

Summary
Objective
To study long‐term survival and mortality among patients with West syndrome.
Methods
The study population included all children born in 1960–1976 and treated for West syndrome in three tertiary care hospitals in Helsinki, Finland. The participants were prospectively followed for five decades for survival. Death data were derived from the National Causes of Death Register of the Population Register Center of Statistics Finland.
Results
During follow‐up, 102 (49%) of 207 patients had died at the mean age of 19 years. The mean overall annual mortality rate was 15.3 per 1,000 patient‐years. The rates ranged from 18.2 per 1,000 after 10 years to 17.2 per 1,000 after 20 years and 15.4 per 1,000 patient‐years after 40 years of follow‐up. One fourth (25%) had died by 17.2 (95% CI 11.8–22.7) years and 50% by 48.6 (95% CI 38.5–NA) years of follow‐up. Etiology at onset was symptomatic in 87% patients and cryptogenic in 13%; 6 of the latter 26 patients later turned out to be symptomatic. The mean annual mortality rate was 3.7 per 1,000 for 4 patients with cryptogenic etiology and 17.6 per 1,000 for those with symptomatic etiology. The hazard of death was fivefold in patients with symptomatic etiology versus cryptogenic etiology. The overall autopsy rate was 73%. Pneumonia was the most frequent cause of death (46%). All patients who died of pneumonia had symptomatic etiology. SUDEP occurred in 10 patients and was the most common epilepsy‐related cause of death (10%).
Significance
Risk of excess death of participants with West syndrome is not limited to early age but continues into adulthood, particularly in those with symptomatic etiology, and leads to death in half the cases at around 50 years of age. Measures should be directed to prevent pneumonia, the most common overall cause, and SUDEP, the most frequent seizure‐related cause, of death.
Keywords: Infantile spasms, Long‐term follow‐up, Mortality in West syndrome, Outcome of West syndrome, Population study of epilepsy
Key Points.
Mortality in West syndrome is high, with 50% of cases of death at around 50 years of age
Mortality remains high throughout life
Symptomatic etiology is fivefold as common a risk factor as cryptogenic etiology
Pneumonia is the leading overall cause of death, and SUDEP is the most common seizure‐related cause
Whereas children with uncomplicated epilepsy are not at higher risk of death than the general population, the mortality risk is many‐fold higher in complicated epilepsy.1 Among the childhood‐onset epilepsies, West syndrome belongs to the high‐mortality category. According to the literature,2, 3 the mortality varied widely between 3% and 30%, largely driven by the duration of follow‐up. A retrospective hospital‐based study of 385 patients of childhood‐onset epilepsy in Japan followed for 10 years or more (no further data given) found a mortality rate of 41% in infantile spasms.4 Two retrospective chart reviews with a follow‐up of 16 and 17 years, respectively, reported a lower mortality rate of 22% each,5, 6 and another paper from Nova Scotia a rate of 25%, based on 20‐year follow‐up of 692 children with generalized epilepsies.7 One large prospective cohort study of children with West syndrome, followed for 25.6 years (mean, range 20–35), showed an overall mortality of 31%.8 In contrast, the present study has a substantially longer follow‐up period, up to middle age, a higher autopsy rate, and examines the etiology of spasms and the immediate circumstances of death. In summary, the previous studies have variable designs, are mostly retrospective, and often are small. These facts prompted us to investigate the occurrence, profile, and covariates of increased mortality, based on death certificates, in a population cohort during a long prospective follow‐up of five decades.
