HNF4α expression in AMPKα2−/− mice (Figure 5C). Consistently, in AMPKα2−/− mice, metformin did not inhibit AngII‐induced TGFβ1 expression at the mRNA or protein level (Figure 5D). Moreover, the area of cardiac fibrosis (Figure 5E) and the mRNA expression levels of collagen I and III (Figure 5F) induced by AngII were not reduced by metformin in AMPKα2−/− mice. Echocardiography demonstrated that E/E’ was not reduced by metformin upon AngII infusion in AMPKα2−/− mice (Supporting Information Fig. S8a). EF% and FS% were also comparable between all groups (Supporting Information Fig. S8b–d). These results suggest that metformin inhibits AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis in an AMPK‐dependent manner.
In summary, HNF4α mediates AngII‐induced TGFβ1 transcript expression and cardiac fibrosis. Metformin targets HNF4α‐TGFβ1 pathway via an AMPK‐dependent pathway (Figure 6).
Figure 6.
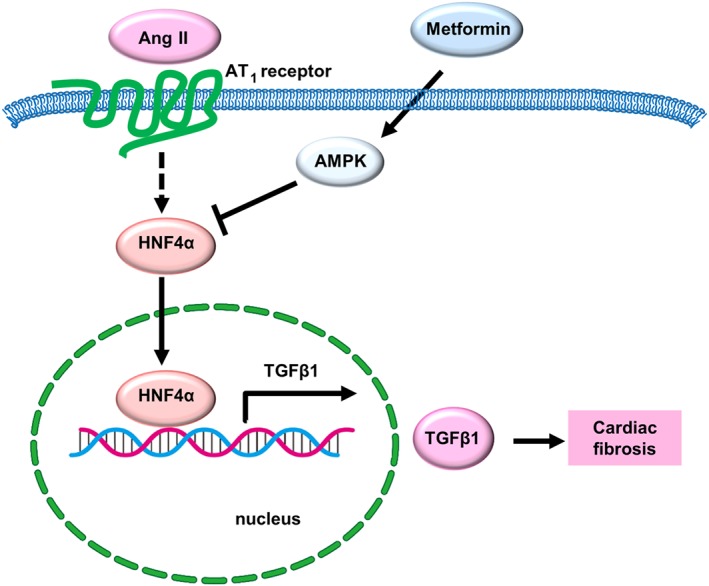
Working model showing how metformin inhibits TGFβ1 expression and cardiac fibrosis. AngII induces TGFβ1 expression by increasing HNF4α protein expression and binding activity. Metformin targets the HNF4α protein and then inhibits TGFβ1 expression via a process dependent on AMPK activation.
Discussion
In the present study, we identified HNF4α as a novel transcription factor targeting TGFβ1 expression and as a novel mediator of AngII‐induced cardiac fibrosis. We demonstrated that metformin inhibits TGFβ1 production by targeting HNF4α in an AMPK‐dependent manner. A working model demonstrating HNF4α as the transcription factor enhancing TGFβ1 expression upon AngII infusion and as a target of metformin to block cardiac fibrosis is illustrated in Figure 6.
The AngII/TGFβ1 network plays a pivotal role in the development of cardiac fibrosis (Rosenkranz, 2004). AngII primarily up‐regulates TGFβ1 expression at the transcriptional level (Wenzel et al., 2001). Previous studies have shown that AngII increases TGFβ1 transcription via the NADPH oxidase‐PKC‐p38‐AP1 pathway and the sterol regulatory element‐binding protein‐1 (SREBP‐1) pathway (Wenzel et al., 2001; Wang et al., 2015). Recently, microRNA has been shown to participate in AngII‐induced TGFβ1 expression. MiR‐29b, which is down‐regulated by AngII, inhibits TGFβ1 expression by directly targeting the TGFβ1 coding region (Zhang et al., 2014). Therefore, AngII can indirectly promote TGFβ1 expression via miR‐29b inhibition. Here, we identified HNF4α as a novel transcriptional factor mediating AngII‐induced TGFβ1 expression and subsequent cardiac fibrosis (Figures 2 and 3).
