

Abstract
Zinc is coreleased with glutamate from excitatory nerve terminals throughout the central nervous system and acutely inhibits N-methyl-d-aspartate (NMDA) receptor activation. Here we report that cultured murine cortical neurons briefly exposed to sublethal concentrations of zinc developed increased intracellular free Na+, phosphorylation of Src kinase at tyrosine 220, and tyrosine phosphorylation of NMDA receptor 2A/2B subunits, in a fashion sensitive to the Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine, PP2. Functionally, this zinc exposure produced a delayed increase in NMDA receptor current in perforated patch but not conventional whole-cell recordings, as well as an increase in NMDA receptor-mediated cell death. These observations suggest that the effect of synaptically released zinc on neuronal NMDA receptors may be biphasic: acute block, followed by Src family kinase-mediated up-regulation of NMDA receptor activity and cytotoxicity.
Zinc is an important trace element functioning in both catalytic and structural capacities in all mammalian cells (1). The central nervous system additionally contains a chelatable pool of zinc located in synaptic vesicles, predominantly within glutamatergic nerve terminals throughout the telencephalon (2, 3). This zinc is released into the synaptic cleft with synaptic activity, reaching concentrations that may routinely exceed 10–20 μM (4) and may approach 300 μM in extreme conditions (5, 6). The precise role of synaptically released zinc is unknown, but acute modulation of the activity of several receptors and channels is likely, including transmitter receptors (glutamatergic, GABAergic, glycinergic, and purinergic), voltage-sensitive cation channels (Na+, K+, and Ca2+), and the Na+/K+-ATPase (7–9). In particular, given its systematic corelease with glutamate, zinc's ability to inhibit N-methyl-d-aspartate (NMDA) receptors may figure prominently in its physiological actions (4, 10, 11). Zinc produces both a voltage-independent reduction of NMDA receptor-gated channel opening frequency and a voltage-dependent flicker block of the channel (12–14). These actions likely correspond to the binding of zinc at high affinity and a low-affinity binding sites on the NMDA receptor (15–20).
Although limited zinc exposure has multiple modulatory effects potentially relevant to a normal signaling function, exposure to higher concentrations or for longer durations can trigger brain cell death, more easily in neurons than astrocytes (21, 22). Zinc toxicity has been postulated to contribute to neuronal loss after prolonged seizures (23), transient global ischemia (24, 25), or traumatic brain injury (26). Zinc-induced neuronal death depends on excessive zinc influx across the plasma membrane via several routes, including voltage- and agonist-gated Ca2+ channels as well as the Na+-Ca2+ exchanger (27–29). Zinc release from intracellular stores or binding sites may also contribute to neurotoxicity (30).
The observation that synaptically released zinc can “translocate” into postsynaptic neurons under pathological conditions (disappear from presynaptic terminals and appear abnormally in postsynaptic cell bodies; refs. 24, 31) raises the intriguing possibility that some translocation may occur physiologically and lead to potentially lasting alterations in postsynaptic neuronal behavior (32). Zinc can interact with many macromolecules and can alter the phosphorylation state and activity of several kinases, including protein kinase C (33–35), calcium/calmodulin-dependent protein kinase II (36, 37), phoshatidylinositol 3-kinase (38), and extracellular signal regulated kinases (39–41). In addition, 0.2–1 mM zinc activated protein kinase in isolated platelet and hippocampal membranes (42, 43), an activity likely mediated primarily by pp60c-src, the cellular homologue of the Rous sarcoma virus-transforming protein (44, 45). Given recent implication of neuronal Src family kinases (SFKs) in brain signaling (46, 47), especially in up-regulating NMDA receptor function (48–50), we explored the possibility that subtoxic zinc exposure might up-regulate NMDA receptors via activation of SFKs.
Methods
Cortical Cell Cultures.
Mixed or near-pure neuronal cortical cell cultures were prepared from fetal mice (15–16 days gestation), as previously described (51). Dissociated cortical cells were plated on established glial cultures in 24-well culture plates or in glass-bottom 35-mm dishes (MatTek, Ashland, MA), or for near-pure neuronal cultures, on plates coated with 50 ng/ml poly(D)-lysine and 2 ng/ml laminin. Plating media consisted of Eagle's MEM (MEM, Earle's salts) supplemented with 5% horse serum/5% FBS/2 mM glutamine/20 mM glucose. In mixed neuronal/glial cell cultures, glial cell replication was inhibited after 5–9 days in vitro (DIV) by addition of 10 μM cytosine arabinoside (Ara C). In near-pure neuronal cultures, Ara C was added at DIV 3. Under these conditions, less than 1% of total cells were astrocytes. Neocortical glial cultures were prepared from 1- to 3-day-old mice and plated on Primaria 24-well culture plates in plating media supplemented with epidermal growth factor (10 ng/ml).
Cell Treatment and Protein Extraction.
Cell cultures were washed three times in Hepes-control salt solution containing (in mM): 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 10 NaOH, 20 Hepes, and 5.5 glucose, pH 7.4, and then exposed to 50 μM ZnCl2 (Zn2+) in the absence or presence of 30- to 60-min pretreatment at 37°C with (1–10 μM) of the SFK inhibitor PP2 (52). After treatment, cell extracts were prepared as described (53) in lysis buffer (1% Nonidet P-40/20 mM Tris⋅Cl, pH 7.5/10 mM EGTA/40 mM β-glycerophosphate/2.5 mM MgCl2/2 mM orthovanadate/1 mM DTT/1 mM PMSF/20 μg/ml aprotinin/20 μg/ml leupeptin) and used for the detection of Src, PLCγ, and Fyn (see below).
