

Abstract
Objective
HCV infection is a leading cause of chronic liver disease and a major indication for liver transplantation. Although direct-acting antivirals (DAAs) have much improved the treatment of chronic HCV infection, alternative strategies are needed for patients with treatment failure. As an essential HCV entry factor, the tight junction protein claudin-1 (CLDN1) is a promising antiviral target. However, genotype-dependent escape via CLDN6 and CLDN9 has been described in some cell lines as a possible limitation facing CLDN1-targeted therapies. Here, we evaluated the clinical potential of therapeutic strategies targeting CLDN1.
Design
We generated a humanised anti-CLDN1 monoclonal antibody (mAb) (H3L3) suitable for clinical development and characterised its anti-HCV activity using cell culture models, a large panel of primary human hepatocytes (PHH) from 12 different donors, and human liver chimeric mice.
Results
H3L3 pan-genotypically inhibited HCV pseudoparticle entry into PHH, irrespective of donor. Escape was likely precluded by low surface expression of CLDN6 and CLDN9 on PHH. Co-treatment of a panel of PHH with a CLDN6-specific mAb did not enhance the antiviral effect of H3L3, confirming that CLDN6 does not function as an entry factor in PHH from multiple donors. H3L3 also inhibited DAA-resistant strains of HCV and synergised with current DAAs. Finally, H3L3 cured persistent HCV infection in human-liver chimeric uPA-SCID mice in monotherapy.
Conclusions
Overall, these findings underscore the clinical potential of CLDN1-targeted therapies and describe the functional characterisation of a humanised anti-CLDN1 antibody suitable for further clinical development to complement existing therapeutic strategies for HCV.
Keywords: HEPATITIS C, ANTIVIRAL THERAPY, TIGHT JUNCTION
Significance of this study.
What is already known on this subject?
The tight junction protein claudin-1 (CLDN1) is an essential HCV entry factor.
Rodent anti-CLDN1 antibodies inhibit HCV infection by disrupting CD81-CLDN1 co-receptor complex formation.
A rodent anti-CLDN1 monoclonal antibody prevented and cured chronic HCV infection in human-liver chimeric mice.
Escape from CLDN1-targeted therapies via other CLDN subtypes has been described in some cell lines.
What are the new findings?
We generated a humanised anti-CLDN1 monoclonal antibody (H3L3) suitable for further clinical development.
H3L3 exhibited pan-genotypic activity against HCV entry into primary human hepatocytes (PHH), irrespective of donor.
There was absent escape in a panel of PHH from 12 donors, indicating that CLDN6 does not function as an entry factor in PHH.
Low expression of other CLDN subtypes on PHH likely precludes escape in the human liver and indicates that escape is likely not clinically relevant in the majority of patients.
How might it impact on clinical practice in the foreseeable future?
These findings enable further clinical development of a CLDN1-targeted therapy to complement existing therapeutic strategies for HCV.
Introduction
Chronic HCV infection is a leading cause of liver cirrhosis and hepatocellular carcinoma worldwide. Although the approval of direct-acting antivirals (DAAs) has revolutionised HCV treatment, treatment failure can result from the selection of DAA-resistant HCV variants.1 2 Occult HCV infection may also persist in patients who achieve sustained virological response.3 Furthermore, not all patient groups respond to therapy.4 5 For example, 10–20% of patients with decompensated cirrhosis do not respond to current DAAs, and treatment is associated with serious adverse effects (10–20%).6 Progressive liver disease associated with chronic HCV infection remains a major indication for liver transplantation. Graft reinfection is universal in patients with detectable HCV RNA at the time of transplantation. The ability of DAAs to prevent liver graft reinfection remains to be determined.7 8 In one study evaluating the efficacy of sofosbuvir and ribavirin during liver transplantation, 35% of patients experienced liver graft reinfection.9 Current DAAs act once infection is already established, whereas inhibitors targeting entry could inhibit de novo infection of the graft.8 Entry inhibitors could also protect HCV-negative transplant recipients from infection by HCV-positive donor organs10 and help to alleviate organ shortage.11
Host-targeting agents (HTAs) offer a promising complementary approach. HCV requires several host factors to establish infection, including cluster of differentiation 81 (CD81),12 scavenger receptor BI (SR-BI),13 CLDN1,14 epidermal growth factor receptor (EGFR),15 Niemann-Pick C1 like 1 (NPC1L1)16 and E-cadherin,17 which are attractive antiviral targets. Antibodies targeting CD81,18 SR-BI19 and CLDN120–23 pan-genotypically inhibit HCV infection in vitro and in vivo, and small molecules targeting EGFR15 and NPC1L116 have similar anti-HCV activities. Several HTAs, including erlotinib,15 ezetimibe16 and the antihistamine chlorcyclizine,24 are approved for other indications and could be repurposed as HCV antivirals. HTAs act synergistically with DAAs25 and prevent emergence of DAA-resistant variants,26 highlighting their potential for combination therapy. Furthermore, as the targets of HTAs are not encoded by highly mutable viral genomes, a higher barrier to resistance is expected.
In the case of CLDN1-targeting HTAs, previous studies in cell lines indicated the possibility of escape via other claudin subtypes, namely CLDN6 and CLDN9.27 28 Indeed, exogenous expression of CLDN9 in CLDN1-deficient Bel7402 hepatoma cells27 and of CLDN6 and CLDN9 in CLDN-null 293T cells27 28 enabled entry of some HCV genotypes.29 CLDN1-specific antibodies only partially inhibited infection by genotypes with broad CLDN tropism (ie, genotypes 1, 3, 4 and 6) in Huh7.5 cells.29 However, the clinical relevance of escape is still unknown and the ability of CLDN1-specific antibodies to block entry into human liver cells without escape remains unclear.30
We previously reported that rat anti-CLDN1 monoclonal antibodies (mAbs) have robust anti-HCV activities.20 31 These antibodies inhibit HCV entry by disrupting CD81-CLDN1 co-receptor complex formation.31 The lead rat anti-CLDN1 mAb (OM-7D3-B3) prevented de novo HCV infection and cleared chronic HCV infection in monotherapy without toxicity in human-liver chimeric mice.21 These promising findings warrant further clinical studies. However, humanisation of this antibody to reduce immunogenicity in humans is critical for any further clinical development.
