

Abstract
Endothelial dysfunction in chronic hypoxia (CH)-induced pulmonary hypertension is characterized by reduced store-operated Ca2+ entry (SOCE) and diminished Ca2+-dependent production of endothelium-derived vasodilators. We recently reported that SOCE in pulmonary arterial endothelial cells (PAECs) is tightly regulated by membrane cholesterol and that decreased membrane cholesterol is responsible for impaired SOCE after CH. However, the ion channels involved in cholesterol-sensitive SOCE are unknown. We hypothesized that cholesterol facilitates SOCE in PAECs through the interaction of Orai1 and stromal interaction molecule 1 (STIM1). The role of cholesterol in Orai1-mediated SOCE was initially assessed using CH exposure in rats (4 wk, 380 mmHg) as a physiological stimulus to decrease PAEC cholesterol. The effects of Orai1 inhibition with AnCoA4 on SOCE were examined in isolated PAEC sheets from control and CH rats after cholesterol supplementation, substitution of endogenous cholesterol with epicholesterol (Epichol), or vehicle treatment. Whereas cholesterol restored endothelial SOCE in CH rats, both Epichol and AnCoA4 attenuated SOCE only in normoxic controls. The Orai1 inhibitor had no further effect in cells pretreated with Epichol. Using cultured pulmonary endothelial cells to allow better mechanistic analysis of the molecular components of cholesterol-regulated SOCE, we found that Epichol, AnCoA4, and Orai1 siRNA each inhibited SOCE compared with their respective controls. Epichol had no additional effect after knockdown of Orai1. Furthermore, Epichol substitution significantly reduced STIM1-Orai1 interactions as assessed by a proximity ligation assay. We conclude that membrane cholesterol is required for the STIM1-Orai1 interaction necessary to elicit endothelial SOCE. Furthermore, reduced PAEC membrane cholesterol after CH limits Orai1-mediated SOCE.
NEW & NOTEWORTHY This research demonstrates a novel contribution of cholesterol to regulate the interaction of Orai1 and stromal interaction molecule 1 required for pulmonary endothelial store-operated Ca2+ entry. The results provide a mechanistic basis for impaired pulmonary endothelial Ca2+ influx after chronic hypoxia that may contribute to pulmonary hypertension.
Keywords: pulmonary hypertension, store-operated calcium entry, stromal interaction molecule 1
INTRODUCTION
Endothelial dysfunction in pulmonary hypertension is characterized by imbalanced production of endothelium-derived vasoconstrictors and vasodilators leading to vasoconstriction, vascular remodeling, and subsequent elevation of pulmonary vascular resistance (11a, 21, 75). The production of many endothelium-derived vasodilators and antimitogenic compounds, including nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factors, is closely regulated by endothelial intracellular Ca2+ levels ([Ca2+]i) (7a, 17, 25, 45, 47). Diminished pulmonary endothelial [Ca2+]i and limited production of endothelium-derived NO are associated with the development of chronic hypoxia (CH)-induced pulmonary hypertension (51, 56). However, the mechanisms of reduced pulmonary endothelial [Ca2+]i after CH exposure are not well investigated.
Endothelial store-operated Ca2+ entry (SOCE) is required for endothelium-dependent vasorelaxation in systemic arteries (28), in part through production of NO (3). SOCE is initiated when a stimulated G protein-coupled receptor activates phospholipase C (PLC), which produces inositol 1,4,5-trisphosphate (IP3) by hydrolyzing phosphatidylinositol bisphosphate (PIP2). IP3, as a second messenger, activates IP3 receptors on the endoplasmic reticulum (ER) membrane, leading to rapid ER Ca2+ release. ER Ca2+ store depletion then activates store-operated cation channels (SOCs), which leads to sustained Ca2+ influx (64). Stromal interaction molecule 1 (STIM1) has been identified as an ER Ca2+ sensor and suggested as the essential link between Ca2+ store depletion and SOC activation (72). As ER Ca2+ levels fall, STIM1 molecules oligomerize on the ER membrane and interact with SOCs on the cell membrane, which include Orai channels (12a). Orai1, one of three isoforms of Orai found in mammalian cells, mediates SOCE when activated by STIM1 (86). The STIM1-Orai1 interaction occurs in cholesterol-rich caveolae on the cell membrane. Interestingly, intact caveolae are required for the activation of Orai1-mediated SOCE in platelets (14). Depletion of membrane cholesterol not only inhibits SOCE but also interrupts STIM1 clustering near the cell membrane (59). However, whether membrane cholesterol directly facilitates STIM1-Orai1 interactions and SOCE activation or indirectly modulates this Ca2+ entry pathway by contributing to lipid raft integrity is not clear.
We have previously reported that decreased endothelial SOCE after CH is associated with diminished membrane cholesterol and that this reduced SOCE is restored by cholesterol supplementation (56, 57, 84). Furthermore, to study the functional role of membrane cholesterol in regulating SOCE, epicholesterol (Epichol) was used to replace native cholesterol. As the enantiomer of cholesterol, Epichol has similar effects on membrane fluidity and lipid domain formation as those of cholesterol but lacks the regulatory influences of cholesterol on ion channel function (22, 65, 83). Substitution of endogenous cholesterol with Epichol mimics the effect of CH in reducing endothelial SOCE while having no effect on caveolar number (84). However, the mechanisms by which reduced endothelial membrane cholesterol limits SOCE after CH are not clear. In the present study, we hypothesized that cholesterol facilitates the interaction of Orai1 with STIM1 to mediate SOCE in pulmonary arterial endothelial cells (PAECs). We tested this hypothesis by examining the effects of either cholesterol or Epichol treatment combined with Orai1 inhibition on SOCE in both freshly isolated PAECs from normoxic and CH rats and in cultured pulmonary microvascular endothelial cells (PMVECs).
METHODS
Animals and CH exposure protocol.
Male Sprague-Dawley rats (200–250 g) were used for in vivo and ex vivo experiments. Rats exposed to CH were placed in a hypobaric chamber with barometric pressure maintained at ≈380 mmHg (inspired Po2 ≈70 mmHg) for 4 wk (84). Age-matched control rats were housed in similar cages under ambient barometric pressure (≈630 mmHg in Albuquerque, NM). The hypobaric chamber was opened 3 times/wk to provide fresh rat chow, water, and clean bedding. All animals were maintained on a 12:12-h light-dark cycle. All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center.
Isolation and preparation of PAECs.
After CH or normoxic exposure, rats were euthanized with pentobarbital sodium (200 mg/kg ip) and the heart and lungs were exposed by midline thoracotomy. The left lung was rapidly excised and placed in ice-cold HEPES buffer solution. Intrapulmonary arteries (third and fourth order, 200- to 400-μm inner diameter) were dissected from the superior region of the left lung, and the parenchymal lung tissue was carefully removed. Arteries were then cut longitudinally and treated with 0.2 mg/ml dithiothreitol and 2 U/ml papain in HEPES buffer for 45 min at 37°C. Vessels were carefully removed from the digestion solution and placed in 1 ml HEPES buffer containing 2 mg/ml BSA. PAEC sheets were then released by gentle trituration with a small-bore fire-polished Pasteur pipette and stored at 4°C. One to two drops of the solution containing freshly isolated rat PAECs were placed on a poly-l-lysine-coated glass coverslip and incubated at 37°C for 30 min before experimentation (84). PAEC sheets collected using this approach were identified by their distinct morphology (57, 84) and uptake of Dil-Ac-LDL (56).
Cell culture.
To complement experiments using freshly isolated PAEC sheets, cultured PMVECs (VEC Technologies) were also used in this study. Preliminary experiments revealed these cells to demonstrate similar cholesterol sensitivity of SOCE as native PAECs and to express functional Orai1 channels. In addition, cultured cells permit the use of genetic manipulation with siRNA and determination of protein-protein interactions (Orai1-STIM1) using a proximity ligation assay (PLA) for better mechanistic analysis of the molecular components of cholesterol-regulated SOCE. PMVECs were maintained in a humidified incubator at 37°C with 5% CO2 in MCDB-131 complete media (VEC Technologies). Cultures were split every 4–6 days, and cells were studied between passages 5 and 10. Depending on the experimental protocol, cells were seeded onto round poly-l-lysine-coated glass coverslips in 6-well plates, 6-well plates coated with attachment factors, or 18-well slides coated with attachment factors.
Preparation of cholesterol and Epichol solutions.
