

Abstract
Post-menopausal osteoporosis is one of the most common bone diseases in women. The aim of the present study was to predict the diagnostic function modules from a differential co-expression gene network in order to enhance the current understanding of the biological processes and to promote the early prevention and intervention of post-menopausal osteoporosis. The diagnostic function modules were extracted from a differential co-expression network by the established protein-protein interaction (PPI) network analysis. First, significant genes were identified from the differential co-expression network, which were regarded as seed genes. Starting from the seed genes, the sub-networks in this disease, referred to as diagnostic function modules, were exhaustively searched and prioritized through a snowball sampling strategy to identify genes to accurately predict clinical outcomes. In addition, crucial function inference was performed for each diagnostic function module. Based on the microarray and PPI data, the differential co-expression network was constructed, which contained 1,607 genes and 4,197 interactions. A total of 110 seed genes were identified, and nine diagnostic modules that accurately distinguished post-menopausal osteoporosis from healthy controls were screened out from these seed genes. The diagnostic modules may be associated with five functional pathways with emphasis on metabolism. A total of nine diagnostic functional modules screened in the present study may be considered as potential targets for predicting the clinical outcomes of post-menopausal osteoporosis, and may contribute to the early diagnosis and therapy of osteoporosis.
Keywords: post-menopausal women, osteoporosis, network, module, pathway
Introduction
Osteoporosis is a common bone disease characterized by reduced bone strength, usually occurring in post-menopausal women and the elderly (1). Post-menopausal osteoporosis (PO) is regarded as a chronic autoimmune and inflammatory disorder, which may lead to destructive fracture and even death (2). The high societal and personal costs of osteoporosis pose challenges to public health and physicians, and of note, most patients with osteoporosis currently remain untreated (3). In addition, ~20% of patients with a fragility require to receive therapy within one year following fracture for reducing future fracture (4). Thus, early diagnostics and early intervention would significantly control the development of PO. However, existing preventive measures and therapies mainly focus on physical activity, avoidance of smoking and excessive alcohol intake, calcium and vitamin D supplementation, and anti-absorption or anabolic pharmacological agents (1,2). Therefore, further understanding the biological processes involved in the development of PO may lead to the discovery of more effective methods to prevent and treat PO.
It has been reported that the progression of various diseases is always associated with the aberrant regulation of a set of genes, and biological networks are widely used to analyse the genetic levels of complex human diseases (5). Network-based approaches are often built on the knowledge of physical or functional interactions between molecules that are usually illustrated as an interaction network (5). It may be used to understand the effects of disease-gene interconnections that have multiple potential biological and clinical applications, and may thus offer better targets for drug development (6,7). Multiple computational methods, including Network-Guided Forests (8) and Support Vector Machine (9), have been applied to identify significant sub-network markers that predict the classification of microarray samples. However, the existing methods are largely heuristic, and the final output and definition of the output sub-networks are ambiguous without any formal topological features. Previous studies have indicated that the use of the sub-network method provides significant genes associated with disease function and may also accurately predict clinical outcomes (10,11). Jordán et al (12) indicated that network methods have an important role in inferring novel disease genes and supporting prediction in pathogenesis studies.
In the present study, to obtain the diagnostic function modules from a differential co-expression network, the crucial genes referred to as seed genes were identified from this network. Subsequently, sub-networks starting from the seed nodes were searched and prioritized by using the snowball sampling strategy. The sub-networks may be used to predict the clinical outcomes, and each sub-network was given a most significant functional inference. In the present study, each sub-network represents a diagnostic functional module, and in those, a number of novel biomarkers for PO with predictive value regarding the phenotypic outcome may be identified, which may contribute to the clinical treatment of PO.
Materials and methods
Microarray data
For performing the bioinformatics analysis on PO, microarray data for PO were retrieved from the Array Express database (http://www.ebi.ac.uk/arrayexpress/) under the accession no. E-GEOD-56116. The microarray data were assessed on the A-AGIL-28-Agilent Whole Human Genome Microarray 4×44K 014850 G4112F (85 colsx532 rows) platform (13). A total of 13 samples were contained in the dataset E-GEOD-56116, comprising those of 10 patients with PO and 3 healthy post-menopausal women, from whom peripheral blood was used to extract the RNA samples. The age range of the disease group and the control group in this dataset was 52–68 and 56–68 years, respectively.
