

Abstract
The C‐type lectin domain family 12, member A (CLEC12A) receptor has emerged as a leukaemia‐associated and cancer stem cell marker in myeloid malignancies. However, a detailed delineation of its expression in normal haematopoiesis is lacking. Here, we have characterized the expression pattern of CLEC12A on the earliest stem‐ and myeloid progenitor subsets in normal bone marrow. We demonstrate distinct CLEC12A expression in the classically defined myeloid progenitors, where on average 39.1% (95% CI [32.5;45.7]) of the common myeloid progenitors (CMPs) expressed CLEC12A, while for granulocyte‐macrophage progenitors and megakaryocyte‐erythroid progenitors (MEPs), the average percentages were 81.0% (95% CI [76.0;85.9]) and 11.9% (95% CI [9.3;14.6]), respectively. In line with the reduced CLEC12A expression on MEPs, functional assessment of purified CLEC12A+/− CMPs and MEPs in the colony‐forming unit assay demonstrated CLEC12A+ subsets to favour non‐erythroid colony growth. In conclusion, we provide evidence that the earliest CLEC12A+ cell in the haematopoietic tree is the classically defined CMP. Furthermore, we show that CLEC12A‐expressing CMPs and MEPs are functionally different than their negative counterparts. Importantly, these data can help determine which cells will be spared during CLEC12A‐targeted therapy, and we propose CLEC12A to be included in future studies of myeloid cancer stem cell biology.
Keywords: CLEC12A protein, haematopoiesis, hMICL, immunophenotyping, myeloid progenitor cells, neoplastic stem cells
1. INTRODUCTION
The transmembrane receptor C‐type lectin domain family 12, member A (CLEC12A) is emerging as a marker of blasts and leukaemic stem cells (LSC) in acute myeloid leukaemia (AML) and by inference as a useful tool in both the diagnostic and follow‐up settings.1, 2, 3, 4, 5 In addition, we have recently shown the relevance of this marker in the context of myelodysplastic syndrome (MDS).6 In the setting of malignant haematology, one of the most appealing features of CLEC12A is its lack of expression on CD34+CD38− cells in normal bone marrow (BM), regenerating BM and G‐CSF‐stimulated peripheral blood stem cells,2 making it a potential treatment target. Recent ventures in this regard include the development of anti‐CD3/anti‐CLEC12A bispecific antibodies.7, 8, 9 The upcoming roles of CLEC12A as a cancer stem cell marker and potential treatment target emphasize the need of knowing the details of its expression pattern on early stem and progenitor cells in healthy individuals.
Although the classical roadmap of haematopoiesis is constantly being revised,10, 11, 12 it is nevertheless providing a powerful tool for understanding the production of mature circulating blood cells and continuously serves as a solid model for studying clonal evolution in the myeloid spectrum of haematological malignancies.13, 14, 15, 16 In this study, we opted to employ the immunophenotypic definition of the haematopoietic stem cell (HSC) as being Lin‐CD34+CD38−CD90+CD45RA−,17, 18, 19 the multipotent progenitor (MPP) as being Lin‐CD34+CD38−CD90−CD45RA− 20 and the human multilymphoid progenitor (MLP, also termed lymphoid‐primed multipotent progenitor) as Lin‐CD34+CD38−CD90−/lowCD45RA+.21 With respect to the Lin‐CD34+CD38+ myeloid progenitors, we distinguished the common myeloid progenitor (CMP), the granulocyte‐macrophage progenitor (GMP) and the megakaryocyte‐erythroid progenitor (MEP) subsets based on their CD123 and CD45RA expression as first described by Manz and colleagues.22
Using these definitions, we have delineated the expression of CLEC12A on normal BM stem‐ and myeloid progenitor cells and evaluated the influence of this marker on the growth of purified CMPs, GMPs and MEPs in the colony‐forming cell assay. Importantly, our results provide evidence that the earliest CLEC12A+ cell in normal haematopoiesis is the classically defined CMP.
2. METHODS
2.1. Bone marrow samples
Bone marrow samples (n = 13) from healthy volunteers were obtained in the setting of BM harvest for allogeneic BM transplantation or donated as part of the anonymous biobank for normal donors in Hemodiagnostic Laboratory, Department of Hematology, Aarhus University Hospital. The biobank is approved by the local Ethics Committee of the Central Denmark Region, and sample collection was performed after informed consent was given. Mononuclear cells (MNCs) were obtained by Lymphoprep (Axis‐Shield plc., Dundee, Scotland) separation according to the manufacturer's instructions, cryopreserved in 10% dimethylsulphoxide and stored in liquid nitrogen.