Solutions were prepared by saturating methyl-β-cyclodextrin (MβCD) with either cholesterol or Epichol, as previously described (11). Briefly, the cyclodextrin-sterol solutions were prepared by the addition of sterols to MβCD (10 mM) in a molar ratio of 1:5 and dissolution in HEPES buffer containing the following (in mM): 150 NaCl, 6 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, and 10 glucose (pH 7.4). Each solution was vortexed and sonicated using a bath sonicator for 10–15 min. The saturated cyclodextrin-sterol solution was then placed in a rotating incubator at 37°C overnight. This stock solution was filtered through a 0.22-μm syringe filter, aliquoted, and stored at −80°C.
Endothelial cell membrane cholesterol manipulation.
Cholesterol supplementation was performed in untreated PAECs or PMVECs by incubation with the cholesterol-MβCD solution for 30 min at 37°C (84). Epichol substitution was similarly achieved by incubating isolated PAECs or cultured PMVECs with the Epichol/MβCD solution under the same conditions. Preliminary studies revealed that, in contrast to effects of membrane cholesterol manipulation in freshly dispersed PAECs from normoxic rats (84), cholesterol supplementation increased SOCE in cultured PMVECs and conferred sensitivity to Epichol substitution (data not shown), suggesting that membrane cholesterol content is reduced under cell culture conditions compared with the native state. Therefore, cultured PMVECs were pretreated with cholesterol-MβCD solution before experimentation to mimic responses of native PAECs.
Ca2+ influx in freshly isolated and cultured pulmonary endothelial cells.
After vehicle, cholesterol, or Epichol treatment, freshly isolated PAEC sheets or cultured PMVECs (passages 5–10) were loaded with fura-2 AM (3 μM and 0.05% pluronic acid) in HEPES buffer for 7 min at room temperature (∼23°C) and washed for 15 min at 37°C. SOCE, depolarization-induced Ca2+ influx, and receptor-operated Ca2+ influx were measured by Mn2+ quenching of fura-2 fluorescence in PAEC sheets or PMVECs as previously described (84). Mn2+ enters the cell as a Ca2+ surrogate and reduces fura-2 fluorescence upon binding to the fluorophore. The preparation was excited at the isosbestic wavelength (360 nm) at 1 Hz (IonOptix Hyperswitch), and emission was recorded at 510 nm with a photomultiplier tube. At this excitation wavelength, fura-2 fluorescence intensity is not influenced by changes in [Ca2+]i, thus providing a measure of Ca2+ influx as reflected by Mn2+ uptake independent of ER Ca2+ release or sequestration or efflux across the cell membrane (31). Fura-2-loaded endothelial cells were superfused with Ca2+-free HEPES buffer in the presence of vehicle or AnCoA4 [20 μM, Orai1 inhibitor (67)] for 5 min and administered the sarco(endo)plasmic reticulum Ca2+-ATPase inhibitor cyclopiazonic acid (CPA; 10 μM) to deplete intracellular Ca2+ stores and activate SOCs. To confirm the specificity of AnCoA4 as an inhibitor of SOCE, KCl (60 mM) or 1-oleoyl-2-acetyl-sn-glycerol (OAG; diacylglycerol analog, 50 μM) was applied in separate experiments to examine the effect of AnCoA4 on depolarization-induced and receptor-operated Ca2+ influx (58), respectively. Ca2+ entry represented by influx of the Ca2+ surrogate Mn2+ was then determined upon addition of extracellular Mn2+ (500 μM) in the continued presence of AnCoA4 or vehicle. SOCE was quantified by the percentage of the Mn2+-quenched fluorescence at 120 s after administration of Mn2+.
Membrane cholesterol content.
We have previously used the fluorescent cholesterol marker filipin to determine the efficacy of cholesterol manipulation protocols in freshly dispersed PAECs (84). To confirm similar effectiveness of cholesterol manipulation in PMVECs, cultures (passage 6) were treated with vehicle, MβCD, cholesterol-MβCD, or Epichol-MβCD at 37°C for 30 min, washed with PBS, and then fixed with 2% paraformaldehyde in PBS for 15 min at room temperature. Endothelial cell membrane cholesterol was detected by incubating cells with filipin III (20 μg/ml, Sigma) for 15 min at room temperature under light-protected conditions, and coverslips were mounted on the slides using mounting media (57). Slides were air dried at 4°C and stored at −20°C until analysis. Samples were imaged by fluorescence confocal microscopy (Zeiss LSM 510 AxioObserver, Göttingen, Germany) using a 405-nm laser (excitation), a 420-nm long-pass filter (emission), and a Plan-Neofluor ×40/1.3 oil objective. Filipin fluorescence intensity was quantified using ImageJ (National Institutes of Health) and calculated as that above threshold assessed using a blank control (filipin-untreated group). Fluorescence of each PMVEC was calculated and averaged to determine mean fluorescence for each treatment group. Cellular cholesterol content was also examined using an Amplex Red cholesterol assay kit (Molecular Probes) following the manufacturer's instructions.
Orai1 siRNA knockdown.
A cocktail of three different Orai1 siRNAs was used (RSS357633-357635, ThermoFisher). Transfection in PMVECs (passages 6–10) was achieved using Lipofectamine (Invitrogen) according to the manufacturer’s instructions. A scrambled sequence (Dharmacon) was used as a nontargeting (NT) control. Cells seeded onto either round glass coverslips (for Ca2+ imaging) or six-well plates (for Western blot analysis) were transfected with 9 μg siRNA in each well and were assayed 72 h after transfection. There were no apparent effects of either siRNA or NT treatments on cell morphology.
Western blot analysis for Orai1.
PMVECs were homogenized in a buffered solution (255 mM sucrose, 10 mM Tris·HCl, 2 mM EDTA, 12 μM leupeptin, 1 μM pepstatin A, and 0.3 μM aprotinin) and centrifuged at 500 g for 5 min at 4°C. Cell lysate protein content was quantified using a NanoDrop (NanoDrop 2000, Thermofisher), and 50 μg of protein were separated by SDS-PAGE (12% Tris-glycine) and transferred onto polyvinylidene fluoride membranes. After being blocked with 5% nonfat milk dissolved in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for 1 h at room temperature, the membrane was probed with primary antibody (1:400, rabbit anti-Orai1, ACC-062, Alomone Laboratories) in TBS-T containing 5% nonfat milk overnight at 4°C. After being washed, the membrane was incubated with secondary antibody (IgG-horseradish peroxidase-conjugated goat anti-rabbit, 1:3,000, Bio-Rad) in TBS-T containing 0.5% nonfat milk for 1 h at room temperature. Anti-β-actin (1:5,000) was used for loading control experiments in which the same membrane probing for Orai1 above was washed and reprobed for β-actin. Detection was performed with the enhanced chemiluminescence reagent (ECL Western Blotting detection reagents, Pierce) and chemiluminescence-sensitive film (GeneMate). All bands of targeted size were quantified by densitometry using ImageJ software.
Duolink PLA.
A STIM1-Orai1 interaction is required for the activation of Orai1 channels and SOCE. To determine whether cell membrane cholesterol regulates this pivotal step in SOCE, the interaction of STIM1 and Orai1 was assessed in PMVECs using the Duolink in situ PLA according to manufacturer’s instructions (Sigma-Aldrich). Briefly, PMVECs were plated on 18-well slides (Ibidi) and grown to 80–90% confluency. PMVECs pretreated with cholesterol or Epichol were then treated with either vehicle or CPA (10 μM, 5 min) before being fixed with 2% paraformaldehyde. PMVECs were incubated with Duolink blocking buffer for 30 min at 37°C and then incubated overnight with rabbit anti-STIM1 (1:250, ab106531, Abcam) and goat anti-Orai1 (1:100, sc-74778, Santa Cruz Biotechnology). Cells were then incubated with anti-rabbit PLUS and anti-goat MINUS PLA probes (1:5) for 1 h at 37°C. Negative controls were completed by 1) omission of primary antibody and 2) incubation with each primary antibody individually. Samples were amplified with Duolink In Situ Detection Reagent Orange (excitation/emission: 554/579 nm, Sigma-Aldrich) for 100 min at 37°C. SYTOX Green (1:5,000, Invitrogen) was used as a nuclear stain. Samples were mounted with Duolink mounting media, and Z-stack images of the PLA interaction were acquired using a confocal microscope (TCS SP5, Leica). The number of puncta per cell was determined using ImageJ.