For the study of NMDA receptor tyrosine phosphorylation, cell extracts were prepared as described by Lin et al. (54). Cells were treated as above, and treatments were stopped by addition of 50 μl of SDS buffer (1% SDS/2 mM sodium orthovanadate/10 μg/ml leupeptin/10 μg/ml aprotinin/1 mM PMSF). The resulting extracts were heated at 90°C for 5 min followed by addition of nine volumes of dilution buffer (1% Nonidet P-40/1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/20 mM Tris⋅HCl, pH 7.4/150 mM NaCl/1 mM EDTA/1 mM EGTA/1 mM sodium orthovanadate/10 μg/ml leupeptin/10 μg/ml aprotinin/1 mM PMSF). Insoluble material was removed by centrifugation at 25,000 × g for 30 min. Protein concentrations were determined by the bicinchoninic acid method (Pierce) by using BSA as standard.
Immunoprecipitation and Western Blotting.
To study tyrosine phosphorylation of Fyn or NR2, 400- to 1,000-μg protein samples were incubated overnight at 4°C with 2 μg of anti-Fyn (Santa Cruz Biotechnology), 3 μl of anti-NR2A, or 2 μg of anti-NR2B antibodies (Chemicon). Immunocomplexes were recovered with the aid of protein A-agarose. Agarose beads were pelleted by centrifugation and washed three times in 1% Nonidet P-40–2 mM orthovanadate in PBS, and once in PBS. The precipitated proteins were resuspended in 35 μl of electrophoresis Laemmli's buffer and heated at 90°C for 5 min. Samples were fractionated on 6% SDS/PAGE and transferred to nitrocellulose membranes (Micron Separations) by using a semidry electrotransfer system (Novablot, Amersham Pharmacia–Pharmacia). Membranes were blocked with 5% milk in TBS-T buffer (20 mM Tris/150 mM NaCl/0.05% Tween 20) and were then incubated with biotinylated antiphosphotyrosine antibody (4G10, Upstate Biotechnology).
To study tyrosine phosphorylation of Src or PLCγ, 10 μg of protein from whole cell lysates were resuspended in Laemmli's sample buffer and proteins resolved in 7% SDS/PAGE and transferred as above. Blots were incubated with phosphorylation site-specific antibodies p-Src215, p-Src418, p-Src527, and p- PLCγ 1:1,000 (BioSource International, Camarillo, CA). Bound antibodies were detected by the enhanced chemiluminescent method (Pierce).
Blots were subsequently stripped and reprobed with specific antibodies directed to Src, Fyn, NR2A, or NR2B (Santa Cruz). For quantification, the intensity of immunoreactive bands obtained in autoradiographic films was measured with an imaging densitometer (Bio-Rad). Phosphotyrosine levels per protein unit ratios were obtained by dividing the phosphotyrosine immunoreactive densitometry values by those obtained for the respective protein redetection blots. Experimental and control conditions were then compared and expressed as percentage of control.
Induction of Neuronal Death and Assessment of Neuronal Injury.
Thirteen to fifteen days of in vitro mixed cortical cultures were exposed to 25 μM NMDA for 5 min in Hepes-control salt solution alone, together with 50 μM zinc, or after 5-min prior exposure to 50 μM zinc. Treatment with the SFK inhibitor PP2 was carried out as described above. After exposure, the cultures were washed several times in MEM supplemented with 20 mM glucose and returned to the incubator. Neuronal cell injury was qualitatively assessed by morphological examination by using bright-field microscopy and quantitatively assessed by propidium iodide fluorescence (55) and comparing it to the near-complete neuronal death without glial death induced by exposing sister cultures to 300 μM NMDA for 24 h.
Electrophysiological Patch-Clamp Recordings of NMDA Receptor Currents.
A glass-bottom 35-mm culture was placed on the stage of an inverted microscope (Nikon), and whole-cell membrane currents were recorded by using an EPC-9 amplifier (List-Electronics, Darmstadt, Germany) and recording electrodes (5–8 MΩ, fire-polished) pulled from Corning Kovar Sealing no. 7052 glass pipettes (PG52151–4, WPI) with a Flaming–Brown instrument (P-80/PC, Sutter Instruments, Novato, CA). The external solution contained (in mM): 120 NaCl, 3 KCl, 2 CaCl2, 10 Hepes, 10 glucose, and 0.5 μM tetrodotoxin. The electrode internal solution contained (in mM): 120 KCl, 2 Na2-ATP, 0.5 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate or -tetraacetic acid, and 10 Hepes. Recordings were performed at room temperature (21–22°C) and at pH 7.3, under continuous bath perfusion at ≈0.2 ml/min.
