

Abstract
Essentials.
Compound heterozygosity causes a VWD2M phenotype in a child with severe bleeding symptoms.
p.P1648fs*45 changes the folding of A2 domain altering VWF binding to GPIbα and type VI collagen.
p.P1648fs*45 was considered as an apparent de novo mutation; AS‐PCR revealed paternal mosaicism.
Bleeding score and DDAVP's response were worse than those seen in VWD2M heterozygous controls.
Background
Type 2M von Willebrand disease (VWD2M) is usually characterized by VWF:RCo/VWF:Ag<0.6 and normal multimeric profile; desmopressin (DDAVP) challenge test commonly shows poor response of VWF:RCo.
Objective
We describe the bleeding tendency and the laboratory phenotype in a patient carrying two heterozygous mutations affecting VWF‐A1 domain and VWF‐A2 domain.
Subjects/methods
A 12‐year‐old patient (O blood group) with severe hemorrhagic tendency was phenotypically and genotypically analyzed; his parents were also studied.
Results
The proband showed decrease FVIII:C, VWF:RCo/VWF:Ag, and VWF:CB6/VWF:Ag ratios, but normal platelet count, VWF:CB1/VWF:Ag ratio, VWFpp and multimeric pattern, suggesting a VWD2M phenotype. The DDAVP challenge test, compared to controls (VWD2M patients with mutations in VWF‐A1 domain), showed lower increase of FVIII:C and VWF:Ag than in heterozygous, but very similar to homozygous control. Two mutations were found in heterozygous and trans presentation: p.Pro1648fs*45 and a novel missense mutation, p.Arg1426Cys. The mother was p.Arg1426Cys heterozygous carrier, with few clinical symptoms. The father was asymptomatic, with no mutations. The p.Pro1648fs*45 was considered an apparent de novo mutation; proband's AS‐PCR revealed mosaicism in the paternal allele. According to the predicted models, p.Arg1426Cys would not be affecting the binding of GPIbα to A1 domain, whereas p.Pro1648fs*45 seems to modify the folding of A2 domain, and in this way, it would affect the binding to GPIbα and type VI collagen. We believe that the combination of these two heterozygous mutations, in a child with O blood group, could result in a defective phenotype enhancer.
Keywords: desmopressin, hemorrhage, mutations, type 2M, von Willebrand disease, von Willebrand factor
1. INTRODUCTION
Von Willebrand disease type 2M (VWD2M) is an autosomal dominant bleeding disorder usually described by laboratory phenotype of VWF:RCo/VWF:Ag<0.6 and normal multimeric profile. Most cases show defective platelet glycoprotein Ibα (GPIbα) binding, revealed by low VWF:RCo. Although VWF:CB/VWF:Ag>0.6 are frequent, abnormal ratios are also described.1, 2 Mutations responsible for VWD2M are mainly clustered in the VWF‐A1 domain.
We describe a patient with a severe bleeding score and VWD2M laboratory phenotype, in which a compound heterozygous state was detected, and his behavior was compared to Caucasian VWD2M controls (heterozygous and homozygous) with mutations in the VWF‐A1 domain.
2. MATERIALS AND METHODS
2.1. Subjects and controls
The proband is a Caucasian 12‐year‐old boy. He was referred due to epistaxis requiring tamponades, bleeding after tooth eruption and extraction, easy bruising, gum bleeding, and bleeding in the central nervous system. After this last episode, he started prophylactic treatment with VWF‐FVIII concentrates (1000 U, twice a week). The bleeding score was calculated according to the ISTH bleeding assessment tool (ISTH‐BAT).3 His 30‐year‐old mother had epistaxis, easy bruising, gum bleeding, and abnormal menstrual score according to the Pictorial Bleeding Assessment Chart4 (PBAC = 192, nv < 185). His 40‐year‐old father and his sister did not show bleeding symptoms. The parents were non‐consanguineous. They gave their written informed consent for the study, in accordance with the declaration of Helsinki. Moreover, approval from the Ethics Committee of the Academia Nacional de Medicina, Buenos Aires, Argentina, was obtained.