Calculations and statistics.
All data are expressed as means ± SE. Values of n refer to the number of animals for experiments using freshly isolated PAECs or to the number of groups as indicated in the figures for other experiments. Percent data were converted to normal distributions by arcsine transformation before parametric analysis. An unpaired t-test, one-way ANOVA, two-way ANOVA, or Kruskal-Wallis H-test were used where appropriate for statistical comparisons. If differences were detected by ANOVA or the Kruskal-Wallis H-test, individual groups were compared with the Student-Newman-Keuls or Dunn’s multiple comparison tests, respectively. P < 0.05 was accepted as statistically significant for all comparisons.
RESULTS
Impaired pulmonary endothelial SOCE after CH is restored by cholesterol supplementation.
The importance of membrane cholesterol in diminished SOCE after CH was confirmed by examining effects of cholesterol supplementation and Epichol substitution on CPA-induced Ca2+ influx in freshly isolated PAECs from control and CH rats using the Mn2+ quenching technique. Although cholesterol treatment did not affect endothelial SOCE in normoxic rats, Epichol substitution greatly inhibited this Ca2+ entry pathway (Fig. 1A). In contrast, cholesterol repletion increased SOCE in PAECs of CH rats (Fig. 1B). Treatment with Epichol did not further attenuate SOCE in PAECs from CH rats compared with vehicle-treated cells (Fig. 1B). These data are shown in Fig. 1C. Exposure to CH significantly blunted SOCE compared with normoxic controls, which was restored by cholesterol supplementation (Fig. 1C). In cells from normoxic animals, treatment with Epichol reduced SOCE, mimicking the effects of CH exposure.
Fig. 1.

Impaired pulmonary endothelial store-operated Ca2+ entry (SOCE) after chronic hypoxia (CH) is restored by cholesterol (Chol) supplementation. Cyclopiazonic acid (CPA)-induced Ca2+ entry was assessed by the Mn2+ quenching technique in freshly isolated pulmonary artery endothelial cell (PAEC) sheets from control and CH rats. Cells were pretreated with vehicle (Veh), Chol, or epicholesterol (Epichol). F, fluorescence intensity at 360-nm excitation; F0, fluorescence intensity at time 0. A: SOCE in PAECs from normoxic (Nor) rats. n = 5–8 animals/group. *P < 0.05 vs. Veh and Chol over the range of 30–120 s. B: SOCE in PAECs from CH rats. n = 4–8 animals/group. *P < 0.05 vs. Veh and Epichol over the range of 60–120 s. C: SOCE in PAECs from each group 120 s after the onset of quenching. *P < 0.05 vs. Nor Veh; #P < 0.05 vs. CH Veh. Values are means ± SE. One-way ANOVA (A and B) or two-way ANOVA (C) followed by the Student-Newman-Keuls test were used to compare between groups.
Orai1 mediates pulmonary endothelial SOCE.
We initially assessed the contribution of Orai1 to pulmonary endothelial SOCE using the Orai1 inhibitor AnCoA4 (67). AnCoA4 was originally identified as an inhibitor of Orai1 using minimal functional domains of Orai1 and STIM1 to screen small-molecule microarrays. This compound was found to directly bind to the COOH-terminus of Orai1 and interfere not only with channel gating but also with the interaction of Stim1 and Orai1. The specificity of AnCoA4 to inhibit SOCE was confirmed by comparing its effectiveness to attenuate SOCE versus other forms of Ca2+ entry not linked to Orai1, including depolarization-induced Ca2+ entry mediated by T-type voltage-gated Ca2+ channels and receptor-operated Ca2+ entry in response to the diacylglycerol analog OAG (58). In PAECs from normoxic rats, AnCoA4 significantly attenuated CPA-induced SOCE without affecting Ca2+ entry to a depolarizing stimulus of KCl (Fig. 2A). AnCoA4 similarly reduced SOCE without affecting receptor-operated Ca2+ influx elicited by OAG (Fig. 2B) in cultured PMVECs, where this pathway of influx is more demonstrable. Together, these data establish the specificity of AnCoA4 as a SOCE-specific inhibitor.
Fig. 2.

Orai1 mediates pulmonary endothelial store-operated Ca2+ entry (SOCE). A: AnCoA4 inhibits SOCE but not depolarization-induced Ca2+ entry in freshly dispersed pulmonary arterial endothelial cells from normoxic rats. F, fluorescence intensity at 360-nm excitation; F0, fluorescence intensity at time 0. B: AnCoA4 attenuates SOCE but not 1-oleoyl-2-acetyl-sn-glycerol (OAG; diacylglycerol analog)-induced Ca2+ entry in cultured pulmonary microvascular endothelial cells. An unpaired t-test was used to compare between groups at 120 s after the onset of quenching. *P < 0.05 vs. vehicle (Veh) cyclopiazonic acid (CPA).
CH impairs Orai1-mediated pulmonary endothelial SOCE: role of membrane cholesterol.
The effect of CH on Orai1-mediated endothelial SOCE was examined using AnCoA4 in freshly isolated PAECs from normoxic and CH rats. Orai1 inhibition significantly reduced endothelial SOCE in PAECs from normoxic rats (Fig. 3, A and B) while having no effect in cells from CH animals (Fig. 3, A and C). However, AnCoA4-sensitive SOCE was restored by cholesterol supplementation in PAECs from CH rats (Fig. 3C), mirroring the efficacy of Orai1 inhibition in cholesterol-treated cells from normoxic rats (Fig. 3B). In contrast, Orai1 inhibition was without effect on Ca2+ influx after Epichol substitution in PAECs from either normoxic rats (Fig. 3B) or CH rats (Fig. 3C).
Fig. 3.

Membrane cholesterol (Chol)-sensitive Orai1 contributes to impaired store-operated Ca2+ entry (SOCE) in pulmonary arterial endothelial cells (PAECs) after chronic hypoxia (CH). A: SOCE was assessed in PAECs from normoxic (Nor) and CH rats pretreated with either the Orai1 inhibitor AnCoA4 or vehicle (Veh). n = 6, Nor Veh; n = 8, CH Veh; n = 5, Nor AnCoA4; n = 4, CH AnCoA4. *P < 0.05 vs. Nor Veh over the range of 40–120 s. F, fluorescence intensity at 360-nm excitation; F0, fluorescence intensity at time 0. B and C: effects of AnCoA4 on SOCE (120 s after the onset of quenching) in freshly dispersed PAECs from normoxic (B) and CH (C) rats after membrane Chol manipulation. *P < 0.05 vs. Veh within groups; #P < 0.05 vs. Veh.
Effect of cholesterol manipulation on membrane cholesterol content in cultured PMVECs.
To further explore the mechanism by which membrane cholesterol regulates Orai1-mediated endothelial SOCE, we focused on cultured PMVECs where genetic approaches and assessment of protein-protein association are technically more feasible. We first examined the effect of cholesterol manipulation on endothelial cell membrane cholesterol levels. Consistent with previous observations in freshly isolated PAECs (84), both MβCD and Epichol treatment significantly reduced filipin fluorescence (Fig. 4, A and B). Interestingly, cholesterol supplementation increased filipin fluorescence in cultured PMVECs (Fig. 4, A and B), which is different from our previous observation in freshly isolated PAECs where cholesterol treatment did not further augment filipin fluorescence (57). We also examined endothelial cholesterol content using an Amplex Red cholesterol assay. Consistently, cholesterol treatment augmented endothelial cholesterol content, whereas MβCD and Epichol treatment reduced native cholesterol content (Fig. 4C). Since these results suggest that cultured cells are relatively cholesterol deplete, PMVECs used in the following experiments were first supplemented with cholesterol before subsequent manipulation to better mimic native cells.
Fig. 4.
