

Abstract
Alveolar soft part sarcoma (ASPS) is a rare form of soft-tissue sarcoma arising from connective tissue. It is most often seen in adolescents and young adults and has high propensity for recurrence and metastasis. Clinically, it mimics haemangioma or arteriovenous malformations. In our case report, an 18-year-old female presented with markedly vascular tumour in the left forearm, for which excision biopsy was done. Histopathological report revealed ASPS. The patient was screened for metastasis. Ultrasound abdomen, computed tomography (CT) chest, CT brain and whole-body skeletal survey was done. The patient was found to have bilateral pulmonary metastasis. The patient was given 6 cycles of chemotherapy with adriamycin, cyclophosphamide and vincristine. There was no locoregional and pulmonary recurrence for 11 months after being treated by excision of the tumour followed by chemotherapy.
KEY WORDS: Alveolar soft part sarcoma, extremity soft part sarcoma, pulmonary metastasis
INTRODUCTION
Alveolar soft part sarcoma (ASPS) is an entity of sarcoma first described by Christopherson et al. in 1952.[1] More commonly seen in children and young adults. ASPS represents <1% of all soft-tissue sarcomas which is more common in females than males. In case of children, it affects the head-and-neck region with predilection to the tongue and orbit. In case of adults, it commonly affects the extremities,[2,3] genital tract, breasts, mediastinum, bladder and gastrointestinal tract.[4,5] ASPS in the forearm is a rare manifestation, only few cases are reported in India.[6] Here, we present a case report of a young girl with ASPS of left forearm.
CASE REPORT
An 18-year-old girl admitted in our department on February 2014 with the complaints of pain and swelling over medial aspect of upper one-third of the left forearm [Figure 1] for the past 6 months, and the mass was gradually increasing in size, for which incisional biopsy was done outside and referred with biopsy report as glomangioma or epithelioid haemangioendothelioma. On examination, there was a palpable firm mass on the medial volar aspect of the left forearm measuring about 7 cm × 5 cm × 3 cm with surgical scar with few dilated veins over the swelling with no bruit. There was no distal neurovascular deficit. Hand functions were normal. Magnetic resonance imaging of the upper forearm showed the mass with high vascularity, involving mainly the muscular plane. The biopsy report showed the possibility of glomangioma or epithelioid haemangioendothelioma, which was a benign condition, and so excision biopsy was done.
Figure 1.
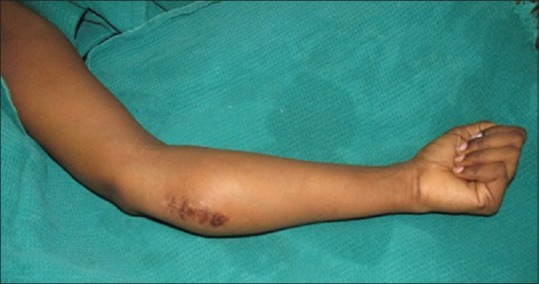
Swelling of left forearm
A lazy 'S' incision was made over the swelling. Highly vascular mass was found encapsulated within the flexor digitorum superficialis (FDS) muscle at the common flexor origin [Figure 2a and b]. The ulnar nerve was found closely adhered to the capsule of the mass and was dissected after dividing its muscular branches [Figure 3]. Mass was excised along with part of FDS muscle up to the musculotendinous junction; the remaining tendinous part was attached to flexor digitorum profundus muscle with adequate tension.
Figure 2.

(a and b) Encapsulated mass with high vascularity
Figure 3.
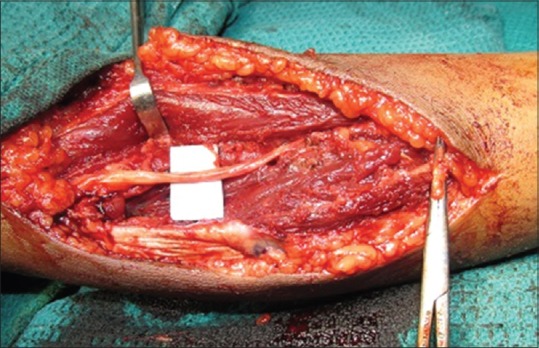
Preserved ulnar nerve
Histopathology examination showed nests of large polygonal cells with abundant eosinophilic cytoplasm having pseudoalveolar pattern [Figure 4a and b], separated by delicate vascular structures. All the findings confirmed ASPS.
Figure 4.