All microarray data and annotation files were downloaded for subsequent analysis. Prior to analysis, standard data pre-processing was performed on the gene expression data using the robust multichip average method (14), the quantiles-based algorithm (15) and the Micro Array Suite 5.0 (MAS 5.0) algorithm (16). The gene expression profile at the probe level was converted into gene symbols and the duplicated symbols were erased. From the microarray dataset, a total of 11,843 genes and their corresponding expression information were obtained.
Differential co-expression network
The present study attempted to identify diagnostic biomarkers from a network approach. For this, the global human protein-protein interactions (PPIs) comprising 1,048,576 interactions were first obtained from the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (http://string-db.org/). A total of 787,896 interactions (covering 16,730 genes) were then obtained by removing feedback loops and duplicated interactions. To map genes from the microarray dataset onto the PPI network, the common ones, including 22,728 interactions (covering 4,985 genes) were extracted out.
Based on the co-expression analysis and differential expression analysis successively, the differential co-expression network was constructed. To characterize the interactions of the PPI network, the Pearson correlation coefficients (PCC) of the PPIs associated with certain diseases may be calculated (17). The PCC is a measure of the correlation between two variables, with assigned values between −1 and +1. In general, the larger the absolute value of the PCC, the greater the intensity of the interaction between the two proteins. In present study, interactions with |PCC|>0.8 were retained to construct the co-expression network. EdgeR (18), a Bioconductor package for differential expression analysis of digital gene expression data, was utilized to assign a weight value to each interaction for weighting the degree of the co-expression and differential expression of the edges. In this case, a differential co-expression network with each edge was assigned a weight value.
Statistical analysis
In the present study, SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) were used to perform the statistical analysis. The P-value that evaluates significant differences was adjusted by the false discovery rate procedure based on the Benjamini and Hochberg method (19). Furthermore, a two-tailed t-test and Fisher's exact test were performed to determine the statistical significance. P<0.05 was considered to indicate a statistically significant difference.
Seed gene prediction
To determine the seed genes from the differential co-expression network, a degree centrality analysis of the network was first performed for obtaining the degree value of each gene. Based on the degree value, a z-score (6) was given for each gene in the adjacency matrix of the network. The significance of genes in the differential co-expression network was measured according to the z-scores. All genes were ranked in descending order according to the z-score values. The top 5% genes were considered as seed genes.
Diagnostic module identification
In the present study, the diagnostic modules were identified with a snowball sampling strategy introduced by Goodman (20), which uses the network structure itself to generate a sample of nodes in the network. From each seed gene, a candidate diagnostic module search was performed by seed gene amplification. In particular, the genes were iteratively involved, thus leading to the largest increase in the prediction accuracy model until the accuracy of the prediction was diminished. To evaluate the accuracy of the prediction, the area under the receiver operating characteristic curve (AUC) was employed as a measure. Modules with AUC=1 and number of genes ≥5 were considered as candidate diagnostic modules.
Subsequently, a permutation test was implemented to determine the statistical significance of these candidate diagnostic modules. First, a stochastic network was constructed based on the null score distribution of the candidate diagnostic modules, and a module search analysis based on the randomized networks was performed according to the above method. Here, each module was completely randomized for 1,000 permutations. Each module was assigned an AUC value in each permutation, and the empirical P-value of a candidate diagnostic module was determined by the probability of the module having a smaller AUC than that of the randomized network. The modules whose adjusted P-values were <0.05 were considered as diagnostic modules.
