

Abstract
Matrix metalloproteinase (MMP)-9 increases in the myocardium with advanced age and after myocardial infarction (MI). Because young transgenic (TG) mice overexpressing human MMP-9 only in macrophages show better outcomes post-MI, whereas aged TG mice show a worse aging phenotype, we wanted to evaluate the effect of aging superimposed on MI to see if the detrimental effect of aging counteracted the benefits of macrophage MMP-9 overexpression. We used 17- to 28-mo-old male and female C57BL/6J wild-type (WT) and TG mice (n = 10–21 mice/group) to evaluate the effects of aging superimposed on MI. Despite similar infarct areas and mortality rates at day 7 post-MI, aging TG mice showed improved diastolic properties and remodeling index compared with WT mice (both P < 0.05). Macrophage numbers were higher in TG than WT mice at days 0 and 7 post-MI, and the post-MI increase was due to elevated cluster of differentiation 18 protein levels (all P < 0.05). RNA sequencing analysis of cardiac macrophages isolated from day 7 post-MI infarcts identified 1,276 statistically different (all P < 0.05) genes (994 increased and 282 decreased in TG mice). Reduced expression of vascular endothelial growth factor A, platelet-derived growth factor subunit A, and transforming growth factor-β3, along with elevated expression of tissue inhibitor of MMP-4, in macrophages revealed mechanisms of indirect downstream effects on fibroblasts and neovascularization. While collagen accumulation was enhanced in TG mice compared with WT mice at days 0 and 7 post-MI (P < 0.05 for both), the post-MI collagen cross-linking ratio was higher in WT mice (P < 0.05), consistent with increased diastolic volumes. Vessel numbers [by Griffonia (Bandeiraea) simplicifolia lectin I staining] were decreased in TG mice compared with WT mice at days 0 and 7 post-MI (P < 0.05 for both). In conclusion, macrophage-derived MMP-9 improved post-MI cardiac wound healing through direct and indirect mechanisms to improve diastolic physiology and remodeling.
NEW & NOTEWORTHY Aging mice with macrophage overexpression of matrix metalloproteinase-9 have increased macrophage numbers 7 days after myocardial infarction, resulting in improved diastolic physiology and left ventricular remodeling through effects on cardiac wound healing.
Keywords: angiogenesis, cardiac wound healing, collagen, left ventricular physiology, RNA sequencing
INTRODUCTION
Each year, one million Americans are diagnosed with myocardial infarction (MI), and the risk for MI increases with age (4, 48). The physiology of the left ventricle (LV) is impaired progressively by aging due to increased myocardial stiffness and by MI due to cardiac remodeling (17, 34, 42, 45). LV stiffness is caused by increased collagen deposition, increased extracellular matrix (ECM) cross-linking, or reduced ECM degradation by matrix metalloproteinases (MMPs) (63). MMPs are a family of zinc-dependent proteases that regulate ECM turnover by processing a wide range of ECM components, including collagen, fibronectin, and laminin (62). In addition, MMP-9 can cleave a number of cytokines, growth factors, and other MMPs, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-8, endothelin-1, vascular endothelial growth factor A (VEGFA), transforming growth factor-β (TGF-β), osteopontin, MMP-2, and MMP-13 (5, 14, 39, 57).
MMP-9 is robustly expressed by macrophages in LV tissue and in neutrophils, endothelial cells, and myocytes (19, 26, 29, 46). MMP-9 concentrations increase with age in various tissues, including plasma and the LV, in humans and in preclinical animal models (7–9, 65, 66). Transgenic (TG) mice overexpressing human MMP-9 only in macrophages have increased age-related LV wall thickness, due to individual cardiomyocyte hypertrophy, and increased inflammation and collagen deposition (58). Adult TG mice show improved LV systolic physiology and reduced inflammation at day 5 post-MI compared with adult wild-type (WT) mice (68), suggesting that the timing, concentration, and setting of MMP-9 lead to divergent mechanisms of response. Because young TG mice showed better outcomes post-MI and aged TG mice showed a worse aging phenotype, we wanted to evaluate the effect of aging in combination with MI to see if the detrimental effect of aging counteracted the benefits of macrophage MMP-9 overexpression. This study also more closely mimics the clinical scenario, where MI patients are typically older.
MATERIALS AND METHODS
Animals.
All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center (50). The mouse colony was maintained in an American Association for Laboratory Animal Care-accredited vivarium. All mice were kept in the same room in a light-controlled environment with a 12:12-h light-dark cycle and had free access to water and standard mouse chow.
We used 17- to 28-mo-old male and female C57BL/6J WT and TG mice to examine macrophage-derived MMP-9 effects in the post-MI aged LV. The TG mouse was generated using human MMP-9 cDNA (2.4 kb) and driven by the scavenger receptor A enhancer/promoter (4.5 kb), as described elsewhere (3, 58, 68). We have previously reported no differences in mouse LV MMP-9 expression between WT and TG mice (63). Total MMP-9 gene expression increased 15 ± 2-fold in post-MI macrophages compared with WT levels.
MI surgery.
The left anterior descending coronary artery was ligated using a well-established MI minimally invasive surgical method (27, 69) to induce permanent occlusion in old WT and TG mice. Mice were checked daily for survival analysis. Autopsies were performed on all MI mice that died, and surviving MI mice were euthanized on day 7 post-MI.
Age-matched old WT and TG mice with no MI surgery at day 0 served as negative controls, and WT day 7 post-MI mice served as positive controls (27). To reduce animal use as recommended by the National Centre for the Replacement, Refinement, and Reduction of Animals in Research (54), the mice used for the day 0 groups and the WT MI group were randomly selected from previously published studies (58, 66, 67). The MI surgeries and tissue collection for day 0 and post-MI groups were performed over the same period of time by the same individuals (A. Yabluchanskiy and H. Toba). For this study, all echocardiograms were analyzed by a single observer (C. A. Meschiari), and all downstream tissue analysis was performed at the same time for all samples and by individuals blinded to groups.
Echocardiography.
Echocardiography was performed using the Vevo 2100 system (VisualSonics) to measure LV anatomy and physiology, as described elsewhere (44, 58, 66, 67). Images were acquired before MI surgery (baseline) and before euthanasia at day 7 post-MI and were acquired from the LV parasternal long-axis B-mode and short-axis M-mode at the midpapillary level.
Tissue harvest.
Mice were euthanized under anesthesia with 5% isoflurane in oxygen. Hearts flushed with cardioplegic solution were arrested in diastole for tissue collection and hearts flushed with saline were used for cell isolation, as previously described (9, 30, 40, 63, 67). The LV and lung were collected and weighed to assess edema. The LV was sliced into apex, middle, and base sections, stained with 1% 2,3,5-triphenyltetrazolium chloride (Sigma, St. Louis, MO) for 5 min, and photographed for infarct area measurements. The LV infarct (LVI) area was calculated using Photoshop (Adobe) and presented as a percentage of the infarct area relative to the total LV area (27). The LVI and remote (LVC) regions were separated and individually snap frozen and stored at −80°C for immunoblot analysis. The LV middle section was fixed in zinc-formalin and used for histological analysis.
Immunohistochemistry and histological staining.