In this context, we humanised the rat anti-CLDN1 antibody into the human IgG4 subtype and functionally characterised the humanised antibody using cell culture models, primary human hepatocytes (PHH), and human-liver chimeric mice. We found that the humanised anti-CLDN1 antibody inhibited entry of all HCV genotypes into PHH without escape, suggesting that escape is not relevant in the human liver, likely due to low expression of alternative CLDN subtypes. The humanised antibody retained its functional activity and its ability to cure chronically infected human-liver chimeric mice in monotherapy. It was also active against DAA-resistant HCV and synergised with current DAAs. Our findings highlight the clinical potential of CLDN1-targeted therapies and pave the way for clinical development of a humanised CLDN1 antibody as an alternative treatment strategy for HCV.
Experimental procedures
Sequencing of the variable region of rat anti-CLDN1 mAb OM-7D3-B3
Total RNA was isolated in pure RNase-free water from 107 hybridoma cells using the Qiagen RNeasy mini kit. Vh and Vl cDNAs were prepared using primers derived from the constant domain (CH1) of rat IgG2b and rat kappa light chain by reverse transcription. cDNAs were amplified using a pool of primers corresponding to the signal sequence region and subsequently cloned into pGem-T Easy vector (Promega). Clones were screened and sequenced. Complementarity-determining regions (CDRs) were identified with reference to other antibody sequences as described.32
Humanisation of rat anti-CLDN1 mAb OM-7D3-B3 and antibody production
Humanisation was performed by CDR grafting as described.33 34 Human germline VH and VL sequences most similar to the rat anti-CLDN1 mAb OM-7D3-B3 in the IMGT database were identified using the IGBLAST tool (http://www.ncbi.nlm.nih.gov/igblast/) (figure 1A). The CDRs of mAb OM-7D3-B3 were engrafted on framework regions of the identified VH and VL human germline sequence. Framework regions of OM-7D3-B3 were compared with rat germline sequences to identify variations that could contribute to antibody specificity. Three variants each of heavy chain (H1, H2, H3) and light chain (L1, L2, L3) were generated (figure 1B). These VH and VL variants were fused in-frame with constant domains of the human IgG4 heavy and light chains, respectively, and cloned into a mammalian expression vector. Nine full-length humanised antibodies were produced by co-transfecting plasmids containing the appropriate heavy and light chain variants into Chinese hamster ovary (CHO) cells. Supernatants were harvested and antibodies purified using the MAbTrap Kit (GE Healthcare). Antibodies were buffer-changed into phosphate-buffered saline (PBS) prior to use.
Figure 1.

Sequence alignment of the variable region of the heavy and the light chains of rat anti-tight junction protein claudin-1 (CLDN1) monoclonal antibody (mAb) OM-7D3-B3. (A) Protein sequences of rat OM-7D3-B3 heavy chain (top) and light chain (bottom) aligned with the nearest rat germline sequence. The most closely matched germline sequences had homology of 86.7% and 90.5% with heavy and light chain, respectively. (B) Sequence alignment of rat OM-7D3-B3 heavy (top) and light (bottom) chain with the nearest human germline sequences. Also shown are the humanised variants of heavy (H1, H2 and H3) and light (L1, L2 and L3) chains that were generated in the process of humanising rat OM-7D3-B3. The complementarity-determining regions (CDRs) are highlighted in yellow and the joining (J)-region is shown in pink. The framework region (FR), CDRs and the J-region are indicated.
Cells and viruses
Huh7.5.1,21 HepG235 and human embryonic kidney 293T36 cells were cultured as described. Production of HCVpp37 bearing envelope glycoproteins of strains H77, HCV-J, JFH1, J8, NIH S52, UKN3A1.28, UKN4.21.16, UKN5.14.4 and UKN6.5.340 has been described previously. Strain gt3SXB1 (genotype 3) was cloned from the serum of a patient chronically infected with HCV and followed at the Strasbourg University Hospitals. E1E2 sequences were amplified and inserted into the phCMV IRES vector using EcoRV restriction sites as described.36 Luciferase reporter HCVcc (Luc-Jc1) and DAA-resistant mutants (Luc-Jc1-A156S and Luc-Jc1-R155K) were produced as described.26 38
Primary human hepatocytes
PHH were isolated from liver tissue obtained from patients undergoing liver resection at the Strasbourg University Hospitals. PHH from 12 donors were isolated and cultured as described.31
Flow cytometry
To evaluate binding of the humanised antibodies to CLDN1 expressed on Huh7.5.1, HepG2 and 293T cells, 2×105 cells were incubated with 20 µg/mL mouse anti-CD81 mAb JS-81 (BD Biosciences), rat anti-CLDN1 mAb OM-7D3-B3, humanised anti-CLDN1 mAb or PBS for 1 hour at room temperature. Cells were washed and incubated with phycoerythrin (PE)-conjugated species-specific secondary antibodies at 4°C for 45 min to allow detection of binding. Cells were subsequently washed and fixed with 2% paraformaldehyde (PFA) prior to analysis by flow cytometry (BD LSRII, 293T cells; FACScan, Huh7.5.1 and HepG2 cells). Delta median fluorescence intensities (ΔMFI) were obtained after subtracting the background fluorescence with isotype control antibody. To evaluate the binding specificity, 293T cells were transfected with either empty vector or pcDNA3.1 encoding human CLDN1. After 48 hours, binding was assessed by flow cytometry (BD LSRII).
We evaluated binding of one humanised antibody clone (H3L3) to PHH, as well as expression of CLDN subtypes on the surface of PHH. Cryopreserved PHHs (from the same donors used in inhibition experiments) were incubated with CLDN-specific mAbs (CLDN1, rat OM-7D3-B3 or humanised H3L3; CLDN6, rat WU-9E1-G2; CLDN9, rat YD-4E9-A2) or isotype control at 20 μg/mL, as described.30 Cells were then stained with PE-conjugated secondary antibody. After washing, cells were fixed with 2% PFA and analysed by flow cytometry (FACScan). In all experiments, Huh7.5.1 cells and PHH were analysed in parallel.
