

Abstract
Acquired immune deficiency syndrome (AIDS), caused by infection with human immunodeficiency virus (HIV), is associated with gastrointestinal disease, systemic immune activation and changes in the gut microbiota. Here, we aim to investigate the gut microbiota patterns of HIV‐infected individuals and HIV‐uninfected individuals in populations from South China. We enrolled 33 patients with HIV (14 participants treated with highly active antiretroviral therapy [HAART] for more than 3 months; the remaining 19 individuals had not received treatment) and 35 healthy controls (HC) for a cross‐sectional comparison of gut microbiota using stool samples. Gut microbial communities were profiled by sequencing the bacterial 16S rRNA genes. Dysbiosis was more common among patients with AIDS compared with healthy individuals. Dysbiosis was characterized by decreased α‐diversity, low mean counts of Bacteroidetes, Faecalibacterium, Prevotella, Bacteroides vulgatus, Dialister and Roseburia inulnivorans, and high mean counts of Proteobacteria, Enterococcus, Streptococcus, Lactobacillus, Lachnociostridium, Ruminococcus gnavus and Streptococcus vestibularis. Increased abundance of Bacilli was observed in homosexual patients. Proteobacteria were higher among heterosexual patients with HIV infections. Tenericutes were higher among patients with history of intravenous drug abuse. Restoration of gut microbiota diversity and a significant increase in abundance of Faecalibacterium, Blautia and Bacteroides were found in patients receiving HAART compared to those who did not receive. HIV infection‐associated dysbiosis is characterized by decreased levels of α‐diversity and Bacteroidetes, increased levels of Proteobacteria and the alterations of gut microbiota correlate with the route of HIV transmission. The imbalanced faecal microbiota of HIV infection is partially restored after therapy.
Keywords: microbiota, dysbiosis, human immunodeficiency virus, acquired immune deficiency syndrome, transmission route, highly active antiretroviral therapy
Introduction
Infection with human immunodeficiency virus (HIV) is a chronic illness characterized by progressive loss of CD4+ T cells and chronic activation of the immune system. An estimated 35.3 million people worldwide are affected with HIV‐1, with more than 2 million new cases since 2012 (UNAIDS 2013). Increasing evidence suggests that the gut microbiome plays a crucial role in HIV transmission and pathogenesis 1, 2, 3. Understanding the interplay between the microbiome and HIV is of great value for developing effective strategies for the prevention and treatment of HIV.
The advancement of sequencing techniques and bioinformatics has allowed researchers to characterize microbial communities in health and disease states including HIV. Direct evidence of alterations in the composition of the gastrointestinal tract microbiome has been reported. Current data indicate increases in commensal bacteria that may be pathogenic (e.g. Pseudomonas) in the faeces of patients with HIV infection and decreases in primarily beneficial commensals such as Lactobacilli and Bifidobacteria 4. These changes suggest that HIV infection increases the risk for intestinal dysbiosis. Mutlu et al. 5 found a less diverse population of commensals in the mucosal microbiome inhabiting the right colon and terminal ileum of patients with HIV compared with control subjects. In addition, levels of bacterial products are increased in the circulation of patients with HIV 6; this can occur even at early stages of HIV infection, before peripheral CD4+ T‐cell depletion reaches levels that can lead to clinically apparent disease and opportunistic infections 4. Prebiotics have been administered to patients with HIV in an effort to improve the composition of the microbiome, resulting in reduced CD4+ T‐cell activation and improved natural killer (NK) cell activity 7.
However, most prior studies on gut microbiota in patients with HIV were performed in Western countries 2, 5, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18. The genetics, ethnic background, environment, dietary habits and lifestyles of such populations differ from those of comparable populations in China 19. Although little is known about alterations of gut microbiota in Chinese patients infected with HIV, Ling et al. researched the issue in Zhejiang province in eastern China. The authors observed that α‐diversity indices did not differ significantly between the healthy controls and patients infected with HIV‐1, while the proportion of Firmicutes/Bacteroidetes increased significantly in HIV‐1‐infected patients 20. Sun et al. 21. conducted research in Shanghai, also located in eastern China. The results showed that the microbiota of individuals infected with HIV had decreased α‐diversity, were enriched for Firmicutes and Proteobacteria at the phylum level and were depleted in families Ruminococcaceae and Lachnospiraceae and class Clostridia. While one study of Chinese populations from seven ethnic groups living in nine provinces found large ethnic and/or geographical differences in gut microbiota among individuals 22. To date, however, a detailed and comprehensive look at the gut microbiome in HIV‐infected individuals from South China has not yet been published. We therefore sought to identify bacterial microbiome patterns in HIV‐infected patients living in South China.