2.2. Flow cytometry
Cryopreserved MNCs were thawed in 37°C water bath and resuspended in RoboSep Buffer (StemCell Technologies, Vancouver, BC, Canada) with 15% heat‐inactivated foetal calf serum (Biochrom, GmbH, Berlin, Germany). Subsequently, cells were stained with the monoclonal antibodies (MoAb) listed in Table 1. Lineage depletion was accomplished by introducing a dump channel with lineage‐MoAbs. Data acquisition was performed on a Navios flow cytometer (Donor 1‐9) (Beckman‐Coulter, Inc., Brea, CA, USA) equipped with three lasers; 405 nm, 488 nm and 638 nm, respectively. Compensation was set using with UltraComp eBeads (eBioscience, San Diego, CA, USA) together with the relevant fluorochrome‐conjugated antibodies. Data were analysed with FlowJo Data Analysis Software, version X (FlowJo, Ashland, OR, USA). For each sample and for each fluorochrome, positive and negative gates were determined by means of fluorescence minus one (FMO) controls.
Table 1.
Applied fluorochrome‐conjugated monoclonal antibodies
| MoAb | Fluorochrome | Clone | #Cat | Company |
|---|---|---|---|---|
| CD45RA | FITC | HI100 | 304106 | BioLegend |
| CLEC12A | PE | HB3 | Own lab | |
| CD38 | ECD | LS198.4.3 | A99022 | BC |
| CD2 | PE‐Cy5 | RPA‐2.10 | 300209 | BioLegend |
| CD3 | PE‐Cy5 | HIT3a | 300309 | BioLegend |
| CD4 | PE‐Cy5 | RPA‐T4 | 300509 | BioLegend |
| CD7 | PE‐Cy5 | CD7‐6B7 | 343110 | BioLegend |
| CD8a | PE‐Cy5 | RPA‐T8 | 301009 | BioLegend |
| CD10 | PE‐Cy5 | HI10a | 312206 | BioLegend |
| CD11b | PE‐Cy5 | ICRF44 | 301307 | BioLegend |
| CD14 | PE‐Cy5 | RMO52 | A07765 | BC |
| CD19 | PE‐Cy5 | HIB19 | 302209 | BioLegend |
| CD20 | PE‐Cy5 | 2H7 | 302307 | BioLegend |
| CD56 | PE‐Cy5 | B159 | 555517 | BD |
| CD235a | PE‐Cy5 | HIR2 | 306605 | BioLegend |
| CD123 | PE‐Cy7 | 6H6 | 306010 | BioLegend |
| CD34 | APC | BIRMA‐K3 | C7238 | DAKO |
| CD90 | BV421 | 5E10 | 328122 | BioLegend |
FITC, fluorescein‐isothiocyanate; PE, phycoerythrin; ECD: PE‐Texas Red PE‐Cy5: phycoerythrin‐cyanine5; PE‐Cy7, phycoerythrin‐cyanine7; APC, allophycocyanin; BV421, brilliant violet 421. BD, BD Biosciences; BC, Beckman‐Coulter.
The gating strategy used for analysing the myeloid progenitor cells is depicted in Figure 1. In short, the lineage negative singlets were defined (not shown) and displayed in a CD34 vs. CD38 plot. Herein, the CD34+CD38+ cell subset was defined (Figure 1A) and the progenitors were further enriched by CD90 negativity (Figure 1B). The CMPs were defined as being Lin‐CD34+CD38+CD90−CD123+CD45RA−, GMPs as Lin‐CD34+CD38+CD90−CD123+CD45RA+ and MEP as Lin‐CD34+CD38+CD90−CD123−CD45RA− (Figure 1C). Next, these three myeloid progenitor subsets were further gated by their CLEC12A expression (Figure 1D). The gating strategy used for analysing CLEC12A expression on HSCs, MPPs and MLPs in the Lin‐CD34+CD38− subset is depicted in Figure S1. The number of analysed events and the corresponding percentages of CLEC12A+ cells are given in Table S1.
Figure 1.