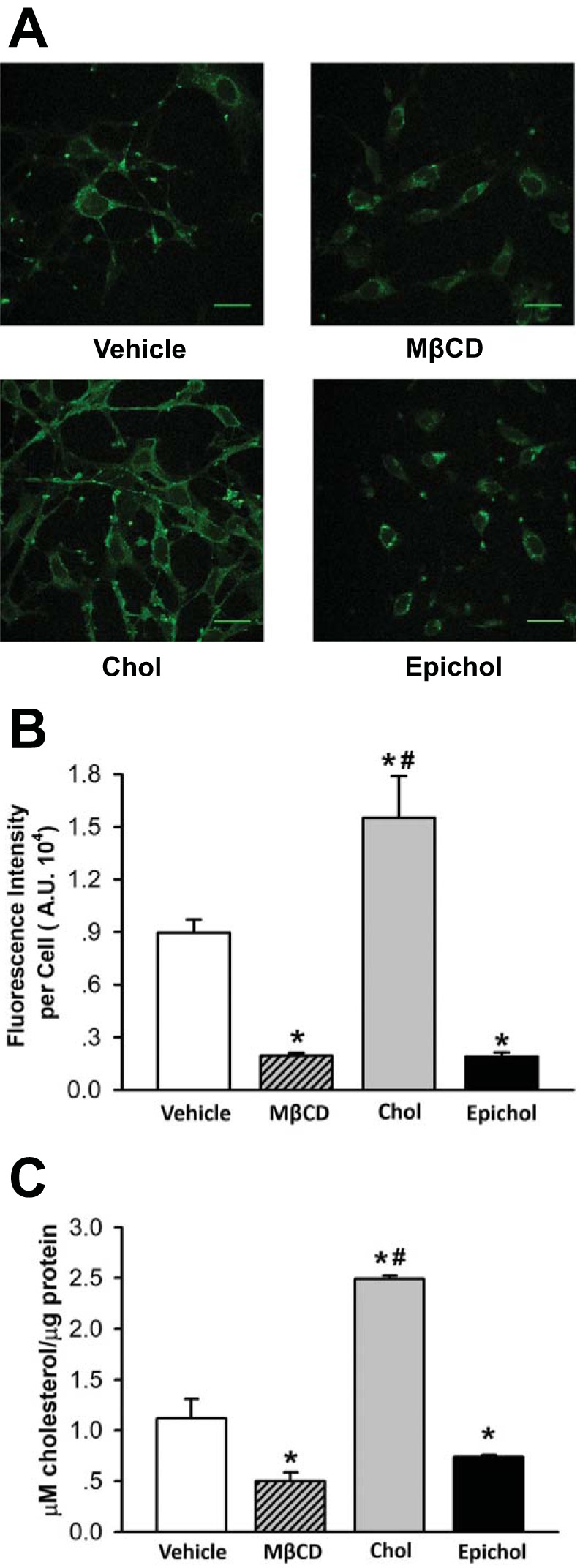
Epicholesterol (Epichol) reduces the endogenous membrane cholesterol (Chol) content of cultured pulmonary microvascular endothelial cells (PMVECs). A: representative images of filipin fluorescence from PMVECs treated with vehicle, methyl-β-cyclodextrin (MβCD), Chol, or Epichol. B: mean filipin fluorescence [in arbitrary units (AU)] in PMVECs from each group. Data were compared by the Kruskal-Wallis H-test and Dunn’s multiple comparison test. n = 5 (filipin fluorescence of 4–16 cells were measured per field of interest and averaged for an n = 1). *P < 0.05 vs. vehicle; #P < 0.05 vs. MβCD and Epichol. C: Amplex Red Chol assay to detect Chol concentration in PMVECs. Data were compared by one-way ANOVA followed by the Student-Newman-Keuls test. n = 3. *P < 0.05 vs. Vehicle; #P < 0.05 vs. MβCD and Epichol.
Epichol substitution reduces SOCE in PMVECs.
SOCE was assessed in cultured PMVECs after cholesterol manipulation. Similar to that observed in native cells, Epichol substitution significantly reduced endothelial SOCE compared with the cholesterol-treated group (Fig. 5).
Fig. 5.

Epicholesterol (Epichol) substitution reduces store-operated Ca2+ entry (SOCE) in pulmonary microvascular endothelial cells pretreated with cholesterol (Chol). n = 5, Chol; n = 5, Epichol; n = 3, time control. F, fluorescence intensity at 360-nm excitation; F0, fluorescence intensity at time 0. *P < 0.05 vs. Chol over the range of 50–120 s.
Orai1 siRNA knockdown inhibits membrane cholesterol-mediated SOCE in PMVECs.
Orai1 siRNA was used as a genetic approach to complement pharmacological inhibition experiments using AnCoA4 and further control for potential off-target effects of the inhibitor. The efficacy of Orai1 siRNA knockdown was confirmed by Western blot analysis. Orai1 siRNA had a moderate but significant effect to reduce Orai1 protein expression compared with the NT control siRNA (Fig. 6, A and B). Consistent with pharmacological Orai1 inhibition in both native and cultured cells (Fig. 2, A and B), Orai1 siRNA knockdown attenuated SOCE in PMVECs (Fig. 6C). Furthermore, Epichol substitution greatly reduced endothelial SOCE of the NT group without affecting that of Orai1 siRNA-treated cells (Fig. 6C). Together, these findings confirm the involvement of Orai1 in cholesterol-sensitive SOCE.
Fig. 6.

Orai1 siRNA knockdown inhibits membrane cholesterol (Chol)-dependent store-operated Ca2+ entry (SOCE) in pulmonary microvascular endothelial cells (PMVECs). A: representative Western blot of Orai1 and β-actin protein bands in PMVECs transfected with Orai1 siRNA or nontargeting (NT) siRNA. F, fluorescence intensity at 360-nm excitation; F0, fluorescence intensity at time 0. B: mean Western blot data of Orai1 expression from NT and Orai1 siRNA-treated PMVECs. Orai1 levels are normalized to those of β-actin. n = 4/group. *P < 0.05 vs. NT. C: SOCE in cultured PMVECs. n = 3–4/group. *P < 0.05 vs. NT + Chol over the range of 60–120 s.
Epichol substitution reduces STIM1-Orai1 interaction in PMVECs.
The role of membrane cholesterol in regulating STIM1-Orai1 interaction was examined in PMVECs. CPA-induced ER store depletion increased STIM1-Orai1 colocalization (i.e., increased the number of red puncta; Fig. 7, A and B). Epichol treatment, however, nearly abolished STIM1-Orai1 colocalization compared with the cholesterol-treated group (Fig. 7, A and B), supporting a role for membrane cholesterol to facilitate the interaction of STIM1 and Orai1 in PMVECs.
Fig. 7.
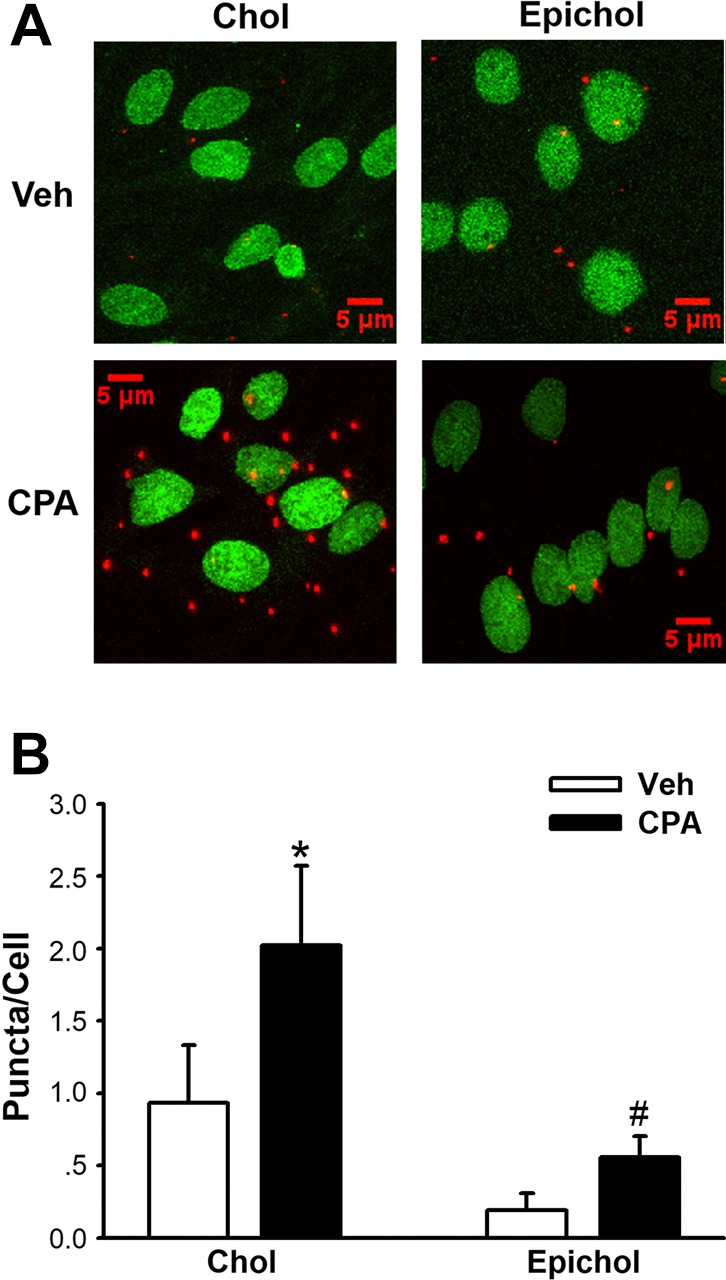
Epicholesterol (Epichol) substitution reduces the stromal interaction molecule 1 (STIM1)-Orai1 interaction in pulmonary microvascular endothelial cells (PMVECs) as assessed by an in situ proximity ligation assay. A: representative images of STIM1-Orai1 interactions (red puncta) in response to cyclopiazonic acid (CPA) in cultured PMVECs with manipulated membrane cholesterol (Chol). Nuclei area labeled with SYTOX (green). B: summarized data of the STIM1-Orai1 interaction expressed as average number of puncta/cell in cultured PMVECs. Chol supplementation increased the CPA-induced STIM1-Orai1 interaction, whereas Epichol substitution significantly inhibited this response. Groups were compared by two-way ANOVA followed by multiple comparisons testing using the Student-Newman-Keuls test. n = 5/group. *P < 0.05 vs. Chol Veh; #P < 0.05 vs. Chol CPA.