The offset potential of the recording pipette was routinely corrected to 0 mV after immersing the tip into the bath. This potential was also checked at the end of experiments and corrected if necessary (usually 0–2 mV); recordings with a potential drift of more than 3 mV were discarded. For perforated patches, gramicidin D (Sigma) was dissolved in DMSO (10 mg/ml) and diluted to a final concentration of 50 μg/ml in the pipette solution; brief voltage steps of −10 mV were applied to monitor changes in input resistance and capacitance for 15–20 min (56, 57). Series resistance compensation was routinely applied during recordings. NMDA current was triggered at a holding potential of −70 mV by 100 μM NMDA plus 0.1 μM glycine delivered by the DAD-12 drug perfusion system (Adams–List, Westbury, NY). Current signals were digitally sampled at 100 μs (10 kHz) and filtered by a 3 kHz 3-pole Bessel filter. Current and voltage traces were displayed and stored on a computer by using a data acquisition/analysis package (pulse, HEKA Electronik, Lambrecht/Pfalz, Germany).
[Na+]i Measurement.
To monitor intracellular Na+ concentrations
([Na+]i), cultures were
loaded with sodium-binding benzofuran isophtalate (SBFI, Molecular
Probes) by incubation with the acetoxymethyl ester at the concentration
of 20 μM for 90 min in a Hepes-buffered solution, the composition of
which was (in mM): 120 NaCl/5.4 KCl/0.8
MgCl2/20 Hepes/15 glucose/1.8
CaCl2/10 NaOH, pH 7.4, at room temperature in
the presence of Pluronic F-127 (0.1%). All experiments were performed
on the stage of an inverted microscope equipped with a 75-W Xenon lamp.
Light was passed through 340- and 380-nm excitation filters mounted on
a filter wheel with a computer controlled filter driver. The light was
then reflected off a dichroic mirror (450 nm) and passed through a
510-nm emission filter. Images were acquired with a cooled
charge-coupled device camera (MicroMAX, Princeton Instruments, Trenton,
NJ) and digitized by using metafluor 4.1 software
(Universal Imaging, Media, PA). Background fluorescence was subtracted
from both (340- and 380-nm) images at the beginning of each experiment.
To calculate [Na+]i, we
performed an in situ calibration according to the protocol
described by Rose and Ransom (58). Cultures were exposed to increasing
concentrations of extracellular Na+ (0–50 mM) in
the presence of gramicidin (3 μM), monensin (10 μM), and ouabain
(0.1 mM) to equilibrate Na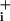 and Na
and Na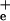 .
.
Results
Brief Zinc Exposure Increased Src Kinase Activity and Stimulated a SFK-Dependent Tyrosine Phosphorylation of NMDA Receptor Subunits NR2A/2B.
Near-pure neuronal cultures were exposed to 50 μM zinc for 5 min, and protein extracts were immediately isolated. No increase in the tyrosine phosphorylation of Fyn kinase (90 ± 19% of control levels; P = 0.636 by using Student t test; Fig. 1A) was produced by this zinc exposure. We next examined the tyrosine phosphorylation of Src by using phosphorylation site-specific Src antibodies targeted at the main regulatory or catalytic sites: tyrosine 418 in human/423 in mouse (the catalytic site), tyrosine 529 in human/534 in mouse (a negative regulatory site), and tyrosine 215 in human/220 in mouse (located in the SH2 protein interaction domain). (The mouse site designation will be used hereafter.) Basal levels of phosphorylation on the different tyrosine sites in sham-treated cells varied, with typically the highest levels of phosphorylation on tyrosine 534, and lower levels on tyrosine 220 or 423 (Fig. 1B). After 5 min of exposure to 50 μM zinc, phosphorylation of tyrosine sites 423 or 534 was unchanged (85 ± 11 and 115 ± 11% of control levels, respectively; P compared with controls was 0.442 in each case, Mann–Whitney Rank Sum Test), but phosphorylation of tyrosine 220 was increased (245 ± 72%; P < 0.001, Mann–Whitney Rank Sum Test). Phosphorylation of tyrosine 220 was zinc concentration-dependent, with enhancement detectable after exposure to only 5 μM zinc (Fig. 1D).
Figure 1.

Brief zinc exposure enhances the phosphorylation of Src kinase. Near pure-neuronal cortical cultures were exposed for 5 min to sham wash (control, Con) or 50 μM zinc (Zn2+) for 5 min in the absence or presence of a 30-min pretreatment with 10 μM PP2, and then protein was immediately extracted. (A) Fyn (Fyn-IP/pY) was immunoprecipitated, and the tyrosine phosphorylation of these kinases was determined by immunoblotting with antiphosphotyrosine antibody (4G10). Blots were stripped and reprobed with anti-Fyn antibody. (B) The tyrosine phosphorylation of Src was examined by Western blot analysis by using phosphorylation site-specific antibodies for Src phosphorylated on tyrosine 220 (pY220), tyrosine 423 (pY423), and tyrosine 534 (pY534). Blots were stripped and reprobed with anti-Src antibody. (C) PLCγ phosphorylation was examined by using a phospho-specific antibody directed against the active (phosphorylated) form of this kinase (phosphotyrosine 783). (D) Protein extracts from cortical neuronal cultures exposed to increasing concentrations of Zn2+ (μM) for 5 min were analyzed by Western blot analysis by using the anti-Src pY220 antibody. All experiments were performed at least three times with consistent results.