Thirteen VWD2M patients, with desmopressin (DDAVP) challenge test, were selected as controls. They carry either homozygous: p.Arg1374Cys (n = 1), or heterozygous mutations: p.Arg1334Gln (n = 1), p.Arg1374Leu (n = 1), p.Arg1374Cys (n = 7), p.Ala1437Thr (n = 2), p.Thr1468Ile (n = 1).
2.2. Sample preparation
Peripheral venous blood was drawn in 3.13% sodium citrate for clotting tests and in 2% EDTA (disodium ethylenediaminetetraacetate dihydrate) for DNA analysis. An anticoagulant with anti‐proteases (3.8% sodium citrate; 60 mmol/L N‐ethylmaleimide, 50 mmol/L EDTA, and 2000 KIU/mL aprotinine), was used to collect samples for multimer analysis.
2.3. Laboratory methods
The following tests were performed: blood platelet count (Plt); bleeding time (BT) (Ivy's method); factor FVIII (FVIII:C) (one‐stage)5; VWF antigen (VWF:Ag) (ELISA)6; ristocetin cofactor activity (VWF:RCo) (aggregometry)7; VWD‐collagen binding (VWF:CB1) (ELISA, type I collagen); VWF:CB6 (ELISA, type VI collagen); VWF propeptide (VWFpp)8 (ELISA); multimer analysis.9 VWF:RCo/VWF:Ag (RCo/Ag), VWF:CB1/VWF:Ag (CB1/Ag), VWF:CB6/VWF:Ag (CB6/Ag), and VWFpp/VWF:Ag (VWFpp ratio) ratios were calculated.
2.4. Biologic response to DDAVP
Blood samples for DDAVP challenge test were obtained before and 90 minutes after the intravenous infusion of DDAVP (0.3 μg/kg body‐weight during 20 minutes). The time courses of FVIII:C, VWF:Ag, and VWF:RCo were analyzed, considering good response when all abnormal parameters reached normal levels.10
Proband's behavior was compared to controls (11 heterozygous VWD2M, mean age of 24.4 ± 14.7‐year‐old, and 1 homozygous patient, aged 7 years); 36% of them had O blood group. BS and BT were lower in heterozygous controls (BS: 4.8 ± 3.5; BT: 7.4 ± 1.7 minutes) than in the homozygous control (BS: 9; BT: 8 minutes).
2.5. Genotypic analysis
Genomic DNA was extracted from peripheral blood using standard methods. Exons 17, 26 to 31, and 52 of VWF gene were analyzed using polymerase chain reaction technique (PCR), and primers designed in our laboratory. Both forward and reverse strands were sequenced by automated sequencing technology (ABI Prism 310 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA). One‐hundred random healthy controls were also screened.
Allele‐specific PCR method (AS‐PCR) was developed to establish if mutations were located in cis or trans position. To achieve this goal, an asymmetric PCR method was optimized. This procedure produced ssDNA fragments of 902 base‐pairs in a single step when primer pairs were used at a 10:1 molar ratio in the presence of genomic DNA. AS‐forward primer (AS‐1426‐Fw‐mut) was designed by using appropriate software to allow the PCR amplification only if the nucleotide at the 3′‐end of the primer complemented the base at the c.4276 position in the C1426‐DNA sample. The PCR reaction tube contained 2.5 μL genomic DNA as template, 1 μL GoTaq DNA polymerase (5 U/μL), 10 μL 10× PCR Buffer, 1 μL dNTP (2 mmol/L mix), 1 μL 25 mmol/L MgCl2, 1 μL 20 μmol/L AS‐forward primer, and 0.1 μL 20 μmol/L 28‐F‐rev as reverse primer.
AS‐1426‐Fw‐mut: 5′‐TGCCAACCTCAAGCAGATCt‐3′.
Primer 28‐F‐rev: 5′‐AAGCCAGGATTAGAACCCGA‐3′, located at intron 28.
PCR cycling: initial denaturation (94°C, 5 minutes); 35 cycles: 94°C, 1 minutes; 63°C, 45 seconds; 72°C, 80 seconds; final extension: 72°C, 10 minutes. The optimum annealing temperatures were determined using a gradient PCR. After the amplification, electrophoresis was performed at 120 V for 30 minutes in 1× tris‐borate‐EDTA buffer on 2.0% agarose gel stained with Sybr‐Safe (3 μL). The amplified PCR products were visualized under UV light. The forward stand was sequenced.