DISCUSSION
Our laboratory has previously shown that reduced membrane cholesterol after CH is associated with impaired SOCE in intrapulmonary artery endothelial cells and that endothelial membrane cholesterol facilitates SOCE through a direct interaction with signaling molecules. However, the mechanism by which membrane cholesterol-regulated endothelial Ca2+ entry via SOCs has not previously been addressed. The goal of the present study was to determine the contribution of membrane cholesterol to Orai1-mediated SOCE in pulmonary endothelial cells. The major findings from this study are that 1) restoration of SOCE by cholesterol supplementation in PAECs from CH rats is sensitive to Orai1 inhibition; 2) both substitution of endogenous membrane cholesterol with its epimer, Epichol, and inhibition of Orai1 by AnCoA4 attenuate SOCE without additive effects in isolated PAECs and cultured PMVECs; 3) Epichol treatment does not further reduce SOCE in PMVECs after Orai1 siRNA knockdown; and 4) membrane cholesterol is required for the interaction of STIM1 and Orai1 in response to ER Ca2+ store depletion in PMVECs. The results from this study suggest that membrane cholesterol directly regulates Orai1-mediated endothelial SOCE by facilitating the interaction of STIM1 and Orai1 and further demonstrate that impaired pulmonary endothelial Ca2+ entry after CH is due to altered membrane cholesterol homeostasis that limits Orai1 activity.
One of the hallmarks of CH-induced pulmonary hypertension is pulmonary arterial endothelial dysfunction. Pulmonary arterial smooth muscle cell contraction and proliferation are regulated by vasoactive factors secreted from the endothelium. The increased production of vasoconstrictors/proliferative factors and decreased synthesis of vasodilatory/antimitogenic factors contribute to enhanced vascular tone and remodeling in CH-induced pulmonary hypertension (71). Production of many endothelium-dependent vasodilators and regulation of membrane potential are largely a function of pulmonary endothelial intracellular Ca2+ levels ([Ca2+]i). The activity of endothelial NO synthase (eNOS) (7a, 17, 53, 62), phospholipase A2 (43, 69), and small- and intermediate-conductance Ca2+-activated K+ channels that are responsible for endothelial cell hyperpolarization upon activation by agonists (25, 45) is regulated by [Ca2+]i. Thus, diminished pulmonary endothelial [Ca2+]i may limit the production of these endothelium-derived vasoactive factors. In CH-induced pulmonary hypertension, for example, posttranslational regulation of eNOS activity is impaired due to reduced agonist-induced Ca2+ influx (51). Our previous work also showed that both basal [Ca2+]i and agonist-induced Ca2+ influx are lower in PAECs from CH rats compared with those of control animals (56, 57). CH similarly inhibits endothelial SOCE, receptor-operated Ca2+ entry, and T-type voltage-gated Ca2+ channel-mediated depolarization-induced Ca2+ influx, which are major components of agonist-induced Ca2+ entry in isolated PAECs (56, 58). These findings suggest that SOCs and T-type voltage-gated Ca2+ channels are important in determining endothelial Ca2+ influx, and impaired Ca2+ entry through these channels may contribute to reduced basal [Ca2+]i in PAECs after CH. Previous studies from our laboratory have suggested that CH inhibits endothelial Ca2+ influx through alterations in membrane lipid domains, which represent key regulatory sites of ion channel function in PAECs (57, 84).
Cholesterol is a polycyclic amphipathic molecule with a polar section consisting of a single β-hydroxyl group that can interact with membrane lipids or proteins through the formation of hydrogen bonds (61). Cholesterol-enriched caveolar microdomains are signal transduction platforms where many ion channels and their regulatory factors reside (71). Membrane cholesterol can inhibit some ion channels by decreasing open probability, unitary conductance, and the number of active channels (38). In contrast, ion channels such as the nicotinic acetylcholine receptor, GABA receptors, epithelial Na+ channels, and transient receptor potential canonical (TRPC) channels are inhibited by removal of membrane cholesterol (4–6, 32, 73), indicating an important role for cholesterol in normal channel function. Consistent with this regulatory function, our previous study (84) showed that membrane cholesterol facilitates major Ca2+ entry pathways in PAECs, including agonist-induced Ca2+ entry, SOCE, and depolarization-induced Ca2+ entry, and that CH impairs endothelial Ca2+ influx by reducing membrane cholesterol levels. However, the mechanisms by which cholesterol regulates endothelial Ca2+ influx are not well investigated.
The present study focused on exploring the ion channel involved in membrane cholesterol-dependent endothelial SOCE, a major component of agonist-induced Ca2+ entry, in the pulmonary circulation. Orai1 and TRPC channels have been reported as putative SOCs (18, 40, 60, 82, 87). Depletion of ER Ca2+ stores causes STIM1 oligomerization (12a, 39), which recruits Orai1 into microdomains and ensures the physical interaction between STIM1 and Orai1, leading to Orai1 activation and Ca2+ influx (86). TRPC1 and TRPC4 channels also localize to caveolar microdomains and regulate endothelial Ca2+ entry in murine PMVECs (50). However, TRPC1-mediated SOCE is less Ca2+ selective compared with Orai1-mediated SOCE (7d) and requires functional Orai1 (7c). Orai1 also interacts with TPRC4 and regulates TRPC1/4 heterotetramer channel activation and Ca2+ selectivity in lung endothelial cells (12). The present study investigated Orai1 as a candidate ion channel in mediating cholesterol-dependent endothelial SOCE. We found that either pharmacological inhibition or gene silencing of Orai1 significantly decreased endothelial SOCE, demonstrating that Orai1 contributes to SOCE in both PAECs and PMVECs. Furthermore, AnCoA4-sensitive SOCE was abolished in PAECs from CH rats, suggesting that the reduction in SOCE after CH results from impaired Orai1 activity. Interestingly, our finding that cholesterol replenishment rescues Ca2+ entry in PAECs from CH rats suggests that altered expression of Orai1 or STIM1 does not explain this deficit but rather regulation of Orai1 activity by the molecular composition of the membrane. Such influences of cholesterol may occur through a direct interaction with Orai1 and STIM1, indirectly through regulation of caveolin-1 or other lipid raft components, or by altering Orai1/STIM1 trafficking and membrane localization.
Although AnCoA4 is characterized as a selective inhibitor of Orai1 (67), whether AnCoA4 similarly inhibits Orai2 or Orai3 is unknown. These related Ca2+ channel isoforms have been implicated as SOCs in a variety of cell types (16, 33, 68, 77) and mediate SOCE when overexpressed with STIM1 in heterologous expression systems (48). However, in contrast to the established contribution of Orai1 to SOCE in pulmonary endothelial cells, store depletion-induced Ca2+ entry is independent of Orai2 and Orai3 in endothelial cells from several vascular beds (30, 74, 85). Furthermore, whether these channels are expressed and play a functional role in PAECs remains to be established. Therefore, while we cannot exclude a possible inhibitory effect of AnCoA4 on Orai2 or Orai3 in the present study, the similar effects of AnCoA4 and Orai1 siRNA to inhibit SOCE are supportive of a contribution of Orai1 to this response.
