

Abstract
Oxidative stress results in mtDNA damage and contributes to myocardial cell death. mtDNA repair enzymes are crucial for mtDNA repair and cell survival. We investigated a novel, mitochondria-targeted fusion protein (Exscien1-III) containing endonuclease III in myocardial ischemia-reperfusion injury and transverse aortic constriction (TAC)-induced heart failure. Male C57/BL6J mice (10–12 wk) were subjected to 45 min of myocardial ischemia and either 24 h or 4 wk of reperfusion. Exscien1-III (4 mg/kg ip) or vehicle was administered at the time of reperfusion. Male C57/BL6J mice were subjected to TAC, and Exscien1-III (4 mg/kg i.p) or vehicle was administered daily starting at 3 wk post-TAC and continued for 12 wk. Echocardiography was performed to assess left ventricular (LV) structure and function. Exscien1-III reduced myocardial infarct size (P < 0.01) at 24 h of reperfusion and preserved LV ejection fraction at 4 wk postmyocardial ischemia. Exscien1-III attenuated TAC-induced LV dilation and dysfunction at 6–12 wk post-TAC (P < 0.05). Exscien1-III reduced (P < 0.05) cardiac hypertrophy and maladaptive remodeling after TAC. Assessment of cardiac mitochondria showed that Exscien1-III localized to mitochondria and increased mitochondrial antioxidant and reduced apoptotic markers. In conclusion, our results indicate that administration of Exscien1-III provides significant protection against myocardial ischemia and preserves myocardial structure and LV performance in the setting of heart failure.
NEW & NOTEWORTHY Oxidative stress-induced mitochondrial DNA damage is a prominent feature in the pathogenesis of cardiovascular diseases. In the present study, we demonstrate the efficacy of a novel, mitochondria-targeted fusion protein that traffics endonuclease III specifically for mitochondrial DNA repair in two well-characterized murine models of cardiac injury and failure.
Keywords: DNA repair, hypertrophic cardiomyopathy, mitochondria-targeted peptide, mitochondrial injury, myocardial infarction
INTRODUCTION
During cardiovascular disease, mitochondrial function and integrity are critical determinants of cell fate (23, 30). Due to its close proximity in mitochondria to the sites of reactive oxygen species (ROS) generation, mitochondrial (mt)DNA is highly susceptible to oxidative damage resulting in oxidation of its bases and sugar/phosphate backbone. Under normal physiological conditions, the mitochondrial base excision repair machinery ameliorates oxidant mtDNA damage; however, prolonged increases in ROS can overwhelm repair machinery (11, 40a). Incomplete repair can lead to an accumulation of abasic sites and strand breaks, further disrupting mtDNA replication and transcription. This leads to heightened oxidative stress (8). The resultant accumulation of mtDNA fragments can induce inflammatory cascades accelerating the progression to heart failure (HF) (34).
Both glycosylase and apurinic/apyrimidinic (AP) lyase activity are required to repair oxidative damage to the mtDNA bases (10, 11). Previous studies have examined the effects of the mitochondrial targeted glycosylase/AP lyases, 8-oxoguanine DNA glycosylase (OGG1) (1, 14, 15, 25, 35), and endonuclease III (Endo III) (20, 21, 38, 46) and reported significant protection against oxidative injury. Stable transfection of these mitochondria-targeted lyases containing either OGG1 or Endo III enhanced cell survival (14, 38), inhibited apoptosis (15, 40), and attenuated mtDNA damage (40) in HeLa cells in response to oxidative insult. Similarly, fibroblasts isolated from OGG1-overexpressing mice are protected against oxidative-induced mitochondrial p53 translocation and cell death, resulting in significantly improved cell vitality (25). These studies have indicated that OGG1 and Endo III specifically targeted to the mitochondria invoke a protective response against oxidative damage.
Additional animal studies have reported protection after treatment with mitochondrial fusion OGG1 or Endo III peptides. Infusion of OGG1 or Endo III protects against ventilator-induced injury in the lungs, with concomitant attenuation of pulmonary inflammation (20, 21). In the present study, we examined the effects of a novel Endo III mitochondria-targeted fusion protein, Exscien1-III, in two in vivo murine models of myocardial injury: myocardial ischemia-reperfusion (MI/R) injury- and pressure overload-induced HF. We hypothesized that enhanced mtDNA repair would protect the myocardium in the setting of MI/R injury and pressure overload-induced HF.
MATERIALS AND METHODS
Proteins.
Exscien1-I and Exscien1-III and their mutants were supplied by Exscien (Mobile, AL).
Experimental animals.
Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and began experimentation at 8–10 wk of age. All experimental protocols were approved by the Animal Care and Use Committee of Louisiana State University Health Sciences Center and conformed with the Guide for the Care and Use of Laboratory Animals. All animals received humane care in compliance with the National Society of Medical Research’s Principles of Laboratory Animal Care and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th ed., Revised 2011).
MI/R protocol and myocardial infarct size determination.
Mice were subjected to 45 min of left coronary artery (LCA) ligation and either 24 h or 4 wk of reperfusion. Surgical procedures and analysis were performed as previously described (16).
Cardiac troponin I assay.
Serum levels of the cardiac-specific isoform of troponin I were assessed at 24 h of reperfusion using an ELISA from Life Diagnostics (West Chester, PA).
TAC protocol.
Cardiac hypertrophy and heart failure were induced via TAC as previously described (2).
Left ventricular structure and function.
Echocardiography was performed using the Visual Sonic Vevo-2100 ultrasound system, as previously described (27). M-mode long-axis views were used to measure interventricular septum thickness at systole (IVSs) and diastole (IVSd), left ventricular (LV) posterior wall thickness at systole (LVPWs) and diastole (LVPWd), and LV chamber diameters at end systole (LVESD) and end diastole (LVEDD). LV ejection fraction (LVEF) was calculated from ECG-gated kilohertz visualization (EKV) analysis.
Histological assessment.