2.6. In silico, alignment, and modeling analysis
The in silico analysis of changes was performed using PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.bii.a-star.edu.sg/), and Mutation Taster (http://www.mutationtaster.org/). The potential effect of p.Arg1426Cys on the destabilization of the VWF protein was analyzed using the I‐mutant 3.0 tool (http://gpcr2.biocomp.unibo.it/cgi/predictors/ accessed August 2017).
The alignment was made using CLC sequence viewer (http://www.clcbio.com) showing part of the wild type VWF and the protein carrying both the p.Arg1426Cys in the VWF‐A1 domain and p.Pro1648fs*45 in the VWF‐A2 domain. To gain graphical comprehension of the interaction between GPIbα and VWF in silico docking simulations were performed; X‐Ray diffraction‐solved structure of the complex GPIbα with the A1 domain of wild type VWF (1SQ0)11 was performed. Using SwissPDBviewer the arginine1426 of the VWF‐A1 domain was substituted by cysteine to create the A1 domain with the missense mutation p.Arg1426Cys. The VWF‐A2 domain was modeled with CPH model 3.2 server (www.cbs.dtu.dk) that used the solved crystal structure of the VWF‐A2 domain (3ZQK.A) as a template.12 The complex structure was obtained using protein‐protein docking server ClusPro2.0 (http://cluspro.bu.edu)13 and PatchDock‐server (http://bioinfo3d.cs.tau.ac.il/PatchDock)14 (Figure 1). The ClusPro prediction was preferred over Patchdock prediction as the PDBsum Procheck (http://www.ebi.ac.uk)15 values were less favorable for the latter. All the models shown had a Ramachandran plot with 98.9% of the residues within the most favored and additionally allowed regions.
Figure 1.
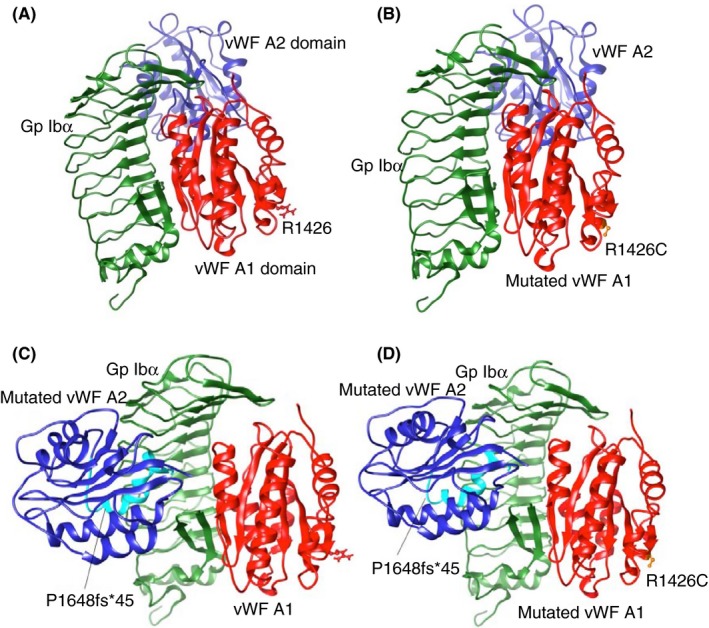
Hypothetical model of platelet glycoprotein Ib‐alpha (GPIbα) with the A1 and A2 domains of von Willebrand factor (VWF). GPIbα (green) with the A1 (red) and A2 (blue) domains of VWF are shown: (A) both A1 and A2 wild type; (B) mutated A1 (p.Arg1426Cys) (mutated residue in orange) and A2 wild type; (C) wild type A1 and mutated A2 (p.Pro1648fs*45) (difference with A2 wild type shown in cyan); (D) mutated A1 (p.Arg1426Cys) and mutated A2 (p.P1648fs*45)
2.7. Statistical analysis
Continuous variables were expressed using mean (X), standard deviation (SD), and minimum (min) and maximum (max) values (ranges).