HNF4α, a member of the nuclear receptor superfamily, plays a key role in liver development and regulates the expression of multiple genes associated with lipid and glucose metabolism (Gonzalez, 2008). In humans, mutation of HNF4α causes maturity onset diabetes of the young (MODY1), which is characterized by deficiency in insulin secretion (Ellard and Colclough, 2006). Most studies of HNF4α have primarily focused on its roles in the liver and in lipid and glucose metabolism. To our knowledge, the present study is the first to identify a role for HNF4α in the heart. Consistent with our results, HNF4α has been shown to potentiate signalling pathway activity downstream of TGFβ1 and to activate Smad2/3 target gene transcription (Mizutani et al., 2011). In contrast to the function of HNF4α in cardiac fibrosis observed in this study, HNF4α has been shown to attenuate hepatic fibrosis in rats by suppressing the epithelial‐mesenchymal transition (Yue et al., 2010). There are two possible reasons for this disparity. Firstly, the inducer of HNF4α is different between models. In Yue's study, dimethylnitrosamine and bile duct ligation were used to induce liver fibrosis. Here, we infused mice with AngII to induce cardiac fibrosis but not liver fibrosis. Secondly, the effects of HNF4α may differ between the cardiovascular system and the hepatic system. Indeed, the function of HNF4α varies among different organs and different physiological conditions (Babeu and Boudreau, 2014). The HNF4α gene contains two distinct promoters (P1 and P2) that drive the expression of 12 known isoforms. The class of P2 isoforms is shorter than the class of P1 isoforms and lacks the cofactor‐interacting domain (Babeu and Boudreau, 2014). These structural differences cause the P1 and P2 isoforms to play distinct roles by interacting with different cofactors and responding to different regulatory signals (Babeu and Boudreau, 2014). The P1 and P2 HNF4α isoforms are differentially expressed between different organs and, therefore, play distinct roles in those organs (Dean et al., 2010). Indeed, our results suggest that P2‐derived HNF4α isoforms are more abundant in the heart but that P1‐derived HNF4α isoforms are more abundant in the liver (Supporting Information Fig. S2b). Although the down‐regulation of HNF4α by siRNA exacerbated hepatic fibrosis in Yue's study, in this study we found that HNF4α siRNA attenuated cardiac fibrosis induced by AngII (Figure 3). These results suggest that HNF4α is antifibrogenic in the liver but profibrogenic in the heart.
Although HNF4α function varies between different organs, metformin is apparently an ideal therapeutic agent to target HNF4α and thus attenuate cardiac fibrosis, as suggested by our results. Multiple findings have suggested that metformin exerts a cardioprotective effect independent of its glucose‐lowering effects (Bromage and Yellon, 2015). The mechanisms underlying its cardioprotective effect include decreased ROS production via inhibition of the mitochondrial respiratory chain, increased NO production due to AMPK activation and inhibition of the TGFβ/Smad pathway (Ladeiras‐Lopes et al., 2015). The effects of metformin are mediated by not only AMPK‐dependent pathways but also AMPK‐independent pathways (El Messaoudi et al., 2013; Ladeiras‐Lopes et al., 2015). Our previous study suggested that metformin inhibits collagen synthesis in CFs by blocking the Smad3 pathway downstream of TGFβ1, independently of AMPK activation (Xiao et al., 2010). Furthermore, our recent study suggested that metformin directly binds with TGFβ1 to target its downstream signalling pathway independently of AMPK activation (Xiao et al., 2016). Here, we further found that metformin inhibited TGFβ1 production by targeting HNF4α in an AMPK activation‐dependent manner. Therefore, metformin can inhibit the TGFβ1 pathway via both AMPK‐dependent and AMPK‐independent processes. Specifically, metformin‐induced inhibition of TGFβ1 production depended on AMPK activation, but its inhibition of the Smad3 signalling pathway downstream of TGFβ1 was independent of AMPK activation. Indeed, here, we also found that metformin had a tendency to decrease cardiac fibrosis in AMPKα2−/− mice, which can be attributed to the AMPK‐independent inhibitory effects of metformin on TGFβ1 downstream of the Smad3 signalling pathway.
In summary, we demonstrated that HNF4α mediates AngII‐induced TGFβ1 transcriptional expression and cardiac fibrosis. Metformin inhibited HNF4α to decrease TGFβ1 production and subsequently attenuate cardiac fibrosis via AMPK activation. These findings reveal a novel antifibrotic mechanism of action of metformin and identify HNF4α as a new potential therapeutic target for cardiac fibrosis.