Cholesterol may modulate the function of membrane proteins via either a direct interaction or through altering the properties of lipid microdomains (38). Several groups have reported that membrane cholesterol depletion by MβCD attenuates SOCE in a variety of cell types (14, 20, 24, 29, 59). Interestingly, however, recent reports by Derler et al. (13) and Pacheco et al. (55) reported cholesterol-binding sites on Orai1 and STIM1, respectively, and demonstrated an inhibitory effect of membrane cholesterol on SOCE in cultured human embryonic kidney 293 cells and rat basophilic leukemia 2H3 cells. Both studies further showed that the cholesterol-Orai1 and cholesterol-STIM1 interaction was attenuated by mutating the cholesterol-binding site of the target protein. Although many studies have used the cholesterol-depleting agents MβCD or filipin to evaluate functional roles for membrane cholesterol, an important limitation of these agents is that they may exert off-target effects by disrupting caveolar stucture, thereby altering biophysical properties of the plasma membrane (2, 61, 84). Thus, these approaches may interrupt SOCE in a nonspecific manner unrelated to direct interaction of membrane cholesterol with ion channels, which may limit the interpretation of how cholesterol regulates SOC. In our present study, we substituted endogenous cholesterol with Epichol, an approach that alters membrane cholesterol content without disrupting caveoli (84), and found that cholesterol is required for Orai1-mediated SOCE in both isolated PAECs and cultured PMVECs. We also showed that Epichol substitution significantly reduced the STIM1-Orai1 interaction, suggesting that membrane cholesterol regulates endothelial SOCE by facilitating this interaction that is required for Orai1 activation. The reason for the conflicting observations of how membrane cholesterol regulates SOCE in our present study and those of other groups (13, 55) is not clear but may be due to heterogeneity between either different cell types or between transfected/cultured cells and native endothelial cells. Additionally, since SOCE is a complex multistep signaling pathway, it is also possible that membrane cholesterol affects components of SOCE differently. Galan et al. (20) and Pani et al. (59) suggested that membrane cholesterol is required for STIM1 oligmerization and subsequent interaction with and activation of Orai1. After the STIM1-Orai1 complex is formed, however, cholesterol may inhibit SOCE through a direct interaction with Orai1 and STIM1 (13, 20, 55). Additional experiments are required to investigate if membrane cholesterol directly regulates endothelial STIM1 clustering, the STIM1-Orai1 interaction, and Orai1 activation in PAECs.
Although our results indicate that membrane cholesterol plays a functional role in Orai1-mediated SOCE in pulmonary endothelial cells, these findings do not preclude a potential role for cholesterol to regulate TRPC channels independent of Orai1 and STIM1. Despite a lack of evidence for direct regulatory cholesterol-binding sites on TRPC channel sequences, the membrane cholesterol sensitivity of TRPC channels has been reported in several cell types (1, 6, 37, 50, 81). For example, the extraction of membrane cholesterol by cyclodextrins impairs TRPC1 signaling processes (6, 42). Although cholesterol may play a pivotal regulatory role in TRPC1-mediated SOCE by regulating membrane structure, direct effects of cholesterol on TRPC functions are possible.
Our study also suggests that loss of membrane cholesterol contributes to impaired endothelial SOCE in CH-induced pulmonary hypertension (84). Due to the limitation of the filipin staining approach to detect oxidized cholesterol (63), the reduced membrane cholesterol after CH is possibly the result of impaired cholesterol de novo synthesis (54), oxidative modification of cholesterol (52), or decreased membrane cholesterol trafficking. Nguyen et al. (54) explored the mechanism by which hypoxia affects de novo cholesterol biosynthesis. They reported that hypoxia induces accumulation of cholesterol biosynthetic intermediates and rapid degradation of HMG-CoA reductase, which contribute to a reduction of cholesterol production. Given the importance of reactive oxygen species to the development of CH-induced pulmonary hypertension (19, 26, 27, 41), endothelial dysfunction in this setting may alternatively result from oxidative modifications to cholesterol that alter membrane lipid domains and interfere with ion channel function.
Based on our findings that CH attenuates pulmonary endothelial Ca2+ entry by depleting membrane cholesterol, it may be predicted that targeted depletion of PAEC cholesterol would minimally or adversely affect the development of pulmonary hypertension in this setting. In apparent contrast to this prediction, HMG-CoA reductase inhibitors (statins) have demonstrated beneficial effects in attenuating the development of CH-induced pulmonary hypertension in animal models (23, 36, 49). However, the mechanisms by which statins mediate this protective influence are thought to be independent of their cholesterol-lowering properties (7b, 46, 70, 78, 80). These include inhibition of RhoA that cannot only decrease vascular smooth muscle contractility (76), proliferation, and migration (79) but also increase eNOS expression and activity (70). Additional evidence suggests that statins increase NO production by inhibiting expression and activity of NADPH oxidase subunits, and thus production of endothelial reactive oxygen species (44, 78, 80), rather than through effects on endothelial cholesterol content. Such nonspecific effects of statins likely result from decreased production of intermediates of the mevalonate pathway that are necessary for posttranslational modification of many signaling proteins (7). In contrast to these animal studies, however, clinical trials of statin therapy in patients with pulmonary arterial hypertension have been largely inconclusive (66).
In conclusion, the present study demonstrates a novel effect of membrane cholesterol to regulate pulmonary endothelial SOCE by facilitating the interaction of STIM1 and Orai1 (Fig. 8). These observations advance our basic understanding of how membrane cholesterol regulates endothelial [Ca2+]i homeostasis and have potentially broader implications for cholesterol-dependent regulation of a wide range of vasoactive and mitogenic pathways. These findings may additionally provide a mechanistic basis to explain the CH-induced diminution of endothelial Ca2+ influx and associated endothelial dysfunction that is central to the pathogenesis of pulmonary hypertension. The challenges of future studies are to identify the potential contribution of reactive oxygen species to decreased endothelial cholesterol after CH, either through direct cholesterol oxidation or impaired sterol trafficking, and the contribution of these responses to increased vasoconstrictor reactivity, arterial smooth muscle mitogenesis, and the development of pulmonary hypertension.
Fig. 8.
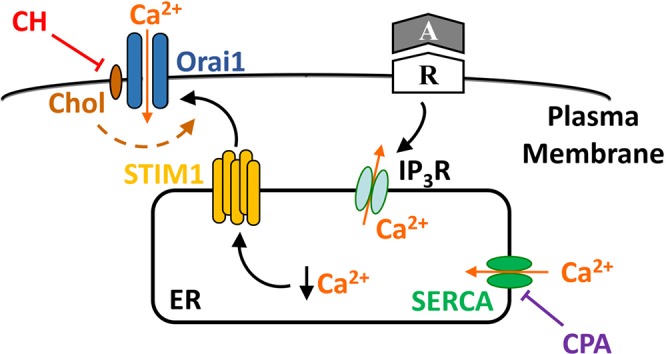
Proposed mechanism by which reduced cell membrane cholesterol (Chol) after chronic hypoxia (CH) attenuates store-operated Ca2+ entry (SOCE) in in pulmonary arterial endothelial cells. Chol facilitates the interaction of stromal interaction molecule 1 (STIM1) and Orai1 in response to depletion of intracellular Ca2+ stores, e.g., by receptor (R)-mediated activation of inositol 1,4,5-trisphosphate receptors (IP3R), or inhibition of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). CH inhibits this mechanism by depletion of cell membrane Chol. ER, endoplasmic reticulum; A, agonist; CPA, cyclopiazonic acid.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-95640 (to B. R. Walker) and R01-HL-132883 and R01-HL-088192 (to T. C. Resta) and by American Heart Association Grant 15GRNT21080001 (to B. R. Walker).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.Z., J.S.N., N.L.J., B.R.W., and T.C.R. conceived and designed research; B.Z. and J.S.N. performed experiments; B.Z. and J.S.N. analyzed data; B.Z., J.S.N., N.L.J., B.R.W., and T.C.R. interpreted results of experiments; B.Z., J.S.N., N.L.J., B.R.W., and T.C.R. prepared figures; B.Z. drafted manuscript; B.Z., J.S.N., N.L.J., B.R.W., and T.C.R. edited and revised manuscript; B.Z., J.S.N., N.L.J., B.R.W., and T.C.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Minerva Murphy, Lindsay Herbert, and Tamara Howard for technical assistance.