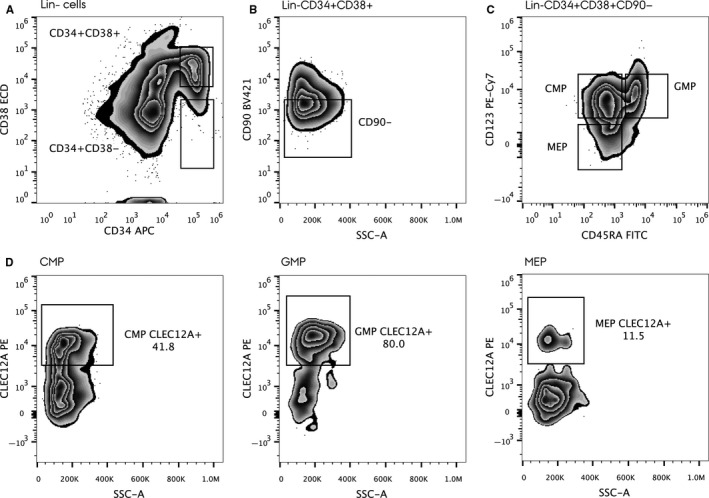
Gating strategy in the myeloid progenitor compartment. Initially, cells were gated in a forward scatter—side scatter plot, and the Lin‐ events were selected (not shown). In the Lin‐ cell population, the CD34+ CD38+ cells were identified (A) and depicted in a CD90 vs. side scatter plot (B). To further enrich for progenitors, the CD90‐ cells were selected and depicted in a CD123 vs. CD45RA plot, whereby the CMP, the GMP and the MEP could be identified (C). These myeloid progenitor subsets were then further gated for their CLEC12A expression (D)
2.3. Fluorescence‐activated cell sorting
Cryopreserved BM MNCs from 6 donors (D1, D2, D10, D11, D12 and D13) were thawed and stained with the MoAbs listed in Table 1. Fluorescence‐activated cell sorting was performed on a BD FACSAria™ III (BD Biosciences) equipped with four lasers; 405 nm, 488 nm, 561 nm and 633 nm, respectively. Using the same gating strategy as described above, the CMPs, GMPs and MEPs were sorted into CLEC12A+/− subsets, respectively. Due to the rarity of the sorted cell populations, post‐sort purity was determined in 21 of 36 cases and reported in Table S2. Flow cytometry data from the sorting experiments (Donor 10‐13) were included in the overall analyses of CLEC12A expression on myeloid progenitors.
2.4. Colony‐forming cell assay
Up to 500 sorted cells from subsets representing CLEC12A+/− CMPs, GMPs and MEPs were seeded in 35‐mm discs in 1 ml of MethoCult H4435 (StemCell Technologies) supplemented with penicillin‐streptomycin (Gibco, Thermo Fischer Scientific Inc.) and allowed to incubate for 14 days at 37°C, in 95% humidity and 5% CO2. Whenever possible, cells were seeded in duplicates. Colony‐forming cells were counted and scored by morphology as burst‐forming unit‐erythroid (BFU‐E), colony‐forming unit‐erythroid (CFU‐E), colony‐forming unit‐granulocyte‐erythroid‐macrophage‐megakaryocyte (CFU‐GEMM), colony‐forming unit‐granulocyte‐macrophage (CFU‐GM), colony‐forming unit‐macrophage (CFU‐M), and colony‐forming unit‐granulocyte (CFU‐G). For the seeded GMPs, a clear distinction between CFU‐GMs, CFU‐Ms and CFU‐Gs was possible and colonies were counted as such. For CMPs and MEPs, the morphology of myeloid colonies was less clear; hence, CFU‐GMs, CFU‐Ms and CFU‐Gs were counted together as myeloid colonies. The cellular composition of each colony type was verified by May‐Grünwald Giemsa staining (Sigma‐Aldrich, St. Louis, MO, USA) of representative colonies cytospun onto slides (n = 1 (donor 1); data not shown).
2.5. Statistical analyses
All calculations were conducted in GraphPad Prism version 6 (GraphPad Software, La Jolla, CA, USA). The D'Agostino & Pearson omnibus test was used to test whether the percentage of CLEC12A+ cells followed a Gaussian distribution. For comparison of the CLEC12A+ cell subsets between CMP, GMP and MEP, Student's unpaired t test was used. The Mann‐Whitney test was applied to test differences in colony formation in CLEC12A+/− progenitor subsets. A two‐sided P‐value < .05 was considered significant.