Methods
The study cohort consisted of all children who were born in 1960–1976 and treated for West syndrome in the Children's Hospital of the University of Helsinki (Finland), the Aurora Hospital in Helsinki, and the Children's Castle Hospital, Helsinki. The inclusion criteria of the original study9 were <2 years of age at onset; typical clinical flexion or extension spasms; and hypsarrhythmia or a variant of hypsarrhythmia on interictal electroencephalogram (EEG). The total, prospectively collected original cohort numbered 214 infants, 107 of whom were from the county of Uusimaa and the remaining 107 from other parts of Finland. For 7 patients, detailed data of death were not available. Thus, the present analyses are based on 207 patients. The study design was described in detail and follow‐up studies were reported previously.9, 10, 11
Cases of death were identified from the National Causes of Death Register of the Population Register Center of Statistics Finland (http://www.tilastokeskus.fi/til/vaerak/index_en.html), which covers all citizens and permanent residents in Finland. The Finnish death register has been shown to be valid and reliable for epidemiological research.12 The information is based on statutory notifications from public authorities and private individuals, including death data. Adrenocorticotropic hormone (ACTH) was standard primary therapy for all patients. Antiepileptic drugs were administered to patients in whom ACTH was contraindicated. Similarly, some severely mentally handicapped patients had other antiepileptic drug therapy. Etiology was defined as cryptogenic if the premorbid neurodevelopment was normal and no abnormalities in clinical status or neuroradiological studies were detected at presentation.11 If such abnormalities were diagnosed before or after onset of spasms, the etiology was considered as being symptomatic. Although the classification of etiology of spasms might have been revised either by clinical course or new neuroimaging studies during follow‐up, for this analysis, the original classification was used. Sudden unexplained death in epilepsy (SUDEP) was defined according to the Annegers criteria.13, 14 Single seizures of longer duration than 30 min and multiple seizures without recovery in between were considered episodes of status epilepticus. Terminal remission was defined as seizure freedom for 1 year or more.
All patients (n = 107) with West syndrome from the county of Uusimaa were treated in one of the aforementioned three hospitals; thus, they represent a population‐based cohort.9 To check the external validity of the results of the present study, we compared the mortality of patients from the cohort from Uusimaa to that of the patients from other parts of the country (n = 107). For the present study, we had the data of 103 (96%) of 107 from the county and 104 (97%) of 107 from outside the county. Of the patients from the county, 45 (44%) and, of those from outside the county, 54 (52%) had died. The difference was not significant (p = 0.267, Fisher's exact test). Accordingly, the risk for death did not relevantly differ according to place of birth, and the results of the study can be considered representative of the Finnish population for the risk of death for West syndrome.
Ethics
For the original study, the design was approved by the ethics committee of the University of Helsinki. The study participants or relatives of study participants or the institutions of the deceased participants were not contacted for the present study. According to Finnish legislation (Personal Data Act 523/1999), approval by an ethical committee or informed consent by study participants is not required for studies based on the register data, including death certificate data that are provided for scientific research purposes.
Statistical analysis
The data are given as n (%), mean (SD), median, and range, or mean mortality rate per 1,000 patient‐years. For comparisons of categorical variables, Fisher's exact test was used. Median survival ages with 95% confidence intervals (CIs) were obtained from life tables. Hazard ratios 9HRs) with 95% CI were calculated from both univariate and multivariate Cox regression models. A p‐value of <0.05 was considered statistically significant. Statistical computations were done using SAS System for Windows, release 9.3 (SAS Institute, Cary, NC, U.S.A.).
Results
The 207 patients were followed for 32.5 years (mean, SD 17.0, median 39.5, range 24–54). Because the onset of West syndrome was before the age of 1 year in all patients, the follow‐up period is virtually the same as the age in years. Until the end of follow‐up, 102 (49%) of 207 patients had died at the mean age of 19 years (Table 1). Seven patients died during the first year of life (Fig. 1).
Table 1.
Duration of follow‐up and age at death (years) in long‐term followed participants with West syndrome
| Characteristic | Follow‐up time | Age at death | ||||||
|---|---|---|---|---|---|---|---|---|
| N | Mean (SD) | Md | Range | N (%) | Mean (SD) | Md | Range | |
| All | 207 | 32.5 (17.0) | 39.5 | 0.24–53.7 | 102 (49) | 19.1 (14.3) | 17.0 | 0.2–51.5 |
| Etiology | ||||||||
| Cryptogenic | 26 | 42.1 (13.4) | 48.5 | 7.6–53.7 | 4 (15) | 21.9 (20.0) | 14.8 | 7.6–50.6 |
| Symptomatic | 181 | 31.1 (17.0) | 38.6 | 0.2–53.1 | 98 (54) | 19.0 (14.1) | 17.0 | 0.2–51.5 |
| Sex | ||||||||
| Male | 126 | 31.7 (17.2) | 38.9 | 0.2–53.7 | 65 (52) | 18.5 (13.6) | 16.1 | 0.2–50.4 |
| Female | 81 | 33.8 (16.7) | 40.7 | 0.4–53.0 | 37 (46) | 20.2 (15.5) | 19.4 | 0.4–51.5 |
| ACTH treatment | ||||||||
| Yes | 161 | 33.7 (16.6) | 40.2 | 0.2–53.7 | 74 (46) | 19.3 (14.4) | 17.2 | 0.2–50.6 |
| No | 46 | 28.4 (18.0) | 27.7 | 1.0–53.3 | 28 (61) | 18.7 (14.2) | 15.8 | 1.0–51.5 |
ACTH, adrenocorticotropic hormone; Md, median; SD, standard deviation.