The middle section of the LV was fixed in zinc-formalin, embedded in paraffin, and sectioned at 5 μm. Anti-Mac-3 antibody (catalog no. CL8943AP, Cedarlane, Burlington, ON, Canada) was used to detect macrophages, and biotinylated Griffonia (Bandeiraea) simplicifolia lectin I (GSL-I; catalog no. B-1105, Vector Laboratories, Burlingame, CA) was used to detect endothelial cells. Heat-mediated antigen retrieval was performed using target retrieval solution (catalog no. S1699, Dako, Carpinteria, CA). Sections were incubated with 3% H2O2 (Sigma) to block endogenous peroxidase activity and then blocked with rabbit serum. Sections were incubated overnight with anti-Mac-3 antibody (1:100 dilution) at 4°C and then incubated for 2 h with rabbit anti-rat IgG and ABC reagent (catalog no. PK-6104, Vector Laboratories) or biotinylated GSL-I (1:50 dilution) for 1 h at room temperature. The HistoMark Black peroxidase kit (Vector Laboratories) was used for positive staining of Mac-3 or GSL-I, and eosin was used as a counterstain. Sections were stained with picrosirius red to determine collagen content. After paraffin removal and tissue rehydration, sections were incubated in 0.2% phosphomolydbic acid, stained with 0.1% sirius red in saturated picric acid, and washed in 0.01 N hydrochloric acid. For image analysis, five random images of each slide were acquired across the LV free wall for day 0 groups or in the infarct region for day 7 post-MI groups. The percentage of the positive-stained area was calculated using Image-Pro software 7.0 (Media Cybernetics).
Protein extraction.
The apex and base were used to extract protein from the infarct region (LVI for day 7) or noninfarcted LV (LVC for day 0). LV tissue was homogenized in PBS (Sigma) with 1× protease inhibitor (Roche, Mannheim, Germany) using the Power Gen 1000 homogenizer (Fisher Scientific). The homogenate was centrifuged at 4,700 rpm for 10 min. The supernatant (soluble protein fraction) was removed and stored at −80°C. The remaining pellet (insoluble protein fraction) was homogenized in Protein Extraction Reagent 4 (Sigma) with protease inhibitor and stored at −80°C. Bradford reagent (Bio-Rad, Hercules, CA) was used to measure the concentration of protein.
Immunoblot analysis.
A total of 10 μg LV protein was loaded on 4–12% SDS-polyacrylamide gels (Bio-Rad), as previously described (30). The insoluble fraction was used to assess collagen type I and collagen type III, and the soluble fraction was used to assess all other proteins. Specific primary antibodies against CD18 (1:1,000 dilution, catalog no. ab157146, Abcam), collagen type I (1:2,000 dilution, catalog no. CL50141AP-1, Cedarlane), collagen type III (1:1,000 dilution, catalog no. CL 50341AP-1, Cedarlane), lysyl oxidase (LOX; 1:2,000 dilution, catalog no. NB110-41568, Novus, Littleton, CO), VEGFA (1:2,000 dilution, catalog no. ab51745, Abcam), and vascular cell adhesion molecule 1 (VCAM-1; 1:1,000 dilution, Novus) were used. Fractions were incubated with primary antibodies overnight at 4°C and with the secondary antibody horseradish peroxidase-labeled anti-rabbit IgG (1:5,000 dilution, catalog no. PI-1000, Vector Laboratories) at room temperature for 1 h. Homogenized spleen, hepatic tumor, purified full-length collagen type I (catalog no. 1066, Chondrex, Redmond, WA), and purified full-length collagen type III (catalog no. CC054, Millipore, Billerica, MA) were used as positive controls. Blots were incubated with ECL Prime Western blot detection reagent (catalog no. RPN2236, GE Healthcare, Little Chalfont, UK) for 5 min, and excess solution was removed. Chemiluminescence was visualized with a digital imaging system (ImageQuant LAS 4000, GE Healthcare). Densitometry of the bands was determined using ImageQuant TL 8.1 analysis software (GE Healthcare). The intensity of every lane was normalized to the densitometry of the total protein for that lane as an internal loading control.
Cardiac fibroblast evaluation.
Six additional 3- to 6-mo-old WT mice were used for cardiac fibroblast isolation. LV infarct tissue was minced into 1- to 2-mm pieces under a sterile laminar flow hood and dissociated into a single cell suspension using collagenase type II (600 U/ml, Worthington Biochemical, Lakewood, NJ) and DNase I (60 U/ml, AppliChem, Darmstadt, Germany) in Hank’s balanced salt solution (HBSS; ThermoFisher, Waltham, MA). After 1 h of incubation at 37°C, with mechanical dissociation applied every 15 min, the cell suspension was centrifuged and resuspended in DMEM supplemented with 10% FBS and 1% antibiotics and plated in 75-mm2 flasks. At passage 2, cells were resuspended in 1 ml DMEM supplemented with 10% FBS and 1% antibiotics and plated in a six-well plate (1.0 × 105 cells/well). At 24 h after being plated, cells were incubated in serum-starved medium (DMEM supplemented with 0.1% FBS and 1% antibiotics) overnight and then in 0.15, 0.5, or 1.0 μg/ml active human MMP-9 (catalog no. PF140, Millipore) for 24 h. Fibroblasts incubated in DMEM with 0.1% FBS and 1% antibiotics served as the negative control, and fibroblasts incubated in DMEM with 10% FBS and 1% antibiotics served as the positive control. RNA was extracted with TRIzol (ThermoFisher) according to the manufacturer’s instructions (41).
Cardiac fibroblast RNA was purified with a Pure Link RNA minikit (ThermoFisher), and concentrations were quantified using a spectrophotometer (NanoDrop ND-1000, ThermoFisher). cDNA was prepared using the High Capacity RNA-to-cDNA kit (ThermoFisher), and quantitative real-time RT-PCR was performed with TaqMan Gene Expression Master Mix (ThermoFisher). Gene expression for collagen type I-α1 (Col1a1; Mm00801666_g1), collagen type III-α1 (Col3a1; Mm01254476_m1), fibronectin 1 (Fn1; Mm01256744_m1), TGF-β1 (Tgfb1, Mm01178820_m1), α-smooth muscle actin (Acta2; Mm00725412_s1), and ets variant 2 (Etv2; Mm00468389_m1) was assessed using FAM dye primers (Thermo Fisher). Hypoxanthine guanine phosphoribosyl transferase 1 (Mm01545399_m1) was used as the reference gene for normalization, as it did not change among groups (P = 0.57 by ANOVA).
Post-MI cardiac macrophage isolation.
Five additional WT and four additional TG 18- to 28-mo-old mice were used for macrophage isolation at day 7 post-MI. Briefly, LV infarct tissue was minced and dissociated as described above. The cell suspension was centrifuged and resuspended in HBSS and applied over preseparation filters (catalog no. 130-041-407, Miltenyi Biotec, Bergisch Gladbach, Germany) to remove nondissociated clumps. After lysis of red blood cells (Blood Cell Lysis Solution, Miltenyi Biotec), the cell suspension was purified to remove neutrophils using the anti-Ly-6G microbead kit (catalog no. 130-092-332, Miltenyi Biotec) in a magnetic MiniMACS column (catalog no. 130-042-201, Miltenyi Biotec). Macrophages were separated from the flow-through using an anti-CD11b microbeads kit (catalog no. 130-049-601, Miltenyi Biotec) in a magnetic MiniMACS column.
RNA sequencing.