Functional analysis
Normally, co-expression genes tend to be involved in similar biological pathways. Thus, the present study investigated the functional pathways of the diagnostic modules. Reactome (http://www.reactome.org/), a confirmed pathway database, was employed to capture biological pathways associated with PO. From Reactome, a total of 1,675 original human pathways were downloaded. Pathways with too many genes may be too complex to understand, while pathways with too few genes may not have sufficient biological content (21); thus the pathways whose number of genes was >100 or <5 were removed from the study. Subsequently, pathway enrichment analysis of genes in PPI networks was performed for each diagnostic module. In the present study, to further investigate the biological functions of the diagnostic modules, data obtained from the Reactome database were entered into Genelibs (genelibs.com/gb/com/index.html) for analysis of enrichment pathways. Genelibs is a data platform that provides a collection of biological information as well as tools for processing and analysis. Therefore, P-values of the pathways were evaluated in Genelibs and significant pathways with adjusted P<0.05 and diagnostic module pathways with the minimum adjusted P-value were screened out.
Results
Differential co-expression network
Prior to analysis, microarray and PPI data were separately obtained from the open-access databases. A total of 11,843 genes were included in the microarray data and 787,896 interactions (covering 16,730 genes) were contained in the PPI network. For mapping genes from the microarray dataset onto the PPI network, the common ones, including 22,728 interactions (covering 4,985 genes), were extracted out. The differential co-expression network was then constructed based on co-expression analysis and differential expression analysis successively, which contained 4,697 interactions and 2,216 nodes. Fig. 1 displays the major network, including 1,607 genes and 4,197 interactions.
Figure 1.

Differential co-expression network constructed based on co-expression analysis and successive differential expression analysis. The major network contained 1,607 genes and 4,197 interactions. The seed genes are highlighted in yellow.
Seed genes
To determine the seed genes, the degree of centrality of the differential co-expression network was analysed and the z-scores were introduced to measure the significance of genes in the network. All genes were ranked in descending order of their z-scores, and the top 5% genes were considered as seed genes. Fig. 2 illustrates the gene distribution by z-scores. A total of 110 seed genes were identified, of which RNA polymerase II subunit I had the highest z-score value (z-score=142.41).
Figure 2.
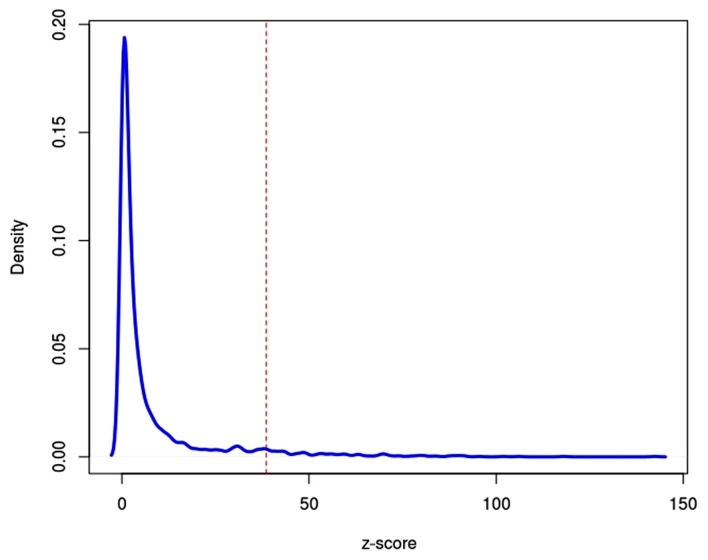
Distribution of z-scores of genes in the differential co-expression network. The integral under the curve to the right of the red dashed linerepresents seed genes with the top 5% z-scores.
Diagnostic modules
Diagnostic modules (sub-networks) with maximum classification accuracy were screened from the differential co-expression network based on the seed genes prediction and diagnostic modules identification. Certain hidden genes that had no significance by themselves, but were clustered in a sub-network module whose genes are highly predictive of the disease status were identified by searching the diagnostic modules by seed gene expansion. Starting from the 110 seed genes, a total of 110 candidate diagnostic modules were identified. Based on the cut off values of AUC=1 and number of genes ≥5, 9 diagnostic modules were identified and named as Modules 1–9 (Fig. 3). The seed genes of these modules were adenylate kinase 6 (AK6), proteasome 26S subunitATPase 5 (PSMC5), TATA-box binding protein associated factor 12 (TAF12), splicing factor 3b subunit 4 (SF3B4), U2 small nuclear RNA auxiliary factor 2 (U2AF2), cleavage and polyadenylation factor I subunit 1 (CLP1), proteasome subunit α5 (PSMA5), NADH:Ubiquinone oxidoreductase complex assembly factor 1 (NDUFAF1) and Y-box binding protein 1 (YBX1), respectively. From Table I, it is apparent that all diagnostic modules have a classification capability of 1, indicating that these diagnostic modules are able to accurately distinguish samples from patients with PO from those from healthy women.