Author contributions
R.F.C. participated in manuscript preparation, designed the experiments and performed most of the experiments and data analysis. H.X. conceived the project, designed the experiments, performed data analysis and wrote the manuscript. Y.Y.Z. managed funding, participated in study design and manuscript preparation. Y.N.F., J.M.W., Y.S., H.L., Q.S., D.L., J.S.Z. and Z.Z.L. collected data. J.M.W. participated in manuscript preparation.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primer sequences used in PCR analysis.
Figure S1 AngII increased the HNF4α protein levels via AT1 receptor in cardiac fibroblasts. (A) Western blot analysis of HNF4α expression in hearts treated with saline or AngII for 7 days. n = 8. (B) CFs were treated with AngII at a dose of 0.01 μM, 0.1 μM or 1 μM for 24 h, and HNF4α protein expression was then determined via western blot analysis, n = 5. (C) Western blot analysis of HNF4α expression in CFs treated with AngII and/or AT1 receptor antagonist losartan (1 μM), n = 5. *P < 0.05, Student's unpaired two‐tailed t‐test was used (A), One‐way ANOVA with the Bonferroni post hoc test was used (B), and Two‐way ANOVA with the Bonferroni post hoc test was used (C).
Figure S2 HNF4α is differentially expressed in the heart and other organs. (A) Western blot analysis of HNF4α in different organs. n = 6. Kruskal–Wallis ANOVA with post hoc Dunn's multiple comparison test was used. (B) Different expression of HNF4α variants derived from P1 or P2 promoters in heart and liver via RT‐PCR.
Figure S3 Metformin improved cardiac diastolic function upon AngII exposure in wild type mice. (A) Left panel: Representative PWD images showing the mitral flow and TD images of the mitral valve ring on the 7th day of AngII infusion in wild type mice. Right panel: E/E’. (B) Representative echocardiograms on the 7th day of AngII or saline infusion in wild type mice. (C) Left ventricular ejection fraction (EF%) on the 7th day of AngII infusion in wild type mice. (D) Left ventricular fractional shortening (FS%) on the 7th day of AngII infusion in wild type mice. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05, NS = not significant. Two‐way ANOVA with the Bonferroni post hoc test (A) or two‐way ANOVA (C and D) was used.
Figure S4 Metformin does not change blood pressure or heart rate in either wild type or AMPKα2−/− mice. Systolic blood pressure (SBP) of (A) wild type mice and (B) AMPKα2−/− mice after 7 days of AngII infusion. (C) Diastolic blood pressure (DBP) of wild type mice and (D) AMPKα2−/− mice after 7 days of AngII infusion. (E) Heart rate of wild type mice after 7 days of AngII infusion. (F) Heart rate of AMPKα2−/− mice after 7 days of AngII infusion. n = 8. Data are expressed as means ± SEM. *P < 0.05, NS = not significant. Two‐way ANOVA with the Bonferroni post hoc test was used (C and D). Welch's ANOVA with post hoc Games‐Howell test was used for the other panels.
Figure S5 Metformin has no effect on fasting blood glucose levels in wild type or AMPKα2−/−mice. (A) Fasting blood glucose levels of wild type mice after 7 days of AngII infusion. (B) Fasting blood glucose levels of AMPKα2−/−mice after 7 days of AngII infusion. n = 7. Data are expressed as means ± SEM. NS = not significant. Two‐way ANOVA was used.
Figure S6 Myocardial AMPK activity was decreased in AMPKα2−/− mice. (A) Western blot analysis of p‐AMPK, AMPK, p‐ACC, ACC and AMPKα2 in wild type and AMPKα2−/− mice after 7 days of AngII infusion. (B) Quantification of the p‐AMPK levels shown in (A). (C) Quantification of the p‐ACC levels shown in (A), n = 8. Data are expressed as means ± SEM. *P < 0.05. Welch's ANOVA with post hoc Games‐Howell test was used.
Figure S7 AMPKα2 knockout exacerbates AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis. (A) Left panel: western blot analysis of HNF4α expression in the heart. The right panel shows quantification of the HNF4α protein levels. (B) Quantitative real‐time PCR analysis of TGFβ1 mRNA expression in heart lysates. (C) The TGFβ1 protein level was determined via elisa. (D) Left panel: representative micrographs of Sirius red‐stained heart sections; the red area represents collagen. Bars =500 μm. Right panel:Quantification of the fibrotic area is expressed as the percentage of the total cardiac area. (E) Collagen I (left panel) and collagen III (right panel) mRNA expression was measured via real‐time PCR analysis. (F) Left panel: representative pulsed wave Doppler (PWD) images across the mitral flow and tissue Doppler (TD) images of the mitral valve ring on the 7th day of AngII infusion in wild type mice. Right panel: E/E’. The E wave and E’ wave are indicated by arrows. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05. Two‐way ANOVA with the Bonferroni post hoc test was used (B and C). Welch's ANOVA with post hoc Games‐Howell test was used for the other panels.