In this study, we examined the gut microbiota of healthy individuals and subjects from South China infected with HIV. We then investigated the relationship between the route of transmission of HIV in composition of the microbiome to identify a universal and specific biomarker for the development of acquired immune deficiency syndrome (AIDS). And we also enrolled HIV‐infected patients treated with highly active antiretroviral therapy (HAART) for ≥3 months to address any microbial differences among untreated HIV‐infected patients, HAART‐treated individuals and healthy controls. This investigation of microbial changes associated with HIV infection has the potential to aid in the prevention and development of therapeutic interventions that could improve many of the pathologic consequences of chronic HIV infection.
Results
Faecal bacterial diversity in patients with HIV infection
Alpha diversity comparison evaluates diversity, particularly taxa richness and evenness, within a habitat (or samples), while β‐diversity compares the similarity or difference in communities between habitats (or samples). We used four α‐diversity indices to compare the richness estimators (PD_whole tree and observed species) and diversity index (Simpson, Shannon index). The values presented represent the means with standard error for each group (Fig. 1). Our results showed that the α‐diversity of faecal microbiota was markedly reduced compared to healthy controls. To reveal the effect of HIV infection on analysis of microbiomes, we used a ß‐diversity comparison (PCA and PCoA), which reveals the similarity of microbiome community structures (Fig. 2). The results of PCA and PCoA showed that the microbiota of patients with HIV infection differed substantially from those of healthy individuals.
Figure 1.
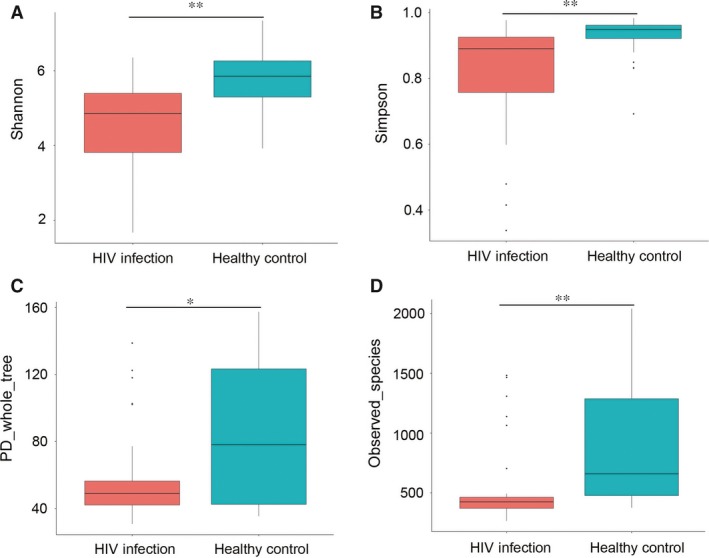
Αlpha diversity is decreased in HIV‐infected individuals. (A) Shannon index, (B) Simpson, (C) PD_whole tree, (D) observed species. *P < 0.05; **P < 0.01.
Figure 2.
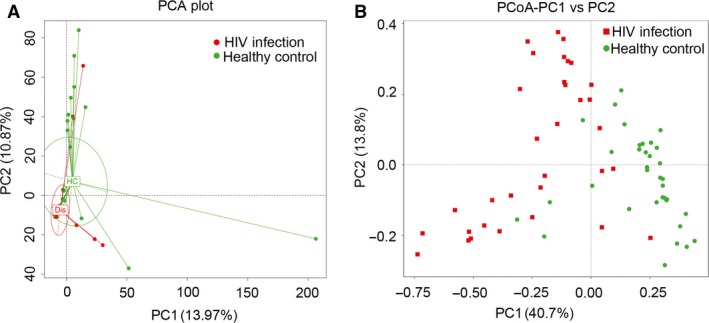
UniFrac‐based principal co‐ordinate analysis (PCoA) (B) and principal component analysis (PCA) (A) showing differences in the clustering of bacterial communities according to HIV infection status. Each dot represents a single faecal sample.