Figure 1.
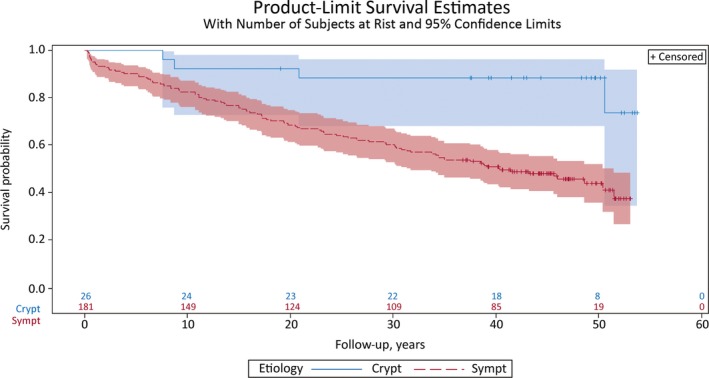
Survival of 207 patients with West syndrome during the 54‐year follow‐up period. The hazard of death is almost fivefold for patients with symptomatic etiology (blue line) compared with those with cryptogenic etiology (red line), p = 0.003. Crypt, cryptogenic; Sympt, symptomatic.
The overall mean annual mortality rate was 15.3/1,000 patient‐years. The risk of death remained high during the follow‐up. Of the 102 deaths, 34 occurred during the first 10 years of follow‐up, 25 during years 11–20, and the remaining 43 after 20 years of follow‐up, with the mortality rates ranging from 18.2/1,000 after 10 years to 17.2/1,000 after 20 years and 15.4/1,000 patient‐years after 40 years of follow‐up. During the total follow‐up period, 25% of the patients had died by 17.2 (95% CI 11.8–22.7) years and 50% by 48.6 (95% CI 38.5–NA) years (NA: upper limit for confidence interval not applicable).
Etiology was symptomatic in 181 (87%) patients and cryptogenic in 26 (13%); 6 of the latter later turned out to be symptomatic. Eight (3.9%) of all patients had tuberous sclerosis. Of the 102 patients who died, only four were of cryptogenic etiology, with mean annual mortality rate 3.7/1,000, whereas it was 17.6/1,000 for those with symptomatic etiology. Within the prenatal symptomatic group, congenital brain malformation and metabolic disorder and familial disease were most common. In addition, both perinatal and postnatal infections were marked risk factors (Table 2).
Table 2.
Etiological classification of patients with West syndrome with regard to term and death
| Etiology | All | Surviving | Dead |
|---|---|---|---|
| Symptomatic | 181 | 82 (45) | 98 (55) |
| Prenatal | 103 | 41 (40) | 62 (60) |
| Congenital brain malformation, tuberous sclerosis, Down syndrome | 38 | 9 (24) | 29 (76) |
| Other familial disease or metabolic disordera | 23 | 9 (39) | 14 (61) |
| Two or more causal comorbidities | 7 | 4 (57) | 3 (43) |
| Unknown prenatal/(perinatal) lesion | 13 | 6 (46) | 7 (54) |
| Unknown prenatal | 23 | 14 (61) | 9 (39) |
| Perinatal | 59 | 32 (54) | 27 (46) |
| Hypoxic‐ischemic insult: birth injury | 21 | 11 (52) | 10 (48) |
| Neonatal hypoglycemia (tested or not tested) | 28 | 19 (68) | 9 (32) |
| Early infection | 10 | 2 (20) | 8 (80) |
| Postnatal | 18 | 9 (50) | 9 (50) |
| Infection | 12 | 4 (33) | 8 (67) |
| Intracranial hemorrhage, anoxia due to aspiration | 5 | 4 (80) | 1 (20) |
| Brain tumor | 1 | 1 | 0 |
| Cryptogenic | 26 | 22 (85) | 4 (15) |
| Total | 207 | 105 (51) | 102 (49) |
Includes deceased participants with inborn error of metabolism NAS (n = 4); PEHO (progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy) (n = 2); Cornelia de Lange syndrome (n = 2); progressive encephalopathy of unknown origin (n = 2); progressive mitochondrial encephalopathy (n = 1); Prader‐Willi syndrome (n = 1); chromosomal anomaly with excessive marker chromosome (n = 1); nonketotic hyperglycinemia (n = 1).