Inhibition of HCV infection
Huh7.5.1 or PHH were preincubated with serially diluted antibodies (control mAb, rat OM-7D3-B3, humanised H3L3, anti-CLDN6 WU-9E1-G2 or combinations thereof) for 1 hour at 37°C and subsequently exposed to HCVcc or HCVpp, respectively. For HCVpp experiments, PHH from several different donors were evaluated. Following treatment, cells were incubated with HCVpp for 4 hours at 37°C. HCVpp entry/HCVcc infection was analysed by measuring intracellular luciferase activity after 72 hours (relative light units (RLU)). Inhibition was expressed as a percentage relative to cells treated with control antibody. The concentration of antibody required for 50% inhibition (IC50) and 90% inhibition (IC90) was calculated by non-linear regression analysis (constrained fit) using GraphPad Prism (V.6.0, GraphPad Software, USA).
Cell–cell transmission assay
The HCV cell–cell transmission assay has been described.39 Huh7.5.1 cells were electroporated with HCV Jc1 RNA (virus-producer cells) and co-cultured with uninfected Huh7.5 target cells stably expressing green fluorescent protein (GFP) in the presence of H3L3, OM-7D3-B3 or isotype control mAb (11 µg/mL). Cell-free transmission was blocked by a neutralising HCV anti-E2 mAb, AP33 (25 μg/mL) as described.15 After 24 hours, cells were fixed with 2% PFA and stained with an NS5A-specific antibody (Virostat, 0.1 μg/mL). De novo infection (GFP and NS5A double-positive cells) was assessed by flow cytometry (BD LSRII). Cell–cell transmission was presented as a percentage of GFP+ NS5A+ infected cells relative to Huh7.5-GFP+ target cells in the presence of HCV anti-E2 mAb AP33.
Assessment of antiviral synergy
Synergy was evaluated as described previously.25 Huh7.5.1 cells were pretreated with H3L3 in combination with sofosbuvir or daclatasvir for 1 hour at 37°C prior to addition of HCVcc (Luc-Jc1) for 4 hours at 37°C. Viral infection was assessed 72 hours later by luciferase activity. Synergy was evaluated using the Prichard and Shipman method.40 Inhibition >20% above the level expected due to additive effects indicates synergy.40
HCV infection and treatment of human-liver chimeric uPA-SCID mice
Experiments were performed according to local ethics committee approval (CREMEAS, project numbers 02014120416254981 (APAFlS#72.02) and 02014120511054408 (APAFIS#74.03)) at the Inserm U1110 animal facility. Severe combined immunodeficient mice homologous for urokinase-type plasminogen activator expression under control of the mouse albumin promoter (uPA-SCID) were engrafted with PHH as described.41 Human-liver chimeric mice were infected with HCVcc (Jc1; genotype 2a) by intraperitoneal injection as described.21 Three weeks after infection, mice were given intraperitoneal injections of 500 μg/mouse of H3L3 or isotype control mAb at weekly intervals for 4 weeks.21 Plasma HCV RNA, human albumin and IgG4 levels were monitored as described.21
Results
Humanisation of anti-CLDN1 antibody OM-7D3-B3
To further the clinical development of the rat anti-CLDN1 mAb OM-7D3-B3 mAb, we humanised it into the human IgG4 subtype. Sequencing of cDNA recovered from hybridoma cells indicated that rat OM-7D3-B3 VH and VL were derived from VH5 (IGHV5-34*01) and Vκ6 (IGKV6S11*01) gene families that underwent rearrangement with the JH2 (IGHJ2*01) and Jκ1 (IGKJ1*01) families of J-segment genes, respectively (figure 1A). To verify that VH and VL sequences were functional, we produced a recombinant full-length IgG2b (parent isotype) antibody in CHO cells. This antibody bound to CLDN1 (see online supplementary figure S1A) and inhibited HCVcc infection (see online supplementary figure S1B), confirming the functionality of the sequences.
gutjnl-2016-312577supp001.pdf (211.1KB, pdf)
We humanised the VH and VL chains by modifying residues in the framework region while keeping the CDRs intact. The most closely matching human Ig germline V sequences were IGHV3-21*01 for VH and IGKV3-15*01 for VL, with corresponding homology of 83.7% and 64.5%, respectively (figure 1A). Guided by these human germline sequences, we generated nine different antibody clones consisting of different combinations of heavy and light chains by CDR engraftment on a human IgG4 backbone (figure 1B), which we termed H1L1, H1L2, H1L3, H2L1, H2L2, H2L3, H3L1, H3L2 and H3L3.
Humanised anti-CLDN1 antibodies bind to CLDN1 and inhibit HCV infection
We tested whether the panel of humanised anti-CLDN1 antibodies bind to CLDN1. As shown in figure 2A, the humanised antibodies bound to CLDN1-expressing Huh7.5.1 and HepG2 cells but not to 293T cells that do not express CLDN1. Like the parental rat antibody, the humanised antibodies likely bind to CLDN1 and prevent formation of the CLDN1-CD81 co-receptor complex, as has been described previously for anti-CLDN1 mAbs.31 We selected clone H3L3, which most potently inhibited HCVpp entry into PHH (see online supplementary figure S2), for further analysis. To investigate the binding specificity, we transfected 293T cells with a plasmid encoding human CLDN1. H3L3 specifically bound to 293T cells overexpressing human CLDN1 but not to the control cells transfected with an empty vector (figure 2B). We next assessed the anti-HCV activity of the humanised antibodies. All nine humanised antibodies potently inhibited HCVcc infection of Huh7.5.1 cells at 25 µg/mL (figure 2D).
Figure 2.