Figure S8 Metformin did not improve cardiac diastolic function upon AngII exposure in AMPKα2−/− mice. (A) Left panel: Representative PWD images showing the mitral flow and TD images of the mitral valve ring on the 7th day of AngII infusion in AMPKα2−/− mice. Right panel: E/E’. (B) Representative echocardiograms on the 7th day of AngII or saline infusion in AMPKα2−/− mice. (C) EF% on the 7th day of AngII infusion in AMPKα2−/− mice. (D) Left ventricular shortening fraction (FS%) on the 7th day of AngII infusion in AMPKα2−/−mice. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05, NS = not significant. Welch's ANOVA with post hoc Games‐Howell test (A) or two‐way ANOVA (C and D) was used.
Acknowledgements
This work was supported by the Natural Science Foundation of China (NSFC, 81530009 to Y.Y.Z.; 81300067 to H.X.); and the National Key Basic Research Programme of the People's Republic of China (2012CB518000 to Y.Y.Z.).
We would like to thank Professor Benoit Viollet (Institute National de la Santé et de la Recherche Médicale U567, Paris) for the generous gift of AMPKα2‐knockout mice.
Chen, R. , Feng, Y. , Wu, J. , Song, Y. , Li, H. , Shen, Q. , Li, D. , Zhang, J. , Lu, Z. , Xiao, H. , and Zhang, Y. (2018) Metformin attenuates angiotensin II‐induced TGFβ1 expression by targeting hepatocyte nuclear factor‐4‐α. British Journal of Pharmacology, 175: 1217–1229. doi: 10.1111/bph.13753.
Contributor Information
Han Xiao, Email: xiaohan@bjmu.edu.cn.
Youyi Zhang, Email: zhangyy@bjmu.edu.cn.
References
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babeu JP, Boudreau F (2014). Hepatocyte nuclear factor 4‐alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol 20: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromage DI, Yellon DM (2015). The pleiotropic effects of metformin: time for prospective studies. Cardiovasc Diabetol 14: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers EE, Pinto YM (2011). Molecular mechanisms that control interstitial fibrosis in the pressure‐overloaded heart. Cardiovasc Res 89: 265–272. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S, Tang JI, Seckl JR, Nyirenda MJ (2010). Developmental and tissue‐specific regulation of hepatocyte nuclear factor 4‐alpha (HNF4‐alpha) isoforms in rodents. Gene Expr 14: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du JH, Xu N, Song Y, Xu M, Lu ZZ, Han C et al. (2005). AICAR stimulates IL‐6 production via p38 MAPK in cardiac fibroblasts in adult mice: a possible role for AMPK. Biochem Biophys Res Commun 337: 1139–1144. [DOI] [PubMed] [Google Scholar]
- Eghbali M (1989). Cellular origin and distribution of transforming growth factor‐beta in the normal rat myocardium. Cell Tissue Res 256: 553–558. [DOI] [PubMed] [Google Scholar]
- El Messaoudi S, Rongen GA, Riksen NP (2013). Metformin therapy in diabetes: the role of cardioprotection. Curr Atheroscler Rep 15: 314. [DOI] [PubMed] [Google Scholar]
- Ellard S, Colclough K (2006). Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity‐onset diabetes of the young. Hum Mutat 27: 854–869. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ (2008). Regulation of hepatocyte nuclear factor 4 alpha‐mediated transcription. Drug Metab Pharmacokinet 23: 2–7. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T (2003). AMP‐activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem 278: 27495–27501. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Tian R (2011). Targeting AMPK for cardiac protection: opportunities and challenges. J Mol Cell Cardiol 51: 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladeiras‐Lopes R, Fontes‐Carvalho R, Bettencourt N, Sampaio F, Gama V, Leite‐Moreira A (2015). Novel therapeutic targets of metformin: metabolic syndrome and cardiovascular disease. Expert Opin Ther Targets 19: 869–877. [DOI] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B (2001). Hepatocyte nuclear factor‐4alpha involved in type 1 maturity‐onset diabetes of the young is a novel target of AMP‐activated protein kinase. Diabetes 50: 1515–1521. [DOI] [PubMed] [Google Scholar]
- Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y et al. (2013). In vivo suppression of microRNA‐24 prevents the transition toward decompensated hypertrophy in aortic‐constricted mice. Circ Res 112: 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani A, Koinuma D, Tsutsumi S, Kamimura N, Morikawa M, Suzuki HI et al. (2011). Cell type‐specific target selection by combinatorial binding of Smad2/3 proteins and hepatocyte nuclear factor 4alpha in HepG2 cells. J Biol Chem 286: 29848–29860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz S (2004). TGF‐beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res 63: 423–432. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M et al. (2004). Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432: 173–178. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group (1998). Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet (London, England) 352: 854–865. [PubMed] [Google Scholar]
- Wang TN, Chen X, Li R, Gao B, Mohammed‐Ali Z, Lu C et al. (2015). SREBP‐1 mediates angiotensin II‐induced TGF‐beta1 upregulation and glomerular fibrosis. J Am Soc Nephrol 26: 1839–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel S, Taimor G, Piper HM, Schluter KD (2001). Redox‐sensitive intermediates mediate angiotensin II‐induced p38 MAP kinase activation, AP‐1 binding activity, and TGF‐beta expression in adult ventricular cardiomyocytes. FASEB J 15: 2291–2293. [DOI] [PubMed] [Google Scholar]
- Wingender E, Karas H, Knuppel R (1997). TRANSFAC database as a bridge between sequence data libraries and biological function. Pac Symp Biocomput : 477–485. [PubMed] [Google Scholar]
- Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M et al. (2010). Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1‐Smad3 signalling pathway. Cardiovasc Res 87: 504–513. [DOI] [PubMed] [Google Scholar]
- Xiao H, Zhang J, Xu Z, Feng Y, Zhang M, Liu J et al. (2016). Metformin is a novel suppressor for transforming growth factor (TGF)‐beta1. Sci Rep 6: 28597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Zhang TP, Tian C, Jia LX, Du J, Li HH (2012). Carboxyl terminus of heat shock protein 70‐interacting protein inhibits angiotensin II‐induced cardiac remodeling. Am J Hypertens 25: 994–1001. [DOI] [PubMed] [Google Scholar]
- Yin M, van der Horst IC, van Melle JP, Qian C, van Gilst WH, Sillje HH et al. (2011). Metformin improves cardiac function in a nondiabetic rat model of post‐MI heart failure. Am J Physiol Heart Circ Physiol 301: H459–H468. [DOI] [PubMed] [Google Scholar]
- Yuan H, Lanting L, Xu ZG, Li SL, Swiderski P, Putta S et al. (2008). Effects of cholesterol‐tagged small interfering RNAs targeting 12/15‐lipoxygenase on parameters of diabetic nephropathy in a mouse model of type 1 diabetes. Am J Physiol Renal Physiol 295: F605–F617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W et al. (2010). Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 59: 236–246. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY (2014). miR‐29b as a therapeutic agent for angiotensin II‐induced cardiac fibrosis by targeting TGF‐beta/Smad3 signaling. Mol Ther 22: 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences used in PCR analysis.
Figure S1 AngII increased the HNF4α protein levels via AT1 receptor in cardiac fibroblasts. (A) Western blot analysis of HNF4α expression in hearts treated with saline or AngII for 7 days. n = 8. (B) CFs were treated with AngII at a dose of 0.01 μM, 0.1 μM or 1 μM for 24 h, and HNF4α protein expression was then determined via western blot analysis, n = 5. (C) Western blot analysis of HNF4α expression in CFs treated with AngII and/or AT1 receptor antagonist losartan (1 μM), n = 5. *P < 0.05, Student's unpaired two‐tailed t‐test was used (A), One‐way ANOVA with the Bonferroni post hoc test was used (B), and Two‐way ANOVA with the Bonferroni post hoc test was used (C).
Figure S2 HNF4α is differentially expressed in the heart and other organs. (A) Western blot analysis of HNF4α in different organs. n = 6. Kruskal–Wallis ANOVA with post hoc Dunn's multiple comparison test was used. (B) Different expression of HNF4α variants derived from P1 or P2 promoters in heart and liver via RT‐PCR.