Figure 3.

Diagnostic modules that accurately distinguish samples with post-menopausal osteoporosis from healthy samples. Yellow nodes represent seed genes. RBBP5, RB binding protein 5; GADD45A, growth arrest and DNA damage inducible α; TADA1, transcriptional adaptor 1; TAF6L, TATA-box binding protein associated factor 6 like; PSMA4, proteasome subunit α4; CCND1, cyclin D1; SELL, selectin L; PSMD5, proteasome 26S subunit, non-ATPase 5; YBX3, Y-box binding protein 3; NDUFA5, NADH:Ubiquinone oxidoreductase complex assembly factor 5; NDUFB3, NADH:Ubiquinone oxidoreductase subunit B3; NDUFB5, NADH:Ubiquinone oxidoreductase subunit B5; SNRPD3, small nuclear ribonucleoprotein D3; CHERP, calcium homeostasis endoplasmic reticulum protein; SNRPA1, small nuclear ribonucleoprotein polypeptide A; SRSF6, serine and arginine rich splicing factor 6; SNRPB2, small nuclear ribonucleoprotein polypeptide B2; NUP107, nucleoporin 107; CD2BP2, CD2 cytoplasmic tail binding protein 2; NCBP2, nuclear cap binding protein subunit 2; CCND2, cyclin D2; NGF, nerve growth factor; CREBBP, CREB binding protein; EP300, E1A binding protein p300; SNRPD3, small nuclear ribonucleoprotein D3.
Table I.
Details of diagnostic modules and functional pathways in post-menopausal osteoporosis.
| Module no. | Seed gene | z-score | Number of genes | Enriched pathway | Adjusted P-value of the pathway |
|---|---|---|---|---|---|
| 1 | AK6 | 69.94 | 15 | Histone acetylation by HATs | 6.86×10−4 |
| 2 | PSMC5 | 58.07 | 16 | Ubiquitin-dependent degradation of cyclin D | 6.55×10−6 |
| 3 | TAF12 | 49.20 | 15 | Ubiquitin-dependent degradation of cyclin D | 2.21×10−9 |
| 4 | SF3B4 | 38.79 | 15 | Degradation of β-catenin by the destruction complex | 5.96×10−6 |
| 5 | U2AF2 | 49.03 | 16 | mRNA splicing | 9.96×10−10 |
| 6 | CLP1 | 48.49 | 15 | Processing of capped intron-containing Pre-mRNA | 3.38×10−9 |
| 7 | PSMA5 | 43.00 | 15 | Histone acetylation by HATs | 3.39×10−5 |
| 8 | NDUFAF1 | 40.49 | 15 | Processing of capped intron-containing pre-mRNA | 3.38×10−9 |
| 9 | YBX1 | 40.19 | 15 | mRNA Splicing | 9.96×10−10 |
AK6, adenylate kinase 6; PSMC5, proteasome 26S subunit ATPase 5; TAF12, TATA box-binding protein-associated factor 12; SF3B4, splicing factor 3B subunit 4; U2AF2, U2 small nuclear ribonucleoprotein auxiliary factor; CLP1, cleavage and polyadenylation factor I subunit 1; PSMA5, proteasome subunit α5; NDUFAF1, NADH-ubiquinone oxidoreductase (complex I); YBX1, Y box binding protein 1; HATs, histone acetyltransferases.