Humanised anti-tight junction protein claudin-1 (CLDN1) monoclonal antibodies (mAbs) specifically bind to CLDN1 and inhibit HCV infection. (A) Flow cytometry analysis of the binding of humanised OM-7D3-B3 anti-CLDN1 mAbs (20 µg/mL) to cell lines. Humanised antibodies specifically bind to Huh7.5.1 and HepG2 cells expressing human CLDN1 but not to 293T cells lacking CLDN1. Binding is expressed as delta median fluorescence intensity (ΔMFI) from one experiment performed in duplicate. (B) H3L3 specifically binds to exogenous CLDN1 expressed on the surface of 293T cells. CLDN-null 293T cells were transfected with either empty vector or CLDN1 prior to staining with isotype control, rat OM-7D3-B3 or humanised H3L3 antibody (20 µg/mL). The ΔMFI from one experiment performed in duplicate is shown. (C) H3L3 binds to primary human hepatocytes (PHH) with similar affinity as the parental rat antibody. PHH were stained with isotype control, rat OM-7D3-B3 or humanised H3L3 antibody (20 µg/mL). The ΔMFI from one experiment performed in duplicate are shown. (D) All humanised anti-CLDN1 mAbs inhibit HCVcc infection. Huh 7.5.1 cells were incubated with different mAbs (25 µg/mL) at 37°C for 1 hour prior to infection with HCVcc. Infectivity was assessed after 72 hours by measuring luciferase activity and is expressed as log relative luciferase units (RLU). (E) H3L3 inhibits entry of HCVpp bearing envelope glycoproteins of strains H77 (genotype 1a) and HCV-J (genotype 1b). PHH were treated with humanised antibody (20 µg/mL) for 1 hour at 37°C prior to infection with HCVpp. Infectivity was measured by luciferase activity after 72 hours and is expressed as RLU. Graphs show results from one experiment performed in duplicate (A–C) or in triplicate (D and E).
We selected the clone H3L3 for detailed studies in PHH, a more physiological context. H3L3 retained the ability of the parental antibody to bind to PHH (figure 2C) and fully inhibit HCVpp genotype 1a and 1b entry into PHH at 20 µg/mL (figure 2E). None of the humanised antibodies exerted cytotoxic effects (see online supplementary figure S3). Taken together, these data indicate that the humanised anti-CLDN1 antibodies are functionally similar to the parental rat anti-CLDN1 mAb.
Humanised anti-CLDN1 H3L3 pan-genotypically and broadly inhibits HCVpp entry into PHH, irrespective of hepatocyte donor
We next evaluated whether the effect of H3L3 was pan-genotypic. PHH were treated with serially diluted H3L3 for 1 hour prior to incubation with HCVpp bearing glycoproteins of all major HCV genotypes. H3L3 pan-genotypically inhibited HCVpp entry into PHH (figure 3). To further assess the clinical relevance of the antibody and evaluate potential donor-to-donor variability, we tested PHH from a panel of 11 additional donors. We focused on one strain of genotype 1b and three strains of HCV genotype 3a as these genotypes have been described to escape CLDN1-targeted therapies in cell lines.29 H3L3 fully inhibited entry of all genotypes in all PHH we tested, irrespective of donor (figure 3, see online supplementary table S1).
Figure 3.

H3L3 pan-genotypically inhibits HCVpp entry into primary human hepatocytes (PHH) without escape. PHH from a panel of donors were treated with serial dilutions of H3L3 (0.001 μg/mL to 100 μg/mL) for 1 hour at 37°C prior to incubation with pseudoparticles bearing glycoproteins of all major genotypes of HCV. HCVpp entry was assessed by measuring luciferase activity after 72 hours. Entry is expressed as a percentage relative to entry into untreated cells. Graphs show one experiment performed in duplicate.
H3L3 inhibits HCVpp entry into PHH without escape via CLDN6 or CLDN9
Given that escape from CLDN1-directed therapies has been reported for certain HCV genotypes in CLDN6-expressing and/or CLDN9-expressing cell lines,27–29 we investigated the physiological relevance of escape in the context of the H3L3 antibody and PHH. We evaluated surface expression levels of CLDN6 and CLDN9 on PHH from four of the donors tested above. In contrast to robust CLDN6 expression in Huh7.5.1 cells, the PHH from these four donors (PHH 218, 235, 283 and S2310) and three additional donors (PHH 297, S1532 and S1533) expressed only very low surface levels of CLDN6, regardless of whether the cells were assessed immediately after isolation (fresh PHH) or following cryopreservation (frozen PHH) (figure 4A). Neither PHH nor Huh7.5.1 cells expressed detectable CLDN9 (figure 4A). Therefore, it is likely that low surface expression levels of CLDN6 in the human liver preclude escape from CLDN1 antibodies in vivo.
Figure 4.

Tight junction protein claudin-6 (CLDN6) is not a functional HCV entry factor in primary human hepatocytes (PHH). (A) PHH express only very low surface levels of CLDN6 and CLDN9. PHH from four donors tested in inhibition experiments and three additional donors were treated with isotype control or CLDN1-specific, CLDN6-specific or CLDN9-specific monoclonal antibodies (20 μg/mL). Labelling was done either immediately following the isolation of PHH (fresh PHH) or using cryopreserved cells (frozen PHH). Expression of CLDN1, CLDN6 and CLDN9 is shown as delta median fluorescence intensity (ΔMFI). (B) Treatment of PHH with a CLDN6-specific mAb (WU-9E1-G2) had no effect on HCVpp infection. PHH were treated with serially diluted WU-9E1-G2 or H3L3, or with serially diluted H3L3 in combination with 20 μg/mL WU-9E1-G2, for 1 hour prior to incubation with HCVpp genotype 1b (HCV-J) or genotype 3a (S52). HCVpp entry was assessed by measuring luciferase activity after 72 hours. (C) Treatment of Huh7.5.1 cells with H3L3 in combination with WU-9E1-G2 inhibited HCVpp entry, in contrast to treatment with H3L3 alone or WU-9E1-G2 alone. Huh7.5.1 cells were treated with serially diluted WU-9E1-G2 or H3L3, or with serially diluted H3L3 in combination with 20 μg/mL WU-9E1-G2, for 1 hour prior to incubation with HCVpp genotype 1b (HCV-J) or genotype 3a (S52). HCVpp entry was assessed by measuring luciferase activity after 72 hours. Graphs show one experiment performed in duplicate (A and B) or two experiments performed in triplicate (C).