Immediately after isolation, RNA was extracted as described above. Total RNA was isolated from the source and checked for quality control measures using a bioanalyzer (Bio-Rad Experion System) for quality, and concentrations were determined using the Qubit fluorometer; RNA quality index scores ≥7.0 were required to proceed. RNA libraries were prepared using the TruSeq Stranded Total RNA with Ribo-Zero Kit, Set A (catalog no. FC-122-2501, Illumina) according to the manufacturer’s instructions. Each sample was prepared using 1 µg total RNA. The resulting cDNA libraries were quantified using the Qubit system (Invitrogen) and checked for quality and size using the Experion DNA 1K chip (Bio-Rad). The fragment size-generated library ranged from 200 to 500 bps with a peak at ~250 bps.
A portion of each library was diluted to 10 nM, and all samples were pooled. The concentrations were verified again via Qubit and further diluted to 4 nM. A total of 5 µl of the 4 nM pooled libraries was diluted and denatured to a final concentration of 1.6 pM. The libraries were sequenced using the NextSeq500 High Output Kit (300 cycles, PE100) on the Illumina NextSeq500 platform. The sequencing reads were automatically uploaded and evaluated for quality using the Illumina BaseSpace Onsite computing platform.
FASTQ sequence files were generated, and analysis was performed using applications available on the BaseSpace Onsite computing platform, including TopHat Alignment (e.g., read mapping to reference genome-USCS-GRCm38/mm10). Clustering and correlation analysis were done using the statistical analysis package in the Metaboanalyst 3.0 software program (http://www.metaboanalyst.ca). Enrichr (http://amp.pharm.mssm.edu/Enrichr/) was used to verify macrophage isolation purity (6, 33). The top 500 genes, by highest average fragments per kilobase of transcript per 1 × 106 mapped reads (FPKM), were entered into the Enrichr software program, and cell type analysis was selected. The top hit, by P value ranking or combined score ranking, was for macrophages, with an adjusted P value of 2.7 × 10−6 and Z-score of −2.44, indicating that the macrophage cell isolation purity was high. The top 50 ranked genes, by P value and fold change, were selected for heat map analysis. Functional network evaluation was performed with ingenuity pathway analysis (Qiagen Bioinformatics).
Statistical analyses.
Values are means ± SE, and data were graphed as individual value plots. Survival rates were analyzed by Kaplan-Meier survival analysis and compared by log-rank test. Rupture rates were analyzed by Fisher’s exact test. Two-group comparisons were analyzed by an unpaired t-test when data were normally distributed around the mean or by a Mann-Whitney test when data were not normally distributed around the mean. Multiple-group comparisons were analyzed using one-way ANOVA followed by the Student-Newman-Keuls test when the Bartlett’s variation test was passed or using the nonparametric Kruskal-Wallis test followed by Dunn’s post hoc test when the Bartlett’s variation test did not pass. P < 0.05 was considered statistically significant. Statistical analyses were performed using Prism7, and power analysis was performed using StateMate 2 (GraphPad).
RESULTS
Macrophage MMP-9 overexpression in aged mice attenuated increases in LV diastolic volumes without affecting day 7 post-MI survival rate, cardiac rupture rate, or infarct area.
Survival rates were not different between WT and TG mice at day 7 post-MI (Fig. 1). Ten of 16 TG mice and 19 of 22 WT mice survived at day 7 post-MI. The power of the survival rate analysis was >80%, which indicates that sample size was large enough to determine significant differences. Cardiac rupture rates were not different between groups [0 of 3 mice (0%) for the WT group and 2 of 6 mice (33%) for the TG group, P = 0.50]. Infarct areas were not different between WT and TG mice (P = 0.067). These results indicate that all mice received similar ischemic injuries.
Fig. 1.
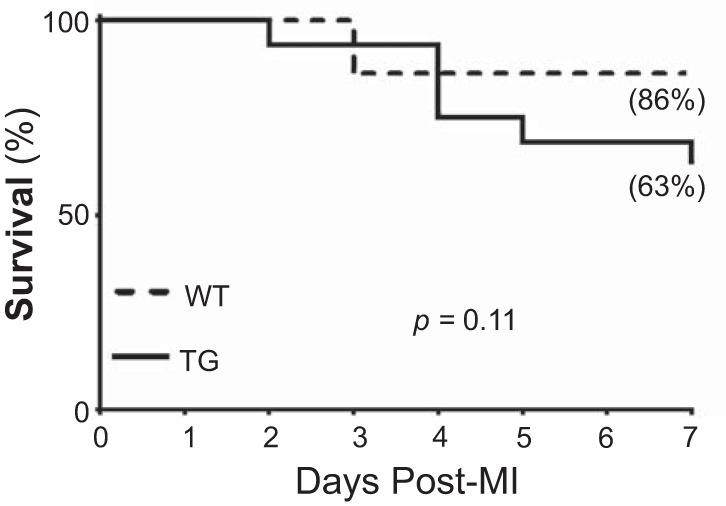
Macrophage matrix metalloproteinase-9 overexpression in transgenic (TG) mice (n = 16) did not affect the day 7 following myocardial infarction (post-MI) survival rate compared with wild-type (WT) mice (n = 22).
LV necropsy and echocardiography variables are shown in Table 1 as raw values and as percent changes from day 0 baseline to day 7 post-MI values. LV mass-to-body weight and LV mass-to-lung weight ratios were increased in WT and TG mice at day 7 post-MI compared with day 0 groups (P < 0.05 for both), consistent with previous reports (27, 68). End-systolic and end-diastolic volumes (LV dilation) were increased in WT and TG mice at day 7 post-MI compared with day 0 controls. Ejection fraction was decreased in WT and TG mice at day 7 post-MI compared with day 0 groups. The LV remodeling index and change in end-diastolic volume were reduced in TG mice compared with WT mice at day 7 post-MI, indicating improved diastolic properties with macrophage MMP-9 overexpression. Ejection fraction and changes in ejection fraction were reduced in TG mice compared with WT mice at day 7 post-MI. The differences were due to changes in end-diastolic volume, since there was no difference in end-systolic volume.
Table 1.
LV necropsy and echocardiography in WT and TG mice at days 0 and 7 following myocardial infarction
|
Day 0 |
Day 7 |
|||
|---|---|---|---|---|
| WT | TG | WT | TG | |
| Number of mice | 21 | 19 | 19 | 10 |
| Sex, male/female | 10/11 | 8/11 | 6/13 | 4/6 |
| Necropsy | ||||
| Body weight, mg | 30 ± 1 | 33 ± 1 | 24 ± 1* | 27 ± 2* |
| LV mass/body weight, mg/g | 2.9 ± 0.1 | 3.0 ± 0.1 | 3.9 ± 0.1* | 3.6 ± 0.2* |
| Lung weight, mg | 148 ± 6 | 155 ± 6 | 256 ± 17* | 291 ± 18* |
| Infarct area, %LV | 43 ± 1 | 48 ± 2 | ||
| Echocardiography | ||||
| End-systolic volume, μl | 22 ± 1 | 21 ± 2 | 107 ± 5* | 107 ± 6* |
| End-diastolic volume, μl | 66 ± 4 | 56 ± 4 | 126 ± 5* | 115 ± 6* |
| Ejection fraction, % | 67 ± 1 | 63 ± 1 | 15 ± 1* | 7 ± 1*# |
| LV remodeling index, μl/g | 0.78 ± 0.05 | 0.60 ± 0.03 | 1.40 ± 0.07* | 1.21 ± 0.07*# |
| %Change from day 0 to day 7 | ||||
| End-systolic volume | 484 ± 46 | 379 ± 37 | ||
| End-diastolic volume | 149 ± 12 | 95 ± 12# | ||
| Ejection fraction | −75 ± 3 | −89 ± 2# | ||
Values are means ± SE. Left ventricular (LV) remodeling index = end-diastolic volume/LV mass. The day 7 percent change was calculated as the percent change from baseline premyocardial infarction values obtained for the same mice. WT, wild-type; TG, transgenic.