Collagen and fibrosis were assessed at the 12-wk end point as previously described (36). To quantify collagen volume fraction (CVF) from the Picrosirus red-stained slides, fluorescent images were obtained (×20) using a Nikon Eclipse (TE2000-U) and processed with NIS Elements Software. CVF was determined by analyzing both interstitial and perivascular collagen, and CVF for each heart was expressed as percent total area and then group averaged (3).
Heart tissues from mouse 277 and mouse 651 were stained with hemotoxylin and eosin. Representative images of cardiomyocytes were obtained (×200) using a Nikon E600 upright microscope and a Micropublisher 3.3 camara (QImaging).
Quantitative PCR and mRNA isolation.
RNA was isolated from the heart tissue of vehicle (Veh)- and Exscien1-III-treated mice at 12 wk post-TAC. One milligram of RNA was transcribed using an I-Script cDNA synthesis kit from Bio-Rad (Hercules, CA). TaqMan primers for p53 and superoxide dismutase 2 (SOD2) from Life Technology were used to amplify quantitative PCR. For real-time quantitative PCR experiments, 18S was used as a housekeeping gene, and values were corrected to 18S. The methode (where CT is threshold cycle) was used for the data analysis of all quantitative PCR results (4).
Western blot analysis.
Western blot analysis was performed as previously described (4), and the following primary antibodies were used: hemagglutinin (HA) tag (Sigma), SOD2 (Cell Signaling), Bax (Abcam), caspase-3 (Cell Signaling), caspase-9 (Cell Signaling), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α; Abcam), GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA), and cytochrome c oxidase subunit IV (Abcam).
Circulating 8-isoprostane measurement.
Plasma levels of 8-isoprostane were measured at 12 wk post-TAC using an ELISA from Cayman Chemical (Ann Arbor, Michigan).
Isolation of subcellular fractions and Western immunoblot analyses.
Subcellular fractions were prepared as previously described (35). In brief, frozen hearts were homogenized in a glass homogenizer with Teflon pestle using homogenization buffer [0.25 M sucrose, 20 mM HEPES-NaOH (pH 7.4), and 1 mM EDTA]. All steps were carried out at 0–4°C, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) was added to all buffers. The homogenate was centrifuged on a cushion containing 0.35 M sucrose at 700 g for 10 min. The fraction around and above the interphase was collected as crude mitochondria and reserved for mitochondrial isolation. The nuclear pellet was suspended in nuclear isolation buffer [0.25 M sucrose, 20 mM HEPES-NaOH (pH 7.4), 25 mM KCl, and 5 mM MgCl2] and purified on a cushion containing 0.8 M sucrose at 3,000 g for 15 min. The nuclear pellet thus obtained was washed with nuclear isolation buffer and suspended in 100 μl RIPA buffer (Cell Signaling Technology, Danvers, MA), incubated for 30 min on ice, and centrifuged at 18,000 g for 15 min. The supernatant was designated the nuclear fraction. The crude mitochondrial fraction, collected as described above, was centrifuged at 18,000 g for 20 min to pellet mitochondria, which were washed in mitochondrial isolation buffer [0.2 M mannitol, 50 mM sucrose, 20 mM HEPES-NaOH (pH 7.4), and 1 mM EDTA] and centrifuged under the same conditions. The resulting supernatant was designated as the cytosolic fraction, and the pellet containing mitochondria was suspended in 100 μl RIPA buffer (Cell Signaling Technology), incubated for 30 min on ice, and centrifuged at 18,000 g for 15 min. This latter supernatant was designated as the mitochondrial fraction. Cytosolic, nuclear, mitochondrial fractions and total lysates were subjected to Western immunoblot analysis as previously described (7) for specific markers and for HA-tagged fusion protein constructs. Antibody against the HA tag (Sigma) was used to determine subcellular distribution of the fusion protein, and antibodies against porin and lamin B (Santa Cruz Biotechnology) were used as markers for mitochondrial and nuclear fractions, respectively. The peroxidase-conjugated secondary antibody was detected by chemiluminescence with SuperSignal West Dura substrate (Thermo Scientific, Rockford, IL) using the Gel Logic 1500 Imaging System (Kodak, Rochester, NY).
Determination of mtDNA damage.
Frozen hearts were powdered with a mortar and pestle, total DNA was isolated, and oxidative mtDNA damage was assessed using previously described methods (7, 18). In brief, purified DNA samples were digested with the restriction enzyme BstXI (New England Biolabs, Beverly, MA). Digested samples were precipitated, dissolved in Tris-EDTA buffer, and quantified using a VersaFluor fluorometer (Bio-Rad) with a Quant-IT Picogreen dsDNA Assay Kit (Life Technologies, Carlsbad, CA). To reveal oxidative base damage, DNA samples were divided into two aliquot parts: one part was treated with formamidopyrimidine glycosylase (Fpg; New England Biolabs), a bacterial DNA repair enzyme that recognizes and cleaves oxidized purines to create apurinic sites and single-strand breaks, whereas the other part was left untreated. Both Fpg-treated and untreated samples were then incubated with 0.1 N NaOH for 15 min at 37°C to cleave the strands at abasic sites or sites of damage to the deoxyribose backbone, mixed with loading dye, and resolved in a 0.6% agarose alkaline gel. After electrophoresis, DNA was vacuum transferred to a nylon membrane (Roche Diagnostics, Sigma) and hybridized with two PCR-generated probes spanning virtually the entire expanse of the mitochondrial genome. The first probe, which hybridized to a 13.1-kb fragment of mtDNA encompassing the common deletion sequence, was generated using mouse mtDNA sequence as the template and the following set of primers: 5′-CTAATTGGATGATGGTATG-3′ for the sense strand and 5′-TATTGTGAGGATTGGAAT-3′ for the antisense strand. The second probe, which hybridized to a 3.2-kb sequence spanning the D-loop transcriptional regulatory region, was generated using the following set of primers: 5′-TACTTATCTTAACCTGAA-3′ for the sense strand and 5′-CATGAATAATTAGCCTTA-3′ for the antisense strand. Both probes were labeled with a DIG-labeled kit (Roche Diagnostics, Sigma). Hybridization bands were detected with Amersham Hyperfilm ECL (GE Healthcare, Piscataway, NJ) and a Gel Logic 1500 Imaging System (Kodak). Changes in Fpg-detectable lesion density for both mtDNA regions were quantified as the fraction of intact mtDNA and calculated as the quotient of hybridization intensities in Fpg-treated and untreated (alkali only) bands. Lesion density values for both mtDNA sequences within each experimental group were pooled for subsequent display and analysis.