Faecal bacterial composition in patients with HIV
We compared community structure among groups. Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria were the most predominant phyla, accounting for 97% of faecal samples, patients with HIV as well as controls (Figs 3A and 4A). The proportion of Proteobacteria increased in HIV‐infected patients compared with controls; the proportion of Bacteroidetes decreased in patients with HIV compared with controls. At the genus and species levels, Enterococcus, Lachnoclostridium, Streptococcus, Lactobacillus, Ruminococcus and Streptococcus vestibularis were most abundant in faecal microbiota in patients with HIV. In contrast, the proportionate representation of Prevotella, Megamonas, Dialister, Ruminiclostridium, Faecalibacterium, Ruminococcus, Lachnospira, Roseburia, Blautia, Bacteroides vulgatus, Bacteroides uniformis, Phascolartobacterium faeclum, Ruminococcus bromii and Bacteroides stercoris was markedly reduced in HIV‐infected patients (Figs 3B and 4B,C).
Figure 3.
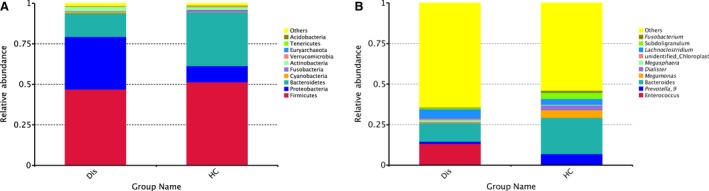
Relative abundance of faecal bacterial taxa (A) at the phylum level and (B) at the genus level. Abbreviations: Dis indicates HIV‐infection; HC indicates Healthy controls.
Figure 4.
Faecal bacterial abundance at the phylum (A), genus (B) and species (C) levels.
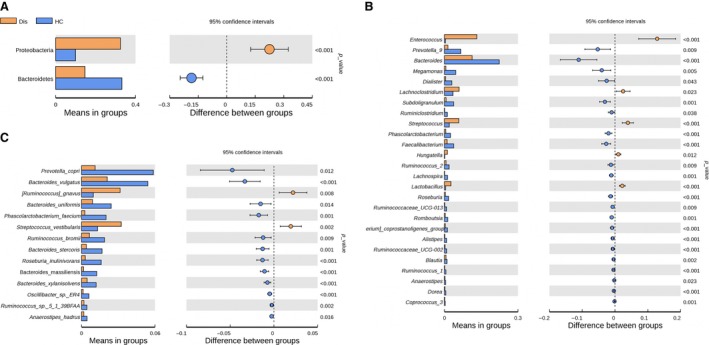
To identify the specific bacterial taxa associated with HIV, we compared the composition of faecal microbiota in healthy controls and patients with HIV using linear discriminant analysis effect size (LEfSe). A cladogram that represents the structure of the faecal microbiota and the predominant bacteria in the healthy control and HIV‐positive patients is shown in Figure 5A. Changes in the composition of faecal microbiota in HIV‐1‐infected samples were also explored using the Mann–Whitney U‐test at different taxon levels. LEfSe analysis revealed 37 discriminative features (LDA score >3, Fig. 5B). Members of Bacteroidetes were enriched in the healthy‐control samples, whereas Firmicutes and Proteobacteria were enriched in the HIV‐positive patient samples. Firmicutes and Proteobacteria could therefore be used as biomarkers to identify patients with HIV infection.
Figure 5.