Pathway analysis
After determining the diagnostic modules, the present study further investigated the functional pathways of these diagnostic modules. Based on the Reactome database, the pathways with number of genes of <5 and >100 were removed, and consequently, a total of 1,002 pathways were obtained. Fisher's exact test was utilized to determine the significance of the pathways, and the pathway with the minimum adjusted P-value was considered as the diagnostic module pathway. Each diagnostic module mapped an important pathway (Table I). Eventually, Modules 1 and 7 were identified to be enriched in histone acetyltransferases (HATs), while Modules 2 and 3 were enriched in ubiquitin-dependent degradation of cyclin D, and Modules 5 and 9 were enriched in mRNA splicing. Furthermore, Modules 6 and 8 were enriched in processing of capped intron-containing Pre-mRNA. The 9 diagnostic modules were associated with 5 pathways, most of which were metabolic signalling pathways.
Discussion
It has been largely accepted that osteoporosis is associated with age as well as environmental and genetic factors, which appear to have an important role in regulating bone mineral density and affect the quantitative ultrasound properties of bone (measures of bone structure components, including broadband ultrasound attenuation and speed of sound), skeletal geometry, bone turnover and the pathogenesis of osteoporosis itself (22,23). In the past few years, numerous studies have been performed among different populations using linkage and association approaches to identify effective biomarkers, including oestrogen receptor 2, tumour necrosis factor-α and interleukin-10, which may increase an individual's susceptibility to PO (24,25). PO remains a difficult problem in the medical field due to the silent symptoms and trivialization by post-menopausal women and doctors (26). To explore markers for classifying different disease states or predicting clinical outcomes, previous and current research focuses on identifying genes that are differentially expressed between different phenotypes of various diseases (27,28). Subsequent analysis has focused on Kyoto Encyclopaedia of Genes and Genomes pathway enrichment assays to uncover the possible enrichment pathways signature of these genes (29). However, gene signatures based solely on expression data are not sufficiently reliable to understand the effects of disease-gene interconnections and disease pathways (30). Therefore, network-based strategies have been largely used because of their broad potential in a variety of biological and clinical applications (31).
In the present study, a differential co-expression network approach was used to identify the diagnostic function modules of PO, which has not been previously assessed for PO, to the best of our knowledge. A dataset associated with PO (E-GEOD-56116) was selected, which is different from those in previous studies. The present study aimed to identify important biomarkers associated with PO for earlier diagnosing and treating PO. To predict the clinical outcomes of PO from a network perspective, diagnostic modules that are functionally associated with numerous differentially expressed genes and contributed to the predictive phenotype of PO were identified. From this network, a total of 9 diagnostic modules were retrieved to accurately distinguish samples of PO patients with those from healthy women. These diagnostic modules involve 5 functional pathways. For instance, Modules 1 and 7 are associated with the histone acetylation through HATs pathway. It is generally accepted that epigenetic regulation has a key role in the oestrogenic differentiation of mesenchymal stem cells. Recent studies have indicated that histone modifications have an important role in the pathological phenotype of defective osteogenesis of bone marrow stromal cells (32,33). Zhang et al (34) revealed that the knockdown of the HAT p300/(CREB binding protein) associated factor significantly reduced bone formation in vivo and in vitro, indicating that histone acetylation may be involved in osteoporosis. The HAT GCN5 was also reported to regulate the osteogenic effect of mesenchymal stem cells in a nuclear factor-κB-dependent way (35). Histone deacetylase inhibitors, including trichostatin A and suberoylanilide hydroxamic acid, have been reported to promote osteoblast maturation by inducing histone acetylation (36,37). Furthermore, Alao et al (38) indicated that a histone deacetylase inhibitor induces ubiquitin-dependent degradation of cyclin D1. In another study by Alao et al (39), a histone deacetylase inhibitor was proven to promote proteasomal degradation of cyclin D1 and repress oestrogen receptor α-dependent transcription. It is well known that oestrogen has essential roles in maintaining bone density and protecting against PO. Furthermore, oestrogen may induce the expression of cyclin D in primary osteoblasts (40). In line with this, in the present study, ubiquitin-dependent degradation of cyclin D was a diagnostic module pathway (Modules 2 and 3). Degradation of β-catenin by the destruction complex is a diagnostic pathway from Module 4. Numerous studies have indicated the association between β-catenin and PO (41–44). In the present study, the diagnostic module-associated pathways were in accordance with the pathological process of PO, which indirectly indicates the feasibility of the application of the present method.