Statistical analysis.
All data in this study are expressed as means ± SE. Differences in data between the groups were compared with Student’s t-test or two-way ANOVA using Prism 6 (GraphPad Software). When statistical significance was found with ANOVA, a Tukey multiple comparison test was performed. P values of <0.05 were considered statistically significant.
RESULTS
Exscien1-III ameliorates myocardial injury after acute MI/R.
Initial experiments were conducted to investigate the effects of Exscien1-III on MI/R. Exscien1-III is a fusion protein containing Endo III glycosylase/AP lyase coupled to a TAT sequence to facilitate cellular uptake, a mitochondrial targeting sequence, a HA tag for immunological localization, and a histidine tail as previously described (Fig. 1A) (21). Mice were subjected to 45 min of LCA ischemia followed by 24 h of reperfusion as shown in the protocol in Fig. 1B. Exscien1-III (4 mg/kg), Veh, or the enzymatically inactive K12Q mutant of Exscien1-III (mExscien1-III) was administered at reperfusion via intracardiac injection into the LV lumen. The extent of myocardial injury was measured after 24 h of reperfusion as shown in Fig. 1, C and D. There was no difference in the area at risk (AAR) per LV among study groups (P = not significant). Mice that received Exscien1-III displayed an infarct size per AAR of 26.4 ± 4.9% compared with Veh-treated mice (44.8 ± 5%), revealing a 41% reduction in infarct size (P < 0.01 vs. Veh). The mutant control peptide mExscien1-III had no effect on infarct size (43.8 ± 5.3% of the AAR) compared with Veh, indicating that the protective actions of Exscien1-III are attributable to its glycosylase/lyase region rather than to a nonspecific region of the protein.
Fig. 1.

Exscien1-III attenuates myocardial ischemic injury. A: schematic construction of Exscien1-III. Exscien1-III consists of a mitochondria targeting sequence (MTS), an endonuclease III protein transduction domain (i.e., DNA glycosylase), a hemagglutinin (HA) tag for immunological localization, a TAT sequence to facilitate cell uptake, and a polyhistidine tail (10-HIS). B: myocardial ischemia-reperfusion (MI/R) experimental protocol. Mice were subjected to 45 min of ischemia and 24 h of reperfusion. Exscien1-III, mExscien1-III, or vehicle (Veh) was administered via intracardiac injection at the time of reperfusion. C: assessment of the myocardial area at risk as a percentage of the left ventricle (AAR/LV) and myocardial infarct as a percentage of the area at risk (INF/AAR) in Exscien1-III-, mExscien1-III-, and Veh-treated animals. D: serum cardiac troponin I in Exscien1-III- and Veh-treated mice. Circles inside bars denote n values per study group.
Circulating levels of troponin I, a marker of myocardial injury, were measured at 24 h postreperfusion (Fig. 1E). Exscien1-III significantly attenuated circulating troponin I by 46% compared with Veh (P < 0.05), further confirming the cardioprotective effects of Exscien1-III.
Exscien1-III preserves LVEF after MI/R injury.
We then examined the potential cardioprotective effects of Exscien1-III on LV function after myocardial ischemia and prolonged reperfusion. As shown in Fig. 2A, mice were subjected to 45 min of LCA ischemia followed by 4 wk of reperfusion. Exscien1-III (4 mg·kg−1·day−1) or Veh was administered via intracardiac injection at the time of reperfusion. Echocardiographic analysis revealed that Exscien1-III attenuated MI/R-induced myocardial dilation with significant (P < 0.05 vs. Veh) preservation in LV dimensions [LVEDD (Fig. 2B) and LVESD (Fig. 2C)] at 1–4 wk after reperfusion. Additionally, Exscien1-III administration resulted in a marked reduction (P < 0.05 vs. Veh) in wall thinning IVSd post-MI throughout the 4-wk reperfusion period (Fig. 2D). Evaluation of LV systolic function (Fig. 2E), as measured by LVEF, revealed a significant preservation of LVEF in Exscien1-III-treated mice compared with Veh-treated mice at 1, 2, and 4 wk post-reperfusion. Taken together, these data provide strong evidence for the sustained cardioprotective properties of Exscien1-III to preserve LV systolic function in the setting of more prolonged MI/R injury.
Fig. 2.

Exscien1-III prevents left ventricular (LV) dilation and preserves LV function after myocardial ischemia (MI). A: experimental protocol for myocardial ischemia and extended reperfusion. Mice were subjected to 45 min of ischemia followed by 4 wk of reperfusion. Exscien1-III or vehicle (Veh) was administered via intracardiac injection at the time of reperfusion. Echocardiography was performed at baseline and at 1, 2, and 4 wk post-MI to assess LV end-diastolic dimension (LVEDD; B), LV end-systolic dimension (LVESD; C), interventricular septum thickness at diastole (IVDd; D), and LV ejection fraction (LVEF; E). *P < 0.05 vs. Veh; **P < 0.01 vs. Veh.
Treatment with Exscien1-III prevents cardiac dilation, preserves LV function, and reduces cardiac hypertrophy after TAC.