REFERENCES
- 1.Alkhani H, Ase AR, Grant R, O’Donnell D, Groschner K, Séguéla P. Contribution of TRPC3 to store-operated calcium entry and inflammatory transductions in primary nociceptors. Mol Pain 10: 43, 2014. doi: 10.1186/1744-8069-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson RG. The caveolae membrane system. Annu Rev Biochem 67: 199–225, 1998. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- 3.Andrews AM, Jaron D, Buerk DG, Barbee KA. Shear stress-induced NO production is dependent on ATP autocrine signaling and capacitative calcium entry. Cell Mol Bioeng 7: 510–520, 2014. doi: 10.1007/s12195-014-0351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 21: 892–905, 2003. doi: 10.1183/09031936.03.00115402. [DOI] [PubMed] [Google Scholar]
- 5.Barrantes FJ. Cholesterol effects on nicotinic acetylcholine receptor: cellular aspects. Subcell Biochem 51: 467–487, 2010. doi: 10.1007/978-90-481-8622-8_17. [DOI] [PubMed] [Google Scholar]
- 6.Bergdahl A, Gomez MF, Dreja K, Xu SZ, Adner M, Beech DJ, Broman J, Hellstrand P, Swärd K. Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ Res 93: 839–847, 2003. doi: 10.1161/01.RES.0000100367.45446.A3. [DOI] [PubMed] [Google Scholar]
- 7.Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering–are they clinically relevant? Eur Heart J 24: 225–248, 2003. doi: 10.1016/S0195-668X(02)00419-0. [DOI] [PubMed] [Google Scholar]
- 7a.Busse R, Mülsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett 265: 133–136, 1990. doi: 10.1016/0014-5793(90)80902-U. [DOI] [PubMed] [Google Scholar]
- 7b.Chen IC, Tan MS, Wu BN, Chai CY, Yeh JL, Chou SH, Chen IJ, Dai ZK. Statins ameliorate pulmonary hypertension secondary to left ventricular dysfunction through the Rho-kinase pathway and NADPH oxidase. Pediatr Pulmonol 52: 443–457, 2017. doi: 10.1002/ppul.23610. [DOI] [PubMed] [Google Scholar]
- 7c.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem 283: 12935–12940, 2008. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d.Cheng KT, Liu X, Ong HL, Ambudkar IS. Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion. Adv Exp Med Biol 704: 731–747, 2011. doi: 10.1007/978-94-007-0265-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res 38: 2264–2272, 1997. [PubMed] [Google Scholar]
- 11a.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 327: 70–75, 1992. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 12.Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, Stevens T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res 110: 1435–1444, 2012. doi: 10.1161/CIRCRESAHA.112.269506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell 21: 1897–1907, 2010. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derler I, Jardin I, Stathopulos PB, Muik M, Fahrner M, Zayats V, Pandey SK, Poteser M, Lackner B, Absolonova M, Schindl R, Groschner K, Ettrich R, Ikura M, Romanin C. Cholesterol modulates Orai1 channel function. Sci Signal 9: ra10, 2016. doi: 10.1126/scisignal.aad7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dionisio N, Galán C, Jardín I, Salido GM, Rosado JA. Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim Biophys Acta 1813: 431–437, 2011. doi: 10.1016/j.bbamcr.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 308: C581–C593, 2015. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Förstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci USA 88: 1788–1792, 1991. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol 3: 121–127, 2001. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 19.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 148: 714–723, 2006. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galan C, Woodard GE, Dionisio N, Salido GM, Rosado JA. Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim Biophys Acta 1803: 1083–1093, 2010. doi: 10.1016/j.bbamcr.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221, 1995. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 22.Gimpl G, Burger K, Fahrenholz F. Cholesterol as modulator of receptor function. Biochemistry 36: 10959–10974, 1997. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- 23.Girgis RE, Li D, Zhan X, Garcia JG, Tuder RM, Hassoun PM, Johns RA. Attenuation of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Heart Circ Physiol 285: H938–H945, 2003. doi: 10.1152/ajpheart.01097.2002. [DOI] [PubMed] [Google Scholar]
- 24.Gwozdz T, Dutko-Gwozdz J, Schafer C, Bolotina VM. Overexpression of Orai1 and STIM1 proteins alters regulation of store-operated Ca2+ entry by endogenous mediators. J Biol Chem 287: 22865–22872, 2012. doi: 10.1074/jbc.M112.356626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinton JM, Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br J Pharmacol 138: 1031–1035, 2003. doi: 10.1038/sj.bjp.0705171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jernigan NL, Walker BR, Resta TC. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 287: L801–L808, 2004. doi: 10.1152/ajplung.00443.2003. [DOI] [PubMed] [Google Scholar]
- 27.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kassan M, Zhang W, Aissa KA, Stolwijk J, Trebak M, Matrougui K. Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflugers Arch 467: 1195–1202, 2015. doi: 10.1007/s00424-014-1556-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato N, Nakanishi M, Hirashima N. Cholesterol depletion inhibits store-operated calcium currents and exocytotic membrane fusion in RBL-2H3 cells. Biochemistry 42: 11808–11814, 2003. doi: 10.1021/bi034758h. [DOI] [PubMed] [Google Scholar]
- 30.Kito H, Yamamura H, Suzuki Y, Yamamura H, Ohya S, Asai K, Imaizumi Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem Biophys Res Commun 459: 457–462, 2015. doi: 10.1016/j.bbrc.2015.02.127. [DOI] [PubMed] [Google Scholar]
- 31.Kwakye GF, Li D, Kabobel OA, Bowman AB. Cellular fura-2 manganese extraction assay (CFMEA). Curr Protoc Toxicol 12: 18, 2011. doi: 10.1002/0471140856.tx1218s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol Pharmacol 70: 1174–1183, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Kwon J, An H, Sa M, Won J, Shin JI, Lee CJ. Orai1 and Orai3 in combination with stim1 mediate the majority of store-operated calcium entry in astrocytes. Exp Neurobiol 26: 42–54, 2017. doi: 10.5607/en.2017.26.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laufs U, Fata V La, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by hmg-coa reductase inhibitors. Circulation 97: 1129−1135, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Lei L, Lu S, Wang Y, Kim T, Mehta D, Wang Y. The role of mechanical tension on lipid raft dependent PDGF-induced TRPC6 activation. Biomaterials 35: 2868–2877, 2014. doi: 10.1016/j.biomaterials.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levitan I, Singh DK, Rosenhouse-Dantsker A. Cholesterol binding to ion channels. Front Physiol 5: 65, 2014. doi: 10.3389/fphys.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou J, Kim ML, Heo W Do, Jones JT, Myers JW, James E, Jr F, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion triggered Ca2+ influx. Curr Biol 15: 1235–1241, 2011. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Singh BB, Ambudkar IS. TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5–S6 region. J Biol Chem 278: 11337–11343, 2003. doi: 10.1074/jbc.M213271200. [DOI] [PubMed] [Google Scholar]
- 41.Liu ZQ, Shan HY. Cholesterol, not polyunsaturated fatty acids, is target molecule in oxidation induced by reactive oxygen species in membrane of human erythrocytes. Cell Biochem Biophys 45: 185–193, 2006. doi: 10.1385/CBB:45:2:185. [DOI] [PubMed] [Google Scholar]
- 42.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem 275: 11934–11942, 2000. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 43.Majed BH, Khalil RA. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev 64: 540–582, 2012. doi: 10.1124/pr.111.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Margaritis M, Sanna F, Antoniades C. Statins and oxidative stress in the cardiovascular system. Curr Pharm Des. In press. doi: 10.2174/1381612823666170926130338. [DOI] [PubMed] [Google Scholar]
- 45.Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol Heart Circ Physiol 285: H1590–H1599, 2003. doi: 10.1152/ajpheart.00376.2003. [DOI] [PubMed] [Google Scholar]
- 46.Mason RP, Walter MF, Jacob RF. Effects of HMG-CoA reductase inhibitors on endothelial function: role of microdomains and oxidative stress. Circulation 109, Suppl 1: II34–II41, 2004. doi: 10.1161/01.CIR.0000129503.62747.03. [DOI] [PubMed] [Google Scholar]