P < 0.05 vs. respective day 0 values;
P < 0.05 vs. WT day 7 following myocardial infarction.
Macrophage MMP-9 overexpression increased pre- and post-MI macrophage numbers in aged mice.
Macrophage numbers were increased in TG mice compared with WT mice at day 0 (Fig. 2A). There were more macrophages in WT and TG mice at day 7 post-MI than at day 0. Macrophage numbers were increased in TG mice compared with WT mice at day 7 post-MI, indicating that macrophage MMP-9 overexpression contributed macrophage numbers during both aging and MI. Protein levels of the leukocyte extravasation facilitator CD18 were not different between WT and TG mice at day 0 (Fig. 2B). The difference in macrophage numbers at day 0 indicates an increase in the resident cardiac macrophage pool that is CD18 independent. CD18 was higher in WT and TG mice at day 7 post-MI than at day 0, and CD18 was increased in TG mice compared with WT mice at day 7 post-MI, consistent with macrophage numbers.
Fig. 2.

Matrix metalloproteinase-9 overexpression increased macrophage cell numbers at day 0 (D0) and day 7 (D7) following myocardial infarction (post-MI). A: left ventricular sections were stained with anti-Mac-3 antibody; arrows depict positive staining. At D7 post-MI, macrophage numbers were increased in wild-type (WT) infarcts compared with D0 controls. The increase in macrophage numbers was greater in post-MI transgenic (TG) than WT control infarcts. B: CD18 was increased in WT and TG infarcts at D7 post-MI compared with D0. CD18 was increased in TG compared with WT infarcts. The spleen was used as a positive control (+). Values are means ± SE and graphed as individual value plots; n = 10–21 mice/group for immunohistochemistry and 7–10 mice/group for immunoblot analysis. *P < 0.05 vs. respective D0 values; #P < 0.05 vs. WT D7; †P < 0.05 vs. WT D0.
Macrophage MMP-9 overexpression reduced proinflammatory macrophage polarization.
RNA sequencing was used to evaluate macrophage polarization. As shown in Fig. 3A, 1,276 of 23,843 genes evaluated were significantly different between WT and TG mice at day 7 post-MI. TG mice showed 994 upregulated genes and 282 downregulated genes compared with WT mice. By ingenuity pathway analysis, the most represented [889 (70%) of the 1,276 genes] was the group of organism injury-related genes. The heat map of the top 50 ranked significantly different genes between WT and TG LVI macrophages, by P value and fold change, and the top 3 ranked upregulated and downregulated genes, by P value and fold change, are shown in Fig. 3B.
Fig. 3.
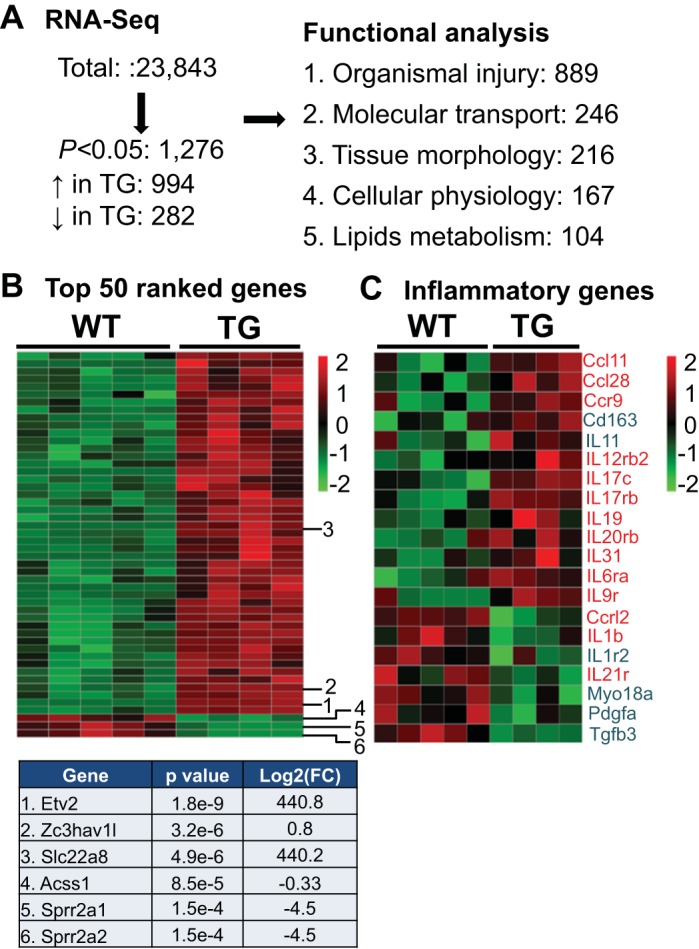
RNA sequencing (RNA-seq) of isolated cardiac macrophages from wild-type (WT) and transgenic (TG) mice at day 7 (D7) following myocardial infarction (post-MI). A: of the 23,843 genes examined, 1,276 genes were different between WT and TG groups (all P < 0.05 by t-test). Ingenuity pathway analysis showed the top 5 most important pathways. B: heat map of the top 50 genes ranked by P value and fold change (FC). Red, upregulation; green, downregulation. Top 3 ranked up- and downregulated genes, by P value and FC, are shown. C: heat map showing proinflammatory (red) and anti-inflammatory (blue) genes differentially expressed in WT and TG mice (all P < 0.05).
We investigated the effects of macrophage MMP-9 overexpression on inflammatory genes (Fig. 3C) because aging and MI are associated with increased inflammation (12, 58, 67). Of the 14 proinflammatory genes that were different between WT and TG mice, 11 genes [chemokine (C-C motif) ligand (Ccl)11, Ccl28, chemokine (C-C motif) receptor 9 (Ccr9), IL-12 receptor-β2 (IL12rb2), IL17c, IL17rb, IL19, IL20rb, IL-31, IL-6 receptor-α (IL6ra), and IL9r] were increased and 3 genes [chemokine (C-C motif) receptor-like 2 (Ccrl2), IL-1β, and IL21r] were decreased in TG mice. Of the six anti-inflammatory genes that were different between WT and TG mice, two genes (CD163 and IL-11) were increased and four genes [IL-1r2, myosin 18A (Myo18A), platelet-derived growth factor subunit A (Pdgfa), and Tgfb3] were decreased in TG mice. Of 26 markers of M1 polarization, IL-1β and prostaglandin-endoperoxide synthase 2 (Ptgs2) levels were decreased in TG mice (18, 31, 67). Of 26 markers of M2 polarization, CD163 was increased and VEGFA and transmembrane protein 26 (Tmem26) were decreased in TG mice.
Compensatory effects on other MMPs are frequently observed with MMP inhibition (28, 38). Of the 20 other MMPs analyzed, no differences were observed in MMP expression between WT and TG macrophages. Tissue inhibitor of metalloproteinase (TIMP)-1, TIMP-2, and TIMP-3 levels were also not different between WT and TG macrophages.
MMP-9 did not directly affect cardiac fibroblast ECM production or activation.