The SH2 domain of Src can bind to phosphorylated tyrosine residues including its own negative regulatory C-terminal tyrosine site, which results in an inactive Src kinase (46). Phosphorylation of tyrosine 220 indicates a displacement of this negative binding and activation of Src kinase (59), as well as availability of the SH2 domain to act as an adapter protein capable of recruiting or activating additional signaling kinases (60, 61). To confirm zinc enhancement of Src activity, we examined the downstream substrate, PLCγ (46). Zinc exposure enhanced the phosphorylation of PLCγ; this phosphorylation was blocked by the specific SFK inhibitor PP2 (Fig. 1C).
To explore the potential effects of zinc-induced Src activation on NMDA receptor phosphorylation and function, we performed a series of experiments in the mixed neuronal and glial cortical cultures that we have used in previous studies of NMDA receptor function and toxicity (62). Besides permitting more direct comparison to these previous studies, we considered the addition of glial cell influences an advantage in moving toward an understanding of intact nervous system behavior.
Mixed cortical cultures were exposed to 50 μM zinc for 5 min; after protein extraction, NR2A and NR2B were immunoprecipitated by using subunit-specific antibodies, and the immunoprecipitates were analyzed by immunoblotting with biotinylated-antiphosphotyrosine antibody (4G10). Basal levels of tyrosine-phosphorylated NR2A (Fig. 2A) and tyrosine-phosphorylated NR2B (Fig. 2B) were detected in sham-treated control cultures. After zinc exposure, the tyrosine phosphorylation of NR2A was increased 408 ± 93% (P = 0.005), and that of NR2B was increased 197 ± 32% (P = 0.013) relative to control cultures by using one-way Anova followed by Student–Neuman–Keuls' multiple comparison test (Fig. 2). The increase in tyrosine phosphorylation of both subunits was reduced by 1-h prior exposure to PP2.
Figure 2.

Brief zinc exposure stimulates SFK-dependent tyrosine phosphorylation of NMDA receptor subunits NR2A/2B. Mixed neuronal/glial cortical cell cultures were exposed to 50 μM zinc for 5 min after pretreatment for 1 h in the absence or presence of the SFK inhibitor PP2 (1 μM). Homogenates from sham-washed controls (Con), zinc-treated (Zn2+), or zinc treatment in the presence of PP2 (+PP2) were extracted and immunoprecipitated with antibodies specific for NR2A (A, NR2A-IP/pY) or NR2B (B, NR2B-IP/pY) and analyzed by immunoblotting with antiphosphotyrosine antibody. The blots were stripped and reprobed with anti-NR2A/2B-specific antibodies (anti-NR2A/NR2B). All experiments were performed at least three times with consistent results.
Zinc Exposure Increased NMDA Receptor Activity.
After obtaining stable whole-cell recordings, 50 μM zinc was added to the bathing medium for 5 min and then washed out. NMDA-induced current was acutely reduced by zinc application, consistent with previous reports (10, 11). However, after washing out zinc, NMDA-induced current recovered gradually toward control current levels (Fig. 3A). Because intracellular signaling components might be dialyzed away by exchange with pipette contents in conventional whole-cell recordings, we then repeated the experiment by using gramicidin perforated patch recordings and observed an enhancement of NMDA current after zinc exposure (Fig. 3B). This up-regulation typically lasted 5–8 min before it faded away (Fig. 3D). Thirty minutes of pretreatment with 10 μM PP2 blocked zinc upmodulation of NMDA current (Figs. 3 C and E).
Figure 3.

Effects of Zn2+ on NMDA current in whole-cell and perforated patch recordings. (A) Whole-cell recording showed that NMDA current (100 μM) was reversibly blocked by 50 μM Zn2+ (5-min bath application). (B) Similar acute Zn2+ block of NMDA current was observed in perforated patch recordings, but after washing out Zn2+, the NMDA current was enhanced compared with baseline. (C) Same as B, but in the presence of 10 μM PP2 in the bath (added 30 min before Zn2+ and maintained throughout the Zn2+ exposure and washout). Ten micromolars PP2 was used to ensure adequate cellular penetration with this shortened exposure time (52). (D) Time course of Zn2+-induced changes in NMDA current in perforated patch recordings after application of 50 μM Zn2+. n = 10 cells; * indicates P < 0.05 compared with baseline by t test. (E) Same as D, but in the presence of 10 μM PP2 in the bath. n = 5 cells.
Zinc Exposure Enhanced NMDA Toxicity.
Exposure of mixed neuronal/glial cortical cultures to 25 μM NMDA for 5 min resulted in an intermediate level of neuronal death (≈20% of the population) over the next 24 h, as assessed by failure to exclude propidium iodide (Fig. 4A). As expected, concurrent addition of 50 μM zinc blocked this NMDA-induced death (63). In contrast, exposure of cells to 25 μM NMDA 4 min after a 5-min pretreatment with 50 μM zinc resulted in increased neuronal death; this zinc enhancement of NMDA neurotoxicity was blocked by a pretreatment with PP2 (Fig. 4 A and B).
Figure 4.