It has been reported that genes in diagnostic modules may be used to effectively predict disease state (43). In the present study, all 9 diagnostic modules displayed good classification performances with AUC=1, indicating that these diagnostic modules were able to accurately distinguish samples from patients with PO from those from healthy women. Clinically, combined detection of genes in diagnostic modules may contribute to the early prediction of PO. Furthermore, diagnostic modules and functional pathways may provide clues for the identification of potential therapeutic targets for PO.
In conclusion, osteoporosis is a common and serious disease among the elderly, particularly in older women. At present, available therapies are only aimed at preventing further bone decay. A better understanding of the biological processes involved in the development of PO may be helpful in its early prevention and intervention. In the present study, 9 diagnostic modules were successfully identified, which were able to accurately distinguish PO from healthy controls. These diagnostic modules are associated with 5 functional pathways, most of which are associated with material metabolism. The results indicated that this module search approach is suitable for the analysis of PO, and these functional diagnostic modules may be considered as potential targets for predicting the clinical outcomes of PO, which is helpful for the early diagnosis and therapy of osteoporosis. As the number of samples in the present study was low, the results obtained only provide a strategy for further exploring the development of PO. In addition, it is still required to validate the results in animal or patient tissue experiments in future studies.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, Lindsay R, National Osteoporosis Foundation Clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25:2359–2381. doi: 10.1007/s00198-014-2794-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black DM, Rosen CJ. Postmenopausal osteoporosis. N Engl J Med. 2016;374:2096–2097. doi: 10.1056/NEJMcp1513724. [DOI] [PubMed] [Google Scholar]
- 3.Kanis J, McCloskey E, Johansson H, Cooper C, Rizzoli R, Reginster J-Y. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2013;24:23–57. doi: 10.1007/s00198-012-2074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haaland DA, Cohen DR, Kennedy CC, Khalidi NA, Adachi JD, Papaioannou A. Closing the osteoporosis care gap-increased osteoporosis awareness among geriatrics and rehabilitation teams. BMC Geriatrics. 2009;9:28. doi: 10.1186/1471-2318-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho DY, Kim YA, Przytycka TM. Chapter 5: Network biology approach to complex diseases. PLoS Comput Biol. 2012;8:e1002820. doi: 10.1371/journal.pcbi.1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Xuan J, Riggins RB, Wang Y, Clarke R. Identifying protein interaction subnetworks by a bagging Markov random field-based method. Nucleic Acids Res. 2013;41:e42–e42. doi: 10.1093/nar/gks951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nie Y, Yu J. Mining breast cancer genes with a network based noise-tolerant approach. BMC Syst Biol. 2013;7:49. doi: 10.1186/1752-0509-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutkowski J, Ideker T. Protein networks as logic functions in development and cancer. PLoS Comput Biol. 2011;7:e1002180. doi: 10.1371/journal.pcbi.1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Shen X, Pan W. Network-based support vector machine for classification of microarray samples. BMC Bioinformatics. 2009;10:S21. doi: 10.1186/1471-2105-10-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang R, Bai Y, Qin Z, Yu T. EgoNet: Identification of human disease ego-network modules. BMC Genomics. 2014;15:314. doi: 10.1186/1471-2164-15-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayati M, Erten S, Chance MR, Koyutürk M. MOBAS: Identification of disease-associated protein subnetworks using modularity-based scoring. EURASIP J Bioinform Syst Biol. 2015;2015:7. doi: 10.1186/s13637-015-0025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordán F, Nguyen T-P, Liu W-c. Studying protein-protein interaction networks: A systems view on diseases. Brief Funct Genomics. 2012;11:497–504. doi: 10.1093/bfgp/els035. [DOI] [PubMed] [Google Scholar]