We further evaluated escape using a CLDN6-specific mAb (WU-9E1-G2). PHH were treated with H3L3 alone or in combination with 20 μg/mL of WU-9E1-G2 prior to incubation with HCVpp genotype 1b (HCV-J) or 3a (NIH S52). If escape in PHH is indeed possible via CLDN6 (even at low expression levels), we expected that addition of a CLDN6-specific antibody would enhance the antiviral potency of H3L3. To address this question, we used a previously described antibody WU-9E1-G2 that specifically binds cell surface CLDN6 with high affinity on Huh7.5.1 cells.30 Combination of H3L3 with WU-9E1-G2 did not affect the inhibition profile for either genotype 1b or 3a in PHH from two different donors (figure 4B). In CLDN6-expressing Huh7.5.1 cells, however, addition of WU-9E1-G2 under the same conditions blocked escape and restored the antiviral effect of H3L3 (figure 4C). Collectively, these data confirm previous findings that HCV can use CLDN6 as an entry factor in some cell lines.27–29 However, low levels of CLDN6 surface expression in PHH largely exclude a functional role during HCV entry, suggesting that escape from CLDN1-targeting therapies is highly unlikely in the human liver.
H3L3 efficiently inhibits HCV infection and spread
We evaluated the dose–response profile of H3L3, using HCVcc (Luc-Jc1; genotype 2a) and Huh7.5.1 cells. H3L3 dose-dependently inhibited HCVcc infection of Huh7.5.1 cells with an IC50 of 0.24 μg/mL, similarly to OM-7D3-B3 (IC50, 0.13 μg/mL) (figure 5A). Furthermore, H3L3 inhibited infection by DAA-resistant HCVcc NS3 mutants Jc1-A156S and Jc1-R155K (IC50, 0.47 and 0.51 μg/mL, respectively), as did OM-7D3-B3 (IC50, 0.23 and 0.36 μg/mL, respectively) (figure 5A). Therefore, both antibodies retained their potency against DAA-resistant HCVcc (figure 5A). Considering the important role of cell–cell transmission in HCV persistence and the demonstrated ability of the rat anti-CLDN1 mAb OM-7D3-B3 to block this route of HCV entry,21 we tested H3L3 in a cell–cell transmission assay in the presence of anti-E2 envelope antibody to inhibit cell-free transmission. As expected, H3L3 efficiently blocked cell–cell transmission of HCV (figure 5B), with similar potency as observed for OM-7D3-B3 (figure 5C).
Figure 5.

Functional characterisation of the humanised anti-tight junction protein claudin-1 (CLDN1) antibody clone H3L3 in cell culture. (A) H3L3 dose-dependently inhibits HCV infection. Huh7.5.1 cells were incubated with rat OM-7D3-B3, humanised H3L3 or isotype control antibodies prior to infection with HCVcc (Jc1 wild type, Jc1-A156S or Jc1-R155K). Infection was assessed by measuring luciferase activity after 72 hours. Results are expressed as log relative luciferase units from three independent experiments performed in triplicate. (B and C) H3L3, like OM-7D3-B3, inhibits HCV cell–cell transmission. Huh7.5.1 cells electroporated with HCV Jc1 RNA (producer cells) were co-cultured with naive Huh7.5.1-GFP cells (target cells) in the presence of control or anti-CLDN1 antibody (11 µg/mL). Co-cultured cells were fixed with paraformaldehyde after 24 hours and stained with an NS5A antibody (B). The extent of cell–cell transmission was determined by calculating percentage of GFP+NS5A+ cells (C). Results from a single experiment performed in duplicate are shown. (D) H3L3 synergises with direct-acting antivirals. Huh7.5.1 cells were pretreated with H3L3 in combination with sofosbuvir (SOF) or daclatasvir (DCV) prior to infection with HCVcc. Infection was assessed after 72 hours by luciferase activity. Synergy (20% above that expected for additive effects, shown in black) was assessed according to the Prichard and Shipman method.40 Results from a representative experiment are shown.
Antiviral synergy is an attractive feature for combination therapies. We used the Prichard method40 to determine whether H3L3 synergises with DAAs, as observed for the parental rat antibody.25 Indeed, combining H3L3 with sofosbuvir or daclatasvir resulted in significant synergistic activity (figure 5D). The increased potency was >20% above that predicted from additive effects, indicating synergy.40
H3L3 cures chronic HCV infection in human-liver chimeric uPA-SCID mice
We next tested the ability of H3L3 to cure human-liver chimeric uPA-SCID mice chronically infected with HCV. Chronically HCV-infected mice were given weekly intraperitoneal injections of 25 mg/kg of H3L3 (n=2) or isotype control human IgG4 mAb (n=1) for 4 weeks. For the control mouse and one H3L3-treated mouse, HCV RNA levels as well as serum human albumin and IgG4 levels were measured for 12 or 13 weeks post-treatment. For the second H3L3-treated mouse, the viral load was monitored for 5 weeks post-treatment until clearance of HCV RNA. Both H3L3-treated mice showed undetectable HCV RNA levels at the end of the study period (figure 6A). Stable human albumin levels in mice confirmed engraftment of human PHH (figure 6B). Increasing serum levels of the antibody (figure 6C) did not affect the levels of human albumin (figure 6B), indicating that H3L3 did not adversely affect liver function. Like the parental rat antibody,21 therefore, H3L3 clears chronic HCV infection in human liver chimeric mice without overt toxicity.
Figure 6.