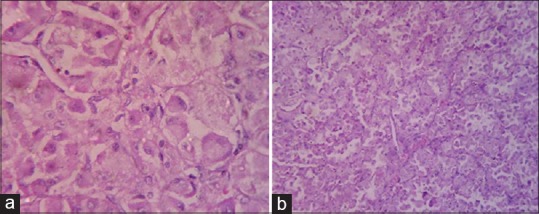
(a and b) Histological view of alveolar soft part sarcoma
The patient was screened for metastasis post-operatively. Computed tomography (CT) chest showed multiple coin-like opacities in both the lung fields, more in lower lobes [Figure 5]. CT brain, CT abdomen and whole-body skeletal survey were normal.
Figure 5.
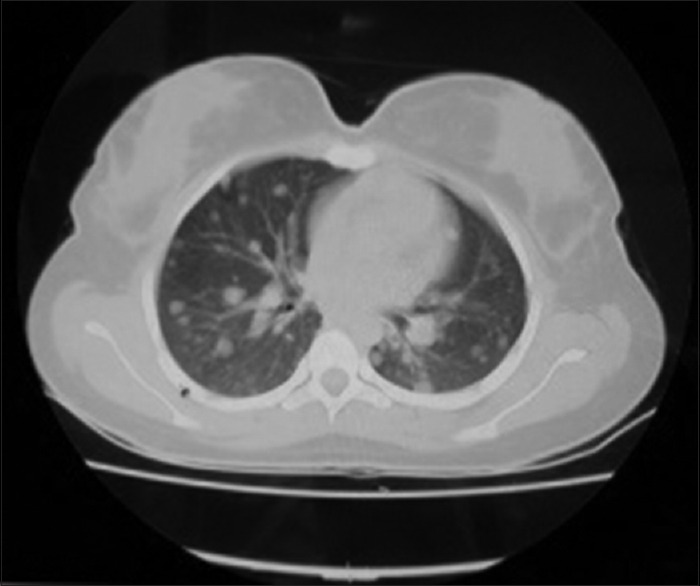
Computed tomography lung shows lung secondaries
After discussion with oncology department (radiation oncology, medical oncology and surgical oncology), we decided to give cycles of chemotherapy and radiotherapy.
The patient received 6 cycles of chemotherapy with adriamycin, cyclophosphamide and vincristine as suggested by the oncologist. The patient was closely followed for 11 months. There was no locoregional recurrence or metastasis elsewhere. Full range of movements in the hand and elbow was possible.
After 11 months, the patient had good functional improvement [Figure 6a and b], due to financial constraints, PET scan and other investigations were not done on subsequent follow-up.
Figure 6.
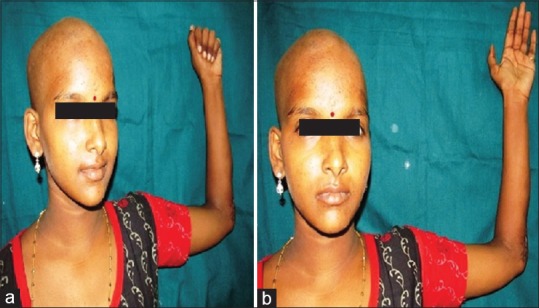
(a and b) Functional status of the hand after 11 months
DISCUSSION
ASPS is one of the rarest forms of soft-tissue sarcoma. This tumour commonly occurs in females, with a male: female ratio of 1:2 and the mean age 22–25. Most common site of origin is lower extremity,[7] then the trunk and the upper limbs. ASPS represents <1% of all soft-tissue sarcomas, and they form one-third of all metastatic soft-tissue sarcomas (33%).[8]
The genetic theory for ASPS[1] is thought to be its origin in chromosomal rearrangement at t (X:17) (p11:Q25), resulting in the ASPL–TFE3 fusion gene.[9] Since women have two X chromosomes, they are more prone for ASPS.
This tumour is slow-growing in nature, but the metastases are more common to the lung followed by bone and brain. Lymph node metastasis is rare.[5] Prognosis of ASPS depends on patient's age, tumour size and the presence of metastasis at the time of diagnosis. Metastases are detected in 20%–25% of the patients at the time of diagnosis.
ASPS are more vascular in nature, because of these prominent vascular structure, the mass shows high-intensity signals in T1- and T2-weighed images[3] and resembles those of haemangioma, but histologically, it shows typical features of ASPS.