Of 95 (95%) of 100 patients who had died and whose intelligence level was known, 85 had an IQ of 80 or lower, and the remaining 10 had an IQ higher than 80. So, 95% needed some kind of assistance in activities of daily living. Furthermore, many patients had a severe spastic tetraplegia and kyphoscoliosis requiring even more assistance.
The underlying condition was mostly a structural brain abnormality of prenatal or perinatal origin (Table 2). A vast majority lived in either an institution for people with mental retardation or a residential care home. Of the 5 (5%) patients with an IQ higher than 80, 4 were in remission and socially competent. The cause of death for each of these 4 patients was SUDEP, acute myeloid leukemia, multiple gliomata, or purulent meningitis. One patient was not in remission and died of pneumonia.
Sufficient data on remission status at death were available for 82 (80%) of 102 patients. During the preceding year, one or more seizures were found in 47 (57%) patients, including 4 with status epilepticus (>30 min), 2 with an eye‐witnessed seizure (≤30 min), and still another likely (but not eye‐witnessed) case of seizure of <30 min as the primary cause of death (Table 3). SUDEP (n = 10) was the most common epilepsy‐related cause of death (Table 3). The mean age at SUDEP was 27.4 years (SD 9.9, median 25, range 9–42). One patient was 9 years of age, while the remaining were adults 21–42 years of age at death. None of the participants who died of SUDEP had witnessed seizures at death. All had mental retardation, except for one who had IQ > 80 and was socially competent (male, age at death 21 years).
Table 3.
Immediate cause of death among 102 patients with West syndrome related to symptomatic etiology of epilepsy
| Cause of death | Alla | Autopsied | Etiology | |||
|---|---|---|---|---|---|---|
| Prenatal | Perinatal | Postnatal | ||||
| Death related to epilepsy | 17 | |||||
| SUDEP | 10 | 9 (90) | 6 (60) | 2 (20) | 2 (20) | |
| Epileptic seizureb | 7 | 4 (57) | 5 (71) | 1 (14) | 1 (14) | |
| Death not related to epilepsy | 85a | |||||
| Pneumonia | 47 | 34 (72) | 29 (62) | 16 (34) | 2 (4) | |
| Other infection | 9 | 7 (78) | 5 (56) | 4 (44) | 0 | |
| Aspiration | 5 | 3 | 5 | 0 | 0 | |
| Vascular | 5 | 3 | 4 | 1 | 0 | |
| Cancer | 3 | 2 | 2 | 0 | 1 | |
| Other | 12 | 10 | 6 | 3 | 3 | |
| Total | 102a | 74 | 62 | 27 | 9 | |
SUDEP, sudden unexplained death in epilepsy.
Includes 4 patients with cryptogenic etiology who died of infection other than pneumonia (1), cancer (1), or other cause of death (2).
Includes 4 cases of status epilepticus, 2 eye‐witnessed cases of seizures of <30 min, and 1 likely case of seizure.
The overall autopsy rate was 73% (74 of 102) of patients. Death was classified as natural in 97 (95%), caused by accident in 4 (4%), and unclear in 1 (1%). In 83%, the immediate cause of death was unrelated to epilepsy. Pneumonia was the most frequent cause of death noted in 47 (46%) of all known causes of death. All 47 patients had symptomatic etiology. Of them, 46 (98%) had mental retardation (severe in 45, and mild in 1). Forty (85%) of 46 patients had structural brain abnormalities, including congenital cerebral malformations (n = 15), microcephaly or cerebral atrophy (13), progressive encephalopathy (5), tuberous sclerosis (3), and isolated cases of other abnormalities (4). Furthermore, 8 of the 40 patients were recorded as cases of combined mental retardation and spastic tetraplegia but without any other congenital disorders. Kyphoscoliosis was often associated.