- 13.Xie LH, Zhao YP, Chen K, Lai YL. Bioinformatics analysis of genes expression profiles of postmenopausal osteoporosis with Kidney Yin deficiency. J Clin Rehab Tissue Engi Res. 2011;15:2833–2837. (In Chinese) [Google Scholar]
- 14.Ma L, Robinson LN, Towle HC. ChREBP• Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem. 2006;281:28721–28730. doi: 10.1074/jbc.M601576200. [DOI] [PubMed] [Google Scholar]
- 15.Rifai N, Ridker PM. Proposed cardiovascular risk assessment algorithm using high-sensitivity C-reactive protein and lipid screening. Clin Chem. 2001;47:28–30. [PubMed] [Google Scholar]
- 16.Pepper SD, Saunders EK, Edwards LE, Wilson CL, Miller CJ. The utility of MAS5 expression summary and detection call algorithms. BMC Bioinformatics. 2007;8:273. doi: 10.1186/1471-2105-8-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benesty J, Chen J, Huang Y, Cohen I. Noise reduction in speech processing. Springer; New York, NY: 2009. Pearson correlation coefficient; pp. 1–4. [DOI] [Google Scholar]
- 18.Robinson MD, McCarthy DJ, Smyth GK. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57:289–300. [Google Scholar]
- 20.Goodman LA. Snowball sampling. Ann Math Statist. 1961;32:148–170. doi: 10.1214/aoms/1177705148. [DOI] [Google Scholar]
- 21.Ahn T, Lee E, Huh N, Park T. Personalized identification of altered pathways in cancer using accumulated normal tissue data. Bioinformatics. 2014;30:i422–i429. doi: 10.1093/bioinformatics/btu449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ralston SH. Genetic control of susceptibility to osteoporosis. J Clin Endocrinol Metab. 2002;87:2460–2466. doi: 10.1210/jcem.87.6.8621. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, Mahendra G, Nagy TR, Ponnazhagan S. Osteogenic differentiation of recombinant adeno-associated virus 2-transduced murine mesenchymal stem cells and development of an immunocompetent mouse model for ex vivo osteoporosis gene therapy. Hum Gene Ther. 2004;15:1197–1206. doi: 10.1089/hum.2004.15.1197. [DOI] [PubMed] [Google Scholar]
- 24.Kotrych D, Dziedziejko V, Safranow K, Sroczynski T, Staniszewska M, Juzyszyn Z, Pawlik A. TNF-alpha and IL10 gene polymorphisms in women with postmenopausal osteoporosis. Eur J Obstet Gynecol Reprod Biol. 2016;199:92–95. doi: 10.1016/j.ejogrb.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 25.Rivadeneira F, van Meurs JB, Kant J, Zillikens MC, Stolk L, Beck TJ, Arp P, Schuit SC, Hofman A, Houwing-Duistermaat JJ, et al. Estrogen receptor beta (ESR2) polymorphisms in interaction with estrogen receptor alpha (ESR1) and insulin-like growth factor I (IGF1) variants influence the risk of fracture in postmenopausal women. J Bone Miner Res. 2006;21:1443–1456. doi: 10.1359/jbmr.060605. [DOI] [PubMed] [Google Scholar]