Fibroblasts have shown distinct and important roles, including ECM remodeling and angiogenesis, in mediating components of the post-MI inflammation, proliferation, and maturation phases (56). Fibroblasts are regulated by cytokines and chemokines secreted in the infarct region, particularly those secreted by macrophages. To determine whether the in vivo effects of MMP-9 overexpression were due to direct effects of MMP-9 on the fibroblast, as well as indirect effects of MMP-9 on the macrophage, isolated cardiac fibroblasts from WT mice at day 0 were stimulated with increasing concentrations (0.15, 0.50, and 1.00 μg/ml) of active human MMP-9.
Expression of Col1a1, Col3a1, Fn1, and Tgfb1 was not different, at any of the MMP-9 concentrations, from the 0.1% FBS negative control cells. mRNA levels of Acta2, a myofibroblast differentiation marker (1), were also not changed by MMP-9 stimulation. The cells were responsive, as Acta2 was increased in the 10% FBS positive control fibroblasts (P < 0.05). Etv2 has been reported to mediate differentiation of fibroblasts to endothelial cells (47), and Etv2 was absent in WT macrophages and significantly elevated in TG macrophages isolated from LVI. Etv2 mRNA levels were not different among fibroblast groups. Together, these results indicate that MMP-9 did not have a direct effect on fibroblast physiology and that the in vivo fibroblast and ECM differences in the TG group are likely mediated via indirect signaling from the macrophages.
Macrophage MMP-9 overexpression stimulated post-MI collagen accumulation and reduced collagen cross-linking in aging mice.
Improved LV diastolic physiology (end-diastolic volume) may be explained by the quality of post-MI ECM remodeling. To assess whether macrophage MMP-9 overexpression had an effect on collagen accumulation at days 0 or 7 post-MI in aging mice, LVs of WT and TG mice were stained with picrosirius red (Fig. 4A). At day 0, collagen content was increased in TG mice compared with WT mice, consistent with our previous independent analysis (58). Collagen deposition was increased in WT and TG mice at day 7 post-MI compared with day 0. Collagen content was increased in TG mice compared with WT mice at day 7 post-MI, reflecting a fibrogenic role of MMP-9 in the aged LV in the presence and absence of MI. This feature would be detrimental in the aging myocardium and beneficial in the scar-forming myocardium.
Fig. 4.

Macrophage matrix metalloproteinase-9 overexpression increased collagen (Col) content at baseline and following myocardial infarction (post-MI). A: left ventricular sections stained with picrosirius red demonstrated increased collagen content in transgenic (TG) mice compared with wild-type (WT) mice at day 0 (D0). The collagen-stained area was increased in WT and TG mice at day 7 (D7) post-MI compared with D0. Collagen content was increased in TG mice compared with WT mice at D7 post-MI. B: procollagen type I levels were decreased in WT mice at D7 post-MI compared with D0. Full-length collagen type I levels were increased in WT and TG mice at D7 post-MI compared with D0. Collagen type I fragment levels were increased in WT mice at D7 post-MI compared with D0 and decreased in TG mice compared with WT mice at D7 post-MI. C: procollagen type III and full-length collagen III levels were increased in WT and TG mice at D7 post-MI compared with D0. Collagen type III fragment levels were decreased in TG mice at D7 post-MI compared with D0. Purified full-length collagen type I and collagen type III were used as positive controls (+). Values are means ± SE and graphed as individual value plots; n = 10–21 mice/group for picrosirius red staining and 7–10 mice/group for immunoblot analysis. *P < 0.05 vs. respective D0 values; #P < 0.05 vs. WT D7; †P < 0.05 vs. WT D0.
Immunoblot analysis for collagen type I and collagen type III was performed to assess macrophage MMP-9 overexpression effects on collagen isoforms. Procollagen I protein decreased in WT mice at day 7 post-MI compared with day 0 (Fig. 4B), and no difference was observed between day 7 post-MI and day 0 in TG mice. Full-length collagen type I was increased in WT and TG mice at day 7 post-MI compared with day 0. Collagen type I fragments were increased in WT mice at day 7 post-MI compared with day 0, and no difference was observed in TG mice between day 7 post-MI and day 0. Procollagen type I and full-length collagen type I levels were not different between WT and TG mice at day 7 post-MI. Collagen type I fragments were decreased in TG mice compared with WT mice at day 7 post-MI. These findings indicate that procollagen type I was cleaved and observed in fragmented forms in WT mice at day 7 post-MI and that cleavage was blunted in TG mice at day 7 post-MI. Protein levels of procollagen type III and full-length collagen type III were increased in WT and TG mice at day 7 post-MI compared with day 0 (Fig. 4C). Collagen type III fragments were decreased in TG mice at day 7 post-MI compared with day 0. Procollagen type III, full-length collagen type III, and collagen type III fragments were not different between WT and TG mice at day 7 post-MI. Together, these results demonstrate that collagen type III cleavage was blunted with MMP-9 overexpression.
Because diastolic volumes were improved in post-MI TG mice, collagen cross-linking was assessed by measuring LOX (Fig. 5A). LOX catalyzes covalent cross-linking between collagen fibers, enhancing their stability (15, 16, 20). Protein levels of LOX were elevated in WT and TG groups at day 7 post-MI compared with day 0, which is consistent with previous studies (20, 63). Collagen cross-linking ratios were calculated by dividing the intensity of LOX bands by the intensity of collagen subtype bands (Fig. 5B). At day 7 post-MI, the procollagen type I cross-linking ratio was reduced in TG mice compared with WT mice, consistent with improved remodeling and diastolic physiology.
Fig. 5.

Macrophage matrix metalloproteinase-9 overexpression reduced following myocardial infarction (post-MI) collagen cross-linking. A: lysyl oxidase (LOX) protein was increased in wild-type (WT) and transgenic (TG) infarcts at day 7 (D7) post-MI compared with day 0 (D0). No differences were observed between WT and TG infarcts at D7 post-MI. B: at D7 post-MI, the procollagen (Pro-Col) type I cross-linking ratio was reduced in TG mice compared with WT mice. Values are means ± SE and graphed as individual value plots; n = 7–10 mice/group. *P < 0.05 vs. respective D0 values; #P < 0.05 vs. WT D7.
Macrophage MMP-9 overexpression reduced vessel numbers and angiogenesis signaling in the post-MI LV in aging mice.
Post-MI angiogenesis is necessary to reestablish blood supply to the infarct region (22). GSL-I staining was performed to assess whether macrophage MMP-9 overexpression affected vessel density at days 0 and 7 post-MI in aging mice (Fig. 6A). At day 0, GSL-I staining was reduced in TG mice compared with WT mice, which was similar to our previously published results (58). At day 7 post-MI, the GSL-I-stained area was reduced in TG mice compared with WT mice, suggesting an antiangiogenic role of MMP-9 in the aging post-MI LV.
Fig. 6.