3. RESULTS AND DISCUSSION
The phenotypic and the molecular data of the proband and his parents are display in Table 1. Proband's results suggested a VWD2M phenotype, whereas mother's results revealed a possible type 1 VWD; father's results were normal.
Table 1.
Phenotypic and molecular data of the proband and his parents
| Patient | Mother | Father | Normal values | ||
|---|---|---|---|---|---|
| Desmopressin | |||||
| Pre | Post | ||||
| BS | 14 | 5 | 0 | Males < 3Females < 5 | |
| Blood group | O | B | A | ||
| Plt (×109/L) | 220 | 224 | 248 | 160 | 150‐400 |
| BT (min) | 5.5 | nd | nd | nd | <4.5 |
| FVIII:C (IU/dL) | 24 | 45 | 90 | 115 | 50‐150 |
| VWF:Ag (IU/dL) | 17 | 34 | 45 | 128 | 50‐150 |
| VWF:RCo (IU/dL) | 9 | 9 | 46 | 101 | 50‐150 |
| RCo/Ag | 0.53 | 0.26 | 1.01 | 0.79 | >0.6 |
| VWF:CB1 (IU/dL) | 17.2 | 24 | nd | nd | 60‐130 |
| CB1/Ag | 1.01 | 0.71 | nd | nd | >0.6 |
| VWF:CB6 (IU/dL) | 4.5 | 8 | nd | nd | 60‐130 |
| CB6/Ag | 0.26 | 0.24 | nd | nd | >0.6 |
| VWFpp (IU/dL) | 27 | nd | nd | nd | 50‐150 |
| VWFpp/Ag | 1.6 | nd | nd | nd | 0.92‐2.14 |
| Multimeric profile | Presence of all the multimeric forms | nd | nd | ||
| Molecular data | c.4276C>T→p.Arg1426Cys c.4944delT→p.Pro1648fs*45 | c.4276C>T→p.Arg1426Cys | No mutations | ||
BS, bleeding score; Plt, platelet count; BT, bleeding time; FVIII:C, factor VIII; VWF:Ag, von Willebrand factor antigen; VWF:RCo, ristocetin cofactor activity; RCo/Ag: VWF:RCo/VWF:Ag; VWF:CB1: VWF‐type I collagen binding; CB1/Ag: VWF:CB1/VWF:Ag; CB6: VWF‐type VI collagen binding; CB6/Ag: VWF:CB6/VWF:Ag; ND: not done.
DDAVP challenge test usually shows poor response of VWF:RCo in VWD2M16; however, poor response is not required for VWD2M diagnosis. The proband, in addition to the absence of response of VWF:RCo, showed FVIII:C, and VWF:Ag that did not reach normal values after DDAVP infusion (poor response to DDAVP). The increase of both FVIII:C and VWF:Ag was lower than the increase observed in heterozygous VWD2M controls. The proband's DDAVP challenge test showed a similar pattern to that seen in the homozygous VWD2M control (Figure 2).
Figure 2.

Desmopressin (DDAVP) challenge test. Comparison of FVIII:C: factor VIII; VWF:Ag: von Willebrand factor antigen; VWF:RCo: ristocetin cofactor activity and VWF:CB1: type I‐collagen‐VWF binding levels; between the compound heterozygous proband and VWD2M controls with missense mutations located at the A1 domain. Before DDAVP infusion:  ; after DDAVP infusion:
; after DDAVP infusion:  . Mutations in VWD2M controls: homozygous state: p.Arg1374Cys (pink); heterozygous state: p.Arg1334Gln (red); p.Arg1374Cys (green); p.Arg1374Leu (yellow); p.Ala1437Thr (light green); p.Thr1468Ile (blue). Mutations in the proband: p.Arg1426Cys and p.Pro1648fs*45 (orange)
. Mutations in VWD2M controls: homozygous state: p.Arg1374Cys (pink); heterozygous state: p.Arg1334Gln (red); p.Arg1374Cys (green); p.Arg1374Leu (yellow); p.Ala1437Thr (light green); p.Thr1468Ile (blue). Mutations in the proband: p.Arg1426Cys and p.Pro1648fs*45 (orange)
Two mutations, in heterozygous state, were found in the exon 28 of the VWF in the proband. A novel missense substitution, c.4276C>T (NM_000552.4) causing the change of an arginine to cysteine at residue 1426 (p.Arg1426Cys) (NP_000543.2; reference ID: rs555366738) in the VWF‐A1 domain was detected, in addition to c.4944delT (p.Pro1648fs*45) in the VWF‐A2 domain. No changes were found in exons 17, 26, 27, 29 to 31, and 52, where several type 2M mutations have been reported. The mother showed the p.Arg1426Cys in heterozygosity; the father did not show mutations in exon 28.