In Cox regression models, the hazard of death was fivefold in patients with symptomatic etiology compared with those with cryptogenic etiology. The result remained in multivariable analysis adjusted for sex and ACTH treatment. The mortality rate was slightly higher among men than among women, and men died at a somewhat younger age, but the differences were nonsignificant (Table 4).
Table 4.
Proportions of dead and mean survival times in 207 patients with West syndrome. Hazard ratios from Cox regression models, calculated for symptomatic versus cryptogenic etiology, male versus female sex, and not treated versus treated with ACTH
| Characteristic | Total | Dead | Median survival time, years | Cox regression | ||||
|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||||
| N | N (%) | Med | 95% CI | HR | 95% CI | HR | 95% CI | |
| All | 207 | 102 (49) | 48.6 | 38.5–NA | – | – | – | – |
| Etiologya | ||||||||
| Symptomatic | 181 | 98 (54) | 40.4 | 31.3–51.5 | 4.7 | 1.8–12.9 | 4.6 | 1.7–12.6 |
| Cryptogenic | 26 | 4 (15) | NA | 50.6–NA | ||||
| Sex | ||||||||
| Male | 126 | 65 (52) | 43.2 | 30.3–NA | 1.2 | 0.8–1.8 | 1.2 | 0.8–1.8 |
| Female | 81 | 37 (46) | 50.6 | 38.8–NA | ||||
| ACTH treatment | ||||||||
| No | 46 | 28 (61) | 27.7 | 16.8–50.4 | 1.5 | 1.0–2.3 | 1.4 | 0.9–2.2 |
| Yes | 161 | 74 (46) | 50.6 | 40.3–NA | ||||
ACTH, adrenocorticotropic hormone; CI, confidence interval; HR, hazard ratio; Med, median.
p = 0.002 in univariate analysis, and p = 0.003 in multivariate analysis.
In Cox regression models adjusted for sex and ACTH treatment, the hazard of death was five times as high among patients with symptomatic etiology as among those with cryptogenic etiology. The mortality rate was nonsignificantly higher among men than among women (Table 4).
Discussion
The strength of this study is that it involves a cohort of patients with West syndrome that was prospectively followed for more than 50 years, with full ascertainment of death and autopsy‐confirmed causes of death in the vast majority of cases. With markedly longer follow‐up than that reported in the literature, the overall mortality is much higher than previously reported (49% vs. 22–31%; see Introductory text). Symptomatic etiology of West syndrome was associated with a fivefold risk of death versus cryptogenic etiology. This compares with a fourfold risk in the population cohort of 688 10‐year‐old children with infantile spasms and Lennox‐Gastaut syndrome.15
In our study, mental and neurologic disability following congenital structural brain malformations, anomalies, infections, and genetic disorders strongly contributed to premature mortality in line with previous reports.1, 14, 16, 17, 18 During 12‐year follow‐up of 120 Swedish children with epilepsy, 11 participants died, 8 (73%) of whom had “neurodeficit,” that is, mental retardation and cerebral palsy, and none of them were in remission.16 According to a Dutch study, 9 (2%) of 472 children died during follow‐up of 5 years or until death. All 9 participants had symptomatic epilepsy.17 During 4,733 person‐years of follow‐up, 13 of 613 children with newly diagnosed epilepsy died. Ten of 13 deaths (77%) were associated with underlying cause of the seizures, and both symptomatic etiology and epileptic encephalopathy were independently associated with mortality.18 Among 2,239 participants followed for more than 300,000 person‐years, mortality was significantly higher in those with complicated epilepsy, whereas participants with uncomplicated epilepsy had no significant excess mortality compared with the general population.1 In a Finnish population study of 245 followed for 40 years,14 60 participants died. Of them, 45 (75%) had symptomatic etiology and mental and/or neurologic disability (IQ <50 in 39 and ≥50 in 6, and an additional cerebral palsy in 24).