Macrophage matrix metalloproteinase-9 overexpression reduced vessel numbers and angiogenesis signaling at baseline and after myocardial infarction (MI). A: left ventricular sections were stained with biotinylated Griffonia (Bandeiraea) simplicifolia lectin I (GSL-I) and quantified. GSL-I was measured in 5 random sections taken from the infarct region across the left ventricular free wall. The percentage of GSL-I-stained area per total area showing reduced vessel numbers in transgenic (TG) mice at day 0 (D0) was compared with that in wild-type (WT) mice at D0. The GSL-I-stained area was reduced in TG mice compared with WT mice at day 7 (D7) post-MI. B: gene expression levels of proangiogenic factors [VEGFA, platelet-derived growth factor subunit A (Pdgfa), VCAM-1, integrin-α5 (Itga5), and tissue inhibitor of metalloproteinase 4 (Timp4)] assessed by RNA sequencing (RNA-seq) in isolated cardiac macrophages. FPKM, fragments per kilobase of transcript per 1 × 106 mapped reads. C: VEGF188 (25 kDa) was increased in WT mice, but not in TG mice, at D7 post-MI compared with D0, and VEGF164 (22 kDa) was decreased in WT and TG mice at D7 post-MI compared with D0 controls. D: VCAM-1 was decreased in WT and TG mice at D7 post-MI compared with D0. Mouse hepatic tumor was used as a positive control (+). Values are means ± SE and graphed as individual value plots; n = 10–21 mice/group for GSL-I staining, 4–5 mice/group for RNA-seq, and 7–10 mice/group for immunoblot analysis. *P < 0.05 vs. respective D0 values; #P < 0.05 vs. WT D7; †P < 0.05 vs. WT D0.
Gene expression levels of the proangiogenic factors VEGFA, Pdgfa, VCAM-1, integrin-α5 (Itga5), and antiangiogenic Timp4 are shown in Fig. 6B. Levels of VEGFA and Pdgfa were reduced and levels of Itga5 were increased in TG macrophages compared with WT macrophages at day 7 post-MI. No difference was observed in VCAM-1 levels in TG macrophages compared with WT macrophages at day 7 post-MI (P = 0.71). Timp4 was increased in TG macrophages compared with WT macrophages at day 7 post-MI. Overall, these results indicate that TG macrophages had an antiangiogenic phenotype, consistent with results from vessel immunohistochemistry.
VEGFA and VCAM-1 protein were assessed by immunoblot analysis in post-MI LV tissue (Fig. 6, C and D). We observed the two alternatively spliced isoforms of VEGFA (70) but not the 16-kDa cleaved form of VEGFA (36). At day 7 post-MI, the 25-kDa band (VEGF188) was increased in the WT group, but not in the TG group, compared with day 0. The 22-kDa band (VEGF164) was decreased in WT and TG mice at day 7 post-MI compared with day 0. Protein levels of VCAM-1 were decreased in WT and TG mice at day 7 post-MI compared with day 0.
DISCUSSION
The goal of our study was to evaluate whether macrophage-specific MMP-9 contributes to post-MI LV remodeling in the aging setting. The most salient findings were that macrophage MMP-9 overexpression resulted in 1) enhanced macrophage numbers and anti-inflammation polarization, 2) increased collagen accumulation with reduced cross-linking, 3) reduced angiogenesis signaling, and 4) improved LV diastolic volume (represented by the percent change in end-diastolic volume from day 0 to day 7 post-MI) and LV remodeling (Fig. 7). Together, these results reveal macrophage-derived MMP-9 as a key mediator of post-MI wound healing in an aged mouse model of MI.
Fig. 7.
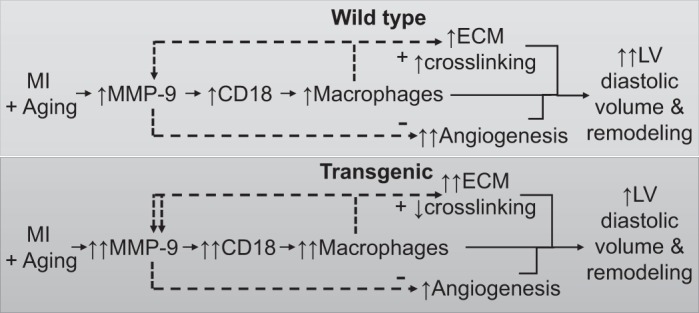
Mechanistic diagram showing hypothesized effects of transgenic macrophage matrix metalloproteinase (MMP)-9 overexpression in the aging and following myocardial infarction (post-MI) left ventricle (LV). In the wild-type LV, MMP-9 is increased by aging and MI, which trigger CD18 expression and macrophage infiltration to further increase MMP-9. Increased inflammation leads to extracellular matrix (ECM) accumulation, collagen cross-linking, and impaired angiogenesis. Together, these changes result in enhanced LV diastolic volume and remodeling. In the transgenic LV, macrophage infiltration and ECM accumulation are increased further, and cross-linking is decreased to improve LV diastolic volume and LV remodeling. +, increase; −, decrease.
Higher plasma MMP-9 is associated with an increased risk of developing post-MI heart failure in patients (64). The mechanisms whereby MMP-9 contributes to post-MI outcomes in the elderly, however, are not fully understood. Our group has used MMP-9 deletion, pharmacological inhibition, and overexpression strategies to demonstrate that MMP-9 is robustly produced by macrophages and plays a key role in ECM remodeling during cardiac aging and in response to MI (28, 58, 66–68). Results from TG and MMP-9-null mice in aging, post-MI, and aging + post-MI studies are compared in Table 2.
Table 2.
Comparative analysis showing effects of MMP-9 on aging, MI, and aging + MI on TG or null mice and their respective WT controls
| Aging | MI | Aging + MI | |
|---|---|---|---|
| Infarct size | NA | TG:↔ (68) Null: ↔ (11, 37) |
TG: ↔ Null: ↔ (67) |
| Survival rate | NA | TG:↔ (68) Null: ↔ (37) |
TG: ↔ Null: ↑ (67) |
| Rupture rate | NA | TG:↔ (68) Null: ↓ (21) |
TG: ↔ Null: ↔ (67) |
| Ejection fraction | TG: * Null: * |
TG: ↑ (68) Null: ↔ (11, 63) |
TG: ↓ Null: * |
| Macrophage numbers | TG: * Null: * |
TG:↔ (68) Null: ↓ (13, 37) |
TG: ↑ Null: ↔ (67) |
| Macrophage M2 polarization | TG: * | TG: ↑ (68) | TG: ↑ |
| Null: ↑ (41) | Null: * | Null: ↑ (67) | |
| Collagen accumulation | TG: ↑ (58) Null: ↓ (8) |
TG:↔ (68) Null: ↓ (13, 63) |
TG: ↑ Null: ↓ (67) |
| Vessel numbers | TG: ↓ (58) Null: ↓ (66) |
TG: * Null: ↑ (37) |
TG: ↓ Null: * |
| Angiogenesis markers | TG: ↓ (58) Null: ↑ (66) |
TG: * Null: ↑ (37) |
TG: ↓ Null: ↑ (67) |
While macrophage numbers were increased in TG mice in both aging and post-MI settings, we did not evaluate macrophage numbers in other macrophage-rich organs, such as the spleen or liver, which may provide additional insights into the roles of macrophage MMP-9 overexpression in the macrophage population. MMP-9 proteolytically activates cytokines and chemokines, including IL-1β, IL-8, chemokine (C-X-C motif) ligand 5 (CXCL5) and TNF-α, which may explain, in part, the increase in macrophage numbers in the TG LV, as these factors are known to drive macrophage infiltration (55, 59–61). Cardiac tissue resident macrophages in young mice exhibit an anti-inflammatory M2 polarization phenotype, suggesting that this phenotype is important for tissue homeostasis (53). Macrophage accumulation is enhanced in senescent mice (7), and cardiac macrophages show altered gene expression and cell physiology with aging (52). Previously and in the present study, we showed that macrophage MMP-9 overexpression in the absence of MI upregulates a proinflammatory profile, resulting in stimulation and acceleration of cardiac aging (58). Macrophages play both destructive and reparative roles in post-MI wound healing, and understanding how particular factors modulate macrophage polarization may drive new therapeutic approaches to promote myocardial repair (25, 49). Here, we observed reduced levels of the anti-inflammatory genes Myo18A, TGF-β, and Pdgfa in TG macrophages. Activation of the macrophage Myo18A receptor is important for tissue repair (2), and macrophage secretion of TGF-β and Pdgfa regulates fibroblast activation (43, 51). Our results indicate that MMP-9 influences both quantity and quality of macrophages in cardiac aging by enhancing inflammation and acting as a profibrotic stimulator.