3. RESULTS
3.1. CLEC12A is differentially expressed on myeloid progenitor subsets
To study at what level in the early human haematopoiesis CLEC12A is expressed, we employed a seven‐colour tube (Table 1) and applied a stringent negative gating strategy based on FMO controls (Figure 1, S1). Consistent with previous observations,2 we did not detect CLEC12A expression on either the HSCs or the MPPs in the Lin‐CD34+CD38− subset (Table S1). For the MLP subset, the number of events was very low, and thus, it cannot formally be ruled out that a fraction of these cells could express CLEC12A (Table S1). The mean percentage of CMPs expressing CLEC12A was 39.1% (95% CI [32.5;45.7]), while for GMPs and MEPs, the CLEC12A+ fractions averaged 81.0% (95% CI [76.0;85.9]) and 11.9% (95% CI [9.3;14.6]), respectively. Hence, the mean percentage of CLEC12A+ CMPs was significantly different from the mean percentage of CLEC12A+ cells in GMPs (P < .0001) and MEPs (P < .0001). Also, the mean CLEC12A+ fractions were significantly different between GMPs and MEPs (P < .0001) (Table S1, Figure 2A). Notably, the CLEC12A+ cells were distributed across the whole CMP and GMP compartments and extended into the MEP compartment (Figure 2B). Taken together, while CLEC12A is a known marker of mature myeloid cells,23, 24 these data reveal that detectable expression of CLEC12A emerges at the level of the CMP, and is abundantly expressed on GMPs and to a lesser extent on the classically defined MEP.
Figure 2.
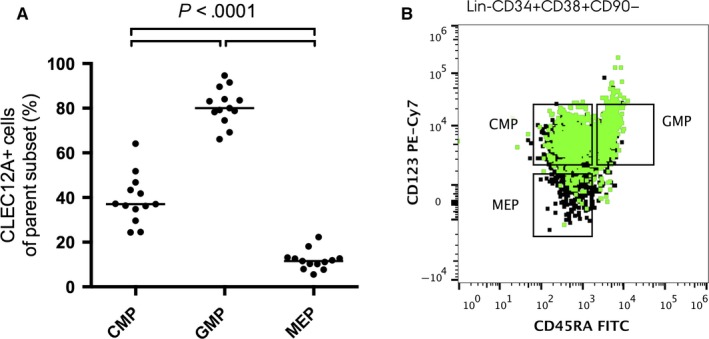
Distribution of CLEC12A+ cells in myeloid progenitors. Percentage of CLEC12A+ cells in CMP, GMP and MEP in BM from 13 healthy donors. Horizontal bars denote the mean. The P‐value applies to all three comparisons (A). Overlay of the CLEC12A+ cell population on the Lin‐CD34+ CD38− CD90− cells displayed in a CD45RA vs. CD123 plot showing the distribution of the CLEC12A+ progenitors (B)
3.2. Different clonogenic potential in CLEC12A positive and negative myeloid progenitors
Next, the clonogenic potential of CLEC12A positive and negative myeloid progenitors was evaluated in the colony‐forming cell assay (Table 2, Figure 3). Being the most immature of the three cell types, the CMPs gave rise to both myeloid, erythroid and CFU‐GEMM colonies in both the CLEC12A+ and CLEC12A− subsets. The total number of colonies was significantly higher in the seeded CLEC12A− CMPs (P = .02). Moreover, significantly fewer erythroid colonies were observed in the seeded CLEC12A+ CMP subset (P = .002), showing that CLEC12A positivity favours a non‐erythroid lineage. For both CLEC12A+/− GMPs, the clonogenic growth was almost exclusively of myeloid origin and no CFU‐GEMMs were evident (Table 2), which is in accordance with the original definition of this cell subset.22 We found no difference neither in the overall clonogenic potential of the CLEC12A+ and CLEC12A− GMPs (P = .31) nor in the distribution of the different myeloid colonies between the CLEC12A+/− GMPs (CFU‐GM (P = .33); CFU‐M (P = .79); CFU‐G (P = .08)). With respect to MEPs, where the expected read out would be colonies of erythroid origin, we found that the CLEC12A+ MEPs predominantly gave rise to myeloid colonies (on average 79.8%) and the number of erythroid colonies was significantly lower among the seeded CLEC12A+ MEP subset compared to the CLEC12A− MEP subset (P = .002), indicating these particular cells to represent a non‐MEP progenitor. The read out of CLEC12A− MEPs was almost exclusively erythroid (on average 96.8%) as would be expected of true MEPs (Table 2). This indicates that adding CLEC12A could be useful in distinguishing true erythroid progenitors from cells with myelomonocytic potential. In conclusion, the CFU‐assays demonstrated heterogeneity in the CMP and MEP progenitor subsets when adding the CLEC12A marker to the sorting scheme with CLEC12A+ progenitors favouring colonies of the myelomonocytic lineage.