The effects of Exscien1-III on the failing heart were then assessed using a TAC model of HF as shown in Fig. 3A. Administration of Exscien1-III or Veh was initiated daily at 3 wk post-TAC and continued for 9 wk. Echocardiography was performed at baseline (3 days before TAC surgery) and every 2 wk post-TAC until the 12-wk end point to assess cardiac remodeling and function. Mice treated with Exscien1-III displayed significant reductions (P < 0.05 vs. Veh) in LV chamber dimensions at end diastole (Fig. 3B) and end systole (Fig. 3C) at week 8 (LVESD: 2.4 ± 0.2 vs. 3.0 ± 0.2 mm and LVEDD: 3.6 ± 0.1 vs. 4.0 ± 0.1) and week 12 (LVESD: 2.5 ± 0.1 vs. 3.4 ± 0.2 mm and LVEDD: 3.7 ± 0.1 vs. 4.3 ± 0.2 mm). Additionally, Exscien1-III administration resulted in an attenuation of TAC-induced LVPWd (Fig. 3D) at both 6 wk (LVPWd: 1.1 ± 0.02 vs. 1.3 ± 0.03 mm) and 8 wk (LVPWd: 1.2 ± 0.03 vs. 1.3 ± 0.04 mm) post-TAC compared with Veh. Exscien1-III preserved LVEF compared with Veh, with significant improvements (P < 0.01) observed as early as 6 wk and continuing on to the 12-wk end point (Fig. 3E).
Fig. 3.

Exscien1-III prevents adverse cardiac remodeling and preserves cardiac function after transverse aortic constriction (TAC). A: experimental protocol for the TAC cardiac failure study. Mice were subjected to TAC surgery, and Exscien1-III or vehicle (Veh) treatment was administered daily via intraperitoneal injection starting 3 wk post-TAC and continued until the 12-wk end point. Echocardiography was performed at baseline and every 2 wk post-TAC to assess left ventricular (LV) end-diastolic dimension (LVEDD; B), LV end-systolic dimension (LVESD; C), LV posterior wall thichness at diastole (LVPWd; D), and LV ejection fraction (LVEF; E). *P < 0.05 vs. Veh; **P < 0.01 vs. Veh.
Postmortem myocardial morphometric data (ventricle/tibia length, atria/tibia length, and lung/tibia length) are shown in Fig. 4, A–C. Mice treated with Exscien1-III showed a significant reduction in TAC-induced cardiac hypertrophy in both ventricles (16% decrease; Fig. 4A) and atria (39% decrease; Fig. 4B), correlating with the echocardiography data. Representative images of hemotoxylin and eosin-stained cardiomyocytes are shown in Fig. 4D. Similarly, there was a significant decrease in the lung-to-tibia length ratio between Exscien1-III- and Veh-treated mice (Fig. 4C).
Fig. 4.

Treatment with Exscein1-III reduces transverse aortic constriction (TAC)-induced hypertrophy. Postmortem morphological assessment of ventricular weight to tibia length (in mg/mm; A), atria weight to tibia length (in mg/mm; B), and lung weight to tibia length (in mg/mm; C) from Exscien1-III- and vehicle (Veh)-treated mice. Circles inside bars denote n values per group. D: representative images of hematoxylin and eosin-stained cardiomyocytes from Exscien1-III- and Veh-treated mice.
Exscien1-III attenuates TAC-induced cardiac mtDNA damage.
Examination of HA expression in whole cell lysates (Fig. 5A) from mouse hearts treated with Exscien1-III revealed a slight accumulation of Exscien1-III in the cytosolic fraction at 12 wk post-TAC. However, Exscien1-III-treated mice displayed an increased presence of the HA tag in the cardiac mitochondrial fraction (Fig. 5B) compared with Veh-treated mice, confirming that a portion of the fusion protein is trafficked to the mitochondria.
Fig. 5.

Exscien1-III is localized in cardiac mitochondria after daily intraperitoneal injections and reduces transverse aortic constriction (TAC)-induced mtDNA damage. A and B: protein expression of the hemagglutinin (HA) tag found on the Exscien1-III fusion peptide measured in total ventricular (A) and mitochondrial tissue fractions (B) isolated from mice treated with Exscien1-III or vehicle (Veh) at 12 wk post-TAC. C: protein expression of HA-tagged Exscien1-III measured in the total cell lysate, cytosol, nuclease, and mitochondria at 6 wk post-TAC. D: fraction of intact mtDNA in the heart measured in mice treated with Exscien1-III or Veh at 6 wk post-TAC or Sham. Circles inside bars denote n values per group. COX IV, cytochrome c oxidase subunit IV.
The additional experiments shown in Fig. 5C demonstrated mitochondrial localization of the HA tag on Exscien1-III in mouse hearts after 6 wk of HF. Finally, administration of Exscien 1 reduced the extent of mtDNA damage at 6 wk after HF (Fig. 5D). The fraction of intact mtDNA was significantly greater (P < 0.01) in mice subjected to TAC surgery and treated with Exscien1-III compared with Veh. The data shown in Fig. 5D also demonstrate that TAC resulted in significant mtDNA damage.
Administration of Exscein1-III reduces myocardial fibrosis.
Histological assessment of interstitial and perivascular collagen via picrosirus red- and Masson’s trichrome-stained sections was performed at 12 wk post-TAC (Fig. 6) to assess myocardial fibrosis. Picrosirius red and Masson’s trichrome staining revealed a significant reduction in collagen deposition after chronic treatment with Exscien1-III, indicating attenuated fibrosis. Representative photomicrographs of heart sections stained with either picrosirius red or Masson’s trichrome are shown in Fig. 6A. Sections showing both interstitial and perivascular collagen are shown in Fig. 6A. Treatment with Exscien1-III resulted in a 58% reduction (P < 0.05) in interstitial collagen deposition as analyzed by the percent CVF (Fig. 6B) of picrosirus red-stained slides. A similar trend was observed with respect to perivascular collagen deposition, as we observed a 43% reduction in Exscien1-III-treated animals (P = 0.05) compared with Veh.
Fig. 6.

Exscien1-III attenuates myocardial fibrosis after transverse aortic constriction (TAC). A: representative images of cardiac sections at 12 wk post-TAC stained for picrosirus red (PSR) or Masson’s trichrome for fibrosis. Assessment of collagen volume fraction was measured via fluorescence quantification of PSR-stained slides. B: schematic construction of Exscien1-III. Exscien1-III consists of a mitochondria targeting sequence (MTS), an endonuclease III protein transduction domain, a hemagglutinin (HA) tag for immunological localization, a TAT sequence to facilitate cell uptake, and a polyhistidine tail (10-HIS), interstitial and perivascular collagen (C). Circles inside bars denote n values per group.