- 26.Alami S, Hervouet L, Poiraudeau S, Briot K, Roux C. Barriers to effective postmenopausal osteoporosis treatment: A qualitative study of patients' and practitioners' views. PLoS One. 2016;11:e0158365. doi: 10.1371/journal.pone.0158365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratford JK, Bentrem DJ, Anderson JM, Fan C, Volmar KA, Marron JS, Routh ED, Caskey LS, Samuel JC, Der CJ, et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010;7:e1000307. doi: 10.1371/journal.pmed.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD, Tsao MS, Penn LZ, Jurisica I. Prognostic gene signatures for non-small-cell lung cancer; Proc Natl Acad Sci; 2009; pp. 2824–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: A network-based approach to human disease. Nat RevGenet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang YX, Sun HL, Liang H, Li K, Fan QM, Zhao QH. Dynamic and distinct histone modifications of osteogenic genes during osteogenic differentiation. J Biochem. 2015;158:445–457. doi: 10.1093/jb/mvv059. [DOI] [PubMed] [Google Scholar]
- 33.Deng P, Chen QM, Hong C, Wang CY. Histone methyltransferases and demethylases: Regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int J Oral Sci. 2015;7:197–204. doi: 10.1038/ijos.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang P, Liu Y, Jin C, Zhang M, Lv L, Zhang X, Liu H, Zhou Y. Histone H3K9 acetyltransferase PCAF is essential for osteogenic differentiation through bone morphogenetic protein signaling and may be involved in osteoporosis. Stem Cells. 2016;34:2332–2341. doi: 10.1002/stem.2424. [DOI] [PubMed] [Google Scholar]
- 35.Zhang P, Liu Y, Jin C, Zhang M, Tang F, Zhou Y. Histone acetyltransferase GCN5 regulates osteogenic differentiation of mesenchymal stem cells by inhibiting NF-κB. J Bone Miner Res. 2016;31:391–402. doi: 10.1002/jbmr.2704. [DOI] [PubMed] [Google Scholar]
- 36.Schroeder TM, Westendorf JJ. Histone deacetylase inhibitors promote osteoblast maturation. J Bone Miner Res. 2005;20:2254–2263. doi: 10.1359/JBMR.050813. [DOI] [PubMed] [Google Scholar]
- 37.Dudakovic A, Evans JM, Li Y, Middha S, McGee-Lawrence ME, van Wijnen AJ, Westendorf JJ. Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. J Biol Chem. 2013;288:28783–28791. doi: 10.1074/jbc.M113.489732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alao JP, Stavropoulou AV, Lam EW, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol Cancer. 2006;5:8. doi: 10.1186/1476-4598-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alao JP, Lam EW, Ali S, Buluwela L, Bordogna W, Lockey P, Varshochi R, Stavropoulou AV, Coombes RC, Vigushin DM. Histone deacetylase inhibitor trichostatin A represses estrogen receptor alpha-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin Cancer Res. 2004;10:8094–8104. doi: 10.1158/1078-0432.CCR-04-1023. [DOI] [PubMed] [Google Scholar]
- 40.Fujita M, Urano T, Horie K, Ikeda K, Tsukui T, Fukuoka H, Tsutsumi O, Ouchi Y, Inoue S. Estrogen activates cyclin-dependent kinases 4 and 6 through induction of cyclin D in rat primary osteoblasts. Biochem Biophys Res Commun. 2002;299:222–228. doi: 10.1016/S0006-291X(02)02640-2. [DOI] [PubMed] [Google Scholar]
- 41.Tian J, Xu XJ, Shen L, Yang YP, Zhu R, Shuai B, Zhu XW, Li CG, Ma C, Lv L. Association of serum Dkk-1 levels with β-catenin in patients with postmenopausal osteoporosis. J Huazhong Univ Sci Technol Med Sci. 2015;35:212–218. doi: 10.1007/s11596-015-1413-6. [DOI] [PubMed] [Google Scholar]
- 42.Urano T, Inoue S. Recent genetic discoveries in osteoporosis, sarcopenia and obesity. Endocr J. 2015;62:475–484. doi: 10.1507/endocrj.EJ15-0154. [DOI] [PubMed] [Google Scholar]
- 43.Wolski H, Drweska-Matelska N, Seremak-Mrozikiewicz A, Lowicki Z, Czerny B. The role of Wnt/β-catenin pathway and LRP5 protein in metabolism of bone tissue and osteoporosis etiology. Ginekol Pol. 2015;86:311–314. doi: 10.17772/gp/2079. [DOI] [PubMed] [Google Scholar]
- 44.Ma C, Shuai B, Shen L, Yang YP, Xu XJ, Li CG. Serum carcinoembryonic antigen-related cell adhesion molecule 1 level in postmenopausal women: Correlation with β-catenin and bone mineral density. Osteoporos Int. 2016;27:1529–1535. doi: 10.1007/s00198-015-3408-3. [DOI] [PubMed] [Google Scholar]