In the vast majority of cases (83%), the immediate cause of death was unrelated to epilepsy, with pneumonia as the most frequent known cause of death. All patients who died of pneumonia had symptomatic West syndrome. Our percentage of deaths due to pneumonia (47%) is well comparable with 50% of Berg et al.1
Most of those patients were bedridden and helpless owing to severe mental retardation, spastic tetraplegia, kyphoscoliosis, and tube feeding. Epilepsy or seizure‐related death, including SUDEP, was uncommon and noted in a minority (17%) of patients. SUDEP was found in 10% of all deceased participants or 2% of those who died before age 16. Much lower percentages of SUDEP of all deaths (<1%) have been presented.1, 19 The wide variation is probably a consequence of different definitions, inclusion criteria, and duration of follow‐up; the longer the follow‐up of childhood‐onset epilepsy, the higher the probability of SUDEPs. Our study with substantially longer follow‐up of childhood‐onset epilepsy supports previously reported percentages of 12–38%.20, 21 There is an ongoing debate about whether SUDEP may occur despite seizure remission. In a long‐term followed population study,14 18 died of SUDEP. Seven (39%) of the 18 patients were in 1‐year remission (unpublished data) and among 11 (61%), seizures were “unlikely” or “none witnessed” at death.14 Among children, nearly all of the mortality is related to an underlying neurologic disorder, not the seizures.22 All but one of our cases of SUDEP (90%) had an underlying neurologic disorder, but none of our cases of SUDEP had witnessed seizures at death. In a long‐term followed study with 60 deaths of 245 participants with childhood‐onset epilepsy,14 10 (43%) of SUDEPs deceased without witnessed seizures at death. In conclusion, the increased rate of death seen in our study of patients with West syndrome was substantial and persisted into adulthood, dispelling the notion that the risk of death is largely limited to childhood or early adulthood.
Although our study is useful because of the very long follow‐up of 50 years and a high autopsy rate of over 70%, limitations exist. We could not assess the role of seizure remission and the effect of drug treatment of West syndrome on mortality. Studies in the literature have shown in general that seizure remission is lowering the risk of death in childhood epilepsy.23 Finally, our study does contribute insight on how to prevent death in West syndrome in the future. The highest mortality risk for people with West syndrome is developing pneumonia. Future efforts to lower the mortality risk need to embrace measures to prevent pneumonia or to treat it more effectively, although entering seizure remission will probably reduce the death rate as in any childhood epilepsy syndrome.
In addition to prevention of pneumonia, epilepsy prevention is an important public health issue.24 Measures to improve prevention of epilepsy need to include earlier consideration of epilepsy surgery; better adherence to medication, possibly with increased nocturnal supervision and more vigorous interaction of the patient postictally; earlier use of rescue mediation; formal status protocols; and education regarding proper positioning during a seizure. Likewise, drowning is a rare cause of seizure‐related death,14 and it is preventable, and persons with epilepsy should be advised to shower rather than to bathe and should be closely supervised when swimming or around water.
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Biography
Matti Sillanpää is former Professor of Child Neurology and present Senior Research Scientist, University of Turku, Turku, Finland.