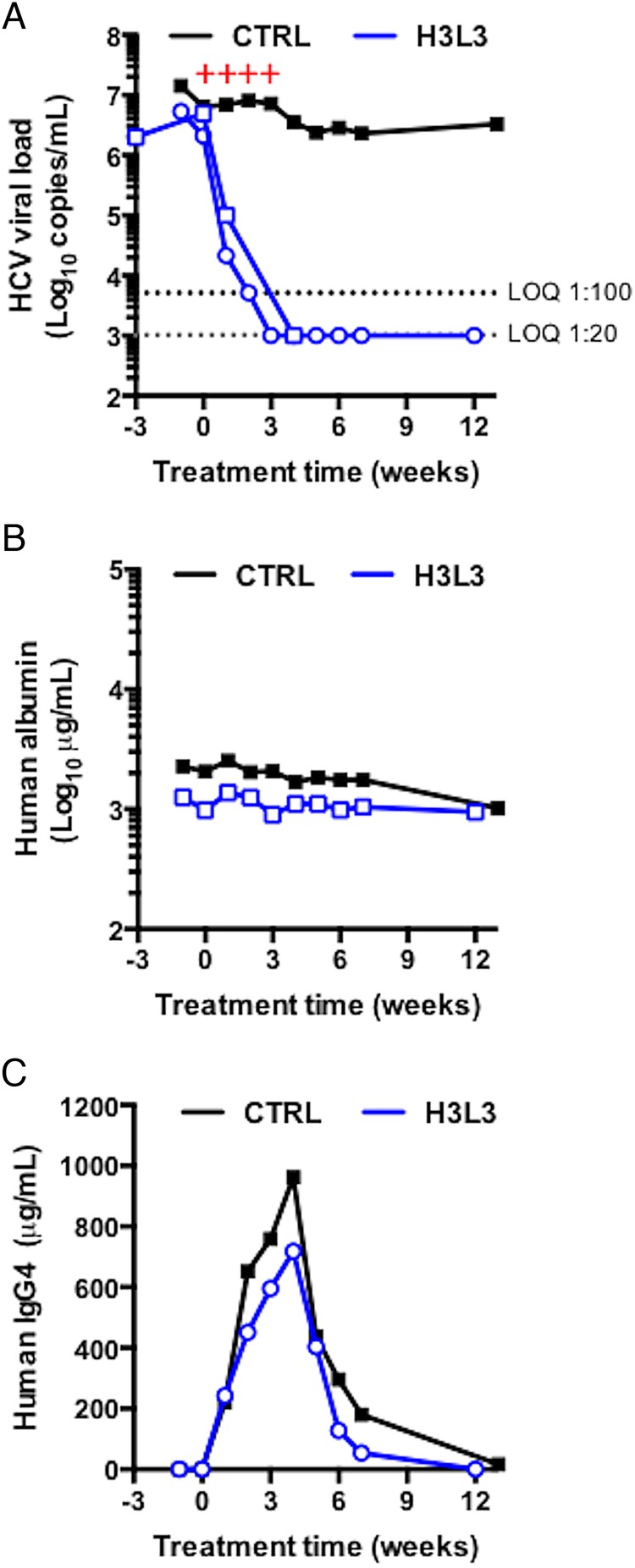
H3L3 cures chronically HCV-infected human-liver chimeric uPA-SCID mice. (A) Two mice chronically infected with HCV Jc1 were treated with 500 µg of H3L3 anti-tight junction protein claudin-1 monoclonal antibody weekly for 4 weeks. As a control, one mouse was similarly treated with 500 μg of isotype control human antibody. Human albumin (B) and IgG4 antibody (C) levels were monitored. Red + symbols indicate the times of antibody treatment. The horizontal dashed line indicates limit of quantification (LOQ).
Discussion
To enable the clinical development of a CLDN1-targeted HCV therapy, we generated humanised anti-CLDN1 antibodies by grafting CDRs of rat anti-CLDN1 mAb OM-7D3-B3 on to the human IgG4 backbone. All of the humanised antibodies bound to CLDN1 and inhibited HCVcc infection with similar potency. We selected one mAb, H3L3, for detailed characterisation. Given that genotype-dependent escape from CLDN1-targeted therapies through CLDN6 and/or CLDN9 has been described in cell lines,27–29 we evaluated the functional relevance of escape from the H3L3 antibody. H3L3 pan-genotypically inhibited HCVpp entry into PHH from up to 12 different donors, without any apparent escape (figure 3), reflecting low surface expression levels of CLDN6 and CLDN9 (figure 4A). This is consistent with the lack of CLDN6 protein expression in liver sections30 and lack of CLDN6 and CLDN9 protein expression in PHH.28 Furthermore, CLDN6 did not appear to play a role in HCV entry into PHH (figure 4B). Therefore, escape from CLDN1-directed therapies such as the H3L3 antibody is likely not relevant in vivo, at least for the majority of patients. Ultimately, clinical trials will be needed to address the clinical relevance of escape.
The humanised anti-CLDN1 antibody demonstrates promising activities that address some limitations of current DAA-based therapies. H3L3 potently inhibited HCVcc infection and HCVpp entry of different genotypes, blocked cell–cell transmission and cured chronically HCV-infected human-liver chimeric uPA-SCID mice in monotherapy. H3L3 has a complementary mechanism of action compared with currently approved DAAs, which results in marked synergy as shown for sofosbuvir and daclatasvir (figure 5D). Such synergy could allow shortening of treatment duration, thus reducing costs and the possibility of side effects. Furthermore, H3L3 is active against DAA-resistant HCV NS3 mutants (figure 5A), another attractive feature for a potential combination therapy. Given that CLDN1 is a host-encoded target, a higher genetic barrier to resistance is expected. In contrast to current DAAs, H3L3 (figure 6A) cured chronically infected human-liver chimeric mice in monotherapy, without resistance or escape. Finally, host-targeting entry inhibitors such as H3L3 are expected to efficiently block HCV de novo infection of the liver graft. Therefore, H3L3 is ideally positioned to address DAA failure in chronic HCV infection and prevent acute liver graft infection.