Pre-zinc exposure enhances NMDA toxicity. (A) Photomicrograph of mixed neuronal/glial cortical cell cultures. Left, bright field; Right, matched propidium iodide fluorescence. Cells exposed to sham wash (Con) or 25 μM NMDA for 5 min, the latter preceded 4 min earlier with 5 min exposure to wash (NMDA), or 50 μM zinc (Zn2+ then NMDA, with or without 30 min pretreatment with 10 μM PP2). (B) Cultures were also treated as above and, in addition, cells were coexposed with 50 μM zinc and 25 μM NMDA. Neuronal death was quantitatively assessed 24 h after treatment by measurement of propidium iodide fluorescence (n = 5 separate experiments). *, P < 0.05 signifies difference from NMDA exposure alone. **, P < 0.05 signifies difference associated with PP2 addition to NMDA toxicity with a zinc pretreatment. (One-way ANOVA followed by Bonferroni t test).
Zinc Induced [Na+]i Increase in Cortical Neurons.
In considering possible mechanisms by which zinc exposure might up-regulate neuronal Src and NMDA receptors, our attention was drawn to earlier studies demonstrating a powerful ability of low micromolar zinc to inhibit the plasma membrane Na+/K+-ATPase (64–66) and to recent studies demonstrating the involvement of Src in the signal-transducing function of the Na+/K+-ATPase (67, 68). Furthermore, in another recent study, it was found that increasing [Na+]i up-regulated NMDA receptor activity, and that this up-regulation was enhanced by Src kinase (69).
Consistent with the hypothesis that zinc might potentiate NMDA receptors via inhibition of the Na+/K+ ATPase, leading to increased [Na+]i and enhanced Src kinase activity, addition of 50 μM Zn2+ to the bathing medium for 5 min elicited a progressive increase in [Na+]i in cortical neurons loaded with SBFI (Fig. 5). The increase in Nai was comparable to that produced by 500 μM ouabain; after washout of zinc, [Na+]i continued to rise, typically exceeding 20 mM (Fig. 5 B and C and data not shown).
Figure 5.

Brief Zn2+ exposure induces an increase in neuronal [Na+]i. (A) In situ calibration of SBFI fluorescence ratio signal (340/380 nm) (see Methods). (B) Pseudocolor images of cortical neurons loaded with SBFI before and at the end of 5-min exposure to 50 μM Zn2+. (C) Time course of change in [Na+]i induced by exposure to Zn2+, in a single experiment (n = 41 neurons, SEM depicted), representative of 4. (D) [Na+]i before and at the end of 5-min exposure to 50 μM Zn2+, in 143 neurons pooled from 4 experiments (* indicates difference at P < 0.05 by t test).
Discussion
We report here that brief exposure to 5–50 μM extracellular zinc, comparable to the concentrations estimated to be released normally into synaptic clefts (4), selectively enhanced the phosphorylation of neuronal Src at tyrosine 220 in the SH2 domain, without affecting tyrosine phosphorylation of Fyn. This phosphorylation was accompanied by increases in Src activity, NMDA receptor phosphorylation, and NMDA receptor function (current and excitotoxicity). Drawing on key earlier studies as well as present measurements of [Na+]i in neurons exposed to zinc, we propose that the ability of zinc to induce this up-regulation of Src activity and NMDA receptor function is mediated by inhibition of plasma membrane Na+/K+ ATPase and elevated [Na+]i. Although low concentrations of zinc can potentiate current mediated by certain homomeric NMDA receptor subunit 1 (NR1) splice variants, zinc potentiation of more physiological heteromeric NR1/NR2 receptors was not previously observed (70, 71).
No change in the phosphorylation of the Src catalytic site tyrosine 423 or the inhibitory site tyrosine 534 was observed after zinc exposure, but this does not exclude enhancement of Src kinase activity upon phosphorylation of tyrosine 220 (59). Indeed, zinc exposure resulted in an SFK-mediated increase in the phosphorylation of the downstream substrate, PLCγ. Furthermore, after phosphorylation of tyrosine 220, Src may act in a kinase-independent fashion (60, 61), perhaps serving as an adapter molecule that recruits or activates other tyrosine kinases (72).
Although further studies will be needed to establish the precise mechanisms by which zinc exposure leads to phosphorylation of Src tyrosine 220 and consequent kinase activation, it is attractive to postulate that this activation is mediated by zinc's known ability to inhibit the Na+/K+ ATPase at micromolar concentrations (64–66), because the pharmacological inhibitor, ouabain, activates Src in cardiac myocytes (67, 68). We confirmed here that zinc exposure leads quickly to a progressive increase in neuronal [Na+]i, consistent with Na+/K+ ATPase inhibition. It has been suggested that the ability of Na+/K+ ATPase inhibition to activate Src may not be mediated by secondary changes in [Na+]i, but rather by altered protein–protein interactions (67, 68).
Whatever the responsible underlying mechanism, once neuronal Src has been activated, a likely consequence is NMDA receptor tyrosine phosphorylation and enhanced activity (73). The idea of Src mediation fits with the observation that zinc-induced NMDA receptor enhancement was observed in perforated patch but not conventional whole cell recordings. Similar washout effects have been observed in studies of other kinase-mediated increases in channel currents (74, 75). Linkage between Src kinase and NMDA receptor up-regulation has also been demonstrated in CA1 hippocampal neurons, subsequent to activation of the focal adhesion kinase CAKβ/Pyk2 (76). We considered the possibility that Pyk2 activation might be involved in zinc-induced Src activation, but we did not detect an increase in phosphorylation of Pyk2 autocatalytic site tyrosine 402 after zinc exposure (unpublished data). Although present data specifically implicate Src kinase in the up-regulation of NMDA receptor activity, we cannot exclude contributions from other SFK members expressed in the central nervous system: Lyn, Lck, and Yes (73).