The novel p.Arg1426Cys was predicted to be deleterious by in silico analysis (PolyPhen‐2 score: 0.998; SIFT score: 0.00). I‐Mutant 3.0 predicted a large decrease of VWF stability for p.Arg1426Cys with a DDG of −0.73 Kcal/mol. In sequence alignment, this residue was shown located in a highly‐conserved region in the mammalian species compared, confirming the deleterious effect of the mutant. It was absent in our 100 healthy controls, therefore, not considered to be a polymorphism in our general population. The global minor allele frequency (MAF) for the C>T change was 0.0002/1, and validated by 1000G Project. The allele frequency informed by ExAC browser was 1.648 × 10−5 (2/121386 alleles) (http://exac.broadinstitute.org Accessed August 2017).
The p.Pro1648fs*45 was found only in the proband, suggesting an apparent de novo presentation. The proband's AS‐PCR showed the absence of both mutations in the same allele, therefore indicating a trans presentation. Considering that the mother only has the p.Arg1426Cys, we assume that the p.Pro1648fs*45 has a paternal origin, possibly due to mosaicism. It is difficult to discriminate whether the mutation occurred at some point during the development of the germ line (in his sperm), or postzygotically (somatic mosaicism), unless DNA from other tissues (hair roots, urine specimen, buccal swap, skin biopsy) were used to see if it is present in his father's somatic cells.17
It has been described2 that several selected VWD2M variants showed normal expression as heterozygous recombinant VWF, which may not reflect plasma VWF in affected patients; whereas homozygous expression of variants was consistent with VWD2A. This inconsistency may result from differences in secretion of VWF, presence of other variants or other coagulation defects that could modify the phenotype. The type 1 VWD p.Arg1379Cys has been described to have a synergistic effect together with p.Ala1377Val (polymorphism in ISTH‐SSC VWF database), determining a VWD2M.18 This phenotype does not appear with p.Ala1377Val alone or with a hypothetical unidentified third mutation located outside the VWF‐A1 domain. Other situations in which a second mutation and the O blood group reinforces the decrease of FVIII:C and VWF have been reported.19, 20
VWF binds types I and III collagen via the VWF‐A3 and VWF‐A1 domains, but it binds sub‐endothelial type VI collagen exclusively via the VWF‐A1 domain21; the latter seems to be important in the VWF‐platelet binding under low shear.22 Reduced binding to type VI collagen has been reported in VWD2M patients in the presence of four mutations located in VWF‐A1 domain.23
Fidalgo et al.1 described a VWD2M patient with heterozygous compound genotype for p.Arg1315Hys associated with type 1 VWD, and another mutation in the splicing site c.7464C>T in exon 44, who presented reduced RCo/Ag and CB6/Ag, but normal CB1/Ag. The proband has shown similar phenotypic characteristics.