Organ shortage is a major challenge facing transplants in all contexts (eg, kidney). In the USA, there are currently >120 000 patients on waiting lists for transplants, yet only approximately 30 000 transplants were performed in 2015.11 This medical burden can be eased by transplantation of organs from HCV-positive patients to HCV-negative recipients, which is no longer overtly contraindicated.42 43 Nonetheless, transplantation of organs from HCV-positive patients to HCV-negative recipients is challenged by de novo HCV infection of the recipient and is often associated with less positive outcomes.10 H3L3 could protect patients receiving HCV-positive organs from HCV infection, which would increase the pool of donor organs. In contrast to DAAs (which are administered postinfection), the humanised anti-CLDN1 antibody could prevent infection of the recipient.
Humanisation of anti-CLDN1 mAb OM-7D3-B3 is the first step towards its clinical development. Any potential anti-receptor antibody will have to be safe and non-immunogenic. In order to avoid destruction of healthy hepatocytes from antibody-mediated effector functions, we humanised anti-CLDN1 mAb OM-7D3-B3 into the IgG4 isotype, which can neither sensitise natural killer cells nor activate the complement system. Thus, IgG4 does not induce antibody-dependent cell-mediated cytotoxicity or complement-mediated lysis of target cells. Although humanisation of the rat antibody is likely to reduce its immunogenicity in humans, further evaluation in an appropriate animal model with functional immune responses will be required. However, rat anti-CLDN1 does not cause any toxicity in immunocompetent mice;21 this is likely also to be the case for the humanised antibody. The humanised antibodies did not exert any cellular toxicity in vitro (see online supplementary figure S3) and we did not observe any overt toxic effects in human-liver chimeric mice (figure 6B).
In conclusion, we successfully humanised a rat anti-CLDN1 mAb without loss of function. This is a critical step for further development of anti-CLDN1 antibodies as an alternative treatment strategy for HCV infection. The humanised antibody has pan-genotypic activity in PHH, without observable escape in PHH from multiple donors, highlighting the clinical potential of CLDN1-targeted therapies such as H3L3. Further evaluation of this antibody in preclinical efficacy and toxicity studies in suitable animal models will pave the way for clinical trials in humans. Overall, H3L3 represents a promising alternative approach for HCV therapy for patients who fail current therapies or to prevent post-transplantation HCV infection.
Acknowledgments
The authors thank Dr R Bartenschlager (University of Heidelberg, Germany) for HCVcc Luc-Jc1 and Jc1 plasmids, Dr. C M Rice (Rockefeller University, New York, USA) and Dr. T Wakita (National Intitute of Infectious Diseases, Tokyo, Japan) for providing HCV JFH1 based expression plasmids, Dr J Bukh (University of Copenhagen, Denmark) for NS3 mutant plasmids, Dr J. Ball (University of Nottingham, UK), Dr R Purcell (NIAID, Bethesda, Maryland, USA) and Dr F-L Cosset (Inserm U1111, Lyon, France) for selected pseudoparticle expression constructs, Dr F Habersetzer (Inserm U1110, Strasbourg, France) for HCV genotype 3-positive patient serum, Dr F Chisari (Scripps Research Institute, La Jolla, California, USA) for Huh7.5.1 cells and Dr A Patel (MRC Virology Unit, Glasgow, UK) for E2-specific mAb AP33 and KalyCell (Plobsheim, France) for providing some of the PHH.
Footnotes
Contributors: CCC and RGT contributed equally. TFB initiated and supervised the study. CCC, RGT, LM, CT, LH, SCD, FX and ER performed the experiments. PP provided essential reagents. CCC, RGT, MBZ and TFB analysed data. CCC, RGT, MZB and TFB wrote the manuscript.
Funding: This study was supported by funding from the European Union (FP7 HEPAMAB GAN 305600, ERC-2014-AdG-HEPCIR, EU H2020 HEPCAR and Interreg IV-Rhin Supérieur-FEDER-Hepato-Regio-Net 2012), the Agence Nationale de Recherche sur le SIDA (ANRS) (2012/318, 2013/108) the Direction Générale de l'Offre de Soins (A12027MS) and the US National Institutes of Health (1U19AI123862-01). This work has been published under the framework of the LABEX ANR-10-LABX-0028_HEPSYS and benefits from funding from the state managed by the French National Research Agency as part of the Investments for the Future programme. CCC is supported by fellowships from the Canadian Institutes of Health Research (201411MFE-338606-245517) and the Canadian Network on Hepatitis C.
Competing interests: TFB is a co-inventor on a patent application on monoclonal anti-claudin1 antibodies for the inhibition of HCV infection (US patent no. 8,518,408; WO2010034812; PCT/EP 08 305 597 0).
Patient consent: Obtained.