In addition to Src-mediated up-regulation of NMDA receptors, the increased [Na+]i resulting from zinc inhibition of the Na+/K+ ATPase may itself also up-regulate NMDA receptors. Yu and Salter (69) found that raising [Na+]i by 30 mM, comparable to the increase observed here after brief exposure to zinc, up-regulated NMDA receptor activity in mammalian neurons. Considering this finding, we suspect that the zinc-induced up-regulation of NMDA receptor current measured here by using perforated patch recordings may be underestimated, as diffusion of intracellular Na+ through gramicidin pores into the patch pipette likely artifactually lowered [Na+]i.
Further study will be necessary to delineate the implications of zinc-induced Src activation and NMDA receptor up-regulation for brain function in normal and abnormal conditions. SFK-induced NMDA receptor up-regulation has been implicated in the induction of long-term potentiation (LTP) after tetanic stimulation of hippocampal Schaffer collateral–CA1 synapses (77, 78); and depletion of endogenous synaptic zinc impaired induction of LTP at mossy fiber-CA3 synapses (79, 80), although contrary findings have been reported (4, 81). In addition, ischemia increases NMDA receptor tyrosine phosphorylation and binding to Src SH2 domains in rat brains (82), and Src-deficient mice exhibit resistance to brain damage after focal ischemia (83). It is attractive to consider that the ability of zinc to enhance NMDA receptor-mediated excitotoxicity demonstrated here could be pathologically synergistic with its native toxicity. Under conditions where excessive amounts of synaptic zinc are released to the extracellular space, some cells may be destroyed by zinc toxicity (23–25), and others might succumb later to enhanced NMDA receptor-mediated calcium overload. However, one could speculatively raise the alternative possibility that under some conditions—perhaps after mild ischemic insults—zinc-induced NMDA receptor up-regulation might play a neuroprotective role by modestly elevating neuronal calcium levels toward a “setpoint” inhibiting apoptosis (84), a fate triggered by exposure to lower levels of toxic zinc exposure.
In summary, we propose that zinc, by inhibiting Na+/K+-ATPase, causes a coincident increase in [Na+]i and activation of Src, resulting in the potentiation of NMDA receptor activity. Thus synaptically released zinc may regulate NMDA receptor activity in a biphasic fashion: acute block mediated by direct interaction with the receptor, followed by a lasting up-regulation of NMDA receptor activity and cytotoxicity, mediated by a signaling cascade involving the Na+/K+ ATPase and Src (Fig. 6).
Figure 6.

Diagram illustrating the proposed pathway depicting the possible biochemical coupling between zinc-mediated inhibition of Na+/K+-ATPase and potentiation of NMDA receptor activity. See text for details [adapted in part from previous studies (67–69)].
Acknowledgments
We thank Aili Cai, Yin Lin, and Ce Wang for expert technical assistance. This work was supported by grants from the Christopher Reeve Paralysis Foundation and National Institute of Neurological Disorders and Stroke (NS 30337) to D.W.C.
Abbreviations
- NMDA
N-methyl-d-aspartate
- NR2
type 2 NMDA receptor
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- SFK
Src family kinase
- SBFI
sodium-binding benzofuran isophtalate
Footnotes
This paper was presented at the Inaugural Arthur M. Sackler Colloquium of the National Academy of Sciences, “Neural Signaling,” held February 15–17, 2001, at the National Academy of Sciences in Washington, DC.
References
- 1.Vallee B L, Falchuck K H. Physiol Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Clausell J, Danscher G. Brain Res. 1985;337:91–98. doi: 10.1016/0006-8993(85)91612-9. [DOI] [PubMed] [Google Scholar]
- 3.Slomianka L, Danscher G, Frederickson C J. Neuroscience. 1990;38:843–854. doi: 10.1016/0306-4522(90)90076-g. [DOI] [PubMed] [Google Scholar]
- 4.Vogt K, Mellor J, Tong G, Nicoll R. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 5.Assaf S Y, Chung S H. Nature (London) 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 6.Howell G A, Welch M G, Frederickson C J. Nature (London) 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 7.Smart T G, Xie X, Krishek B J. Prog Neurobiol. 1994;42:393–441. doi: 10.1016/0301-0082(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 8.Harrison N L, Gibbons S J. Neuropharmacology. 1994;33:935–952. doi: 10.1016/0028-3908(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 9.Frederickson C J. Int Rev Neurobiol. 1989;30:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- 10.Peters S, Koh J-Y, Choi D W. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- 11.Westbrook G L, Mayer M L. Nature (London) 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 12.Mayer M L, Vyklicky L, Westbrook G L. J Physiol. 1989;415:329–350. doi: 10.1113/jphysiol.1989.sp017724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christine C W, Choi D W. J Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Legendre P, Westbrook G L. J Physiol. 1990;429:429–449. doi: 10.1113/jphysiol.1990.sp018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams K. Neurosci Lett. 1996;215:9–12. doi: 10.1016/s0304-3940(96)12924-4. [DOI] [PubMed] [Google Scholar]