- 47.Michiels C, Arnould T, Knott I, Dieu M, Remacle J. Stimulation of prostaglandin synthesis by human endothelial cells exposed to hypoxia. Am J Physiol Cell Physiol 264: C866–C874, 1993. [DOI] [PubMed] [Google Scholar]
- 48.Moccia F, Zuccolo E, Soda T, Tanzi F, Guerra G, Mapelli L, Lodola F, D’Angelo E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front Cell Neurosci 9: 153, 2015. doi: 10.3389/fncel.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murata T, Kinoshita K, Hori M, Kuwahara M, Tsubone H, Karaki H, Ozaki H. Statin protects endothelial nitric oxide synthase activity in hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 25: 2335–2342, 2005. doi: 10.1161/01.ATV.0000186184.33537.48. [DOI] [PubMed] [Google Scholar]
- 50.Murata T, Lin MI, Stan RV, Bauer PM, Yu J, Sessa WC. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J Biol Chem 282: 16631–16643, 2007. doi: 10.1074/jbc.M607948200. [DOI] [PubMed] [Google Scholar]
- 51.Murata T, Sato K, Hori M, Ozaki H, Karaki H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem 277: 44085–44092, 2002. doi: 10.1074/jbc.M205934200. [DOI] [PubMed] [Google Scholar]
- 52.Murphy RC, Johnson KM. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J Biol Chem 283: 15521–15525, 2008. doi: 10.1074/jbc.R700049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakane M, Mitchell J, Förstermann U, Murad F. Phosphorylation by calcium calmodulin-dependent protein kinase II and protein kinase C modulates the activity of nitric oxide synthase. Biochem Biophys Res Commun 180: 1396–1402, 1991. doi: 10.1016/S0006-291X(05)81351-8. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol Chem 282: 27436–27446, 2007. doi: 10.1074/jbc.M704976200. [DOI] [PubMed] [Google Scholar]
- 55.Pacheco J, Domingu L, Bohórquez-herná A, Asanov A, Vaca L. OPEN A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci Rep 6: 1–16, 2016. doi: 10.1038/srep29634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paffett ML, Naik JS, Resta TC, Walker BR. Reduced store-operated Ca2+ entry in pulmonary endothelial cells from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 293: L1135–L1142, 2007. doi: 10.1152/ajplung.00432.2006. [DOI] [PubMed] [Google Scholar]
- 57.Paffett ML, Naik JS, Riddle MA, Menicucci SD, Gonzales AJ, Resta TC, Walker BR. Altered membrane lipid domains limit pulmonary endothelial calcium entry following chronic hypoxia. Am J Physiol Heart Circ Physiol 301: H1331–H1340, 2011. doi: 10.1152/ajpheart.00980.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paffett ML, Riddle MA, Kanagy NL, Resta TC, Walker BR. Altered protein kinase C regulation of pulmonary endothelial store- and receptor-operated Ca2+ entry after chronic hypoxia. J Pharmacol Exp Ther 334: 753–760, 2010. doi: 10.1124/jpet.110.165563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB. Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J Biol Chem 283: 17333–17340, 2008. doi: 10.1074/jbc.M800107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev 85: 757–810, 2005. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 61.Parpal S, Karlsson M, Thorn H, Strålfors P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem 276: 9670–9678, 2001. doi: 10.1074/jbc.M007454200. [DOI] [PubMed] [Google Scholar]
- 62.Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA 88: 10480–10484, 1991. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pörn MI, Slotte JP. Localization of cholesterol in sphingomyelinase-treated fibroblasts. Biochem J 308: 269–274, 1995. doi: 10.1042/bj3080269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Putney JW Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. Mechanisms of capacitative calcium entry. J Cell Sci 114: 2223–2229, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J 83: 3211–3222, 2002. doi: 10.1016/S0006-3495(02)75323-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rysz-Górzynska M, Gluba-Brzózka A, Sahebkar A, Serban MC, Mikhailidis DP, Ursoniu S, Toth PP, Bittner V, Watts GF, Lip GY, Rysz J, Catapano AL, Banach M. Efficacy of statin therapy in pulmonary arterial hypertension: a systematic review and meta-analysis. Sci Rep 6: 30060, 2016. doi: 10.1038/srep30060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sadaghiani AM, Lee SM, Odegaard JI, Leveson-Gower DB, McPherson OM, Novick P, Kim MR, Koehler AN, Negrin R, Dolmetsch RE, Park CY. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem Biol 21: 1278–1292, 2014. doi: 10.1016/j.chembiol.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 68.Saliba Y, Keck M, Marchand A, Atassi F, Ouillé A, Cazorla O, Trebak M, Pavoine C, Lacampagne A, Hulot JS, Farès N, Fauconnier J, Lompré AM. Emergence of Orai3 activity during cardiac hypertrophy. Cardiovasc Res 105: 248–259, 2015. doi: 10.1093/cvr/cvu207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seid JM, MacNeil S, Tomlinson S. Calcium, calmodulin, and the production of prostacyclin by cultured vascular endothelial cells. Biosci Rep 3: 1007–1015, 1983. doi: 10.1007/BF01121027. [DOI] [PubMed] [Google Scholar]
- 70.Shiga N, Hirano K, Hirano M, Nishimura J, Nawata H, Kanaide H. Long-term inhibition of RhoA attenuates vascular contractility by enhancing endothelial NO production in an intact rabbit mesenteric artery. Circ Res 96: 1014–1021, 2005. doi: 10.1161/01.RES.0000165483.34603.91. [DOI] [PubMed] [Google Scholar]
- 71.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res 49: 549–560, 2000. [PubMed] [Google Scholar]
- 72.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 13: 549–565, 2012. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sooksawate T, Simmonds MA. Influence of membrane cholesterol on modulation of the GABA(A) receptor by neuroactive steroids and other potentiators. Br J Pharmacol 134: 1303–1311, 2001. doi: 10.1038/sj.bjp.0704360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol 81: 510–526, 2012. doi: 10.1124/mol.111.074658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159: 1925–1932, 1999. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 76.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389: 990–994, 1997. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 77.Vaeth M, Yang J, Yamashita M, Zee I, Eckstein M, Knosp C, Kaufmann U, Karoly Jani P, Lacruz RS, Flockerzi V, Kacskovics I, Prakriya M, Feske S. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat Commun 8: 14714, 2017. doi: 10.1038/ncomms14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wagner AH, Köhler T, Rückschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 20: 61−69, 2000. [DOI] [PubMed] [Google Scholar]
- 79.Walker J, Undem C, Yun X, Lade J, Jiang H, Shimoda LA. Role of Rho kinase and Na+/H+ exchange in hypoxia-induced pulmonary arterial smooth muscle cell proliferation and migration. Physiol Rep 4: e12702, 2016. doi: 10.14814/phy2.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wassmann S, Laufs U, Müller K, Konkol C, Ahlbory K, Bäumer AT, Linz W, Böhm M, Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 22: 300–305, 2002. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 81.Weerth SH, Holtzclaw LA, Russell JT. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium 41: 155–167, 2007. doi: 10.1016/j.ceca.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 82.Wu X, Babnigg G, Villereal ML. Functional significance of human Trp1 and Trp3 in store-operated Ca2+ entry in HEK-293 cells. Am J Physiol Cell Physiol 278: 526–536, 2000. doi: 10.1152/ajpcell.2000.278.3.C526. [DOI] [PubMed] [Google Scholar]
- 83.Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 39: 843–849, 2000. doi: 10.1021/bi992543v. [DOI] [PubMed] [Google Scholar]
- 84.Zhang B, Naik XJ, Jernigan NL, Walker BR, Resta TC. Reduced membrane cholesterol limits pulmonary endothelial Ca2+ entry after chronic hypoxia. Am J Physiol Heart Circ Physiol 312: H1176−H1184, 2017. doi: 10.1152/ajpheart.00097.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou MH, Zheng H, Si H, Jin Y, Peng JM, He L, Zhou Y, Muñoz-Garay C, Zawieja DC, Kuo L, Peng X, Zhang SL. Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J Biol Chem 289: 29446–29456, 2014. doi: 10.1074/jbc.M114.578492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou Y, Wang X, Wang X, Loktionova NA, Cai X, Nwokonko RM, Vrana E, Wang Y, Rothberg BS, Gill DL. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat Commun 6: 8395, 2015. doi: 10.1038/ncomms9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Lückhoff A, Schultz G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 16: 1189–1196, 1996. doi: 10.1016/S0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]