Table 2.
Colony distribution of cultured myeloid progenitor subsets
| CFU type | CLEC12A− subset | CLEC12A+ subset | P‐value | ||
|---|---|---|---|---|---|
| Median number of colonies/100 cells plated (range) | % | Median number of colonies/100 cells plated (range) | % | ||
| CMP | |||||
| Total | 32.9 (24.4‐46.0) | 100 | 25.6 (13.7‐31.2) | 100 | .02 |
| CFU‐GEMM | 1.3 (0.6‐2.8) | 4.0 | 0.45 (0‐2.2) | 2.3 | .18 |
| Myeloid | 15.1 (12.8‐29.2) | 48.4 | 16.6 (8.3‐20.2) | 66.5 | .90 |
| Erythroid | 16.5 (10.1‐25.6) | 47.6 | 8.8 (3.0‐9.3) | 31.2 | .002 |
| GMP | |||||
| Total | 15.8 (9.3‐23.7) | 100 | 19.8 (16.9‐23.3) | 100 | .31 |
| CFU‐GEMM | 0.0 (0.0‐0.0) | 0.0 | 0.0 (0.0‐0.0) | 0.0 | ‐ |
| Myeloid | 15.8 (9.3‐23.4) | 99.8 | 19.8 (16.9‐23.1) | 99.9 | .31 |
| Erythroid | 0.0 (0.0‐0.3) | 0.2 | 0.0 (0.0‐0.2) | 0.1 | >.99 |
| MEP | |||||
| Total | 32.5 (16.5‐39.2) | 100 | 24.5 (15.2‐38.9) | 100 | .79 |
| CFU‐GEMM | 0.0 (0.0‐0.2) | 0.1 | 0.0 (0.0‐0.0) | 0 | >.99 |
| Myeloid | 0.7 (0.5‐1.8) | 3.2 | 20.2 (15.2‐29.6) | 79.8 | .002 |
| Erythroid | 31.5 (15.7‐38.6) | 96.8 | 6.5 (0.0‐13.3) | 20.2 | .002 |
CMP, common myeloid progenitor; GMP, granulocyte‐macrophage progenitor; MEP, megakaryocyte‐erythroid progenitor; CFU, colony forming unit.
Figure 3.
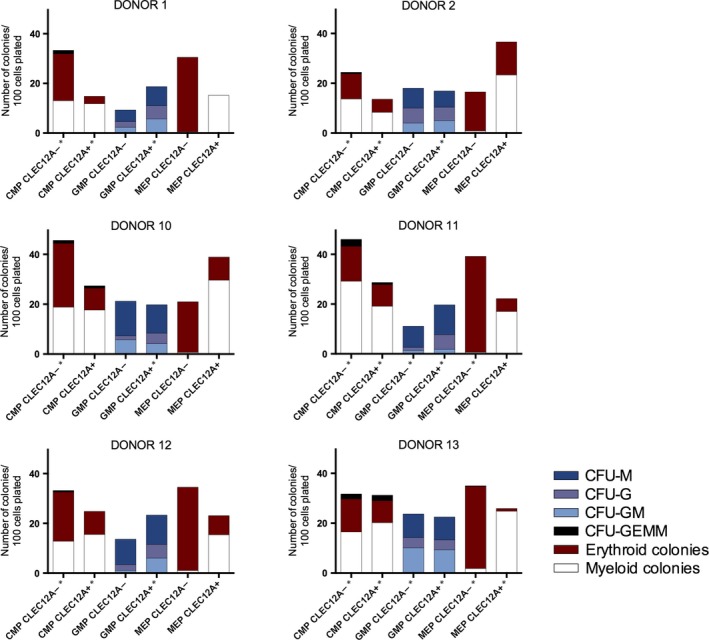
Colony‐forming cell assays on CLEC12A+/− CMPs, GMPs and MEPs from 6 normal donors. Asterisk (*) denotes mean number of colonies from plates seeded in doublets.
4. DISCUSSION
This data set confirms the observation of normal CD34+CD38− cells lacking the CLEC12A marker (Table S1, Figure S1), and by including a detailed delineation of the myeloid CD34+CD38+ progenitors, we have extended the seminal findings by van Rhenen et al.2 We demonstrated the consistently distinct expression of CLEC12A at the level of the classically defined CMPs, GMPs and MEPs with the marker emerging at the level of the CMP, being abundantly expressed on the GMP subset and to a lesser extent on the phenotypically defined MEPs. By investigating the CLEC12A positive and negative progenitors in the colony‐forming cell assay, we furthermore provided evidence of functional differences particularly in the CMP and MEP subsets, where CLEC12A positivity favoured colony growth of myelomonocytic lineage.
Importantly, our findings could add to the deeper understanding of CLEC12A as a blast‐ and cancer stem cell marker in the spectrum of myeloid neoplasms. Acute myeloid leukaemia has to a large extent served as a model for studying disease‐propagating cells,25 and while the first publications indicated the LSC to immunophenotypically resemble the normal CD34+CD38− HSC,26, 27 recent research has indicated most cases of AML to propagate from the progenitor compartment with functional LSC activity in cells resembling the MLP16 and for CD34‐ AML even more mature progenitors/precursors.15, 28 In addition, in low‐risk myelodysplastic syndrome (MDS), the cancer stem cells have been shown to reside in the HSC compartment,13 although studies on high‐risk MDS indicate the MDS‐propagating cell to arise in the MPP or GMP compartment, much resembling AML.29, 30 In this context, the likely usefulness of CLEC12A as a marker of the leukaemic blasts and/or stem cells could be linked to the fact that the level of differentiation arrest occurs in a cell type naturally expressing this marker. By inference—regarding CLEC12A as a marker of myeloid progenitors and their downstream progeny—the presence of CLEC12A on AML blasts and stem cells could indicate a myeloid progenitor/precursor disease, while CLEC12A− AML could correspond to a more immature stem cell disease originating from a cell at the level of the CLEC12A− CMP or upstream. In accordance with this, CLEC12A expression on AML blasts is highly correlated with CD34 negativity,1 which is often associated with a more mature subtype of AML.15 Whether CLEC12A expression on leukaemic blasts has prognostic impact in CD34+ AML remains to be elucidated. Future studies are warranted to explore this in detail and also to examine whether the aberrant expression of CLEC12A on CD34+CD38− cells shown in some cases of AML2, 4 and MDS6 is due to an up‐regulation of CLEC12A in these early stem/progenitor cells or a down‐regulation of CD38 in the leukaemic CLEC12A+ cells.
Our data impact on the notion of CLEC12A as a druggable protein in myeloid malignancies, where the first open‐label, first‐in‐human phase 1 study of a bispecific CLEC12A‐CD3 antibody has been initiated in Europe (EudraCT number 2015‐003704‐23). Thus, while such a treatment will most likely spare the earliest haematopoietic stem and progenitor cells (the Lin‐CD34+CD38− cell subset) and thereby not affect long‐term haematopoiesis, this study provides evidence that approximately 60% of CMPs together with progenitors capable of maintaining the erythroid and—although not addressed in this work—megakaryocyte lineages will also remain unaffected by such approaches. On the other hand, given that more than 80% of GMPs would be expected to be eradicated, it cannot be excluded that a pronounced and long‐lasting depletion of functional neutrophils would follow. While CLEC12A is expressed on mature eosinophilic‐ and basophilic granulocytes,24, 31 our study did not include a functional evaluation of the eosinophil and basophil potential of the CLEC12A+/− myeloid progenitors; hence, a prediction of how CLEC12A directed therapy would affect these cell types cannot be made. In addition, our results cannot formally rule out the possibility that CLEC12A could be expressed at low levels intracytoplasmic in possible myeloid primed early CD34+CD38− haematopoietic stem‐ and progenitor cells. In fact, novel insights into human haematopoiesis question the existence of true oligo‐ and bipotent progenitors.32 Nevertheless, as new targeted treatment strategies directed towards surface proteins are continuously evolving, most recently supplemented with the exciting development of chimeric antigen receptor (CAR)‐engineered T cells (CAR‐T cells) directed against CLEC12A,33, 34 detailed knowledge of the surface expression of this receptor remains important in a translational setting.