Exscien1-III treatment attenuates oxidative stress by increasing expression of the mitochondrial antioxidant manganese SOD2.
The circulating 8-isoprostane level (markers of oxidative stress) was significantly lower in mice treated with Exscien1-III than in those treated with Veh (P < 0.05; Fig. 7A). Manganese SOD2 is an antioxidant enzyme found on the inner mitochondrial membrane that inhibits cell necrosis and has a role in apoptosis (40b). Examination of SOD2 mRNA (P < 0.05; Fig. 7B) and protein (P < 0.05; Fig. 7C) expression in LV homogenates at 12 wk after TAC revealed that Exscien1-III administration resulted in a significant increase compared with Veh. Furthermore, Exscien1-III-treated mice had higher mitochondrial SOD2 protein expression (P < 0.05; Fig. 7D) compared with mice that received Veh. PGC-1α has an important role in mediating mitochondrial biogenesis and metabolism (17) as well as mitochondrial antioxidants (32). Examination of PGC-1α in cardiac mitochondrial lysates (Fig. 7E) did not reveal significant changes.
Fig. 7.
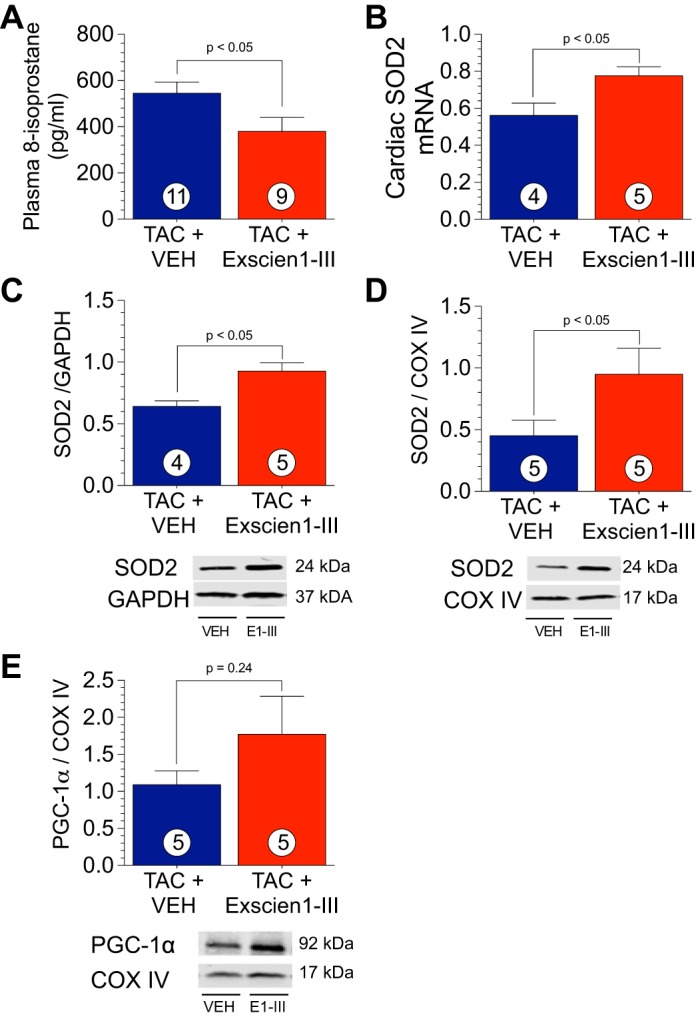
Exscien1-III increases superoxide dismutase (SOD)2. A: levels of total circulating 8-isoprostane in plasma from mice treated with Exscien1-III or vehicle (Veh) at 12 wk after transverse aortic constriction (TAC). B: mRNA levels of SOD2 in ventricular homogenates from mice treated with Exscien1-III or Veh at 12 wk post-TAC. C and D: protein expression of SOD2 was measured in total ventricular homogenates (C) and cardiac mitochondrial fraction (D). E: protein expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) was measured in the cardiac mitochondrial fraction. Circles inside bars denote n values per group. COX IV, cytochrome c oxidase subunit IV.
Exscien1-III reduces apoptotic markers.
Apoptosis is a major component in exacerbating HF (9, 24). We next examined mitochondrial apoptotic signaling markers. In response to treatment with Exscien1-III, we observed reduced p53 mRNA (P < 0.05; Fig. 8A) and Bax protein expression (P < 0.05; Fig. 8B) in mitochondrial lysates at 12 wk post-TAC compared with mice that received Veh. Furthermore, cleaved caspase-3 (Fig. 8C) and cleaved caspase-9 (Fig. 8D) were decreased (P < 0.05) in Exscien1-III-treated mice.
Fig. 8.
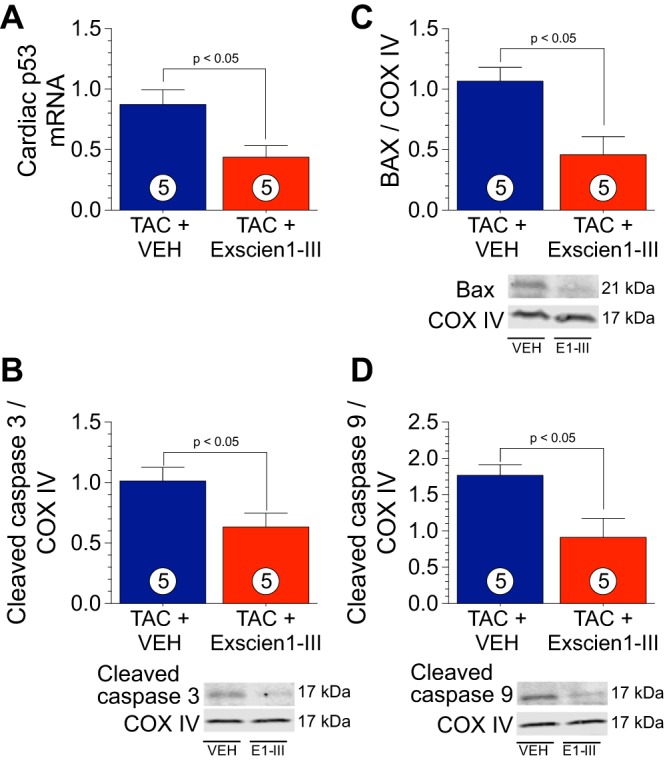
Exscien1-III reduced mitochondrial apoptotic markers. A: mRNA levels of p53 in ventricular homogenates from mice treated with Exscien1-III or vehicle (Veh). B−D: protein expression of Bax (B), cleaved caspase-3 (C), and cleaved caspase-9 (D) in cardiac mitochondrial fractions from mice treated with Exscien1-III or Veh at 12 wk after transverse aortic constriction (TAC). Circles inside bars denote n values per group. COX IV, cytochrome c oxidase subunit IV.