References
- 1. Berg AT, Nickels K, Wirrell EC, et al. Mortality risks in new‐onset childhood epilepsy. Pediatrics 2013;132:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeavons PM, Bower BD. Infantile spasms. A review of the literature and a study of 112 cases In Bax MCO, MacKeith RC. (Eds) Clinics in developmental medicine, no. 15. London: Heinemann Medical Books, 1964:8–25; 82. [Google Scholar]
- 3. Gastaut H, Soulayrol R, Roger J, et al. L'encéphalopathie myoclonique infantile ave hypsarythmie de west. Paris: Masson; 1964. [DOI] [PubMed] [Google Scholar]
- 4. Kurokawa T, Fung KC, Hanai T, et al. Mortality and clinical features in cases of death among epileptic children. Brain Dev 1982;4:321–325. [DOI] [PubMed] [Google Scholar]
- 5. Jeavons PM, Bower BD, Dimitrakoudi M. Long‐term prognosis of 150 cases of “West syndrome”. Epilepsia 1973;14:153–164. [DOI] [PubMed] [Google Scholar]
- 6. Matsumoto A, Watanabe K, Negoro T, et al. Long‐term prognosis after infantile spasms: a statistical study of prognostic factors in 200 cases. Dev Med Child Neurol 1981;23:51–65. [DOI] [PubMed] [Google Scholar]
- 7. Camfield P, Camfield C. Long‐term prognosis for symptomatic (secondarily) generalized epilepsies: a population‐based study. Epilepsia 2007;48:1128–1132. [DOI] [PubMed] [Google Scholar]
- 8. Riikonen R. Long‐term outcome of West syndrome: a study of adults with a history of infantile spasm. Epilepsia 1996;37:367–372. [DOI] [PubMed] [Google Scholar]
- 9. Riikonen R, Donner M. Incidence and aetiology of infantile spasms from 1960–1976: a population study in Finland. Dev Med Child Neurol 1979;21:333–343. [DOI] [PubMed] [Google Scholar]
- 10. Riikonen R, Amnell G. Psychiatric disorders in children with earlier infantile spasms. Dev Med Child Neurol 1981;23:747–760. [DOI] [PubMed] [Google Scholar]
- 11. Riikonen R. Epidemiological data of west syndrome in Finland. Brain Rev 2001;23:539–541. [DOI] [PubMed] [Google Scholar]
- 12. Lahti RA, Penttilä A. The validity of death certificates: routine validation of death certification and its effects on mortality statistics. Forensic Sci Int 2001;115:15–32. [DOI] [PubMed] [Google Scholar]
- 13. Annegers JF. United States perspective on definitions and classifications. Epilepsia 1997;38(Suppl 11):9–12. [DOI] [PubMed] [Google Scholar]
- 14. Sillanpää M, Shinnar S. SUDEP and other causes of mortality in childhood onset epilepsy. Epilepsy Behav 2013;28:249–255. [DOI] [PubMed] [Google Scholar]
- 15. Autry AR, Trevathan E, Van Naarden Braun K, et al. Increased risk of death among children with Lennox‐Gastaut syndrome and infantile spasms. J Child Neurol 2010;25:441–447. [DOI] [PubMed] [Google Scholar]
- 16. Brorson LO, Wranne L. Long‐term prognosis in childhood epilepsy: survival and seizure prognosis. Epilepsia 1987;28:324–330. [DOI] [PubMed] [Google Scholar]
- 17. Callenbach PMC, Westendorp RGJ, Geerts AT, et al. Mortality risk in children with epilepsy: the Dutch study of epilepsy in childhood. Pediatrics 2001;107:1259–1263. [DOI] [PubMed] [Google Scholar]
- 18. Berg AT, Lin J, Ebrahimi N, et al. Modeling remission and relapse in pediatric epilepsy: application of a Markov process. Epilepsy Res 2004;60:31–40. [DOI] [PubMed] [Google Scholar]
- 19. Ackers R, Besag FM, Hughes E, et al. Mortality rates and causes of death in children with epilepsy prescribed antiepileptic drugs: a retrospective cohort study using the UK General Practice Research Database. Drug Saf 2011;34:403–413. [DOI] [PubMed] [Google Scholar]
- 20. Grønborg S, Uldall P. Mortality and causes of death in children referred to a tertiary epilepsy center. Eur J Paediatr Neurol 2014;18:66–71. [DOI] [PubMed] [Google Scholar]
- 21. Harvey AS, Nolan T, Carlin JB. Community‐based study of mortality in children with epilepsy. Epilepsia 1993;34:597–603. [DOI] [PubMed] [Google Scholar]
- 22. Camfield P, Camfield C. Sudden unexpected death in people with epilepsy: a pediatric perspective. Semin Pediatr Neurol 2005;12:10–14. [DOI] [PubMed] [Google Scholar]
- 23. Sillanpää M, Shinnar S. Long‐term mortality in childhood‐onset epilepsy. N Engl J Med 2010;363:2522–2529. [DOI] [PubMed] [Google Scholar]
- 24. Sillanpää M, Gissler M, Schmidt D. Efforts in epilepsy prevention in the last 40 years: lessons from a large nationwide study. JAMA Neurol 2016;73:390–395. [DOI] [PubMed] [Google Scholar]