Based on our findings showing a highly differentiated expression of CLEC12A on the classically defined myeloid progenitors, we propose CLEC12A could be a helpful marker to add in future detailed studies of the early human haematopoiesis. The technical advances in flow cytometry allowing the addition of more surface markers combined with novel RNA sequencing techniques and functional assays at the single cell level have opened new doors to study haematopoiesis.35 Indeed, Kawamura et al recently applied CLEC12A together with CD64 to study the early monocytic differentiation at the level of GMPs.36 In further support of adding CLEC12A in the studies of human haematopoiesis, Drissen et al studied murine myeloid differentiation pathways performed with RNA sequencing of 63 single pre‐granulocyte‐macrophage cells, and interestingly, CLEC12A turned up as a potential subpopulation classifier being one of the 55 genes showing differential expression by cluster analysis and proved to be mutually exclusive with GATA1.37 In this study, the GATA1+ (and thereby CLEC12A−) cells gave rise to mast cells and eosinophils, while the GATA1− (CLEC12A+) cells gave rise to monocytes and neutrophils—cells that are also known to express CLEC12A in humans.23, 24, 37 Interestingly, by adding the surface markers CD71 and BAH‐1 to the established sorting schemes of myeloid progenitors, a study by Notta and colleagues found the originally defined CMPs to be quite heterogeneous and to consist of unipotent myeloid and erythroid progenitors with very sparse megakaryocyte activity.11 In line with this, our data indicate that adding CLEC12A as a de‐selection marker in the sorting scheme of MEPs could purify the early erythroid subset further.
In conclusion, we here show the differentiated expression of CLEC12A at the level of myeloid progenitors and provide evidence that the earliest CLEC12A+ cell in the haematopoietic tree is the classically defined CMP. While this knowledge is readily translational in the setting of CLEC12A‐targeted treatment, we also suggest that the marker is continuously explored in basic research studying the early myeloid differentiation.
AUTHOR CONTRIBUTIONS
M.B., L.N., P.B.v.K.N., P.W. and P.H. designed the study. M.B., L.L.H. and L.N. performed the experiments. M.B. and L.N. analysed the data. M.B. drafted the manuscript and received input and critical reviews from L.N., P.B.v.K.N., P.H., P.W. and A.S.R.. P.H. provided the financial background for the study. All authors read and approved the final manuscript.
CONFLICT OF INTERESTS
The authors confirm that there is no conflict of interests.
Supporting information
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Professor Gordon D. Brown, MRC Centre for Medical Mycology, University of Aberdeen, for providing the anti‐CLEC12A hybridoma. The Danish Cancer Society and The Meyer Foundation supported the study.
Bill M, van Kooten Niekerk PB, Woll PS, et al. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC12A‐related cancer stem cell biology. J Cell Mol Med. 2018;22:2311–2318. https://doi.org/10.1111/jcmm.13519
Peter Hokland and Line Nederby contributed equally to this work.
REFERENCES
- 1. Larsen HØ, Roug AS, Just T, Brown GD, Hokland P. Expression of the hMICL in acute myeloid leukemia‐a highly reliable disease marker at diagnosis and during follow‐up. Cytometry B Clin Cytom. 2012;82:3‐8. [DOI] [PubMed] [Google Scholar]
- 2. van Rhenen A, van Dongen GAMS, Kelder A, et al. The novel AML stem cell associated antigen CLL‐1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659‐2666. [DOI] [PubMed] [Google Scholar]
- 3. van Rhenen A, Moshaver B, Kelder A, et al. Aberrant marker expression patterns on the CD34 + CD38‐ stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700‐1707. [DOI] [PubMed] [Google Scholar]
- 4. Terwijn M, Zeijlemaker W, Kelder A, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE. 2014;9:e107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roug AS, Larsen HØ, Nederby L, et al. hMICL and CD123 in combination with a CD45/CD34/CD117 backbone ‐ a universal marker combination for the detection of minimal residual disease in acute myeloid leukaemia. Br J Haematol. 2014;164:212‐222. [DOI] [PubMed] [Google Scholar]
- 6. Toft‐Petersen M, Nederby L, Kjeldsen E, et al. Unravelling the relevance of CLEC12A as a cancer stem cell marker in myelodysplastic syndrome. Br J Haematol. 2016;175:393‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao X, Singh S, Pardoux C, et al. Targeting C‐type lectin‐like molecule‐1 for antibody‐mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95:71‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu H, Zhou Q, Deshmukh V, et al. Targeting human C‐type lectin‐like molecule‐1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew Chem Int Ed Engl. 2014;53:9841‐9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leong SR, Sukumaran S, Hristopoulos M, et al. An anti‐CD3/anti‐CLL‐1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129:609‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woolthuis CM, Park CY. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood. 2016;127:1242‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Notta F, Zandi S, Takayama N, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016;351:aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Görgens A, Radtke S, Möllmann M, et al. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 2013;3:1539‐1552. [DOI] [PubMed] [Google Scholar]