DISCUSSION
mtDNA is a critical target for oxidative stress. Significant damage to mtDNA induces a cascade of events, amplifying cellular oxidative damage and triggering cellular dysfunction and apoptosis, which ultimately results in organ failure. Recent studies have revealed that mitochondria-targeted DNA repair enzymes possessing glycosylase/AP lyase activity enhance cellular survival during prolonged periods of oxidative stress (20, 21). In the present study, Exscien1-III, a recently developed mitochondria-targeted DNA glycosylase/AP lyase peptide, displayed strong cardioprotective actions in two different models of cardiovascular disease (i.e., MI/R- and TAC-induced HF). In the setting of MI/R, significant cardioprotection was observed after 24 h and 4 wk of reperfusion when Exscien1-III was given at reperfusion. In a murine model of cardiac hypertrophy and HF, Exscien1-III administration preserved cardiac function and prevented maladaptive hypertrophy and remodeling. We also investigated the effects of a formulation of Exscien1-III (Endo III) lacking the HA tag (Fig. 9A) and observed very similar cardioprotective effects (Fig. 9B). Finally, myocardial infarct size was significantly reduced when a mitochondria-targeted fusion protein containing OGG1 was administered to mice subjected to MI/R injury (Fig. 10, A and B), providing further support for this novel therapeutic strategy in the setting of acute MI/R injury.
Fig. 9.

A simplified version of Exscien1-III, which does not contain the hemagglutinin (HA) tag, also attenuates myocardial ischemia-reperfusion (MI/R) injury. Mice were subjected to 45 min of ischemia and 24 h of reperfusion. Exscien1-III or vehicle (Veh) was administered via intracardiac injection at the time of reperfusion. A: schematic construction of Exscien1-III without the HA tag. B: data for the myocardial area at risk (AAR) as a percentage of the left ventricle (AAR/LV) and infarct size as a percentage of the area at risk (INF/AAR) in Exscien1-III without HA tag- and Veh-treated animals.
Fig. 10.

8-Oxoguanine DNA glycosylase (OGG1) attenuates myocardial ischemic injury and reduces myocardial infarct size. Mice were subjected to 45 min of ischemia and 24 h of reperfusion. OGG1, mOGG1, or vehicle (Veh) was administered via intracardiac injection at the time of reperfusion. A: data for the myocardial area at risk as a percentage of the left ventricle (AAR/LV) and infarct size as a percentage of the area at risk (INF/AAR) in OGG1-, mOGG1-, and Veh-treated animals. B: serum cardiac troponin I in OGG1- and Veh-treated mice. Circles inside bars denote n values per group.
Previous studies have shown that mitochondrial targeted delivery of OGG1 and Endo III have significant protection against oxidative damage (14, 20, 21, 38, 46). OGG1 is a mammalian enzyme that targets oxidized purines, whereas Endo III, derived from bacteria, targets oxidized pyrimidines (46). Transfection with mitochondria-targeted OGG1 or Endo III in HeLa cells increases repair of oxidative lesion by enhancing mitochondrial lyase activity (8) and improves cellular vitality in response to oxidative stress (14, 38). Similarly, OGG1 also reduces oxidative damage and inhibits apoptosis in pulmonary artery endothelial cells (15, 40). Furthermore, fibroblasts isolated from OGG1 knockout mice have attenuated mitochondrial function and ATP synthesis with increased apoptosis after oxidative insult (1). Moreover, overexpression of OGG1 blocks oxidative-induced mtDNA damage and prevents mitochondrial p53 translocation and apoptosis (25). These finding provide the foundation for the hypothesis that the use of AP lyases is a viable therapeutic strategy for the treatment of cardiac failure either caused by myocardial infarction or pressure overload.
Mitochondria-targeted fusion proteins containing either OGG1 (13) or Endo III (14), identical to the peptides used in the present study, attenuate mechanical lung injury in mice subjected to high peak inflation pressure ventilation with reductions in pulmonary inflammation and oxidative mtDNA damage (20, 21). Similarly, in response to a single treatment with OGG1 at reperfusion, we found that OGG1 significantly reduced infarct size and circulating levels of cardiac troponin (Fig. 10, A and B). Likewise, Yang et al. (46) have previously reported that Endo III protects the rat heart after MI/R injury. Although the mechanism for cardioprotection has not been fully elucidated, previous studies have shown that the uptake of Exscien1-III into the heart’s mitochondria occurs as early as 20 min after injection (46). Additionally, in that recent study (46), Yang et al. found higher mitochondrial levels of Exscien1-III in the areas of the myocardium subjected to ischemia and reperfusion rather than those areas that were continuously perfused, suggesting that Exscein1-III may target those mitochondria that are exposed to oxidative damage.