Histologically, the tumour has peculiar, orderly arranged epithelioid-appearing tumour cells separated by delicate fibrovascular septae. ASPS can be easily differentiated from its histopathological mimics by demonstrating cytological uniformity, lack of nuclear atypia and paucity of mitotic figures in ASPS.[6]
Treatment
For primary tumour, extensive resection of the mass (R0 resection: Complete resection with no microscopic residual tumour) and adjuvant regional radiotherapy is considered as the most effective treatment of ASPS. The management algorithm for soft-tissue sarcoma [Flow chart 1] is shown below.[10]
Flowchart 1.
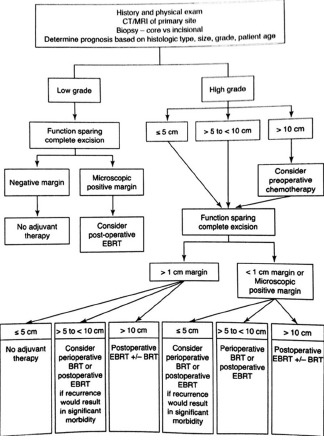
Management algorithm for soft-tissue sarcoma
The local recurrence rate reportedly ranges from 10% to 30% after the removal of the mass and an adjunctive radiation treatment could be beneficial to prevent a relapse. Combination of surgery with radiotherapy and chemotherapy may prolong the mean survival time. There are reports regarding excision of pulmonary metastases providing a good prognosis along with radiotherapy or chemotherapy. In the literature, 5-year overall survival was reported as 83%. The presence of lung metastases is not a contraindication for surgery in patients with ASPS. The reason for favourable prognosis with this tumour can be explained by the slow-growing and indolent nature of this disease.
CONCLUSION
ASPS are one of the rarest soft-tissue tumours, which rarely involve upper extremity. To the best of our knowledge, very few cases of ASPS has been reported from India. Characteristic histological features would facilitate an early diagnosis. Long-term follow-up is needed to find out the recurrence of a tumour or metastasis. Because of its rarity of occurrence in the upper extremity, this case has been reported.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Christopherson WM, Foote FW, Jr, Stewart FW. Alveolar soft-part sarcomas; structurally characteristic tumors of uncertain histogenesis. Cancer. 1952;5:100–11. doi: 10.1002/1097-0142(195201)5:1<100::aid-cncr2820050112>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 2.Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY, et al. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989;63:1–3. doi: 10.1002/1097-0142(19890101)63:1<1::aid-cncr2820630102>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 3.Suh JS, Cho J, Lee SH, Shin KH, Yang WI, Lee JH, et al. Alveolar soft part sarcoma: MR and angiographic findings. Skeletal Radiol. 2000;29:680–9. doi: 10.1007/s002560000285. [DOI] [PubMed] [Google Scholar]
- 4.Fanburg-Smith JC, Miettinen M, Folpe AL, Weiss SW, Childers EL. Lingual alveolar soft part sarcoma; 14 cases: Novel clinical and morphological observations. Histopathology. 2004;45:526–37. doi: 10.1111/j.1365-2559.2004.01966.x. [DOI] [PubMed] [Google Scholar]
- 5.Pang LM, Roebuck DJ, Griffith JF, Kumta SM, Metreweli C. Alveolar soft-part sarcoma: A rare soft-tissue malignancy with distinctive clinical and radiological features. Pediatr Radiol. 2001;31:196–9. doi: 10.1007/s002470000388. [DOI] [PubMed] [Google Scholar]
- 6.Sarkar P, Mukherjee S, Saha ML, Biswas RS. Alveolar soft part sarcoma: A rare diagnosis. Indian J Dermatol. 2013;58:244. doi: 10.4103/0019-5154.110873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidi V, Fragandrea I, Hatzipantelis E, Kyriakopolous C, Papanikolaou A, Bandouraki M, et al. Alveolar soft – Part sarcoma of the extremity: A case report. Hippokratia. 2008;12:251–3. [PMC free article] [PubMed] [Google Scholar]
- 8.Perry JR, Bilbao JM. Metastatic alveolar soft part sarcoma presenting as a dural-based cerebral mass. Neurosurgery. 1994;34:168–70. [PubMed] [Google Scholar]
- 9.Stockwin LH, Vistica DT, Kenney S, Schrump DS, Butcher DO, Raffeld M, et al. Gene expression profiling of alveolar soft-part sarcoma (ASPS) BMC Cancer. 2009;9:22. doi: 10.1186/1471-2407-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.John R. Goldblum, Sharon W. Weiss, Andrew L. Folpe. Enzinger and Weiss' Soft Tissue Tumors. US: Elsevier Health; 2008. Malignant soft tissue tumors of uncertain type; pp. 1182–9. [Google Scholar]