The native state of VWF‐A1 domain is highly flexible; platelet adhesion under shear flow is highly dependent on it.24 Certain mutations can induce changes in thermodynamic and kinetic stability, resulting in the local misfolding of the VWF‐A1 domain in secondary structures that affects GP1bα binding, and results in loss‐of‐function phenotype (VWD2M).25 The p.Arg1426Cys mutation would not affect the binding of GPIbα to VWF‐A1 domain, because is not in the zone of contact; nevertheless, it could affect the binding to other molecules. The p.Pro1648fs*45 determines the position of the VWF‐A2 domain, close to the leucine rich repeats rather than on the concave part of GPIbα as seen in the model obtained with wild type VWF‐A2 domain interaction. This mutation seems to affect the GPIbα‐VWF binding, as clashes were found between them which do not appear in the wild type VWF‐A2 domain. Therefore, this mutation may correspond to a misfolding of the VWF‐A2 structure, which would affect its function in direct or indirect way; it may hinder VWF secretion or function. These observations, plus the fact that the in silico result for I‐Mutant 3.0 predicted a large decrease of VWF stability for p.Arg1426Cys, allow us to hypothesize that the presence of p.Arg1426Cys and p.Pro1648fs*45 may cause destabilization of the VWF‐A1 domain and misfolding of VWF‐A2 domain, resulting in the reduced binding of the double‐mutant to GPIbα and to type VI collagen.
Compound heterozygosity would result in a severe clinical phenotype, similar to a homozygous VWD2M behavior, despite it is not the case. Heterozygous p.Arg1426Cys in proband's mother suggests possible type 1 VWD, and p.Pro1648fs*45 has been described as responsible for type 1 VWD. Therefore, there are some inconsistencies between proband's genotype and phenotype. The bleeding history and the VWD2M laboratory phenotype, including the poor response to DDAVP, seen in the proband would be the result of a synergistic effect. We believe that the combination of the two heterozygous mutations could modify the expression of VWF and result in a defective phenotype enhancer, as was described for other mutations.2, 19, 20
AUTHOR CONTRIBUTIONS
A. I. Woods and M. A. Lazzari were responsible for the study concept and design, analysis of data, and wrote the manuscript. A. I. Woods performed VWF:RCo, VWF:CB6, and genotypic assays, and was responsible for collecting data. J. Paiva and A. C. Kempfer performed the multimeric profile, VWF:CB1, and VWFpp assays. D.M. Primrose made the modeling and alignment analysis. A. N. Blanco was responsible for FVIII:C and VWF:Ag assays. A. Sánchez‐Luceros was responsible of recruitment patients and the maintenance of clinical records. M. A. Lazzari gave final approval of the version to be published. All authors have revised the draft and approved the final manuscript.
RELATIONSHIP DISCLOSURE
None of the authors have any disclosures relevant to this paper.
ACKNOWLEDGEMENTS
The authors are grateful to Maria Marta Casinelli for her excellent technical assistance.
Woods AI, Paiva J, Kempfer AC, et al. Combined effects of two mutations in von Willebrand disease 2M phenotype. Res Pract Thromb Haemost. 2018;2:162–167. 10.1002/rth2.12067
Funding information
This work was supported by CONICET, (National Scientific and Technical Research Council) Fundación René Barón, and Academia Nacional de Medicina, Buenos Aires, Argentina
REFERENCES
- 1. Fidalgo T, Oliveira A, Silva Pinto C, et al. VWF collagen (types III and VI)‐binding defects in a cohort of type 2M VWD patients – a strategy for improvement of a challenging diagnosis. Haemophilia. 2017;23:e143–7. [DOI] [PubMed] [Google Scholar]
- 2. Doruelo AL, Haberichter SL, Christopherson PA, et al. Clinical and laboratory phenotype variability in type 2M von Willebrand disease. J Thromb Haemost. 2017;15:1559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodeghiero F, Tosetto A, Abshire T, et al.; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group . ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–5. [DOI] [PubMed] [Google Scholar]
- 4. Janssen CA, Scholten PC, Heintz AP. A simple visual assessment technique to discriminate between menorrhagia and normal menstrual blood loss. Obstet Gynecol. 1995;85:977–82. [DOI] [PubMed] [Google Scholar]