Chronic increases in mitochondrial ROS during cardiac injury have a cataclysmic effect on mtDNA, accelerating the progression of HF; however, there is a paucity of data regarding whether enhanced mtDNA repair during the development of HF can preserve function. A previous study has shown that cardiac restricted overexpression of OGG1 reduced cardiac fibrosis without preserving function (43). However, in the present study, we demonstrate that delayed therapy with Exscien1-III daily administration starting at 3 wk post-TAC attenuated TAC-induced LV dilation, wall thickening, and hypertrophy. Moreover, Exscien1-III treatment preserved cardiac function starting at 6 wk post-TAC. Examination of myocardial levels of Exscien1-III using the HA tag in the present study revealed increased mitochondrial HA present in Exscien1-III treated mice and negligible cytosolic localization.
ROS plays an integral role in regulating signaling cascades that can promote myocardial remodeling. In cardiac myocytes, ROS can differentially activate hypertrophic factors (28) that are key regulators of extracellular matrix remodeling, including matrix metalloproteinases (6, 41, 44) and lysyl oxidase (42). Dai et al. demonstrated that targeting mitochondrial ROS, either by administration of mitochondrial targeted antioxidants (12) or by overexpressing mitochondrial catalase (13), protects against TAC-induced cardiac remodeling and dysfunction. mtDNA repair during chronic periods of oxidative stress has been shown to have beneficial protective effects against ROS (19) and inhibit those pathways that lead to pathological remodeling. Formation of cardiac fibrosis is a hallmark of pathological cardiac remodeling. Cardiac fibrosis can be divided into three categories: interstitial fibrosis, perivascular fibrosis, and replacement fibrosis (36). In the TAC-induced HF model, interstitial and perivascular fibrosis are the prominent types of fibrosis due to the chronic pressure overload (36). In the present study, administration of Exscien1-III attenuated the TAC-induced cardiac fibrotic response with a reduction of both interstitial and perivascular collagen deposition. Further study is needed examine the exact mechanism of action by which Exscien1-III attenuated fibrosis.
SOD2, found on the inner mitochondrial membrane, protects against oxidant-induced mitochondrial damage and dysfunction. Genetic deletion of SOD results in dilated cardiomyopathy (31) and mitochondrial injury and neurodegeneration (29), leading to embryonic or neonatal lethality. Heterozygous SOD2−/+ mice are prone to increased mitochondrial oxidative damage that is correlated with reduced mitochondrial function as well as an increased release of apoptogenic factors (26, 40b). Overexpression of mitochondrial SOD2 reduces infarct size after MI/R injury potentially through the scavenging of free radicals (5). In the present study, we observed increases in myocardial SOD2 mRNA and protein expression in mice treated with Exscien1-III at 12 wk post-TAC.
In congestive HF patients, cardiomyocyte apoptosis is mediated by the proapoptotic Bax protein in the regions bordering old infarcts (33). Cardiac hypertrophy and HF results in increased cardiomyocyte apoptosis through Bax signaling (9). In neurodegenerative diseases, the activation of p53 or proapoptotic Bax signaling is induced through increases in mitochondrial ROS and mtDNA damage (45). In response to Exsecien1-III treatment, we observed reduced cardiac p53 mRNA and decreased Bax protein expression in mitochondria at 12 wk post-TAC. Further examination of downstream Bax signaling revealed reduced mitochondrial protein expression of caspase-3 and caspase-9 in the hearts of Exscien1-III-treated mice, indicating that the absolute amount of cardiomyocytes entering apoptosis was reduced by this therapy.
In summary, Exscien1-III significantly abrogated myocardial infarct size, attenuated cardiac troponin release, and preserved LV function after acute MI/R injury. Daily administration of Exscien1-III starting at 3 wk post-TAC significantly preserved LV function and attenuated TAC-induced hypertrophy, myocardial fibrosis, and remodeling. Exscien1-III increased mitochondrial antioxidant expression and reduced apoptotic markers in TAC mice. These data indicate that Exscien1-III may prove to be an efficacious therapeutic agent for the treatment of HF and other cardiovascular disease.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants 5-R01-HL-092141, 5-R01-HL-093579, 1-R01-HL-11657, and 1-U24-HL-094373 (to D. J. Lefer).
DISCLOSURES
D. J. Lefer serves as an unpaid consultant to Exscien Inc. (Mobile, AL) and holds stock in Exscien. M. N. Gillespie and G. L. Wilson serve as consultants to Exscien Inc. (Mobile, AL) and hold stock in Exscien..
AUTHOR CONTRIBUTIONS
J.M.B., M.R., M.N.G., G.L.W., and D.J.L. conceived and designed research; J.M.B., Z.L., C.L.O., D.J.P., H.O., S.B., O.M.G., and M.R. performed experiments; J.M.B., Z.L., C.L.O., D.J.P., H.O., S.B., O.M.G., M.R., and M.N.G. analyzed data; J.M.B., Z.L., C.L.O., H.O., S.B., O.M.G., M.R., M.N.G., and D.J.L. interpreted results of experiments; J.M.B., Z.L., C.L.O., and D.J.L. prepared figures; J.M.B. drafted manuscript; J.M.B., Z.L., C.L.O., and D.J.L. edited and revised manuscript; D.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Craig Zibilich for technical support during the course of the study. We thank Dr. Luis Marrero and the Morphology and Imaging Core at Louisiana State University Health Sciences Center for performing hematoxylin and eosin staining on heart tissues.