Figure S3 Metformin improved cardiac diastolic function upon AngII exposure in wild type mice. (A) Left panel: Representative PWD images showing the mitral flow and TD images of the mitral valve ring on the 7th day of AngII infusion in wild type mice. Right panel: E/E’. (B) Representative echocardiograms on the 7th day of AngII or saline infusion in wild type mice. (C) Left ventricular ejection fraction (EF%) on the 7th day of AngII infusion in wild type mice. (D) Left ventricular fractional shortening (FS%) on the 7th day of AngII infusion in wild type mice. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05, NS = not significant. Two‐way ANOVA with the Bonferroni post hoc test (A) or two‐way ANOVA (C and D) was used.
Figure S4 Metformin does not change blood pressure or heart rate in either wild type or AMPKα2−/− mice. Systolic blood pressure (SBP) of (A) wild type mice and (B) AMPKα2−/− mice after 7 days of AngII infusion. (C) Diastolic blood pressure (DBP) of wild type mice and (D) AMPKα2−/− mice after 7 days of AngII infusion. (E) Heart rate of wild type mice after 7 days of AngII infusion. (F) Heart rate of AMPKα2−/− mice after 7 days of AngII infusion. n = 8. Data are expressed as means ± SEM. *P < 0.05, NS = not significant. Two‐way ANOVA with the Bonferroni post hoc test was used (C and D). Welch's ANOVA with post hoc Games‐Howell test was used for the other panels.
Figure S5 Metformin has no effect on fasting blood glucose levels in wild type or AMPKα2−/−mice. (A) Fasting blood glucose levels of wild type mice after 7 days of AngII infusion. (B) Fasting blood glucose levels of AMPKα2−/−mice after 7 days of AngII infusion. n = 7. Data are expressed as means ± SEM. NS = not significant. Two‐way ANOVA was used.
Figure S6 Myocardial AMPK activity was decreased in AMPKα2−/− mice. (A) Western blot analysis of p‐AMPK, AMPK, p‐ACC, ACC and AMPKα2 in wild type and AMPKα2−/− mice after 7 days of AngII infusion. (B) Quantification of the p‐AMPK levels shown in (A). (C) Quantification of the p‐ACC levels shown in (A), n = 8. Data are expressed as means ± SEM. *P < 0.05. Welch's ANOVA with post hoc Games‐Howell test was used.
Figure S7 AMPKα2 knockout exacerbates AngII‐induced HNF4α expression, TGFβ1 expression and cardiac fibrosis. (A) Left panel: western blot analysis of HNF4α expression in the heart. The right panel shows quantification of the HNF4α protein levels. (B) Quantitative real‐time PCR analysis of TGFβ1 mRNA expression in heart lysates. (C) The TGFβ1 protein level was determined via elisa. (D) Left panel: representative micrographs of Sirius red‐stained heart sections; the red area represents collagen. Bars =500 μm. Right panel:Quantification of the fibrotic area is expressed as the percentage of the total cardiac area. (E) Collagen I (left panel) and collagen III (right panel) mRNA expression was measured via real‐time PCR analysis. (F) Left panel: representative pulsed wave Doppler (PWD) images across the mitral flow and tissue Doppler (TD) images of the mitral valve ring on the 7th day of AngII infusion in wild type mice. Right panel: E/E’. The E wave and E’ wave are indicated by arrows. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05. Two‐way ANOVA with the Bonferroni post hoc test was used (B and C). Welch's ANOVA with post hoc Games‐Howell test was used for the other panels.
Figure S8 Metformin did not improve cardiac diastolic function upon AngII exposure in AMPKα2−/− mice. (A) Left panel: Representative PWD images showing the mitral flow and TD images of the mitral valve ring on the 7th day of AngII infusion in AMPKα2−/− mice. Right panel: E/E’. (B) Representative echocardiograms on the 7th day of AngII or saline infusion in AMPKα2−/− mice. (C) EF% on the 7th day of AngII infusion in AMPKα2−/− mice. (D) Left ventricular shortening fraction (FS%) on the 7th day of AngII infusion in AMPKα2−/−mice. Data are expressed as means ± SEM of 8 mice per group. *P < 0.05, NS = not significant. Welch's ANOVA with post hoc Games‐Howell test (A) or two‐way ANOVA (C and D) was used.