Cardiac inflammation triggers growth factors and release of profibrotic cytokines, which regulates fibrillar collagens and deposition of other ECM proteins in the aging and post-MI LV (10, 32). Our results extend previous findings and indicate a cumulative effect of macrophage MMP-9 in the aged LV (58). Collagen deposition is robustly increased post-MI (27), and MMP-9 deletion reduced the collagen accumulation at day 7 in post-MI adult mice (63). Our group reported no difference in collagen content between adult TG and WT mice at day 5 post-MI (68). While an infarct scar that is too stiff leads to diastolic heart failure, an infarct scar that is too compliant will lead to LV dilation and even cardiac rupture (24, 63). The mechanical strength and stiffness of collagen derive from proper collagen fiber cross-linking. Macrophage MMP-9 overexpression did not change collagen cross-linking with aging but reduced post-MI collagen cross-linking.
Angiogenesis is an essential component of post-MI wound healing (23), and MMP-9 impairs angiogenesis in aging and post-MI LV settings (37, 66). MMP-9 deletion improves angiogenesis and endothelial dysfunction in aging mice (66), and macrophage MMP-9 overexpression reduces vessel numbers in aging mice (58). We further confirmed this finding, showing reduced levels of GSL-I staining in aging TG mice at day 0. VEGFA is a key promoter of angiogenesis, in that it induces endothelial cell growth, migration, and tube formation through VEGF receptors on endothelial cells (35).
While end-diastolic volume, LV remodeling index, and collagen cross-linking were reduced in TG mice and point to beneficial effects, there was also evidence that MMP-9 overexpression generated detrimental effects, such as reduced ejection fraction and angiogenesis. Macrophage MMP-9 overexpression reduced VEGFA and Pdgfa gene expression by macrophages, indicating that macrophage MMP-9 impairs angiogenesis and may also negatively affect post-MI wound healing. Future studies are warranted to determine if MMP-9 overexpression at days 0–1 post-MI only and days 1–5 post-MI only would distinguish beneficial and detrimental effects. Additional studies evaluating sex differences would provide details on MMP-9 effects on male versus female mice.
In conclusion, macrophage-derived MMP-9 improved post-MI remodeling through direct and indirect mechanisms on cardiac wound healing and diastolic properties. Our results provide evidence that selective therapies for macrophage-derived MMP-9 may reduce specific features of post-MI adverse remodeling.
GRANTS
This work was supported by National Institutes of Health Grants HL-075360, HL-129823, HL-051971, GM-104357, GM-114833, GM-115428, GM-103476, and GM-103328 and by Veterans Affairs Office of Research and Development Biomedical Laboratory Research and Development Service Grant 5I01BX000505.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association, the National Institutes of Health, or the Veterans Administration.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.A.M., M.J., R.P.I., A.Y., H.T., and M.R.G. performed experiments; C.A.M., M.J., R.P.I., A.Y., H.T., M.R.G., and M.L.L. analyzed data; C.A.M., M.J., R.P.I., A.Y., H.T., and M.L.L. interpreted results of experiments; C.A.M. and M.L.L. prepared figures; C.A.M. and M.L.L. drafted manuscript; C.A.M., M.J., R.P.I., A.Y., H.T., M.R.G., and M.L.L. approved final version of manuscript; M.J., R.P.I., A.Y., H.T., M.R.G., and M.L.L. edited and revised manuscript; H.T. and M.L.L. conceived and designed research.
REFERENCES
- 1.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol 57: 376–379, 2011. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouchery T, Harris NL. Specific repair by discerning macrophages. Science 356: 1014, 2017. doi: 10.1126/science.aan6782. [DOI] [PubMed] [Google Scholar]
- 3.Cabrera S, Gaxiola M, Arreola JL, Ramírez R, Jara P, D’Armiento J, Richards T, Selman M, Pardo A. Overexpression of MMP9 in macrophages attenuates pulmonary fibrosis induced by bleomycin. Int J Biochem Cell Biol 39: 2324–2338, 2007. doi: 10.1016/j.biocel.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 4.Carro A, Kaski JC. Myocardial infarction in the elderly. Aging Dis 2: 116–137, 2011. [PMC free article] [PubMed] [Google Scholar]
- 5.Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol 42: 113–185, 2007. doi: 10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- 6.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128, 2013. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, Chou YM, Lindsey ML, Jin YF. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet 4: 455–462, 2011. doi: 10.1161/CIRCGENETICS.111.959981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin YF, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res 96: 444–455, 2012. doi: 10.1093/cvr/cvs275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLeon-Pennell KY, de Castro Brás LE, Iyer RP, Bratton DR, Jin YF, Ripplinger CM, Lindsey ML. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol 76: 218–226, 2014. doi: 10.1016/j.yjmcc.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML. Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing. JCI Insight 2: 94207, 2017. doi: 10.1172/jci.insight.94207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ Cardiovasc Genet 9: 14–25, 2016. doi: 10.1161/CIRCGENETICS.115.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48: 504–511, 2010. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106: 55–62, 2000. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2: 161–174, 2002. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 15.El Hajj EC, El Hajj MC, Ninh VK, Bradley JM, Claudino MA, Gardner JD. Detrimental role of lysyl oxidase in cardiac remodeling. J Mol Cell Cardiol 109: 17–26, 2017. doi: 10.1016/j.yjmcc.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 16.El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med (Maywood) 241: 539–549, 2016. doi: 10.1177/1535370215616511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrari AU, Radaelli A, Centola M. Aging and the cardiovascular system. J Appl Physiol 95: 2591–2597, 2003. doi: 10.1152/japplphysiol.00601.2003. [DOI] [PubMed] [Google Scholar]
- 18.Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV, Karpov RS, Ryabov V. Macrophage activation and polarization in post-infarction cardiac remodeling. J Biomed Sci 24: 13, 2017. doi: 10.1186/s12929-017-0322-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest 118: 3012–3024, 2008. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González-Santamaría J, Villalba M, Busnadiego O, López-Olañeta MM, Sandoval P, Snabel J, López-Cabrera M, Erler JT, Hanemaaijer R, Lara-Pezzi E, Rodríguez-Pascual F. Matrix cross-linking lysyl oxidases are induced in response to myocardial infarction and promote cardiac dysfunction. Cardiovasc Res 109: 67–78, 2016. doi: 10.1093/cvr/cvv214. [DOI] [PubMed] [Google Scholar]
- 21.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 22.Hilfiker-Kleiner D, Landmesser U, Drexler H. Molecular mechanisms in heart failure: focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol 48: 56–66, 2006. doi: 10.1016/j.jacc.2006.07.007. [DOI] [Google Scholar]
- 23.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56: 549–580, 2004. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 24.Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng 7: 223–253, 2005. doi: 10.1146/annurev.bioeng.7.060804.100453. [DOI] [PubMed] [Google Scholar]