REFERENCES
- 1.Bacsi A, Chodaczek G, Hazra TK, Konkel D, Boldogh I. Increased ROS generation in subsets of OGG1 knockout fibroblast cells. Mech Ageing Dev 128: 637–649, 2007. doi: 10.1016/j.mad.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhushan S, Kondo K, Polhemus DJ, Otsuka H, Nicholson CK, Tao YX, Huang H, Georgiopoulou VV, Murohara T, Calvert JW, Butler J, Lefer DJ. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide-mediated cytoprotective signaling. Circ Res 114: 1281–1291, 2014. doi: 10.1161/CIRCRESAHA.114.301475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradley JM, Cryar KA, El Hajj MC, El Hajj EC, Gardner JD. Exposure to diesel exhaust particulates induces cardiac dysfunction and remodeling. J Appl Physiol 115: 1099–1106, 2013. doi: 10.1152/japplphysiol.00343.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley JM, Islam KN, Polhemus DJ, Donnarumma E, Brewster LP, Tao YX, Goodchild TT, Lefer DJ. Sustained release nitrite therapy results in myocardial protection in a porcine model of metabolic syndrome with peripheral vascular disease. Am J Physiol Heart Circ Physiol 309: H305–H317, 2015. doi: 10.1152/ajpheart.00163.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 30: 2281–2289, 1998. doi: 10.1006/jmcc.1998.0789. [DOI] [PubMed] [Google Scholar]
- 6.Cho A, Graves J, Reidy MA. Mitogen-activated protein kinases mediate matrix metalloproteinase-9 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 20: 2527–2532, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Chouteau JM, Obiako B, Gorodnya OM, Pastukh VM, Ruchko MV, Wright AJ, Wilson GL, Gillespie MN. Mitochondrial DNA integrity may be a determinant of endothelial barrier properties in oxidant-challenged rat lungs. Am J Physiol Lung Cell Mol Physiol 301: L892–L898, 2011. doi: 10.1152/ajplung.00210.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cline SD. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim Biophys Acta 1819: 979–991, 2012. doi: 10.1016/j.bbagrm.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G, de Rienzo A, Roncarati R, Trimarco B, Lembo G. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 99: 3071–3078, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem 272: 25409–25412, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Croteau DL, Stierum RH, Bohr VA. Mitochondrial DNA repair pathways. Mutat Res 434: 137–148, 1999. doi: 10.1016/S0921-8777(99)00025-7. [DOI] [PubMed] [Google Scholar]
- 12.Dai DF, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58: 73–82, 2011. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 93: 79–88, 2012. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtdna repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am J Physiol Lung Cell Mol Physiol 283: L205–L210, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem 275: 37518–37523, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA 104: 15560–15565, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (pgc-1) regulatory cascade in cardiac physiology and disease. Circulation 115: 2540–2548, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Grishko V, Solomon M, Wilson GL, LeDoux SP, Gillespie MN. Oxygen radical-induced mitochondrial DNA damage and repair in pulmonary vascular endothelial cell phenotypes. Am J Physiol Lung Cell Mol Physiol 280: L1300–L1308, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Grishko VI, Rachek LI, Spitz DR, Wilson GL, LeDoux SP. Contribution of mitochondrial DNA repair to cell resistance from oxidative stress. J Biol Chem 280: 8901–8905, 2005. doi: 10.1074/jbc.M413022200. [DOI] [PubMed] [Google Scholar]
- 20.Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Potter BJ, Wilson GL, Gillespie MN, Parker JC. Mitochondrial-targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator-induced lung injury in intact mice. Am J Physiol Lung Cell Mol Physiol 304: L287–L297, 2013. doi: 10.1152/ajplung.00071.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Wilson GL, Gillespie MN, Parker JC. Mitochondrial targeted endonuclease III DNA repair enzyme protects against ventilator induced lung injury in mice. Pharmaceuticals (Basel) 7: 894–912, 2014. doi: 10.3390/ph7080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88: 529–535, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Kang PM, Izumo S. Apoptosis and heart failure: A critical review of the literature. Circ Res 86: 1107–1113, 2000. doi: 10.1161/01.RES.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 25.Kim SJ, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT, Weitzman S, Bohr VA, Kamp DW. Mitochondria-targeted ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem 289: 6165–6176, 2014. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA 98: 2278–2283, 2001. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127: 1116–1127, 2013. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol 35: 615–621, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 93: 9782–9787, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: Ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 33: 1065–1089, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11: 376–381, 1995. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 32.Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal 13: 1011–1022, 2010. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T, Fujiwara H. Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation 94: 1506–1512, 1996. doi: 10.1161/01.CIR.94.7.1506. [DOI] [PubMed] [Google Scholar]
- 34.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pastukh VM, Gorodnya OM, Gillespie MN, Ruchko MV. Regulation of mitochondrial genome replication by hypoxia: The role of DNA oxidation in D-loop region. Free Radic Biol Med 96: 78–88, 2016. doi: 10.1016/j.freeradbiomed.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piek A, de Boer RA, Silljé HH. The fibrosis-cell death axis in heart failure. Heart Fail Rev 21: 199–211, 2016. doi: 10.1007/s10741-016-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ, Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail 6: 1077–1086, 2013. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rachek LI, Grishko VI, Alexeyev MF, Pastukh VV, LeDoux SP, Wilson GL. Endonuclease III and endonuclease VIII conditionally targeted into mitochondria enhance mitochondrial DNA repair and cell survival following oxidative stress. Nucleic Acids Res 32: 3240–3247, 2004. doi: 10.1093/nar/gkh648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase Ogg1 defends against oxidant-induced mtdna damage and apoptosis in pulmonary artery endothelial cells. Free Radic Biol Med 50: 1107–1113, 2011. doi: 10.1016/j.freeradbiomed.2010.10.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 5: 145–152, 2006. [DOI] [PubMed] [Google Scholar]
- 40b.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol 281: H1422–H1432, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B, Quiroga C, Pedrozo Z, Munoz JP, Garcia L, Castro PF, Lavandero S. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep 14: 532–539, 2012. doi: 10.1007/s11906-012-0305-4. [DOI] [PubMed] [Google Scholar]
- 42.Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem Biophys Res Commun 413: 370–375, 2011. doi: 10.1016/j.bbrc.2011.08.109. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. Am J Physiol Heart Circ Physiol 301: H2073–H2080, 2011. doi: 10.1152/ajpheart.00157.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie Z, Singh M, Singh K. Differential regulation of matrix metalloproteinase-2 and-9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1beta. J Biol Chem 279: 39513–39519, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Yang JL, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst) 7: 1110–1120, 2008. doi: 10.1016/j.dnarep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang XM, Cui L, White J, Kuck J, Ruchko MV, Wilson GL, Alexeyev M, Gillespie MN, Downey JM, Cohen MV. Mitochondrially targeted endonuclease iii has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol 110: 3, 2015. doi: 10.1007/s00395-014-0459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]