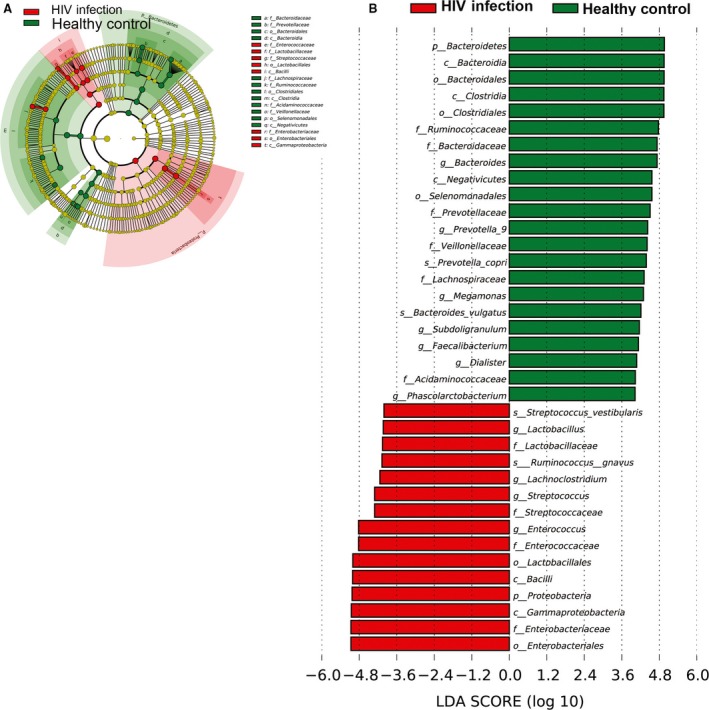
Taxonomic differences between the faecal microbiota of HIV‐infected patients and healthy controls by LEfSe and LDA. (A) LEfSe results for the bacterial communities, (B) cladogram using the LDA model results for the bacterial hierarchy.
Effects of HIV transmission route on faecal microbiota
HIV patients were divided into three groups according to transmission route: heterosexual, homosexual or intravenous drug abuse (IDA). We then analysed alterations of the gut microbiota among these three groups. Results (Fig. 6) showed that several microbial signatures in the faecal microbiota differed among homosexual HIV‐infected patients, intravenous drug abusers with HIV and healthy individuals. Bacilli, Lactobacillales and Enterococcaceae were significantly more abundant in the faeces of HIV‐infected patients infected by homosexual transmission, whereas Enterobacteriales, Enterobacteriaceae, Lachnospiraceae, Streptococcaceae and Lactobacillaceae were significantly more abundant in faecal samples from HIV‐infected patients infected through IDA. Provotella, Lachnoclostridium, Phascolarctobacterium and Parabacteroides were markedly more abundant in heterosexual as compared with homosexual patients (Fig. 6D).
Figure 6.
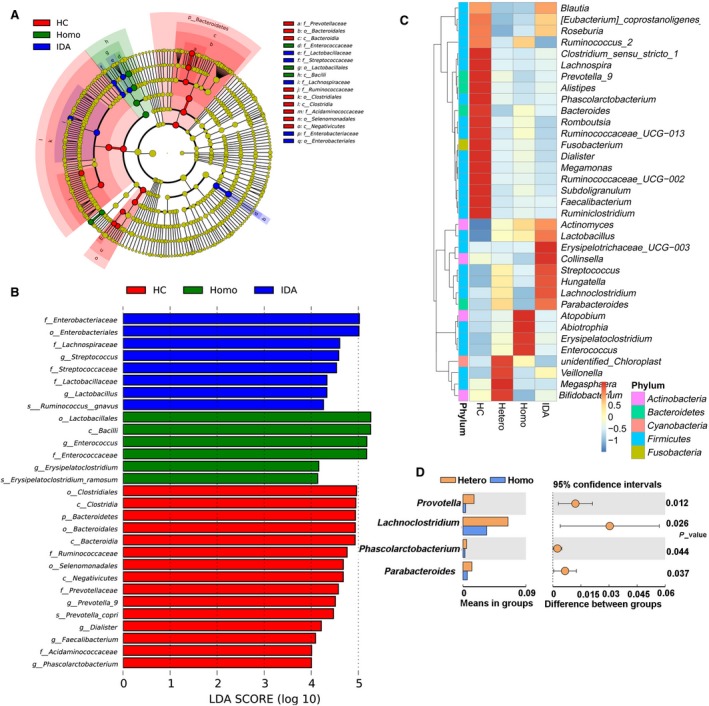
Effects of HIV transmission route on faecal microbiota in patients with HIV. (A) PCA plot, (B) relative abundance at the phylum level, (C) LEfSe results among HIV‐infected patients infected by different routes of transmission and (D) relative abundance bacteria with significance between heterosexual (Hetero) and homosexual (Homo) subjects. Abbreviations: HC, Healthy controls; Homo, Homosexual; Hetero, heterosexual; IDA, intravenous drug abuse.