- 16.Chen N, Moshaver A, Raymond L A. Mol Pharmacol. 1997;51:1015–1023. doi: 10.1124/mol.51.6.1015. [DOI] [PubMed] [Google Scholar]
- 17.Paoletti P, Ascher P, Neyton J. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traynelis S F, Burgess M F, Zheng F, Lyuboslavsky P, Powers J L. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi Y B, Lipton S A. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- 20.Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, Neyton J. Neuron. 2000;25:683–694. doi: 10.1016/s0896-6273(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 21.Choi D W, Yokoyama M, Koh J. Neuroscience. 1988;24:67–79. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- 22.Dineley K E, Scanlon J M, Kress G J, Stout A K, Reynolds I J. Neurobiol Dis. 2000;7:310–320. doi: 10.1006/nbdi.2000.0303. [DOI] [PubMed] [Google Scholar]
- 23.Sloviter R S. Brain Res. 1985;330:150–153. doi: 10.1016/0006-8993(85)90017-4. [DOI] [PubMed] [Google Scholar]
- 24.Tonder N, Johanson F F, Fredrickson C J, Zimmer J, Diemer N H. Neurosci Lett. 1990;109:247–252. doi: 10.1016/0304-3940(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 25.Koh J-Y, Suh S W, Gwag B J, He Y Y, Hsu C Y, Choi D W. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 26.Suh S W, Chen J W, Motamedi M, Bell B, Listiak K, Pons N F, Danscher G, Frederickson C J. Brain Res. 2000;852:268–273. doi: 10.1016/s0006-8993(99)02095-8. [DOI] [PubMed] [Google Scholar]
- 27.Sensi S L, Canzoniero L M T, Yu S P, Ying H S, Koh J-Y, Kerchner G A, Choi D W. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sensi S L, Yin H Z, Carriedo S G, Rao S S, Weiss J H. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canzoniero L M T, Turetsky D M, Choi D W. J Neurosci. 1999;19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aizenman E, Stout A K, Hartnett K A, Dineley K E, McLaughlin B, Reynolds I J. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 31.Frederickson C J, Hernandez M D, McGinty J F. Brain Res. 1989;480:317–321. doi: 10.1016/0006-8993(89)90199-6. [DOI] [PubMed] [Google Scholar]
- 32.Weiss J H, Koh J-Y, Christine CW, Choi D W. Nature (London) 1989;338:212. doi: 10.1038/338212b0. [DOI] [PubMed] [Google Scholar]
- 33.Murakami K, Whiteley M K, Routtenberg A. J Biol Chem. 1987;262:13902–13906. [PubMed] [Google Scholar]
- 34.Csermely P, Szamel M, Resch K, Somogyi J. J Biol Chem. 1988;263:6487–6490. [PubMed] [Google Scholar]
- 35.Noh K-M, Kim Y H, Koh J-Y. J Neurochem. 1999;72:1609–1616. doi: 10.1046/j.1471-4159.1999.721609.x. [DOI] [PubMed] [Google Scholar]
- 36.Weinberger R P, Rostas J A. J Neurochem. 1991;57:605–614. doi: 10.1111/j.1471-4159.1991.tb03791.x. [DOI] [PubMed] [Google Scholar]
- 37.Lengyel I, Fieuw-Makaroff S, Hall A L, Sim A T R, Rostas J A P, Dunkley P R. J Neurochem. 2000;75:594–605. doi: 10.1046/j.1471-4159.2000.0750594.x. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Jung Y, Kim D, Koh H, Chung J. J Biol Chem. 2000;275:25979–25984. doi: 10.1074/jbc.M001975200. [DOI] [PubMed] [Google Scholar]
- 39.Hansson A. Arch Biochem Biophys. 1996;328:233–238. doi: 10.1006/abbi.1996.0168. [DOI] [PubMed] [Google Scholar]
- 40.Samet J M, Graves L M, Quay J, Dailey L A, Devlin R B, Ghio A J, Wu W, Bromberg P A, Reed W. Am J Physiol. 1998;275:L551–L558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- 41.Park J A, Koh J-Y. J Neurochem. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- 42.Fidnick D, Presek P. FEBS Lett. 1988;235:51–56. doi: 10.1016/0014-5793(88)81232-8. [DOI] [PubMed] [Google Scholar]
- 43.Vener A V, Loeb J. FEBS Lett. 1992;303:261–264. doi: 10.1016/0014-5793(92)80534-n. [DOI] [PubMed] [Google Scholar]
- 44.Brugge J S, Erikson R L. Nature (London) 1977;316:346–348. doi: 10.1038/269346a0. [DOI] [PubMed] [Google Scholar]
- 45.Purcchio A F, Erikson E, Brugge J S, Erikson R L. Proc Natl Acad Sci USA. 1978;75:1567–1571. doi: 10.1073/pnas.75.3.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown M T, Cooper J A. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- 47.Thomas S M, Brugge J S. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y T, Salter M W. Nature (London) 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 49.Kohr G, Seeburg P H. J Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen S J, Leonard J P. J Neurochem. 1996;67:194–200. doi: 10.1046/j.1471-4159.1996.67010194.x. [DOI] [PubMed] [Google Scholar]