- 5. Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol. 1961;36:212–9. [DOI] [PubMed] [Google Scholar]
- 6. Taylor LD. The application of the biotin‐avidin system to the von Willebrand factor antigen immunoassay. Thromb Haemost. 1988;59:251–4. [PubMed] [Google Scholar]
- 7. Macfarlane DE, Stibbe J, Kirby EP, Zucker MB, Grant RA, McPherson J. Letter: a method for assaying von Willebrand factor (ristocetin cofactor). Thromb Diath Haemorrh. 1975;34:306–11. [PubMed] [Google Scholar]
- 8. Borchiellini A, Fijnvandraat K, ten Cate JW, et al. Quantitative analysis of von Willebrand factor propeptide release in vivo: effect of experimental endotoxemia and administration of 1‐deamino‐8‐d‐arginine vasopressin in humans. Blood. 1996;88:2951–8. [PubMed] [Google Scholar]
- 9. Farias C, Kempfer AC, Blanco A, Woods AI, Lazzari MA. Visualization of the multimeric structure of von Willebrand factor by immunoenzymatic stain using avidin‐peroxidase complex instead of avidin‐biotin peroxidase complex. Thromb Res. 1989;53:513–8. [DOI] [PubMed] [Google Scholar]
- 10. Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM‐1VWD. Blood. 2008;111:3531–9. [DOI] [PubMed] [Google Scholar]
- 11. Dumas JJ, Kumar R, McDonagh T, et al. Crystal structure of the wild‐type von Willebrand factor A1‐glycoprotein Ibalpha complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J Biol Chem. 2004;279:23327–34. [DOI] [PubMed] [Google Scholar]
- 12. Jakobi AJ, Mashaghi A, Tans SJ, Huizinga EG. Calcium modulates force sensing by the von Willebrand factor A2 domain. Nat Commun. 2011;2:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein‐protein docking. Nat Protoc. 2017;12:255–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nielsen M, Lundegaard C, Lund O, Petersen TN. CPHmodels‐3.0 – remote homology modeling using structure guided sequence profiles. Nucleic Acids Res. 2010;38:W576–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29:221–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Michiels JJ, Smejkal P, Penka M, et al. Diagnostic differentiation of von Willebrand disease types 1 and 2 by von Willebrand factor multimer analysis and DDAVP challenge test. Clin Appl Thromb Hemost. 2017;23:518–31. [DOI] [PubMed] [Google Scholar]
- 17. Armaroli A, Trabanelli C, Scotton C, et al. Paternal germline mosaicism in collagen VI related myopathies. Eur J Paediatr Neurol. 2015;19:533–6. [DOI] [PubMed] [Google Scholar]
- 18. Pagliari MT, Baronciani L, Stufano F, et al. von Willebrand disease type 1 mutation p.Arg1379Cys and the variant p.Ala1377Val synergistically determine a 2M phenotype in four Italian patients. Haemophilia. 2016;22:e502–11. [DOI] [PubMed] [Google Scholar]
- 19. Hickson N, Hampshire D, Winship P, et al. MCMDM‐1VWD and ZPMCB‐VWD study groups. von Willebrand factor variant p.Arg924Gln marks an allele associated with reduced von Willebrand factor and factor VIII levels. J Thromb Haemost. 2010;8:1986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Woods AI, Kempfer AC, Sánchez‐Luceros A, Calderazzo JC, Grosso SH, Lazzari MA. Clinical profile of the association of p. R1205H and p.R924Q in a patient with von Willebrand's disease. Haemophilia. 2013;19:e180–1. [DOI] [PubMed] [Google Scholar]
- 21. Flood VH, Gill JC, Christopherson PA, et al. Comparison of type I, type III and type VI collagen binding assays in diagnosis of von Willebrand disease. J Thromb Haemost. 2012;10:1425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ross JM, McIntire LV, Moake JL, Rand JH. Platelet adhesion and aggregation on human type VI collagen surfaces under physiological flow conditions. Blood. 1995;85:1826–35. [PubMed] [Google Scholar]
- 23. Larsen DM, Haberichter SL, Gill JC, Shapiro AD, Flood VH. Variability in platelet‐ and collagen‐binding defects in type 2M von Willebrand disease. Haemophilia. 2013;19:590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tischer A, Campbell JC, Machha VR, et al. Mutational constraints on local unfolding inhibit the rheological adaptation of von Willebrand factor. J Biol Chem. 2016;291:3848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tischer A, Madde P, Moon‐Tasson L, Auton M. Misfolding of vWF to pathologically disordered conformations impacts the severity of von Willebrand disease. Biophys J. 2014;107:1185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]