- 25.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 114: 266–282, 2014. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwata M, Pillai M, Ramakrishnan A, Hackman RC, Deeg HJ, Opdenakker G, Torok-Storb B. Reduced expression of inducible gelatinase B/matrix metalloproteinase-9 in monocytes from patients with myelodysplastic syndrome: correlation of inducible levels with the percentage of cytogenetically marked cells and with marrow cellularity. Blood 109: 85–92, 2007. doi: 10.1182/blood-2006-05-020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer RP, de Castro Brás LE, Cannon PL, Ma Y, DeLeon-Pennell KY, Jung M, Flynn ER, Henry JB, Bratton DR, White JA, Fulton LK, Grady AW, Lindsey ML. Defining the sham environment for post-myocardial infarction studies in mice. Am J Physiol Heart Circ Physiol 311: H822–H836, 2016. doi: 10.1152/ajpheart.00067.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyer RP, de Castro Brás LE, Patterson NL, Bhowmick M, Flynn ER, Asher M, Cannon PL, Deleon-Pennell KY, Fields GB, Lindsey ML. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J Mol Cell Cardiol 100: 109–117, 2016. doi: 10.1016/j.yjmcc.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iyer RP, Jung M, Lindsey ML. MMP-9 signaling in the left ventricle following myocardial infarction. Am J Physiol Heart Circ Physiol 311: H190–H198, 2016. doi: 10.1152/ajpheart.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol 185: 198–208, 2015. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado JD, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS One 10: e0145342, 2015. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 57: 195–202, 2003. doi: 10.1016/S0753-3322(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 33.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97, 2016. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. II. The aging heart in health: links to heart disease. Circulation 107: 346–354, 2003. doi: 10.1161/01.CIR.0000048893.62841.F7. [DOI] [PubMed] [Google Scholar]
- 35.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell 130: 691–703, 2007. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol 169: 681–691, 2005. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM Jr, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol 290: H232–H239, 2006. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- 38.Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, Crawford J, Black S, Libby P, Mitchell PG, Lee RT. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation 105: 753–758, 2002. doi: 10.1161/hc0602.103674. [DOI] [PubMed] [Google Scholar]
- 39.Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol 91: 134–140, 2016. doi: 10.1016/j.yjmcc.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Brás LE. A novel collagen matricryptin reduces left ventricular dilation post-myocardial infarction by promoting scar formation and angiogenesis. J Am Coll Cardiol 66: 1364–1374, 2015. doi: 10.1016/j.jacc.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Y, Chiao YA, Clark R, Flynn ER, Yabluchanskiy A, Ghasemi O, Zouein F, Lindsey ML, Jin YF. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc Res 106: 421–431, 2015. doi: 10.1093/cvr/cvv128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma Y, de Castro Brás LE, Toba H, Iyer RP, Hall ME, Winniford MD, Lange RA, Tyagi SC, Lindsey ML. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch 466: 1113–1127, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML. Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Trends Pharmacol Sci 38: 448–458, 2017. doi: 10.1016/j.tips.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 110: 51–61, 2016. doi: 10.1093/cvr/cvw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115: 888–895, 2007. doi: 10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 46.Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. Geroscience 39: 7–18, 2017. doi: 10.1007/s11357-017-9959-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T, Kimura A, Sasaki K, Yasukawa H, Yoshimura A. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc Natl Acad Sci USA 112: 160–165, 2015. doi: 10.1073/pnas.1413234112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics−2015 update: a report from the American Heart Association. Circulation 131: e29–e322, 2015. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 49.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121: 2437–2445, 2010. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.National Research Council; Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research; National Academies Press . Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies, 2011, p. xxv. [Google Scholar]
- 51.Pierce GF, Mustoe TA, Altrock BW, Deuel TF, Thomason A. Role of platelet-derived growth factor in wound healing. J Cell Biochem 45: 319–326, 1991. doi: 10.1002/jcb.240450403. [DOI] [PubMed] [Google Scholar]
- 52.Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, Wang L, Rosenthal NA. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY) 6: 399–413, 2014. doi: 10.18632/aging.100669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7: e36814, 2012. doi: 10.1371/journal.pone.0036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prescott MJ, Lidster K. Improving quality of science through better animal welfare: the NC3Rs strategy. Lab Anim (NY) 46: 152–156, 2017. doi: 10.1038/laban.1217. [DOI] [PubMed] [Google Scholar]
- 55.Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1β by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1β processing. J Immunol 161: 3340–3346, 1998. [PubMed] [Google Scholar]
- 56.Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 70: 74–82, 2014. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17: 463–516, 2001. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D’Armiento J, Lindsey ML. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am J Physiol Heart Circ Physiol 312: H375–H383, 2017. doi: 10.1152/ajpheart.00633.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by amino-terminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood 96: 2673–2681, 2000. [PubMed] [Google Scholar]
- 60.Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem 270: 3739–3749, 2003. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- 61.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol 82: 1375–1381, 2007. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 62.Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol 48: 222–272, 2013. doi: 10.3109/10409238.2013.770819. [DOI] [PubMed] [Google Scholar]
- 63.Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML, Han HC. Building a better infarct: modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J Mol Cell Cardiol 85: 229–239, 2015. doi: 10.1016/j.yjmcc.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner DR, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail 12: 66–72, 2006. doi: 10.1016/j.cardfail.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 65.Webb CS, Bonnema DD, Ahmed SH, Leonardi AH, McClure CD, Clark LL, Stroud RE, Corn WC, Finklea L, Zile MR, Spinale FG. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: relation to left ventricular remodeling. Circulation 114: 1020–1027, 2006. doi: 10.1161/CIRCULATIONAHA.105.600353. [DOI] [PubMed] [Google Scholar]
- 66.Yabluchanskiy A, Ma Y, Chiao YA, Lopez EF, Voorhees AP, Toba H, Hall ME, Han HC, Lindsey ML, Jin YF. Cardiac aging is initiated by matrix metalloproteinase-9-mediated endothelial dysfunction. Am J Physiol Heart Circ Physiol 306: H1398–H1407, 2014. doi: 10.1152/ajpheart.00090.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yabluchanskiy A, Ma Y, DeLeon-Pennell KY, Altara R, Halade GV, Voorhees AP, Nguyen NT, Jin YF, Winniford MD, Hall ME, Han HC, Lindsey ML. Myocardial infarction superimposed on aging: MMP-9 deletion promotes M2 macrophage polarization. J Gerontol A Biol Sci Med Sci 71: 475–483, 2016. doi: 10.1093/gerona/glv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zamilpa R, Ibarra J, de Castro Brás LE, Ramirez TA, Nguyen N, Halade GV, Zhang J, Dai Q, Dayah T, Chiao YA, Lowell W, Ahuja SS, D’Armiento J, Jin YF, Lindsey ML. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J Mol Cell Cardiol 53: 599–608, 2012. doi: 10.1016/j.yjmcc.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zamilpa R, Zhang J, Chiao YA, de Castro Brás LE, Halade GV, Ma Y, Hacker SO, Lindsey ML. Cardiac wound healing post-myocardial infarction: a novel method to target extracellular matrix remodeling in the left ventricle. Methods Mol Biol 1037: 313–324, 2013. doi: 10.1007/978-1-62703-505-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao T, Zhao W, Chen Y, Ahokas RA, Sun Y. Vascular endothelial growth factor (VEGF)-A: role on cardiac angiogenesis following myocardial infarction. Microvasc Res 80: 188–194, 2010. doi: 10.1016/j.mvr.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]