- 51.Rose K, Goldberg M P, Choi D W. In: In Vitro Biological Methods. Methods in Toxicology. Tyson C A, Frazier J M, editors. San Diego: Academic; 1993. pp. 46–60. [Google Scholar]
- 52.Hanke J H, Gardner J P, Dow R L, Changelian P S, Brissette W H, Weringer E J, Pollok B A, Connelly P A. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 53.Behrens M M, Strasser U, Koh J Y, Gwag B J, Choi D W. Neuroscience. 1999;94:917–927. doi: 10.1016/s0306-4522(99)00212-2. [DOI] [PubMed] [Google Scholar]
- 54.Lin S Y, Wu K, Len G W, Xu J L, Levine E S, Suen P C, Mount H T, Black I R. Brain Res Mol Brain Res. 1999;70:18–25. doi: 10.1016/s0169-328x(99)00122-9. [DOI] [PubMed] [Google Scholar]
- 55.Trost L C, Lemasters J J. Anal Biochem. 1994;220:149–153. doi: 10.1006/abio.1994.1311. [DOI] [PubMed] [Google Scholar]
- 56.Kyrozis A, Reichling D B. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- 57.Akaike N. Prog Biophys Mol Biol. 1996;65:251–264. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- 58.Rose C R, Ransom B R. J Physiol. 1996;491:291–305. doi: 10.1113/jphysiol.1996.sp021216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stover D R, Furet P, Lydon N B. J Biol Chem. 1996;271:12481–12487. doi: 10.1074/jbc.271.21.12481. [DOI] [PubMed] [Google Scholar]
- 60.Kaplan K B, Swedlow J R, Morgan D O, Varmus H E. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 61.Schlaepfer D D, Broome M A, Hunter T. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi D W. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 63.Koh J, Choi D W. J Neurosci. 1988;8:2164–2171. doi: 10.1523/JNEUROSCI.08-06-02164.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donaldson J, St-Pierre T, Minnich J, Barbeau A. Can J Biochem. 1971;49:1217–1224. doi: 10.1139/o71-175. [DOI] [PubMed] [Google Scholar]
- 65.Hexum T D. Biochem Pharmacol. 1974;23:3441–3447. doi: 10.1016/0006-2952(74)90347-5. [DOI] [PubMed] [Google Scholar]
- 66.Gettelfinger D M, Siegel G J. J Neurochem. 1978;31:1231–1237. doi: 10.1111/j.1471-4159.1978.tb06247.x. [DOI] [PubMed] [Google Scholar]
- 67.Haas M, Askari A, Xie Z. J Biol Chem. 2000;275:27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 68.Liu J, Tian J, Haas M, Shapiro J I, Askari A, Xie Z. J Biol Chem. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 69.Yu X M, Salter M W. Nature (London) 1998;396:469–474. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]
- 70.Hollmann M, Boulter J, Maron C, Beasley L, Sullivan J, Pecht G, Heinemann S. Neuron. 1993;10:943–954. doi: 10.1016/0896-6273(93)90209-a. [DOI] [PubMed] [Google Scholar]
- 71.Zheng X, Zhang L, Durand G M, Bennet M V L, Zukin R S. Neuron. 1994;12:811–818. doi: 10.1016/0896-6273(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 72.Schwartzberg P L. Oncogene. 1998;17:1463–1468. doi: 10.1038/sj.onc.1202176. [DOI] [PubMed] [Google Scholar]
- 73.Salter M W. Biochem Pharmacol. 1998;56:789–798. doi: 10.1016/s0006-2952(98)00124-5. [DOI] [PubMed] [Google Scholar]
- 74.Carrol R C, Nicoll R, Malenka R C. J Neurophysiol. 1998;80:2797–2800. doi: 10.1152/jn.1998.80.5.2797. [DOI] [PubMed] [Google Scholar]
- 75.Kurata S, Ishikawa K, Iijima T. Pharmacology. 1999;59:21–33. doi: 10.1159/000028302. [DOI] [PubMed] [Google Scholar]
- 76.Huang Y-Q, Lu W-Y, Ali D W, Pelkey K A, Pitcher G M, Lu Y M, Aoto H, Roder J C, Salter M W, MacDonald J F. Neuron. 2001;29:484–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- 77.Lu Y M, Roder J C, Davidow J, Salter M W. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- 78.Huang C-C, Hsu K-S. J Physiol. 1999;520:783–796. doi: 10.1111/j.1469-7793.1999.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Budde T, Minta A, White J A, Kay A R. Neuroscience. 1997;79:347–358. doi: 10.1016/s0306-4522(96)00695-1. [DOI] [PubMed] [Google Scholar]
- 80.Lu Y-M, Taverna F A, Tu R, Ackerley C A, Wang Y-T, Roder J. Synapse. 2000;38:187–197. doi: 10.1002/1098-2396(200011)38:2<187::AID-SYN10>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 81.Xie X, Smart T G. Pflügers Arch. 1994;427:481–486. doi: 10.1007/BF00374264. [DOI] [PubMed] [Google Scholar]
- 82.Takagi N, Cheung H H, Bissoon N, Teves L, Wallace M C, Gurd J W. J Cereb Blood Flow Metab. 1999;19:880–888. doi: 10.1097/00004647-199908000-00007. [DOI] [PubMed] [Google Scholar]
- 83.Paul R, Zhang Z G, Eliceiri B P, Jiang Q, Boccia A D, Zhang R L, Chopp M, Cheresh D A. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- 84.Koike T, Martin D P, Johnson E M. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]