Partial restoration of gut microbiota in HIV‐infected patients after HAART
We also enrolled 14 HIV‐infected patients treated with HAART for ≥3 months to identify any microbial differences among untreated HIV‐infected patients, HAART‐treated individuals and healthy controls. The results showed that microbial diversity (as measured with the Shannon index) was increased after HAART; these effects were most apparent as increased levels of Bacteroides, Blautia and Faecalibacterium (Fig. 7). Levels of bacterial taxa such as Rminccoccaceae, Ruminiclostridium, Eubacterium_coprostanoligenes, Lachnospira and Roseburia showed no change. Thus, despite the efficacy of HAART, the faecal microbiota of HIV‐1‐infected patients was not completely restored after therapy.
Figure 7.
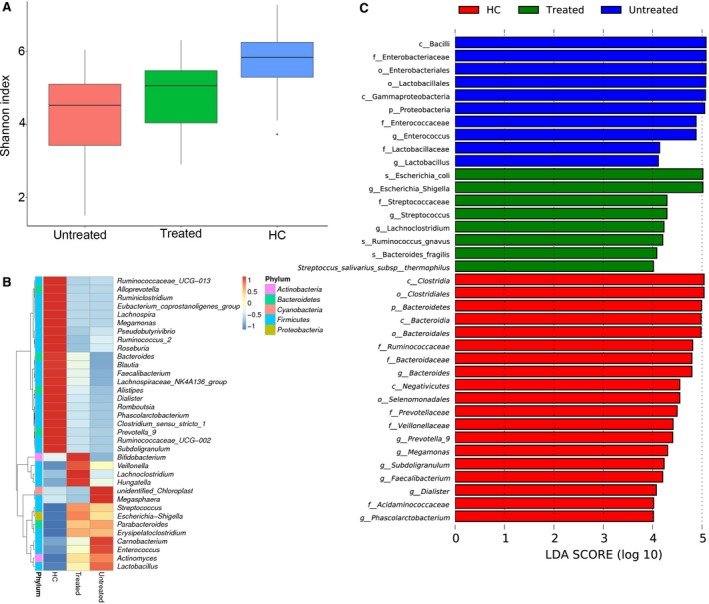
Effects of HAART on gut microbiota in patients with HIV. (A) Bacterial diversity by Shannon index. (B) Heatmap showing abundance distribution of the OTUs identified as key variables among HC, HAART‐treated and untreated HIV individuals. (C) LDA results for the bacterial hierarchy.
Discussion
The present study is the first to compare gut microbiota in HIV‐infected patients with those of age‐ and gender‐matched healthy individuals in a population from South China using sequencing technologies. Our data show that the intestinal bacterial communities in HIV‐infected patients are less diverse and distinct from those of non‐HIV controls. In patients with HIV, the gut microbiota typically included higher proportions of potentially pathogenic microbes such as Proteobacteria, Enterococcus, Lachnoclostridium, Streptococcus, Lactobacillus and Ruminococcus. The gut microbiota of patients with HIV, compared with those of controls, contained lower levels of Bacteroidetes, Prevotella, Megamonas, Dialister, Ruminiclostridium, Faecalibacterium, Ruminococcus, Lachnospira, Roseburia, Blautia, Bacteroides vulgatus, Bacteroides uniformis, Phascolartobacterium faeclum, Ruminococcus bromii and Bacteroides stercoris.
Changes to the gut microbiota during infection with HIV are influenced by factors such as study population, race, geography, dietary habits, lifestyle, sex, age, sample type and treatment. The complexity of the issue has led to methodological discrepancies between studies that make it difficult to compare results. Dillon et al. 2. observed an increased abundance of the phylum Proteobacteria in HIV‐infected individuals was only noted with mucosal samples, suggesting that luminal samples may miss potentially pathogenic alterations. However, in the present study, we also observed an increased abundance of the phylum Proteobacteria in HIV‐infected patients with faecal samples. Samples were collected from the mid‐stream stool in the present study, which is less convenient than swabbing, but may retain the signal of microbial changes better, because of the biogeographic heterogeneity in the stool.
Several studies have found that α‐diversity was significantly decreased in untreated HIV‐infected patients 9, 14. While Ling et al. 20 showed no difference in bacterial alpha diversity of gut microbiota between HIV‐infected patients and healthy controls, another study found a clear decrease in overall bacterial diversity after effective HAART 5, 8. In this study, HIV‐infected individuals had lower levels of Prevotella, a genus of Gram‐negative anaerobic bacteria previously grouped in the genus Bacteroides and Bacteroides. These findings are inconsistent with previous studies, although previous studies largely focused on populations from Western countries 2, 5, 9, 13. This variation in composition of the gut microbiota might reflect differences in diet or host genetic background between Chinese and Western populations. The Western diet is high in fat and calories, while Chinese diets typically include low levels of fat, sugar and meat, which may have significant effects on the gut microbiota. Kashyap et al. 26 have suggested that different dietary patterns are strongly associated with gut microbiota enterotypes. Lozupone et al. 8. have shown that diets high in fat and protein and low in carbohydrates and fibre diets are correlated with the loss of beneficial bacteria in HIV‐infected patients.
This study also provided a comparison of gut microbiota among HIV‐infected patients infected by different transmission routes to identify key biomarkers that might be associated with HIV infection. In the largest study to date examining the gut microbiota of HIV‐infected individuals, Noguera‐Julian et al. 14. found that a high Prevotella/low Bacteroides enterotype in stool specimens was highly associated with men who have sex with men (MSM) behaviour, regardless of HIV‐1 infection status. This may explain the perceived association between this enterotype and HIV infection status in prior studies that did not control for sexual behaviour. In the present study, several microbial signatures in the faecal microbiota differed among homosexual HIV‐infected patients, intravenous drug abusers with AIDS and healthy individuals. Although our results were not completely consistent with previous studies, this is the first attempt, to the best of our knowledge, to compare microbiota communities among Chinese individuals infected with HIV through different routes of transmission. This study shows that gut microbiota may be used to help prevent and treat HIV.
A few limitations to the present study should be acknowledged. In an observational study such as this, it is not possible to untangle the causal relationship between gut microbiota and HIV. As samples were collected from patients who had already been diagnosed with HIV, changes in the gut microbiota may have been a cause or a consequence of HIV. HIV infection disrupts host–microbe interactions such as the mucosal immune response, which may have confounded our results. Secondly, the sample size for this study, especially the subgroups of HIV‐infected individuals stratified by transmission route, were rather small. In addition, because of the lack of data on follow‐up, we could not evaluate the gut microbial changes that may be associated with effective antiretroviral therapy.
In summary, our study reveals alterations in the gut microbiome and dysbiosis in HIV‐infected patients from South China. These findings may aid the establishment of principles guiding HIV management. Our results confirm that the gut microbiome contains promising biomarkers for the non‐invasive evaluation of routes of HIV transmission. The identification of microbiota associated with specific routes of HIV transmission represents one step towards establishing a set of microbiota‐based biomarkers for the assessment of HIV. Prospective studies are needed to evaluate the long‐term consequences of alterations of the microbiota that accompany HIV infection, the host's immune response and HIV treatment.
Conflicts of interest
None declared.
Author contributions
ZYL involved in design of the study, recruitment of patients, statistical analysis and interpretation of the data, and drafting of the article; OZT involved in recruitment of patients, statistical analysis and interpretation of the data; TXP revised the article; XHM involved in sample collection and DNA extraction; WXF, LK, HJ, DYL, ZYJ and WH involved in interpretation of the data and revision of the article; CY involved in interpretation of the data and revision of the article; NYQ involved in concept and design of the study, interpretation of the data and revision of the article.
Methods
Subjects
This study was performed at the Institute for Infectious Diseases, Guangzhou No. 8 People's Hospital, Guangzhou Medical University, China. During the period from March through October 2015, 33 patients with HIV‐infected and 35 healthy controls (HC) were enrolled for cross‐sectional comparison of gut microbiota. HIV infection was diagnosed according to standard protocols. Age‐ and gender‐matched healthy volunteers were recruited from the adjacent community (Table 1).
Table 1.
Baseline clinical characteristics of the patients
| Treatment‐naïve HIV (N = 19) | HIV with highly active antiretroviral therapy (N = 14) | Healthy controls (N = 35) | |
|---|---|---|---|
| Sex (female/male) | 5/14 | 5/9 | 15/20 |
| Age (years; mean ± S.D.) | 42.74 ± 14.54 | 43.21 ± 16.26 | 38.66 ± 8.66 |
| CD4 count (cell/μl) | 54.89 ± 68.70 | 171.78 ± 135.76 | ND |
| Viral load (HIV‐1 RNA copies/ml, mean ± S.D.) | 312091.93 ± 395676.31 | 174412.62 ± 409758.89 | ND |
| Transmission, no. | |||
| Intravenous Drug Abuse (IDA) | 1 | 2 | ND |
| Heterosexual (Hetero) | 14 | 12 | ND |
| Homosexual (Homo) | 4 | 0 | ND |
ND, not done.
Among the HIV‐infected participants, 14 participants had been treated with highly active antiretroviral therapy (HAART) for more than 3 months; the remaining 19 individuals had not received treatment. Exclusion criteria were age <18 years, use of antibiotics or probiotics over the previous 4 weeks, history of inflammatory bowel disease (IBD), evidence of hepatitis B or C virus infection, other known chronic disease and pregnancy or breastfeeding status. Viral load and peripheral blood CD4+ and CD8+ cell counts were determined with standard methods 11.
Faecal sample collection and extraction of genomic DNA
When the participants were initially examined at the hospital, approximately 2 g of a fresh faecal sample (from mid‐stream stool) was collected in a sterile plastic cup and stored in a refrigerator. Samples to be used for bacterial genomic DNA extraction were transferred immediately to the laboratory and stored at −80℃. Total genome DNA from samples was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Dusseldorf, Germany). DNA concentrations were measured with a NanoDrop 2000 BioAnalyser at 260 nm (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Remaining samples were stored at −20°C before polymerase chain reaction (PCR).
PCR amplification and Illumina sequencing
We used bar‐coded V4‐515F 5′ GTGCCAGCMGCCGCGGTAA 3′ and V4‐806R 5′ GGACTACHVGGGTWTCTAAT 3′ primers to amplify bacterial 16S rRNA V4 fragments. All PCRs were carried out with Phusion® High‐Fidelity PCR Master Mix (New England Biolabs, Beverly, MA, USA). PCR products were combined with the same volume of 1× loading buffer (containing SYB green) prior to electrophoresis on 2% agarose gel for detection. Samples with a bright strip between 400 and 450 bp were selected for subsequent experiments. Sequencing libraries were generated using the TruSeq DNA PCR‐Free Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer's recommendations, and index codes were added. Library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific, Carlsbad, CA, USA) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina HiSeq 2500 platform.
Data analysis
Paired‐end reads were assigned to samples based on their unique barcodes and truncated by cutting off the barcode and primer sequence. Paired‐end reads were merged using FLASH 23, which was designed to merge paired‐end reads when at least some of the reads overlap the read generated from the opposite end of the same DNA fragment. Splicing sequences are called raw tags. Sequences were analysed using the QIIME 24 software package. In‐house Perl scripts were used to analyse α‐diversity within samples and β‐diversity among samples. First, reads were filtered by QIIME quality filters. Then, we used pick_de_novo_otus.py to pick operational taxonomic units (OTUs) by making an OTU table. Sequences with ≥97% similarity were assigned to the same OTUs. A representative sequence for each OTU was screened for further annotation. We picked a representative sequence for each OTU and used the RDP classifier 25 to annotate taxonomic information for each representative sequence. α‐diversity was applied to analyse the complexity of species diversity for a sample through four indices, including observed species, Shannon, PD‐whole tree and Simpson. Beta diversity on both weighted and unweighted UniFrac was calculated with QIIME software (version 1.7.0). Cluster analysis was preceded by principal component analysis (PCA), which was applied to reduce the dimension of the original variables using the FactoMineR package and ggplot2 package in R software (version 2.15.3). Principal co‐ordinate analysis (PCoA) was performed to obtain principal co‐ordinates and visualize complex, multi‐dimensional data. A distance matrix of weighted or unweighted UniFrac among samples obtained previously was transformed to a new set of orthogonal axes, in which the maximum variation factor is demonstrated by first principal co‐ordinate, the second maximum by second principal co‐ordinate and so on. PCoA analysis was displayed using the WGCNA package, stat packages and ggplot2 package in R software (version 2.15.3). Statistical analysis was performed with t‐test, MetaStat, LEfSe and Anosim.
Ethics statement
The Ethics Committee of Guangzhou No. 8 People's Hospital, Guangzhou Medical University, approved this study. Patients included in the study had signed informed consent.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (NSFC 81700487) and the National Clinical Key Institute Foundation of Chinese Health and Family Planning Ministry (Grant No. 2013‐544).
Funding source: The authors declare no competing financial interests.
References
- 1. Cohen CR, Lingappa JR, Baeten JM, et al Bacterial vaginosis associated with increased risk of female‐to‐male HIV‐1 transmission: a prospective cohort analysis among African couples. PLoS Med. 2012; 9: 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dillon SM, Lee EJ, Kotter CV, et al An altered intestinal mucosal microbiome in HIV‐1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014; 7: 983–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doerflinger SY, Throop AL, Herbst‐Kralovetz MM. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species‐specific manner. J Infect Dis. 2014; 209: 1989–99. [DOI] [PubMed] [Google Scholar]
- 4. Gori A, Tincati C, Rizzardini G, et al Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J Clin Microbiol. 2008; 46: 757–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mutlu EA, Keshavarzian A, Losurdo J, et al A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 2014; 10: e1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brenchley JM, Price DA, Schacker TW, et al Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006; 12: 1365–71. [DOI] [PubMed] [Google Scholar]
- 7. Gori A, Rizzardini G, Van't Land B, et al Specific prebiotics modulate gut microbiota and immune activation in HAART‐naive HIV‐infected adults: results of the “COPA” pilot randomized trial. Mucosal Immunol. 2011; 4: 554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lozupone CA, Rhodes ME, Neff CP, et al HIV‐induced alteration in gut microbiota: driving factors, consequences, and effects of antiretroviral therapy. Gut Microbes. 2014; 5: 562–70. [DOI] [PubMed] [Google Scholar]
- 9. McHardy IH, Li X, Tong M, et al HIV infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome. 2013; 1: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vujkovic‐Cvijin I, Dunham RM, Iwai S, et al Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013; 5: 193ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lozupone CA, Li M, Campbell TB, et al Alterations in the gut microbiota associated with HIV‐1 infection. Cell Host Microbe. 2013; 14: 329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nowak P, Troseid M, Avershina E, et al Gut microbiota diversity predicts immune status in HIV‐1 infection. AIDS. 2015; 29: 2409–18. [DOI] [PubMed] [Google Scholar]
- 13. Yu G, Fadrosh D, Ma B, et al Anal microbiota profiles in HIV‐positive and HIV‐negative MSM. AIDS. 2014; 28: 753–60. [DOI] [PubMed] [Google Scholar]
- 14. Noguera‐Julian M, Rocafort M, Guillen Y, et al Gut microbiota linked to sexual preference and HIV infection. EBioMedicine. 2016; 5: 135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang L, Poles MA, Fisch GS, et al HIV‐induced immunosuppression is associated with colonization of the proximal gut by environmental bacteria. AIDS. 2016; 30: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dinh DM, Volpe GE, Duffalo C, et al Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. 2015; 211: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dillon SM, Lee EJ, Kotter CV, et al Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T‐cell activation in untreated HIV‐1 infection. Mucosal Immunol. 2016; 9: 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez‐Santiago J, Gianella S, Massanella M, et al Gut Lactobacillales are associated with higher CD4 and less microbial translocation during HIV infection. AIDS. 2013; 27: 1921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ng SC, Tang W, Ching JY, et al Incidence and phenotype of inflammatory bowel disease based on results from the Asia‐pacific Crohn's and colitis epidemiology study. Gastroenterology. 2013; 145: 158–65 e2. [DOI] [PubMed] [Google Scholar]
- 20. Ling Z, Jin C, Xie T, et al Alterations in the fecal microbiota of patients with HIV‐1 infection: an observational study in A Chinese population. Sci Rep. 2016; 6: 30673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Y, Ma Y, Lin P, et al Fecal bacterial microbiome diversity in chronic HIV‐infected patients in China. Emerg Microbes Infect. 2016; 5: e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang J, Guo Z, Xue Z, et al A phylo‐functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J. 2015; 9: 1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27: 2957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caporaso JG, Kuczynski J, Stombaugh J, et al QIIME allows analysis of high‐throughput community sequencing data. Nat Methods. 2010; 7: 335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q, Garrity GM, Tiedje JM, et al Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007; 73: 5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kashyap PC, Marcobal A, Ursell LK, et al Genetically dictated change in host mucus carbohydrate landscape exerts a diet‐dependent effect on the gut microbiota. Proc Natl Acad Sci USA. 2013; 110: 17059–64. [DOI] [PMC free article] [PubMed] [Google Scholar]