Ethics approval: Strasbourg University Hospitals (CPP 10-17).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 2016;64:486–504. 10.1016/j.jhep.2015.09.011 [DOI] [PubMed] [Google Scholar]
- 2.Pawlotsky JM. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016;151:70–86. 10.1053/j.gastro.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 3.Elmasry S, Wadhwa S, Bang BR, et al. . Detection of occult hepatitis C virus infection in patients who achieved a sustained virologic response to direct-acting antiviral agents for recurrent infection after liver transplantation. Gastroenterology 2016; doi: 10.1053/j.gastro.2016.11.002. [Epub ahead of print 9 Nov 2016] 10.1053/j.gastro.2016.11.002 [DOI] [Google Scholar]
- 4.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013;368:1907–17. 10.1056/NEJMra1213651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zoulim F, Liang TJ, Gerbes AL, et al. . Hepatitis C virus treatment in the real world: optimising treatment and access to therapies. Gut 2015;64:1824–33. 10.1136/gutjnl-2015-310421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curry MP, O'Leary JG, Bzowej N, et al. . Sofosbuvir and Velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med 2015;373:2618–28. 10.1056/NEJMoa1512614 [DOI] [PubMed] [Google Scholar]
- 7.Chung RT, Baumert TF. Curing chronic hepatitis C--the arc of a medical triumph. N Engl J Med 2014;370:1576–8. 10.1056/NEJMp1400986 [DOI] [PubMed] [Google Scholar]
- 8.Felmlee DJ, Coilly A, Chung RT, et al. . New perspectives for preventing hepatitis C virus liver graft infection. Lancet Infect Dis 2016;16:735–45. 10.1016/S1473-3099(16)00120-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curry MP, Forns X, Chung RT, et al. . Sofosbuvir and ribavirin prevent recurrence of HCV infection after liver transplantation: an open-label study. Gastroenterology 2015;148:100–7.e1. 10.1053/j.gastro.2014.09.023 [DOI] [PubMed] [Google Scholar]
- 10.Englum BR, Ganapathi AM, Speicher PJ, et al. . Impact of donor and recipient hepatitis C status in lung transplantation. J Heart Lung Transplant 2016;35:228–35. 10.1016/j.healun.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.“Organ Procurement and Transplant Data” 2016; Retrieved October 1, 2016 http://optn.transplant.hrsa.gov/data/
- 12.Pileri P, Uematsu Y, Campagnoli S, et al. . Binding of hepatitis C virus to CD81. Science 1998;282:938–41. 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 13.Scarselli E, Ansuini H, Cerino R, et al. . The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 2002;21:5017–25. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans MJ, von Hahn T, Tscherne DM, et al. . Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007;446:801–5. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 15.Lupberger J, Zeisel MB, Xiao F, et al. . EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 2011;17:589–95. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sainz B Jr, Barretto N, Martin DN, et al. . Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 2012;18:281–5. 10.1038/nm.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Q, Sodroski C, Lowey B, et al. . Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc Natl Acad Sci USA 2016;113:7620–5. 10.1073/pnas.1602701113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meuleman P, Hesselgesser J, Paulson M, et al. . Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008;48:1761–8. 10.1002/hep.22547 [DOI] [PubMed] [Google Scholar]
- 19.Vercauteren K, Van Den Eede N, Mesalam AA, et al. . Successful anti-scavenger receptor class B type I (SR-BI) monoclonal antibody therapy in humanized mice after challenge with HCV variants with in vitro resistance to SR-BI-targeting agents. Hepatology 2014;60:1508–18. 10.1002/hep.27196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fofana I, Krieger SE, Grunert F, et al. . Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 2010;139:953–64. 10.1053/j.gastro.2010.05.073 [DOI] [PubMed] [Google Scholar]
- 21.Mailly L, Xiao F, Lupberger J, et al. . Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat Biotechnol 2015;33:549–54. 10.1038/nbt.3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paciello R, Urbanowicz RA, Riccio G, et al. . Novel human anti-Claudin 1 monoclonal antibodies inhibit HCV infection and may synergize with anti-SRB1 mAb. J Gen Virol 2015;97:82–94. 10.1099/jgv.0.000330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita M, Iida M, Tada M, et al. . Discovery of anti-claudin-1 antibodies as candidate therapeutics against hepatitis C virus. J Pharmacol Exp Ther 2015;353:112–18. 10.1124/jpet.114.217653 [DOI] [PubMed] [Google Scholar]
- 24.He S, Lin B, Chu V, et al. . Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci Transl Med 2015;7:282ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao F, Fofana I, Thumann C, et al. . Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 2015;64:483–94. 10.1136/gutjnl-2013-306155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao F, Fofana I, Heydmann L, et al. . Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog 2014;10:e1004128 10.1371/journal.ppat.1004128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng A, Yuan F, Li Y, et al. . Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 2007;81:12465–71. 10.1128/JVI.01457-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meertens L, Bertaux C, Cukierman L, et al. . The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J Virol 2008;82:3555–60. 10.1128/JVI.01977-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haid S, Grethe C, Dill MT, et al. . Isolate-dependent use of claudins for cell entry by hepatitis C virus. Hepatology 2014;59:24–34. 10.1002/hep.26567 [DOI] [PubMed] [Google Scholar]
- 30.Fofana I, Zona L, Thumann C, et al. . Functional analysis of claudin-6 and claudin-9 as entry factors for hepatitis C virus infection of human hepatocytes by using monoclonal antibodies. J Virol 2013;87:10405–10. 10.1128/JVI.01691-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krieger SE, Zeisel MB, Davis C, et al. . Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 2010;51:1144–57. 10.1002/hep.23445 [DOI] [PubMed] [Google Scholar]
- 32.Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol 1991;147:1709–19. [PubMed] [Google Scholar]
- 33.Jones PT, Dear PH, Foote J, et al. . Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986;321: 522–5. 10.1038/321522a0 [DOI] [PubMed] [Google Scholar]
- 34.Verhoeyen M, Milstein C, Winter G. Reshaping human antibodies: grafting an antilysozyme activity. Science 1988;239:1534–6. 10.1126/science.2451287 [DOI] [PubMed] [Google Scholar]
- 35.Mee CJ, Harris HJ, Farquhar MJ, et al. . Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol 2009;83:6211–21. 10.1128/JVI.00246-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pestka JM, Zeisel MB, Blaser E, et al. . Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci USA 2007;104:6025–30. 10.1073/pnas.0607026104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fafi-Kremer S, Fofana I, Soulier E, et al. . Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J Exp Med 2010;207:2019–31. 10.1084/jem.20090766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koutsoudakis G, Kaul A, Steinmann E, et al. . Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 2006;80:5308–20. 10.1128/JVI.02460-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witteveldt J, Evans MJ, Bitzegeio J, et al. . CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol 2009;90:48–58. 10.1099/vir.0.006700-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prichard MN, Shipman C Jr. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 1990;14:181–205. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 41.Mercer DF, Schiller DE, Elliott JF, et al. . Hepatitis C virus replication in mice with chimeric human livers. Nat Med 2001;7:927–33. 10.1038/90968 [DOI] [PubMed] [Google Scholar]
- 42.Scalea JR, Barth RN, Munivenkatappa R, et al. . Shorter waitlist times and improved graft survivals are observed in patients who accept hepatitis C virus+ renal allografts. Transplantation 2015;99:1192–6. 10.1097/TP.0000000000000479 [DOI] [PubMed] [Google Scholar]
- 43.Reese PP, Abt PL, Blumberg EA, et al. . Transplanting hepatitis C-positive kidneys. N Engl J Med 2015;373:303–5. 10.1056/NEJMp1505074 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
gutjnl-2016-312577supp001.pdf (211.1KB, pdf)