- 13. Woll PS, Kjällquist U, Chowdhury O, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25:794‐808. [DOI] [PubMed] [Google Scholar]
- 14. Chesnais V, Arcangeli M‐L, Delette C, et al. Architectural and functional heterogeneity of hematopoietic stem/progenitor cells in non‐del(5q) myelodysplastic syndromes. Blood. 2017;129:484‐496. [DOI] [PubMed] [Google Scholar]
- 15. Quek L, Otto GW, Garnett C, et al. Genetically distinct leukemic stem cells in human CD34‐ acute myeloid leukemia are arrested at a hemopoietic precursor‐like stage. J Exp Med. 2016;213:1513‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP‐like and GMP‐like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19:138‐152. [DOI] [PubMed] [Google Scholar]
- 17. Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune‐deficient mice. Proc Natl Acad Sci USA. 1997;94:5320‐5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Conneally E, Cashman J, Petzer A, Eaves C. Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho‐myeloid repopulating activity in nonobese diabetic‐scid/scid mice. Proc Natl Acad Sci USA. 1997;94:9836‐9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lansdorp PM, Sutherland HJ, Eaves CJ. Selective expression of CD45 isoforms on functional subpopulations of CD34 + hemopoietic cells from human bone marrow. J Exp Med. 1990;172:363‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585‐593. [DOI] [PubMed] [Google Scholar]
- 22. Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99:11872‐11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bakker ABH, van den Oudenrijn S, Bakker AQ, et al. C‐type lectin‐like molecule‐1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004;64:8443‐8450. [DOI] [PubMed] [Google Scholar]
- 24. Marshall ASJ, Willment JA, Lin H‐H, Williams DL, Gordon S, Brown GD. Identification and characterization of a novel human myeloid inhibitory C‐type lectin‐like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J Biol Chem. 2004;279:14792‐14802. [DOI] [PubMed] [Google Scholar]
- 25. Wang JCY, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494‐501. [DOI] [PubMed] [Google Scholar]
- 26. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645‐648. [DOI] [PubMed] [Google Scholar]
- 27. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 28. Taussig DC, Miraki‐Moud F, Anjos‐Afonso F, et al. Anti‐CD38 antibody‐mediated clearance of human repopulating cells masks the heterogeneity of leukemia‐initiating cells. Blood. 2008;112:568‐575. [DOI] [PubMed] [Google Scholar]
- 29. Dimitriou M, Woll PS, Mortera‐Blanco T, et al. Perturbed hematopoietic stem and progenitor cell hierarchy in myelodysplastic syndromes patients with monosomy 7 as the sole cytogenetic abnormality. Oncotarget. 2016;7:72685‐72698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rouault‐Pierre K, Smith AE, Mian SA, et al. Myelodysplastic syndrome can propagate from the multipotent progenitor compartment. Haematologica. 2017;102:e7‐e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toft‐Petersen M, Stidsholt Roug A, Plesner T, Ebbesen L, Brown GD, Nederby L. The CLEC12A receptor marks human basophils: Potential implications for minimal residual disease detection in acute myeloid leukemia. Cytometry B Clin Cytom. https://doi.org/10.1002/cyto.b.21540. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 32. Moignard V, Göttgens B. Dissecting stem cell differentiation using single cell expression profiling. Curr Opin Cell Biol. 2016;43:78‐86. [DOI] [PubMed] [Google Scholar]
- 33. Tashiro H, Sauer T, Shum T, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C‐type lectin‐like molecule 1. Mol Ther. 2017;25:2202‐2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laborda E, Mazagova M, Shao S, et al. Development of A chimeric antigen receptor targeting C‐type lectin‐like molecule‐1 for human acute myeloid leukemia. Int J Mol Sci. 2017;18:E2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Etzrodt M, Endele M, Schroeder T. Quantitative single‐cell approaches to stem cell research. Cell Stem Cell. 2014;15:546‐558. [DOI] [PubMed] [Google Scholar]
- 36. Kawamura S, Onai N, Miya F, et al. Identification of a human clonogenic progenitor with strict monocyte differentiation potential: a counterpart of mouse cMoPs. Immunity. 2017;46:835‐848. e4. [DOI] [PubMed] [Google Scholar]
- 37. Drissen R, Buza‐Vidas N, Woll P, et al. Distinct myeloid progenitor‐differentiation pathways identified through single‐cell RNA sequencing. Nat Immunol. 2016;17:666‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials