

Abstract
The incidence of Crohn's disease is increasing in many Asian countries, but considerable differences in genetic susceptibility have been reported between Western and Asian populations. This study aimed to fine‐map 23 previously reported Crohn's disease genes and identify their interactions in the Chinese population by Illumina‐based targeted capture sequencing. Our results showed that the genetic polymorphism A>G at rs144982232 in MST1 showed the most significant association (P = 1.78 × 10−5; odds ratio = 4.87). JAK2 rs1159782 (T>C) was also strongly associated with Crohn's disease (P = 2.34 × 10−4; odds ratio = 3.72). Gene‐gene interaction analysis revealed significant interactions between MST1 and other susceptibility genes, including NOD2, MUC19 and ATG16L1 in contributing to Crohn's disease risk. Main genetic associations and gene‐gene interactions were verified using ImmunoChip data set. In conclusion, a novel susceptibility locus in MST1 was identified. Our analysis suggests that MST1 might interact with key susceptibility genes involved in autophagy and bacterial recognition. These findings provide insight into the genetic architecture of Crohn's disease in Chinese and may partially explain the disparity of genetic signals in Crohn's disease susceptibility across different ethnic populations by highlighting the contribution of gene‐gene interactions.
Keywords: Crohn's disease, fine mapping, gene‐gene interactions, MST1, next‐generation sequencing
1. INTRODUCTION
Crohn's disease is one of the two major forms of inflammatory bowel diseases (IBD) characterized by chronic and relapsing inflammation in the gastrointestinal tract. Crohn's disease has long been thought to be uncommon in Asian countries. However, the incidence of Crohn's disease has been rapidly increasing in Asian countries as seen in many recent epidemiological studies.1, 2 In this regard, China has the highest incidence of IBD in Asia within the Asia‐Pacific Crohn's and Colitis Epidemiologic Study Group.1
Genetic susceptibility, gut microbiota and environmental factors act synergistically in the pathogenesis of Crohn's disease. Although more than 140 susceptibility loci of Crohn's disease in Caucasians have been identified by genome‐wide association studies (GWASs) and meta‐analyses,4, 5, 6, 7, 8, 9 considerable differences in genetic susceptibility to Crohn's disease have been reported between Western and Asian populations. Moreover, the heritability of Crohn's disease in Asian populations has not been fully explained.5, 6 In particular, the well‐established Caucasian Crohn's disease susceptibility genes, such as NOD2, ATG16L1 and PTPN, showed a lack of association in the Asian populations.6, 8, 10, 11, 12, 13, 14, 15 Inconsistent results on IL23R and IRGM were also reported.16, 17 Recent genetic studies in Korean and Japanese populations further revealed new Crohn's disease susceptibility loci (eg rs11235604 in ATG16L2 and rs7329174 in ELF1) that were not significantly associated with disease status in Western populations.18, 19, 20 This may be in part related to heterogeneity in effect size (eg TNF‐SF15 and ATG16L), differences in risk allele frequency in some of the loci (eg CARD15/NOD2) or altered gene‐microbiota and gene‐gene interactions across different populations.21 Collectively, these findings underpinned different genetic architectures in different ethnicities in determining genetic risk for Crohn's disease.
The impact of new loci underlying susceptibility to Crohn's disease cannot be determined until causal variants are identified by fine mapping via directed sequencing. Moreover, it is imperative to determine whether Crohn's disease susceptibility genes identified in Europeans are also associated with disease state in non‐European ancestry populations.22 To address whether genes previously reported in Caucasian populations contribute to Crohn's disease in the Chinese population and their effect sizes, we performed fine‐mapping analysis using next‐generation targeted capture sequencing. Moreover, as interactions among multiple genes could impact on the patients’ disease phenotype, we aimed to identify interactions among the targeted captured genes to provide insight into the genetic of Crohn's disease.
2. METHODS
2.1. Study participants
Crohn's disease patients of Han Chinese ethnicity and healthy individuals were recruited at the Prince of Wales Hospital, Hong Kong, and the First Affiliated Hospital of Sun Yat‐Sen University, Guangzhou. Both hospitals are geographically located in the Guangdong Province of China. Inclusion criteria of cases included (i) age >18 years old and (ii) diagnosis of Crohn's disease established in accordance with clinical, radiological, endoscopic and histological features criteria. Inclusion criteria of controls included (i) age >18 years and (ii) asymptomatic individuals participating in colonoscopy screening or healthy volunteers or students from the Chinese University of Hong Kong. The study was prospectively reviewed and approved by the Joint CUHK‐NTEC Clinical Research Ethics Committee and the clinical ethics committee of Sun Yat‐Sen University. All participants had provided written informed consents. Controls were excluded if they had previously been diagnosed with IBD or if they had one or more first‐ or second‐degree relative with IBD. Clinical phenotype data were collected and stored in a database: age, sex, family history, smoking history, surgery and date of first surgery, extraintestinal diseases, disease location and behaviour (Montreal classification).
2.2. Targeted gene capture and next‐generation sequencing
The chip‐based gene capture technology coupled with next‐generation sequencing was employed for comprehensive genotyping of 23 Crohn's disease susceptibility genes and their promoters. Genomic DNA was extracted from blood lymphocytes (250 μL) of Crohn's disease patients and healthy individuals using Gentra Puregene Blood Kit (Gentra Systems, Inc., Minneapolis, MN) and stored at −20°C in the Prince of Wales Hospital. Twenty micrograms of genomic DNA from each sample was sheared by nebulizer (Roche Applied Science, Hong Kong) to fragments around 500 bp. After ligation with linkers at both ends, small fragments <300 bp were removed with AMPure DNA purification beads (Agencourt, Beverly, MA). The linker‐ligated DNA fragments were then hybridized to the custom‐designed NimbleGen Sequence Capture 2.1M Array and the enriched captured DNA fragments were eluted from the array and amplified by ligation‐mediated PCR. Quantitative PCR was used to estimate the magnitude of enrichment. Twenty‐three genes captured for next‐generation sequencing are listed as follows: PTPN22, IL23R, ITLN1, ATG16L1, PTGER4, MST1, IRGM, IL12B, CDKAL1, CCR6, JAK2, TNFSF15, ZNF365, NKX2‐3, C11orf30, LRRK2, MUC19, NOD2, ORMDL3, STAT3, PTPN2, ICOSLG and VDR.
The captured DNA fragments were first randomly ligated by DNA ligase to sizes ranged from 1 to 8 kb, then sheared to 200 bp on average, and finally ligated with Illumina‐compatible adapters and subject to standard library preparation. Resulting DNA libraries were sequenced on Illumina 2000 with the target sequencing depth of 50×, which was more than sufficient for genotyping heterozygous loci with high confidence. Real‐time image analysis and base calling were performed by the Genome Analyzer Pipeline version 1.3.1 using standard parameters. Reads with ≥ 12‐bp adapter or linker sequences or reads <29 bp, or with >6 missing bases, or 40 continuous identical bases were discarded. SOAP aligner was used to align the remaining reads to the human reference genome (human NCBI Build 36) with maximum two mismatches. Only unique matched reads were retained. A Bayesian statistics‐based algorithm was used for base calling.
2.3. ImmunoChip data set
The design and genotyping of the ImmunoChip have been previously described.21 In brief, the ImmunoChip is an Illumina Infinium microarray comprising 196 524 single nucleotide polymorphisms (SNPs) and small indel markers selected based on results from GWASs of 12 different immune‐mediated diseases. The ImmunoChip enables replication of all nominally associated SNPs (P < .001) from the index GWAS scans and fine mapping of 186 loci associated at genome‐wide significance with at least 1 of the 12 index immune‐mediated diseases. The chip also contains around 3000 SNPs added as part of the Wellcome Trust Case Control Consortium 2 (WTCCC2) project replication phase. The genotype data were extracted for 531 Hong Kong Chinese subjects on the ImmunoChip data set. Quality control was performed as described.21 The cohort includes 235 controls and 531 IBD cases, including 388 patients with Crohn's disease.
2.4. Statistical analysis
The SNPs from targeted sequencing had low to rare minor allele frequencies (MAFs). The sequence kernel association test (SKAT) is an effective method to detect association of the sequencing data to disease phenotypes.23, 24 The method uses a linear mixed model and performs variance component score test.24 For epistasis evaluation, a robust W‐test was used to evaluate SNP‐SNP interactions.25 The W‐test is testing for the difference in genotype distributions formed by a SNP pair in case and control groups. The test follows a chi‐squared distribution of which the degrees of freedom is bootstrap‐estimated from the data. Therefore, the method is able to correct for bias in distributions due to complicated genetic architecture and return robust estimates.25 The SKAT and W‐test were conducted using R packages.23, 25 The LocusZoom tool was used to draw SNPs Manhattan plot in a specific region and provided a detailed view of the P‐value distribution within a gene.26 A SNP or an interaction pair was significant if its P‐value was smaller than Bonferroni‐corrected alpha of 5%. Expression quantitative trait loci (eQTL) analysis was carried out using the Genotype‐Tissue Expression database.27
2.5. Power calculation
The power of an association study depends on the sample size, effect size of a variant and its allele frequency. Assuming findings from a previously validated SNP (rs2241880) in ATG16L1 with an odds ratio of 0.69 and a minor allele frequency of 45%, our study had 86.2% power to detect such a variant with an α‐error rate of 5%. Alternatively, our study had at least 80% power to detect a variant with odds ratio of 1.5 at a MAF of 20%.
3. RESULTS
3.1. Patient characteristics, quality control and SNP calling
A total of 262 patients with Crohn's disease and 323 controls were included. The mean age was 43.6 and 55.9 years in the case and control groups, respectively. About half of the subjects were female (45.9%). Table S1 summarizes the basic characteristics of the cases and controls. DNA samples were collected from all patients for targeted capture sequencing, generating a genotype data set of 2046 SNPs. Four subjects were excluded because of empty data files. Targeted capture of all DNA samples was completed with an average sequencing depth (on target) of >50 and a coverage of >99.7% (Table S2). When calling the genotype, missing value was coded if genotype quality was less than 20. Quality control of the genotype data was conducted, and we excluded samples whereby (i) the percentage of missing genotypes was greater than 5%, (ii) SNPs had no variance, and (iii) P‐values of test on Hardy‐Weinberg equilibrium (HWE) were smaller than 0.05 after Bonferroni correction28 (Table S3).
3.2. Novel associations of MST1 rs144982232 with Crohn's disease
Sequence kernel association test analysis identified one locus, namely the rs144982232 in MST1 (A>G, P = 1.78 × 10−5, odds ratio = 4.87), which was significantly associated with Crohn's disease after controlling for multiple testing by Bonferroni method (Tables 1 and S6). The susceptibility to the disease was 4.87 higher for individuals with a G allele at this locus than those with an A allele (odds ratio = 4.87). The regional association plot of MST1 is shown in Figure 1. There were 26 SNPs sequenced in this gene in our fine‐mapping study. The distribution of three genotypes (GG, AA and GA) at MST1 rs144982232 was in accordance with HWE. Two more SNPs showing strong effect were rs1159782 in JAK2 (T>C, P = 2.34 × 10−4, odds ratio = 3.72; Figure 2) and rs2111234 in NOD2 (G>A, P = 7.59 × 10−4, odds ratio = 5.09). The large odds ratios indicated a strong risk effect in the MST1, JAK2 and NOD2 gene to Crohn's disease.
Table 1.
Top 10 SNPs identified by SKAT among 23 Crohn's disease susceptibility genes. The total number of SNPs is 2046, and the Bonferroni‐corrected significance threshold is 2.4 × 10−5
| Rank | Chr | Pos | SNP | P‐value | Gene | Description | MAF | Odds ratio | 95% CI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Chr3 | 49723141 | rs144982232 | 1.78E‐05 | MST1 | A>G | 0.038 | 4.87 | 2.25, 10.54 |
| 2 | Chr9 | 5078117 | rs1159782 | 2.34E‐04 | JAK2 | T>C | 0.053 | 3.72 | 2.03, 6.82 |
| 3 | Chr16 | 50734033 | rs2111234 | 7.59E‐04 | NOD2 | G>A | 0.034 | 5.09 | 2.24, 11.58 |
| 4 | Chr9 | 117569046 | rs7848647 | 1.46E‐03 | TNFSF15 | T>C | 0.448 | 0.69 | 0.55, 0.87 |
| 5 | Chr10 | 64418656 | rs7915131 | 1.34E‐03 | ZNF365 | C>T | 0.343 | 0.68 | 0.54, 0.87 |
| 6 | Chr1 | 114377148 | rs1970559 | 3.29E‐03 | PTPN22 | T>C | 0.041 | 0.39 | 0.21, 0.72 |
| 7 | Chr1 | 114396955 | rs2476602 | 4.09E‐03 | PTPN22 | G>A | 0.039 | 0.38 | 0.20, 0.73 |
| 8 | Chr10 | 64426056 | rs4746516 | 2.03E‐03 | ZNF365 | G>T | 0.200 | 0.63 | 0.47, 0.84 |
| 9 | Chr9 | 5069837 | rs7869668 | 3.29E‐03 | JAK2 | G>A | 0.417 | 1.44 | 1.15, 1.82 |
| 10 | Chr10 | 64418089 | rs10822044 | 4.33E‐03 | ZNF365 | T>C | 0.231 | 0.68 | 0.52, 0.89 |
MAF, minor allele frequencies; SKAT, sequence kernel association test; SNP, single nucleotide polymorphisms.
Figure 1.
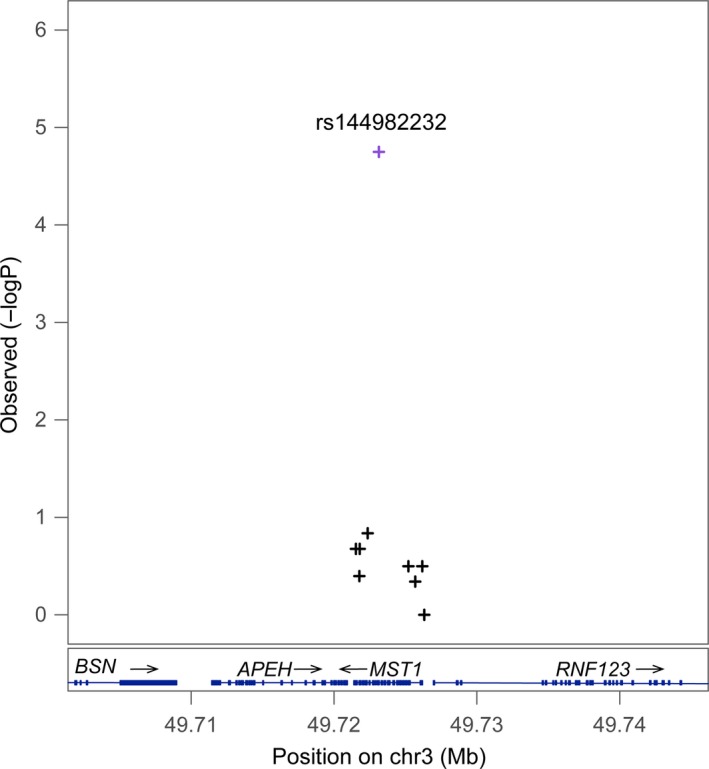
Regional association plot of MST1. The A>G polymorphism at rs144982232 corresponding to synonymous H425H increased the risk for Crohn's disease in the Chinese population (P = 1.78E‐05; odd ratios: 4.87). Grey colour indicates that the information of linkage disequilibrium (r 2 values) for the points was not available in reference genome
Figure 2.
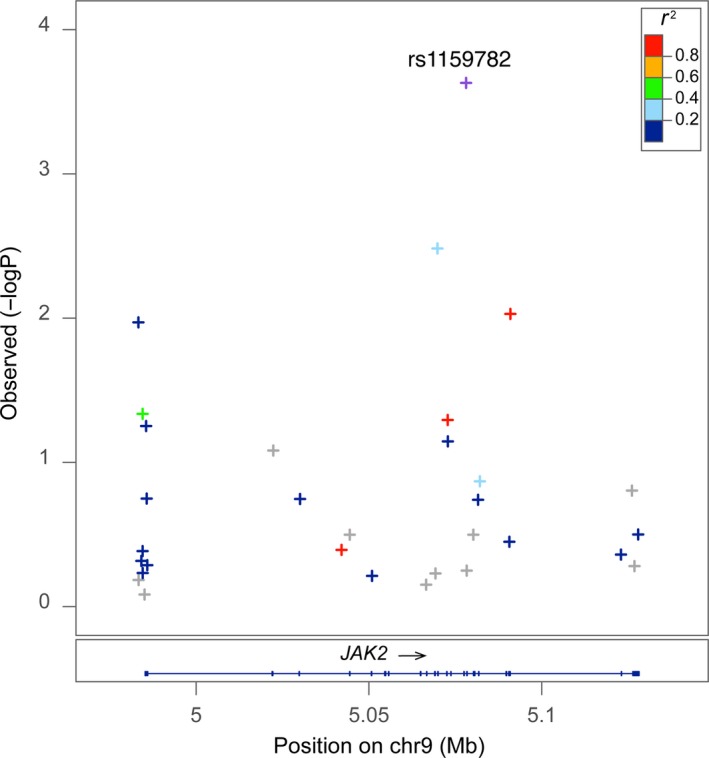
Regional association plot of JAK2. Subjects with allele T>C at rs1159782 had a higher risk for Crohn's disease (P = 2.34E‐04, odds ratio: 3.72). The r 2 was estimated by the LocusZoom software from HapMap Phase II JPT + CHB population. It measures the linkage disequilibrium of each SNP with the most significant SNP
3.3. Interactions of MST1 with MUC19, JAK2, VDR and other Crohn's disease susceptibility genes
A robust and powerful epistasis analysis tool known as W‐test was performed to detect SNP‐SNP interactions.25 From the total 2046 SNPs in this study, 202 SNPs had P‐value less than 0.1 and passed the first‐stage filtering. Among these SNPs, 20 301 SNP pairs were formed, and a total number of 95 pairs had P‐values passed the Bonferroni‐corrected significance level at 2.46 × 10−6. Top interacting pairs identified are shown in Tables 2 and S7. The significant SNP pairs producing an 18‐gene interaction network are visualized in Figure 3, in which MST1, MUC19, JAK2 and VDR play central roles. Interestingly, except MST1, none of the 18 genes showed significant main effect. The top pairs include interactions of MST1‐JAK2 (A>G at rs144982232, T>C at rs1159782, P = 9.44 × 10−11, odds ratio = 4.34), MST1‐NOD2 (A>G at rs144982232, G>A at rs2111234, P = 1.79 × 10−9, odds ratio = 4.69) and MST1‐MUC19 (A>G at rs144982232, T>C at rs116937891, P = 1.01 × 10−8, odds ratio = 4.89). For the interaction pair MST1‐JAK2, the susceptibility to Crohn's disease was 4.34 higher for genotypes with allele G at rs144982232 in MST1 or with allele C at rs1159782 in JAK2 than those with allele A at rs144982232 and T at rs1159782 (odds ratio = 4.34).
Table 2.
Top 20 SNP‐SNP interactions among 95 significant pairs in 23 genes identified by W‐test. A P‐value < 2.46 × 10−6 was considered statistically significant
| Rank | SNP1 | Chr | Position1 | Gene1 | SNP1 description | SNP2 | Chr | Position2 | Gene2 | SNP2 description | OR | 95% CI | MAF1 | MAF2 | P‐value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs1159782 | Chr9 | 5078117 | JAK2 | T>C | 4.34 | 2.50, 7.52 | 0.04 | 0.05 | 9.44E‐11 |
| 2 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs2111234 | Chr16 | 50734033 | NOD2 | G>A | 4.69 | 2.55, 8.64 | 0.04 | 0.03 | 1.79E‐09 |
| 3 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs116937891 | Chr12 | 40815261 | MUC19 | T>C | 4.89 | 2.48, 9.62 | 0.04 | 0.01 | 1.01E‐08 |
| 4 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs11564247 | Chr12 | 40821478 | MUC19 | T>C | 5.71 | 2.74, 11.91 | 0.04 | 0.01 | 1.34E‐08 |
| 5 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs80205770 | Chr12 | 40823230 | MUC19 | A>G | 3.50 | 2.05, 5.98 | 0.04 | 0.03 | 2.15E‐08 |
| 6 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs56191322 | Chr1 | 114362437 | PTPN22 | A>G | 5.12 | 2.53, 10.37 | 0.04 | 0.01 | 3.06E‐08 |
| 7 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs191850264 | Chr12 | 40920180 | MUC19 | G>A | 4.99 | 2.46, 10.12 | 0.04 | 0.01 | 3.11E‐08 |
| 8 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs2289473 | Chr2 | 234182025 | ATG16L1 | C>T | 4.64 | 2.35, 9.17 | 0.04 | 0.01 | 4.11E‐08 |
| 9 | rs2111234 | Chr16 | 50734033 | NOD2 | G>A | rs1159782 | Chr9 | 5078117 | JAK2 | T>C | 3.82 | 2.16, 6.76 | 0.03 | 0.05 | 5.76E‐08 |
| 10 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs12370083 | Chr12 | 40816185 | MUC19 | C>A | 4.99 | 2.46, 10.12 | 0.04 | 0.01 | 6.68E‐08 |
| 11 | rs6687620 | Chr1 | 67648460 | IL23R | T>C | rs144982232 | Chr3 | 49723141 | MST1 | A>G | 4.99 | 2.46, 10.12 | 0.01 | 0.04 | 6.73E‐08 |
| 12 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs2229829 | Chr12 | 48238607 | VDR | G>T | 4.64 | 2.35, 9.17 | 0.04 | 0.01 | 8.16E‐08 |
| 13 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs11564248 | Chr12 | 40820570 | MUC19 | C>T | 4.86 | 2.39, 9.86 | 0.04 | 0.01 | 1.14E‐07 |
| 14 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs2291282 | Chr17 | 40498565 | STAT3 | T>C | 3.81 | 2.12, 6.84 | 0.04 | 0.02 | 1.22E‐07 |
| 15 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs9837520 | Chr3 | 49722356 | MST1 | G>A | 3.01 | 1.81, 5.02 | 0.04 | 0.03 | 1.23E‐07 |
| 16 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs78930461 | Chr2 | 234201767 | ATG16L1 | A>G | 5.46 | 2.50, 11.90 | 0.04 | 0.003 | 1.27E‐07 |
| 17 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | Chr12 | 48238682 | VDR | G>A | 5.62 | 2.58, 12.23 | 0.04 | 0.003 | 1.36E‐07 | |
| 18 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs7487333 | Chr12 | 40812148 | MUC19 | C>T | 3.58 | 2.02, 6.34 | 0.04 | 0.02 | 1.53E‐07 |
| 19 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | rs12601611 | Chr17 | 40497828 | STAT3 | C>T | 5.78 | 2.66, 12.56 | 0.04 | 0.01 | 1.63E‐07 |
| 20 | rs144982232 | Chr3 | 49723141 | MST1 | A>G | Chr5 | 40679674 | PTGER4 | G>C | 5.12 | 2.44, 10.75 | 0.04 | 0.01 | 1.96E‐07 |
MAF, minor allele frequencies; SNP, single nucleotide polymorphisms.
Figure 3.
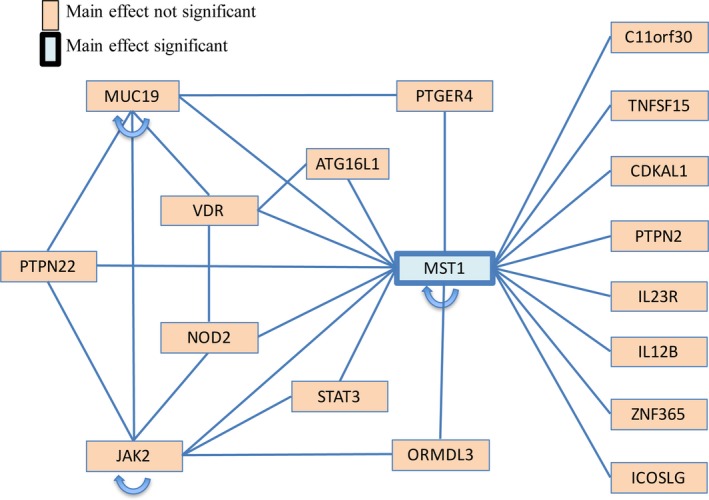
Gene‐gene interaction network visualizing the results of W‐test. MST1 had extensive interactions with other Crohn's disease susceptibility genes. JAK2,NOD2,MUC19 and VDR also interacted widely. An arrow indicates interactions between two or more SNPs within the same gene
3.4. MST1, JAK2, MUC19 and VDR acted in concert with NOD2 to alter risk for Crohn's disease
From the regional association plots, rs144982232 in MST1, rs1159782 in JAK2, rs11564247 in MUC19 (Figure S1) and rs11574129 in VDR (Figure S2) were the most significant SNPs within the genes, but none of these SNPs was associated with gene expression by eQTL analysis (data not shown). Further analysis found that all of these four SNPs interacted with rs2111234 in NOD2 directly or indirectly. Most interestingly, this polymorphic variant of NOD2 was strongly associated with altered NOD2 gene expression in multiple tissue types, including whole blood and liver (Figure 4), indicating that MST1, JAK2, MUC19 and VDR have synergistic reinforcing action in Crohn's disease development in the Chinese population through interacting with NOD2.
Figure 4.
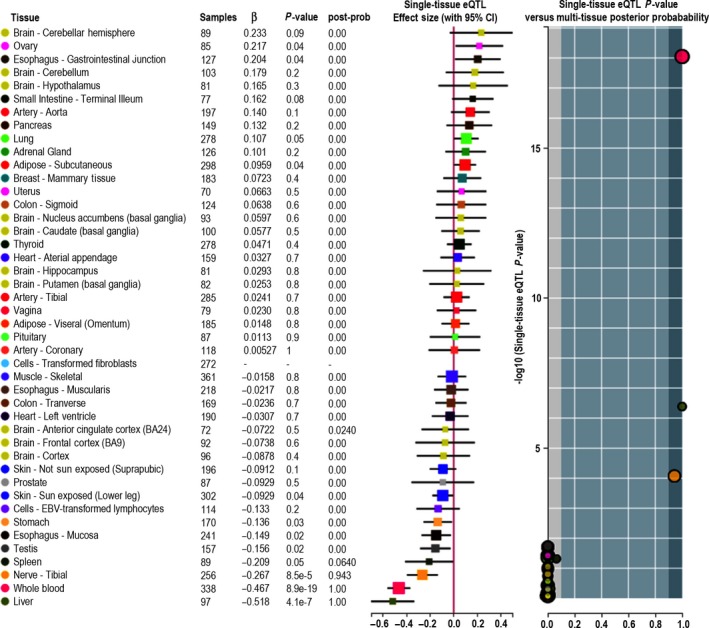
eQTL analysis revealing the association between NOD2 rs2111234 genotypes and NOD2 mRNA expression in multiple tissue types
3.5. Validation of main genetic associations and gene‐gene interactions using ImmunoChip data set
We performed validation analysis of the main genetic effects and gene‐gene interactions identified by target capture sequencing using data from the Hong Kong Chinese ImmunoChip data set comprising 235 controls and 531 patients with IBD, including 388 patients with Crohn's disease, In the ImmunoChip data set, there was no MUC19 gene marker. Therefore, its main and interaction could not be calculated. For MST1, after quality control, there was only 1 SNP in the validation data set, which was restrictive for our analysis. For main effects, JAK2, TNFSF15, ZNF365 and PTPN22 showed consistent small P‐values in the original sequencing data as well as the validation with IBD and Crohn's disease data sets (Table S4). For gene‐gene interactions, the top pair MST1‐JAK2 had significant P‐values in the original and validation data set (original: P = 9.44 × 10−11; validation IBD: P = 7.28 × 10−5; validation Crohn's disease: P = 4.81 × 10−3). The remaining three pairs that exhibited consistent small P‐values were NOD2‐JAK2, IL23R‐MST1 and MST1‐PTGER4 (Table S5).
4. DISCUSSION
Although the prevalence and incidence of Crohn's disease are higher in Western countries, they continue to rise in Asia, especially in China. It is anticipated that the number of cases of IBD in Asia might overtake that of the Western world by 2025.29 In this study, we fine‐mapped 23 known Crohn's disease susceptibility genes to identify the causal variants and delineate the relative contribution of these variants to Crohn's disease in the Chinese population. Identification of causal variants is key to understanding the molecular mechanism by which disease susceptibility genes contribute to pathogenesis as well as formulating novel therapeutic strategies. A major advantage of using targeted capture sequencing for fine mapping is the directed focus on genes of interest. Therefore, unlike GWAS, our study was not restricted by the conventional genome‐wide significance threshold as a result of fewer multiple testing.
In the targeted captured regions, the synonymous SNP (ie rs144982232) in MST1 was most significantly associated with Crohn's disease in the Chinese population. Although synonymous SNPs have long been regarded as inconsequential as the primary sequence of the protein is retained, studies have demonstrated that synonymous SNPs can affect mRNA splicing, stability and structure as well as protein folding.30 Through in silico RNA folding prediction (http://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Fold/Fold.html), we found that although the SNP A>G at rs144982232 did not significantly affect the secondary structure of MST1 mRNA, the corresponding nucleotide change occurred in the loop region of the predicted RNA stem‐loop structure (Figure S3), which is implicated in the control of RNA‐protein complex formation.31, 32
MST1, also known as MSP, is involved in regulating the innate immune response to infections and cellular stress. It binds to the receptor RON to trigger macrophage chemotaxis and activation.31 MST1 is a serum protein that circulates in the blood as an inactive single‐chain precursor (pro‐MST1) comprising two chains, α and β.34 MST1 was first recognized as a Crohn's disease risk gene (odds ratio = 1.20) in a GWAS where a single non‐synonymous SNP rs3197999 corresponding to the amino acid substitution R689C in the β‐chain was identified.35 The mechanism of this potential causal variant for Crohn's disease was controversial.36, 37 One study showed that R689C polymorphism had no impact on the ability of MST1 to bind to or signal through RON, whereas carriers of the 689C polymorphism had lower concentrations of MST1 in their serum, which could possibly increase Crohn's disease risk.36 However, another study showed that the affinity to RON of MST1 with the 689C polymorphism was approximately 10‐fold lower than that of the wild‐type MST1 and the thermal stability of the mutant MST1 was slightly lower than that of wild‐type MST1.37 However, rs3197999 did not show up as a significant SNP in our study. Instead, the SNP A>G at rs144982232 showed the strongest association with Crohn's disease in our cohort (odds ratio = 4.87; P = 1.78 × 10−5). Early GWASs showed that with the exception of NOD2, the typical effect size of Crohn's disease susceptibility locus was modest (odds ratio < 1.3).38, 39 Herein, we reported for the first time a new Crohn's disease susceptibility SNP with a high odds ratio in the Chinese population. Moreover, our data suggested that different causal variants of MST1 might be operative in the Western and the Chinese populations. Nevertheless, it is important to note that our ImmunoChip data set did not cover MST1 rs144982232 and its association with Crohn's disease has to be consolidated with an independent Chinese cohort.
The genetic heterogeneity between East Asians and Europeans at alleles of large effect could be exemplified by NOD2. The contribution of NOD2 rare variants to risk and site of Crohn's disease was well studied and explained in Caucasians, with 4 mutations (P268S, R702W, G908R and 3020insC) showing the strongest association.40, 41, 42 However, a previous study demonstrated no association of NOD2 R702W and G908R with IBD in Chinese patients.43 In line with previous genetic studies of Crohn's disease in Asians,1 a recent study showed that the three coding variants in NOD2 in Europeans did not exist in East Asians.21 Furthermore, no SNP within NOD2 even attained a suggestive evidence of association in the East Asian cohort, indicating that different genetic factors are operative in the Western and East Asian populations to contribute to Crohn's disease. All these findings prompted us to examine whether NOD2 could interact with other genes to influence Crohn's disease risk in the Chinese cohort. In this study, by SKAT analysis and W‐test, even though NOD2 SNPs individually were not significantly associated with Crohn's disease, co‐occurrence of NOD2 rs2111234 and MST1 rs144982232, JAK2 rs1159782 or VDR rs11574129 attained a significant association. eQTL analysis further substantiated this discovery by showing the strong association between NOD2 rs2111234 and NOD2 gene expression, especially in whole blood and spleen. It is tantalizing to postulate that when combined with other IBD SNPs, NOD2 SNPs could synergistically influence the risk for Crohn's in the Chinese population. It also suggests that the pathogenesis of IBD, in both the West and the East, is likely to be driven by the interplay of an abnormal immune response to gut microbes.
Another example of genetic heterogeneity in different ethnic groups in Crohn's disease pathogenesis is JAK2. A recent meta‐analysis demonstrated that JAK2 rs10758669 was significantly associated with Crohn's disease in Caucasians but not Asians.44 Consistently, rs10758669 did not exhibit association with disease status in our cohort. In contrast, another SNP rs1159782 was fine‐mapped to be the most strongly associated SNP in JAK2 with an odds ratio of 3.72. Given that many JAK inhibitors for IBD are now undergoing phase 3 trials,45 it is hopeful that JAK inhibition will benefit this subset of patients with genetic susceptibility in JAK2.
From epistasis analysis, we found the most connected gene with other SNPs that synergistically conferred risks to Crohn's disease was MST1. In particular, MST1 rs144982232 showed interactions with other IBD genes, including JAK2, NOD2, ATG16L, VDR and STAT3, indicating a more complicated role of MST1 in Crohn's disease pathogenesis. Among these pairs, the top significant interactions were with JAK2, NOD2 and MUC19. To this end, MST1‐JAK2 interaction has also been identified in ulcerative colitis,46 another major form of IBD. NOD2 polymorphisms could also modulate innate immune response47 whereas MUC19 deficiency could impair mucus production,48 both of which are important for mucosal barrier function and the control of subsequent invasion of commensals or opportunistic pathogens. The co‐involvement of NOD2, JAK2, MUC19 and MST1 in mucosal defence and inflammation in Crohn's disease therefore deserves further study.
Another noteworthy observation is that VDR was centred by MUC19, MST1, ATG16L1 and NOD2, which synergistically contributed to Crohn's disease risk. The association between VDR and Crohn's disease has been supported by multiple studies.49, 50 VDR, which codes for vitamin D receptor, is engaged in NOD2 gene transcription and signalling through NOD2 to induce expressions of β‐defensin 2 and cathelicidin.51 Variants or deletion of VDR may also change the microbiota and reduce the host defence through diminishing the production of microbicidal peptides as well as ATG16L1.52, 53 However, the interaction network among genes of interest that leads to Crohn's disease needs to be elucidated in‐depth in future studies.
Taken together, our data suggested that a novel locus in MST1 is involved in Crohn's disease in the Chinese population. Interactions between MST1 and other Crohn's disease susceptibility genes also contribute to disease risk. Future research should focus on resequencing and fine‐mapping analysis to identify causal variants in other Crohn's disease susceptibility genes. Further insights into how different risk alleles interact with each other in different ethnic populations may unravel the complex genetic and environmental influence on IBD and contribute to our understanding of disease pathogenesis.
CONFLICT OF INTEREST
The authors confirm that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
WKKW designed the study; WKKW, SHW, MHW and SCN managed the project; RS, YT and MHW conducted the bioinformatic analysis; ZZ and SCN collected clinical specimens; all authors analysed the results; WKKW, RS, ZT and YT wrote the manuscript; SHW, MHW and SCN revised the manuscript and managed the project.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the Health and Medical Research Fund (01120426), Shenzhen Science and Technology Programme (JCYJ20150630165236956, JCYC20140905151710921) and Natural Science Foundation of Guangdong Province (2015A030313886). The study sponsor has no role in the study design in the collection, analysis and interpretation of data.
Wu WKK, Sun R, Zuo T, et al. A novel susceptibility locus in MST1 and gene‐gene interaction network for Crohn's disease in the Chinese population. J Cell Mol Med. 2018;22:2368–2377. https://doi.org/10.1111/jcmm.13530
WKKW, RS, TZ and YT contributed equally.
Contributor Information
Sunny H. Wong, Email: wonghei@cuhk.edu.hk
Maggie H. Wang, Email: maggiew@cuhk.edu.hk
Siew C. Ng, Email: siewchienng@cuhk.edu.hk.
REFERENCES
- 1. Ng SC, Tang W, Ching JY, et al. Asia–Pacific Crohn's and Colitis Epidemiologic Study (ACCESS) Study Group . Incidence and phenotype of inflammatory bowel disease based on results from the Asia‐pacific Crohn's and colitis epidemiology study. Gastroenterology. 2013;145:158‐165. e2. [DOI] [PubMed] [Google Scholar]
- 2. Prideaux L, Kamm MA, De Cruz PP, Chan FK, Ng SC. Inflammatory bowel disease in Asia: a systematic review. J Gastroenterol Hepatol. 2012;27:1266‐1280. [DOI] [PubMed] [Google Scholar]
- 3. Ng SC, Zeng Z, Chen M, et al. Incidence and phenotype of inflammatory bowel disease from 2012‐2013 across 9 countries in Asia: results from the 2012 access inception cohort. Gastroenterology. 2015;148:S466‐S466. [Google Scholar]
- 4. Duerr RH, Taylor KD, Brant SR, et al. A genome‐wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461‐1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franke A, McGovern DP, Barrett JC, et al. Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jostins L, Ripke S, Weersma RK, et al. Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rioux JD, Xavier RJ, Taylor KD, et al. Genome‐wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14:3499‐3506. [DOI] [PubMed] [Google Scholar]
- 10. Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002;123:86‐91. [DOI] [PubMed] [Google Scholar]
- 11. Leong RW, Armuzzi A, Ahmad T, et al. NOD2/CARD15 gene polymorphisms and Crohn's disease in the Chinese population. Aliment Pharmacol Ther. 2003;17:1465‐1470. [DOI] [PubMed] [Google Scholar]
- 12. Ng SC, Tsoi KK, Kamm MA, et al. Genetics of inflammatory bowel disease in Asia: systematic review and meta‐analysis. Inflamm Bowel Dis. 2012;18:1164‐1176. [DOI] [PubMed] [Google Scholar]
- 13. Yamazaki K, Onouchi Y, Takazoe M, Kubo M, Nakamura Y, Hata A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn's disease in Japanese patients. J Hum Genet. 2007;52:575‐583. [DOI] [PubMed] [Google Scholar]
- 14. Yamazaki K, Takahashi A, Takazoe M, et al. Positive association of genetic variants in the upstream region of NKX2‐3 with Crohn's disease in Japanese patients. Gut. 2009;58:228‐232. [DOI] [PubMed] [Google Scholar]
- 15. Yang SK, Park M, Lim J, et al. Contribution of IL23R but Not ATG16L1 to Crohn's disease susceptibility in Koreans. Inflamm Bowel Dis. 2009;15:1385‐1390. [DOI] [PubMed] [Google Scholar]
- 16. Kim SW, Kim ES, Moon CM, et al. Genetic polymorphisms of IL‐23R and IL‐17A and novel insights into their associations with inflammatory bowel disease. Gut. 2011;60:1527‐1536. [DOI] [PubMed] [Google Scholar]
- 17. Moon CM, Shin DJ, Kim SW, et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm Bowel Dis. 2013;19:106‐114. [DOI] [PubMed] [Google Scholar]
- 18. Yang SK, Hong M, Zhao W, et al. Genome‐wide association study of Crohn's disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63:80‐87. [DOI] [PubMed] [Google Scholar]
- 19. Yang SK, Hong M, Choi H, et al. Immunochip analysis identification of 6 additional susceptibility loci for Crohn's disease in Koreans. Inflamm Bowel Dis. 2015;21:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamazaki K, Umeno J, Takahashi A, et al. A genome‐wide association study identifies 2 susceptibility loci for Crohn's disease in a Japanese population. Gastroenterology. 2013;144:781‐788. [DOI] [PubMed] [Google Scholar]
- 21. Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McGovern DP, Kugathasan S, Cho JH. Genetics of inflammatory bowel diseases. Gastroenterology. 2015;149:1163‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee S, Abecasis GR, Boehnke M, Lin X. Rare‐variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare‐variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang MH, Sun R, Guo J, et al. A fast and powerful W‐test for pairwise epistasis testing. Nucleic Acids Res. 2016;44:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics. 2010;26:2336‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. GTEx Consortium . The Genotype‐Tissue Expression (GTEx) project. Nat Genet, 2013;45:580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laurie CC, Doheny KF, Mirel DB, et al. Quality control and quality assurance in genotypic data for genome‐wide association studies. Genet Epidemiol. 2010;34:591‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720‐727. [DOI] [PubMed] [Google Scholar]
- 30. Hunt R, Sauna ZE, Ambudkar SV, Gottesman MM, Kimchi‐Sarfaty C. Silent (synonymous) SNPs: should we care about them? Methods Mol Biol. 2009;578:23‐39. [DOI] [PubMed] [Google Scholar]
- 31. Kramarova TV, Antonicka H, Houstek J, Cannon B, Nedergaard J. A sequence predicted to form a stem‐loop is proposed to be required for formation of an RNA‐protein complex involving the 3′UTR of beta subunit F0F1‐ATPase mRNA. Biochim Biophys Acta. 2008;1777:747‐757. [DOI] [PubMed] [Google Scholar]
- 32. Battle DJ, Doudna JA. The stem‐loop binding protein forms a highly stable and specific complex with the 39 stem‐loop of histone mRNA. RNA. 2001;7:123‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yao HP, Zhou YQ, Zhang R, Wang MH. MSP‐RON signalling in cancer: pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13:466‐481. [DOI] [PubMed] [Google Scholar]
- 34. Wang MH, Skeel A, Leonard EJ. Proteolytic cleavage and activation of pro‐macrophage‐stimulating protein by resident peritoneal macrophage membrane proteases. J Clin Invest. 1996;97:720‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wellcome Trust Case Control Consortium . Genome‐wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kauder SE, Santell L, Mai E, et al. Functional consequences of the macrophage stimulating protein 689C inflammatory bowel disease risk allele. PLoS ONE. 2013;8:e83958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gorlatova N, Chao K, Pal LR, et al. Protein characterization of a candidate mechanism SNP for Crohn's disease: the macrophage stimulating protein R689C substitution. PLoS ONE. 2011;6:e27269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu JZ, Anderson CA. Genetic studies of Crohn's disease: past, present and future. Best Pract Res Clin Gastroenterol. 2014;28:373‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cuthbert AP, Fisher SA, Mirza MM, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology. 2002;122:867‐874. [DOI] [PubMed] [Google Scholar]
- 41. Folwaczny C. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Z Gastroenterol. 1998;36:863‐866.9795417 [Google Scholar]
- 42. Cavanaugh J; IBD International Genetics Consortium . International collaboration provides convincing linkage replication in complex disease through analysis of a large pooled data set: Crohn disease and chromosome 16. Am J Hum Genet. 2001;68:1165‐1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li M, Gao X, Guo CC, Wu KC, Zhang X, Hu PJ. OCTN and CARD15 gene polymorphism in Chinese patients with inflammatory bowel disease. World J Gastroenterol. 2008;14:4923‐4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang JX, Song J, Wang J, Dong WG. JAK2 rs10758669 polymorphisms and susceptibility to ulcerative colitis and Crohn's disease: a meta‐analysis. Inflammation. 2014;37:793‐800. [DOI] [PubMed] [Google Scholar]
- 45. Danese S, Grisham M, Hodge J, Telliez JB. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310:G155‐G162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Festen EA, Stokkers P, Wapenaar MC, et al. Genetic and mucosal expression analysis shows important roles for JAK2 and MST1 in the pathogenesis of ulcerative colitis and identifies more susceptibility loci. Gastroenterology. 2009;136:A38‐A38. [Google Scholar]
- 47. Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32:597‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ananthakrishnan AN, Cagan A, Cai T, et al. Common genetic variants influence circulating vitamin D levels in inflammatory bowel diseases. Inflamm Bowel Dis. 2015;21:2507‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ardesia M, Ferlazzo G, Fries W. Vitamin D and inflammatory bowel disease. Biomed Res Int. 2015;2015:470805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang TT, Dabbas B, Laperriere D, et al. Direct and indirect induction by 1, 25‐dihydroxyvitamin D3 of the NOD2/CARD15‐defensin β2 innate immune pathway defective in Crohn disease. J Biol Chem. 2010;285:2227‐2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Verway M, Behr MA, White JH. Vitamin D, NOD2, autophagy and Crohn's disease. Exp Rev Clin Immunol. 2010;6:505‐508. [DOI] [PubMed] [Google Scholar]
- 53. Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64:1082‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials