

Abstract
Signaling of the prototypical G protein–coupled receptor (GPCR) rhodopsin through its cognate G protein transducin (Gt) is quenched when arrestin binds to the activated receptor. Although the overall architecture of the rhodopsin/arrestin complex is known, many questions regarding its specificity remain unresolved. Here, using FTIR difference spectroscopy and a dual pH/peptide titration assay, we show that rhodopsin maintains certain flexibility upon binding the “finger loop” of visual arrestin (prepared as synthetic peptide ArrFL-1). We found that two distinct complexes can be stabilized depending on the protonation state of E3.49 in the conserved (D)ERY motif. Both complexes exhibit different interaction modes and affinities of ArrFL-1 binding. The plasticity of the receptor within the rhodopsin/ArrFL-1 complex stands in contrast to the complex with the C terminus of the Gt α-subunit (GαCT), which stabilizes only one specific substate out of the conformational ensemble. However, Gt α-subunit binding and both ArrFL-1–binding modes involve a direct interaction to conserved R3.50, as determined by site-directed mutagenesis. Our findings highlight the importance of receptor conformational flexibility and cytoplasmic proton uptake for modulation of rhodopsin signaling and thereby extend the picture provided by crystal structures of the rhodopsin/arrestin and rhodopsin/ArrFL-1 complexes. Furthermore, the two binding modes of ArrFL-1 identified here involve motifs of conserved amino acids, which indicates that our results may have elucidated a common modulation mechanism of class A GPCR–G protein/-arrestin signaling.
Keywords: arrestin, Fourier transform IR (FTIR), G protein, G protein-coupled receptor (GPCR), rhodopsin, biased signaling, functional selectivity
Introduction
Unlike other class A G protein–coupled receptors (GPCRs)2 the photoreceptor rhodopsin contains the light-sensitive cofactor retinal as a ligand, which is covalently bound to the opsin apoprotein. Upon light-induced cis/trans isomerization of retinal and subsequent alterations within the binding pocket, the signal propagates via GPCR-conserved interhelical networks toward the cytoplasmic surface of the receptor, where major structural rearrangements take place. The most prominent structural change is the rotational outward tilt of transmembrane helix (TM) 6, which opens a cytoplasmic binding crevice and thus constitutes the main prerequisite for binding and activation of signaling via the G protein transducin (1, 2). Even though first identified in rhodopsin, for many other GPCRs homologous structural changes have been shown to exist, and a common structural and functional framework for GPCRs is being developed (3).
Binding of arrestins to their cognate receptors blocks G protein signaling and is, for most GPCRs, connected to distinct G protein–independent signaling pathways (4). The interaction of arrestin occurs via initial “prebinding” to the phosphorylated receptor (5) followed by binding of the arrestin “finger loop” (6, 7) which recognizes the active receptor conformation (8). The necessity of prebinding is circumvented in the naturally occurring arrestin splice variant p44 lacking the C-tail (9, 10).
Crystal structures of active GPCRs in complex with G protein or arrestin-1 (visual arrestin) have been reported, revealing the overall architecture of the interacting binding partners (11, 12). Fig. 1A shows the binding interface of a constitutively active, thermostable, and unphosphorylated opsin mutant (E3.28Q/M6.40Y and N2C/N282C), bound to the finger loop of preactivated arrestin-1 (3A murine arrestin; substitutions, L374A, V375A, F376A, residues 10–392) as determined by serial femtosecond X-ray crystallography (12). The M763 backbone carbonyl of the arrestin finger loop forms a hydrogen bond to N8.47 of the receptor (d = 3.0 Å). The M76 side chain has however not been resolved in this crystal structure indicating substantial flexibility. On the receptor side, substituted M6.40Y forms a stable hydrogen bond to highly conserved R3.50 (3.1 Å), resulting in the strong constitutive activity observed for this mutant (13, 14). A more recently published structure of the rhodopsin/arrestin complex shows an almost identical arrangement of the two binding partners and also reveals the hydrogen bond between R3.50 and M6.40Y (5). Interestingly, this hydrogen bond stabilizes the R3.50 side chain in a conformer too far away from highly conserved Y5.58 to make a direct interaction (3.9 Å). This link has been suggested as crucial for stabilization of the active receptor conformation (2, 15) and Gt activation (16). Further examination of the structure suggests that the changed hydrogen bond network, because of the activating M6.40Y mutation, prevents the R3.50 guanidinium group from forming intermolecular bonds with the arrestin finger loop. However, the interaction between R3.50 and the finger loop is present in the crystal structure of native light-activated rhodopsin in complex with the arrestin finger loop peptide ArrFL-1 (Fig. 1B). In this crystal structure, distances are such that two strong intermolecular hydrogen bonds between R3.50-G75 (2.7 Å) and the backbones of K8.48-G76 (3.0 Å) are formed, and also an additional intramolecular R3.50-Y5.58 (3.2 Å) bond is within range of hydrogen bonding (17).
Figure 1.

Crystal structures presenting the rhodopsin/arrestin-1 finger loop binding interface. A, crystal structure of a fusion protein of constitutively active, thermostable human opsin (orange, substitutions: E3.28Q/M6.40Y and N2C/N282C) with preactivated mouse visual arrestin (purple). B, crystal structure of light-activated native bovine rhodopsin (orange) and ArrFL-1 (purple), an 11-mer peptide derived from the arrestin-1 finger loop (67YGQEDIDVMGL77). PDB IDs are 4ZWJ and 4PXF for A and B, respectively. Parts of TM6 were omitted for clarity (asterisk).
In addition to the perturbation which might be introduced by stabilizing inserts or mutations, crystal structures represent only a single low energy conformation stabilized by the crystal lattice. GPCRs in situ, however, exist in a variety of different conformations in equilibrium whose populations are dependent on environmental variables, the characteristics of intracellular/extracellular binding partners, and bound ligand (3, 18–20). In the case of light-activated rhodopsin, the agonist-bound equilibrium conformations have been identified as “Metarhodopsin” states, each characterized by a specific arrangement of crucial amino acids and their connecting hydrogen bond networks. These “microswitches” (21) are built on residues conserved among GPCRs and thus corresponding conformational states can be delineated for other receptors (Scheme 1).
Scheme 1.
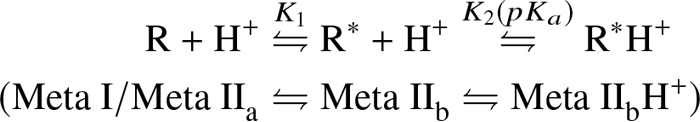
The agonist-bound receptor conformations are either active (R*) or inactive (R) conformations referring to whether or not TM6 outward tilt has occurred. Final proton uptake at the open binding crevice neutralizes E3.49 from the conserved (D)ERY motif (22), which results in the pH independence of the conformational equilibria for the E3.49Q mutant (23). Concomitantly, TM5 is stabilized in an inward position, thereby hindering TM6 to adopt its inactive inward position (2, 16). In native membranes, proton uptake occurs with an apparent pKa of 7.5, hence a significant amount of agonist-bound receptor exists in the deprotonated active R* conformation (22, 24) and binding of arrestin to R* could play an important role during signal shutoff, G protein-independent (arrestin) signaling, or receptor internalization in non-rhodopsin GPCRs (25).
In the present study, we employ a dual pH/peptide titration assay, site-directed mutagenesis, and FTIR double difference spectroscopy to answer the following questions on arrestin-receptor interaction: (i) Which of the receptor conformations shown in Scheme 1 interact with the arrestin finger loop, and do the resulting protein complexes exhibit distinct binding modes? (ii) To what extent is R3.50 involved in binding the finger loop? This is of special interest because R3.50 is one of the highest conserved amino acids within the class A of GPCRs and its mutation is known to cause autosomal dominant retinitis pigmentosa (26, 27). (iii) Finally, we hope to resolve the conflicting pictures of the rhodopsin/finger loop–binding interface provided by X-ray crystallographic studies (Fig. 1).
Full-length arrestins are known to undergo substantial conformational changes upon binding to the active receptor (8, 10, 29). Such changes interfere with identifying receptor changes when monitored by FTIR spectroscopy, making unambiguous band assignments challenging (30). Therefore, to allow more straightforward and unambiguous interpretation of our data, we made use of ArrFL-1, a synthetic peptide derived from the finger loop of arrestin-1 (rod visual arrestin, 67YGQEDIDVMGL77) (31). The finger loop constitutes the key interaction site of arrestins with their cognate GPCRs (32) and ArrFL-1 has been shown to compete with full-length arrestin for the same receptor-binding site (17). This makes ArrFL-1 a robust and powerful tool to investigate binding of the arrestin finger loop to rhodopsin.
Results
ArrFL-1/pH dual titration assay
In Fig. 2A, normalized FTIR difference spectra of native rhodopsin in urea-washed membranes are shown. The difference spectra were calculated from spectra recorded before and after photoactivation at pH values between 6.0 and 9.1. From such difference spectra, fingerprints of active and inactive receptor conformations can be derived (33). A good measure of the amount of active species is the positive difference band at 1744 cm−1, which has been assigned to the reorganization of a hydrogen bond network connecting TM3 and TM5 occurring during receptor activation (34). The intensity increase at this frequency as the pH is lowered indicates that the active conformation is stabilized by proton uptake and formation of R*H+ (cf. Scheme 1).
Figure 2.

FTIR difference spectra of rhodopsin light activation in the native membrane and at different pHs are recorded to illuminate the different agonist-bound receptor conformations in equilibrium (Scheme 1). A, lowering the bulk pH causes E3.49 protonation and disruption of the E3.49-R3.50 ionic lock (23), which leads to stabilization of the active R*H+ conformation and an intensity increase of difference bands indicating activating structural changes. The band at 1744 cm−1 is a suitable monitor of the active conformations R* and R*H+ and it is isolated from other changing absorbance bands. B, addition of 10 mm ArrFL-1 leads to several additional difference bands (e.g. 1659 cm−1). It also stabilizes the active conformation, because even at high pH the intensity of the 1744 cm−1 marker band reflects predominantly active conformation formed. C, evaluation of 1744 cm−1 intensity changes as a function of pH and ArrFL-1. With increasing ArrFL-1 concentration an upshift of the apparent pKa of proton uptake and an increase of the alkaline end point level are observed, which indicates stabilization of R* and R*H+ because of ArrFL-1 binding. For each pH value two datasets have been acquired independently and averaged, the deviation is shown as error bar.
A plot of the normalized intensity change at 1744 cm−1 versus pH is shown in Fig. 2C (θ(1744 cm−1), blue symbols). At pH 6 the receptor molecules are quantitatively present in the protonated active conformation (R*H+). Receptor deprotonation, occurring with an apparent pKa of 7.5, releases the receptor into an equilibrium between R and R* states (24). At the alkaline end point of the titration (approx. pH 9) about 35% of light-activated rhodopsin exists in the deprotonated conformation (R*), whereas the remaining 65% exists as inactive R. It is important to emphasize that proton uptake is not the cause for TM6 outward movement but is instead its consequence (35, 36). Hence formation of the cytoplasmic binding crevice for G protein and arrestin interaction occurs already in R*, although catalytic activity toward the G protein requires the fully accomplished binding site of the protonated R*H+ state (37). A finding which is in agreement with the C-terminal peptide of the Gt α-subunit stabilizing exclusively the protonated R*H+ conformation (38).
In the presence of 10 mm ArrFL-1 peptide, several additional bands appear in the difference spectrum which reflect binding-induced structural changes in both binding partners, as well as newly formed specific interactions between them (Fig. 2B). Additionally, binding of ArrFL-1 peptide leads to a pronounced stabilization of the active conformations as indicated by the overall more consistent intensity of FTIR difference bands in the amide regions (amide I around 1650 cm−1, amide II around 1550 cm−1) and above 1700 cm−1 (protonated side chain carboxyls), even at high pH. Evaluation of θ values at 1744 cm−1 reveals a slight upshift of the apparent pKa and a significant increase of the alkaline end point level, i.e. the amount of the deprotonated active conformation R* (Fig. 2C, green). This trend is clearly dependent on the ArrFL-1 concentration: At 20 mm ArrFL-1 the apparent pKa is around 8.1 and the alkaline end point level is approximately 70% (Fig. 2C, magenta).
To globally evaluate the stabilizing effect of increasing peptide concentration on the equilibria of receptor species and to determine the ArrFL-1 binding constants to the R* and R*H+ conformations we extended Scheme 1 by two more receptor species representing the ArrFL-1 interacting conformations (Scheme 2).
Scheme 2.
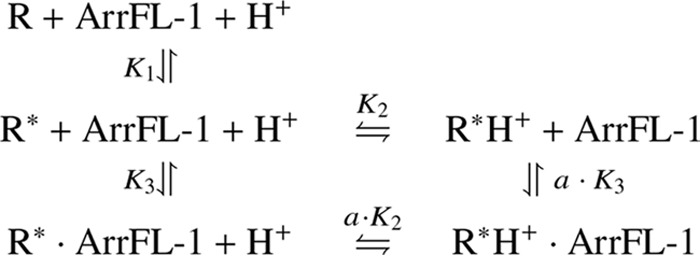
The connecting equilibrium constants and respective binding affinities to R* and R*H+ can be deduced from a global fit of this model to all data points in Fig. 2C (colored lines) (see “Experimental Procedures” or Ref. 38 for in-depth description of the fitted model). The calculated binding constants for ArrFL-1 binding to the two active receptor conformations R* and R*H+ are KD (R*) = K3 = 6 ± 1 mm and KD (R*H+) = a· K3 =3 ± 2 mm, respectively (compare KD = 350 μm for the Gα C-terminal peptide GαCT (38)).
Peptide-binding spectra
ArrFL-1 binding leads to the occurrence of additional difference bands overlapping those reflecting receptor activation (e.g. the positive band at 1659 cm−1) (Fig. 2B). To be able to directly compare the modes of interaction between the arrestin finger loop and the deprotonated or protonated receptor species (R*·ArrFL-1 and R*H+·ArrFL-1), we calculated the so-called peptide-binding spectra (PBS), which have been used repeatedly to characterize conformational changes and new molecular interactions formed because of peptide binding (38, 39). FTIR difference spectra were measured in the presence and absence of 20 mm ArrFL-1 peptide (Δ(w/ ArrFL-1) and Δ(w/o ArrFL-1), gray and black lines in Figs. 3 and 4, respectively). As for all difference spectra shown in this manuscript, data analysis was performed using a combination of singular value decomposition (SVD) and global analysis (40), which allows determination of pure difference spectra of the light-induced transition without any contribution of photoproduct decay. This approach led to extremely high signal to noise ratio and reproducibility, thus allowing the interpretation of all bands, which appear clearly above the noise level (see supporting information for assessment of signal to noise ratio). Subsequently, the PBS were calculated as the difference between the two difference spectra, i.e. the double difference: Δ(with ArrFL-1) minus Δ(without ArrFL-1). It is noteworthy that the PBS at high pH were also corrected for residual amounts of the inactive R conformation present in the sample. For this the inactive conformation was stabilized at low temperature and high pH; the respective difference spectra are given as supporting information.
Figure 3.
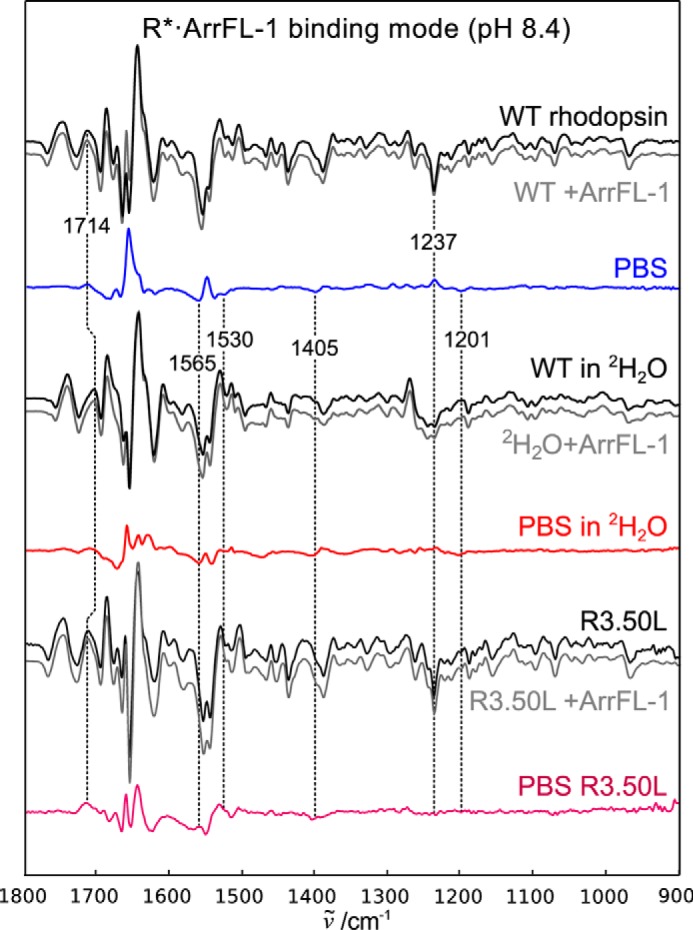
Peptide-binding spectra of the deprotonated binding mode R*·ArrFL-1. Difference spectra of wildtype rhodopsin (WT) in native disk membranes were recorded in the absence (black) and presence (gray) of 20 mm ArrFL-1 peptide. The resulting double difference (PBS) is shown in blue and reflects structural changes because of ArrFL-1 binding to the deprotonated receptor. Identical experiments were performed after H2O/2H2O buffer exchange and R3.50L mutation, the resulting PBS are shown in red and purple, respectively.
Figure 4.
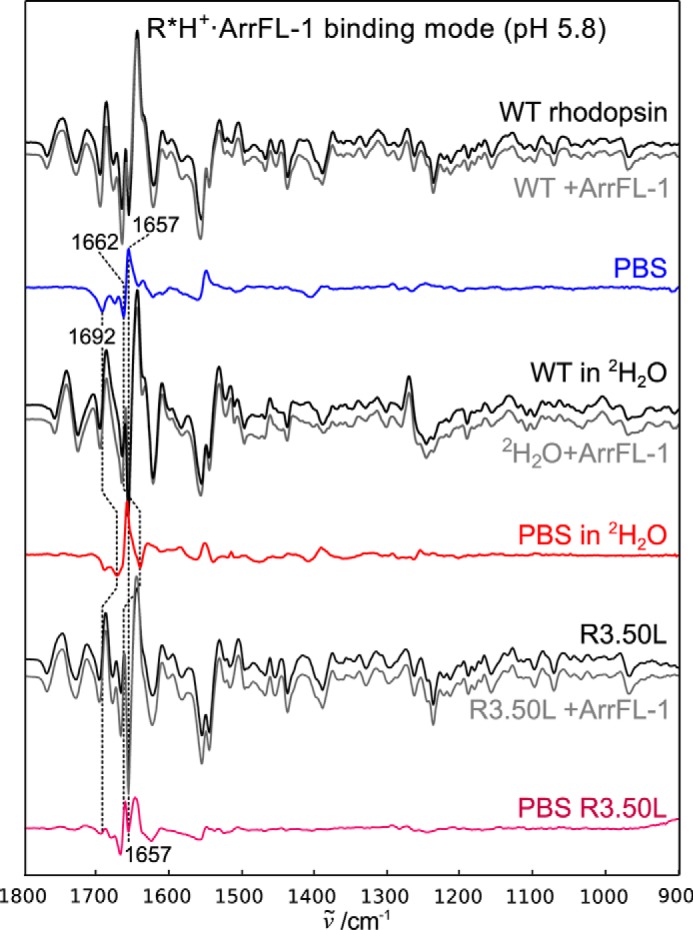
The R*H+·ArrFL-1 binding mode stabilized at low pH. Difference spectra recorded in the absence of ArrFL-1 peptide are shown in black, in the presence of 20 mm ArrFL-1 in gray. The resulting PBS are shown in blue (WT), red (in 2H2O), or purple (R3.50L mutant). Note the difference in band pattern compared with the deprotonated complex. The small effect of 2H2O on the positive 1657 cm−1 indicates that this band is a structurally sensitive amide I band.
We followed two strategies to decompose the PBS and assign specific FTIR difference bands to their molecular origins: First, H2O/2H2O buffer exchange was conducted, which induces isotopic shifts of hydrogen-coupled molecular vibrations. For example, the intense coupled CN3H5+vibration of arginine side chains absorbing between 1695 and 1650 cm−1 (in H2O) is known to experience considerable frequency downshifts or even decouples and vanishes entirely upon sample deuteration. In contrast, C=O stretching vibrations of the protein backbone (amide I), also located in this region, exhibit only minor isotopic shifts (41). Additionally, we investigated the effect of R3.50L substitution to obtain site-specific information on involvement of this key amino acid for arrestin binding, and to resolve the apparent discrepancy in the crystal structures (cf. Fig. 1). R3.50L mutation, just like E3.49Q mutation, completely abolishes the pH dependence of light-activated rhodopsin (23), therefore the PBS recorded at high pH did not have to be corrected for any contribution of inactive R.
In Fig. 3 the PBS and underlying difference spectra recorded under high pH conditions are shown. Under these conditions E3.49 is still deprotonated. Binding of Gα C-terminal peptide to R*H+ has been shown to stabilize one specific substrate out of the receptor ensemble and the concomitant structuring of the peptide leads to a distinct negative band around 1530 cm−1 (38, 39), likely caused by hydrophobic interaction between the peptide and the inner faces of TM5 and TM6 (2). We observe a very similar spectral feature in the PBS of R*·ArrFL-1 (Fig. 3, blue line), which suggests that there is a hydrophobic stabilization of ArrFL-1 occurring in the deprotonated binding mode, similar to what has been observed for GαCT. The negative 1530 cm−1 feature is absent in the PBS recorded in 2H2O (red line) supporting the notion that this band is a structurally sensitive amide II band of the flexible peptide in the unbound state (38, 41). 2H2O also leads to decreased intensity in the 1695–1650 cm−1 region (arginine vibrations, see above). Because the PBS of the R*·ArrFL-1 complex is strongly influenced by R3.50L substitution, we conclude that R3.50 is taking part in the deprotonated binding mode.
ArrFL-1 binding to deprotonated R* furthermore leads to protonation of a carboxylate as clearly indicated by the positive ν(COOH) band at 1714 cm−1, which experiences the characteristic 10 cm−1 downshift in 2H2O and negative ν(COO−) corresponding bands at 1565 cm−1 and 1405 cm−1. Because E3.49 is still deprotonated and bound to R3.50 under these conditions, we attribute these difference bands to E6.30, which is liberated from R3.50 with formation of R* facilitating the activating outward tilt of TM6. This corresponds well to the R*·ArrFL-1-specific bands at 1237 and 1201 cm−1, which have already been tentatively assigned to the formation of a hydrogen bond between K5.66 and E6.30 (16). Formation of the K5.66-E6.30 hydrogen bond in R*·ArrFL-1 is consistent with the formation of the TM5/TM6 helix pair stabilized by hydrophobic contacts to ArrFL-1.
The most prominent differences observed for the protonated R*H+·ArrFL-1 complex are the two additional negative bands at 1692 and 1662 cm−1 (Fig. 4, blue line). These bands appear to be very sensitive to H2O/2H2O exchange (red line) and vanish completely upon R3.50L substitution (purple line), which again suggests R3.50 as the origin of these bands (symmetric and asymmetric guanidinium vibration, respectively). Because of proton uptake in R*H+, the R3.50 side chain is liberated from the intrahelical salt-bridge to E3.49 and forms a new hydrogen bond to Y5.58 (16, 42). This finding is in agreement with the crystal structure of the R*H+·ArrFL-1 complex, which shows this interaction and also the free η-nitrogen of R3.50 forming a strong hydrogen bond to the arrestin finger loop (Fig. 1B) (17). The strongest band in the PBS of R*H+·ArrFL-1 is the positive band at 1657 cm−1, whose intensity increases in 2H2O, probably because of the deuteration-induced shift of the negative arginine band at 1662 cm−1. The origin of this intense amide I band is likely a solvent-exposed near α-helical conformation, similar to what has been described for the C terminus of the Gt α-subunit. We attribute this band to the arrestin finger loop rather than the receptor, in agreement with crystal structures of rhodopsin·ArrFL-1, and an earlier NMR study describing a partially structured arrestin finger loop when bound to rhodopsin (31). This coil-to-helix transition is missing upon R3.50L replacement, which again highlights the importance of the R3.50 interaction for binding and recognition of ArrFL-1.
Other intermolecular hydrogen bonds may occur between backbone residues of ArrFL-1 and e.g. helix 8, as suggested by the crystal structure (cf. Fig. 1); however, these interactions are buried in the amide I region and thus not reliably detected in this dataset (43).
Discussion
Despite existing crystal structures of active GPCRs in complex with G protein and arrestin, many open questions remain regarding the mechanism of how GPCRs couple to their cognate G proteins and arrestins. Interconversion between different receptor conformations takes place on the microsecond to millisecond timescale, thus knowing the affinities to cytoplasmic binding partners is crucial for an understanding of G protein–dependent and –independent signaling. In its native lipid environment, rhodopsin exhibits several distinct and well-characterized conformations, representing an archetypical system for the investigation of GPCR-mediated signaling (44). The equilibrium populations are susceptible to the bound ligand or cytoplasmic binding partners such as G protein or arrestin. Moreover, E3.49, part of the conserved (D)ERY motif, takes up a proton in the final steps of receptor activation, thus making proton concentration another variable to explore the conformational landscape (23, 35, 37). The present study aimed to exploit this pH dependence of receptor activation to elucidate the binding promiscuity of the arrestin key binding site, ArrFL-1, and its interaction with light-activated rhodopsin.
ArrFL-1 promiscuity
In contrast to GαCT, which stabilizes R*H+ exclusively, the key binding site of visual arrestin (finger loop, ArrFL-1) binds the activated receptor also in the absence of proton uptake. Binding to the protonated R*H+ receptor conformation occurs with an affinity of KD (R*H+) = 3 ± 2 mm. The relatively low affinity of ArrFL-1 indicates the influences of other binding interfaces between the arrestin holoprotein and active rhodopsin outside the ArrFL-1 sequence to achieve the high-affinity complex, in accordance with results from alanine scanning (45) or serial femtosecond X-ray crystallography (5, 12). The affinity for formation of the deprotonated R* complex is even lower (KD (R*) = 6 ± 1 mm) and binding leads to the emergence of distinct FTIR difference bands. This corroborates the existence of two distinct binding modes and indicates that arrestin-1 can distinguish between different receptor conformations.
Characterization of the two different ArrFL-1 complexes
FTIR peptide-binding spectra recorded under R* or R*H+ favoring conditions provide final evidence for the existence of two distinct modes of ArrFL-1 binding. Both complexes appear to involve highly conserved R3.50, which is also a key residue during binding and activation of the G protein α-subunit (46). However, the binding spectrum of the deprotonated complex indicates that R* exhibits a more open and more flexible conformation than R*H+. Deprotonated R* seems to lack the hydrogen bond between K5.66 and E6.30, an important feature, which otherwise locks the activating inward and outward tilts of TM5 and TM6, respectively (3). Our FTIR-binding spectra strongly suggest that hydrophobic contacts of ArrFL-1 to the inner face of TM5, very similar to the interactions established by GαCT (”hydrophobic patch”), are stabilizing this hydrogen bond in the absence of proton uptake and the concurrent R3.50-Y5.58 interaction.
The promiscuity of ArrFL-1 toward different active receptor conformations is in sharp contrast to the highly selective binding of the G protein α-subunit to R*H+ and shows similar specificity as binding of the G protein γ-subunit. For comparison, the respective binding constants for the G protein–derived synthetic peptides to R*H+ have been reported as 330 μm (GαCT) and 4.2 mm (GγCT), respectively (38), meaning that lower specificity yields a lower affinity complex. Like GαCT, both ArrFL-1 complexes involve a hydrogen bond to the guanidinium side chain of key amino acid R3.50. According to earlier studies investigating the final steps of rhodopsin activation, proton uptake is linked to an inward stabilization of TM5 (2, 16, 38) and a decrease in conformational flexibility (24, 47). We therefore propose that the more open and more flexible deprotonated R* conformation might allow a deeper penetration of ArrFL-1 into the cytoplasmic binding crevice formed by TM3, TM5, and TM6, leading to the observed stabilization of the peptide by contacts with inner surface of TM5 or TM6. Instead, E3.49 protonation is likely to induce finger loop helix formation as reflected in our PBS of R*H+·ArrFL-1.
Mechanistic implications and concluding remarks
Protonation of the highly conserved carboxylic acid side chain at position 3.49 is known to be critical for the catalytic activity of light-activated rhodopsin toward the inhibitory G protein transducin (37). This is in accordance with the finding that binding of the key binding site of transducin, GαCT, selectively stabilizes the protonated R*H+ conformation (38). However, the promoting effect of low pH has also been described for the β2-adrenergic receptor and its activity toward the stimulating G protein Gs (48). In the present report we investigated how protonation of E3.49 affects binding of light-activated rhodopsin to the key binding site of visual arrestin (ArrFL-1). We show that ArrFL-1, in sharp contrast to GαCT, is promiscuous, as it binds to both R* and R*H+ receptor conformations. Each of these two ArrFL-1 complexes exhibits different affinities and employs distinct modes of binding, which we were able to describe in detail. How far the two binding modes described here are connected to different conformations of holo-arrestin remains to be elucidated. Our findings correlate well with a biochemical study of the type-1 angiotensin receptor, which has shown that receptor binding to β-arrestin is less specific than its interaction with the Gq α-subunit. β-arrestin binding occurred in the presence of all peptide agonists, whereas efficient Gq binding was limited to a small subset of the ligands studied (49).
Rhodopsin activation has been described extensively, facilitated by natural abundance and its photochemical properties, which provide an ideal system for biophysical and biochemical characterization. Still, rhodopsin is considered “different” from GPCRs activated by diffusible ligands. This is mostly because rhodopsin possesses only one endogenous ligand (retinal), which is moreover covalently bound. However, it has been shown that the Schiff base linkage of all-trans-retinal is surprisingly unstable, allowing the agonist to exchange with external ligand (50). Furthermore, a variety of retinal analogues have been synthesized and investigated on their functional properties. In particular, the selective (de-)methylation of the retinal polyene chain and its influence on the conformational equilibria of rhodopsin has been studied in great detail. These studies have identified how different ligand scaffolds are able to steer the receptor along its conformational landscape, consequently affecting the conformational equilibrium and the pKa of proton uptake (51–54).
Taken together, the combination of our results with studies of other GPCRs suggests that the R* and R*H+ conformations are part of a general structural and functional framework of GPCR signaling, modulated by proton uptake at residue 3.49. This implies that rhodopsin, despite its specific ligand properties, can be used as model system for studying GPCR functional selectivity. Even more, studies on retinal analogues could greatly improve our molecular understanding of functionally selective, diffusible ligands, which are able to bias activity toward either G protein or arrestin in other, “common” GPCRs.
Experimental procedures
Sample preparation
Rhodopsin membranes were prepared from frozen retinae (W.L. Lawson Co., Omaha, NE) as described earlier (28). Briefly, samples of native urea-stripped rod outer segments membranes suspended in buffered solution (40 μl, buffer A: 20 mm BTP, 100 mm NaCl, 1 mm MgCl2) are pH adjusted by adding small drops of concentrated acid or base to the wall of the sample tube and then mixing quickly using a vortex. The sample is measured using a pH meter coupled to a microprobe. Note that these steps must be conducted in the dark under dim red light. The membranes are pelleted by centrifugation at 10,000·× g. The pellet, ∼1.5 μl in volume and containing about 2.5 nm rhodopsin, is transferred with a small spatula to a BaF2 sandwich cuvette (3 μm optical path length). The loaded cuvette is placed in the sample chamber of the FTIR spectrometer (Bruker ifs66v/s, Ettlingen, Germany), and the sample undergoes an equilibration period of at least 2 h while the sample chamber is evacuated (p < 5 mbar). Data acquisition is performed in time-resolved rapid-scan mode with quasi-logarithmic time base (Δt between 171 ms and 30 s). Light-activation of rhodopsin is achieved by illumination with three orange LEDs (λmax = 595 nm) for 10 s, which quantitatively activates the rhodopsin in the sample (16). For experiments using peptide, the membrane pellet is resuspended with 40 μl peptide solution (of desired concentration in buffer A) and pH is adjusted before second concentration to obtain the sample pellet.
Double titration assay, determination of binding constants
The pH/ArrFL-1 titration data were evaluated according to Elgeti et al. (38). Briefly, difference spectra were calculated from time-resolved RapidScan data using the singular value decomposition and global analysis tools to obtain pure difference spectra without contribution of decay products as described earlier (40). Subsequently, all difference spectra were normalized to the intensities in the 1050–970 cm−1 region, where all light-activated conformations exhibit identical absorbance changes. The intensity of the 1744 cm−1 difference band was then evaluated in a pH-dependent manner for all ArrFL-1 concentrations. For each pH value at least two independent datasets have been acquired and averaged; the deviation is given as error bar. To evaluate the binding constants the pH/ArrFL-1 titration data were subjected a global nonlinear least squares fit to the function:
| (1) |
derived in Ref. 38. Receptor concentration cR was determined to 1.6 mm by UV-visible spectroscopy using the extinction coefficient of rhodopsin at 500 nm (ϵ500 = 40,600 m−1 cm−1).
Peptide-binding spectra
Peptide-binding spectra (PBS) were determined as following: First, difference spectra in the absence and presence of 20 mm ArrFL peptide were recorded. In a second step the two difference spectra were subtracted (with peptide minus without peptide) to obtain the PBS. To calculate this double difference spectrum both difference spectra have to be normalized; we chose the 1050–970 cm−1 region where mostly chromophore vibrations absorb. This second step is slightly more difficult for the PBS of the R*-ArrFL complex, because at high pH a significant amount of inactive R (Meta I) is present in the presence and absence of ArrFL. To subtract the amount of inactive R (Meta I) we calculated the following double difference spectrum: Δ(w/ArrFL) − (A·Δ(w/o ArrFL) + B·Δ(Meta I)) The prefactors A and B were determined by a least squares fit in the region 1010–945 cm−1, where Meta I exhibits a specific positive band (cf. Fig. S1).
Author contributions
M. E. conceived the experiments, M. E. and R. K. conducted the experiments, M. S. and P. S. provided the rhodopsin mutant R3.50L and ArrFL-1 peptide, M. E. and R. K. analyzed the results, F. J. B. and K. P. H. supervised the study, M. E. wrote the manuscript with contributions from A. S. R., P. W. H., M. S., P. S., and K. P. H. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
We acknowledge Margaux Kreitman for critically reading the manuscript. M. E. is furthermore grateful to Dr. Wayne L. Hubbell for financial support during the final stage of this project.
This work was supported by the Deutsche Forschungsgemeinschaft EL 779 (to M. E.), SFB 1078 (to F. J. B. and P. S.), Deutsche Forschungsgemeinschaft Cluster of Excellence “Unifying Concepts in Catalysis” Research Field D3/E3–1 (to P. S.), and SFB 740 to (P. W. H., P. S., and K. P. H.), and the European Research Council TUDOR (to K. P. H.). The authors declare that they have no conflicts of interest with the contents of this article.
Note that the amino acid numbering in the holo-arrestin sequence is shifted by one compared with ArrFL-1.
- GPCR
- G protein–coupled receptor
- TM
- transmembrane helix
- PBS
- peptide-binding spectra.
References
- 1. Farrens D. L., Altenbach C., Yang K., Hubbell W. L., and Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin., Science 274, 768–770 10.1126/science.274.5288.768 [DOI] [PubMed] [Google Scholar]
- 2. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H.-W. W., Hofmann K. P., and Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation, Nature 455, 497–502 10.1038/nature07330 [DOI] [PubMed] [Google Scholar]
- 3. Hofmann K. P., Scheerer P., Hildebrand P. W., Choe H.-W., Park J. H., Heck M., and Ernst O. P. (2009) A G protein-coupled receptor at work: the rhodopsin model., Trends Biochem. Sci. 34, 540–552 10.1016/j.tibs.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 4. Shenoy S. K., and Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction., Trends Pharmacol. Sci. 32, 521–533 10.1016/j.tips.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou X. E., He Y., de Waal P. W., Gao X., Kang Y., Van Eps N., Yin Y., Pal K., Goswami D., White T. A., Barty A., Latorraca N. R., Chapman H. N., Hubbell W. L., Dror R. O., Stevens R. C., Cherezov V., Gurevich V. V., Griffin P. R., Ernst O. P., Melcher K., and Xu H. E. (2017) Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 170, 457–469.e13 10.1016/j.cell.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sommer M. E., Smith W. C., and Farrens D. L. (2005) Dynamics of arrestin-rhodopsin interactions: Arrestin and retinal release are directly linked events. J. Biol. Chem. 280, 6861–6871 10.1074/jbc.M411341200 [DOI] [PubMed] [Google Scholar]
- 7. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., and Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 10.1073/pnas.0600733103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schleicher A., Kühn H., and Hofmann K. P. (1989) Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry 28, 1770–1775 10.1021/bi00430a052 [DOI] [PubMed] [Google Scholar]
- 9. Pulvermüller A., Maretzki D., Rudnicka-Nawrot M., Smith W. C., Palczewski K., and Hofmann K. P. (1997) Functional differences in the interaction of arrestin and its splice variant, p44, with rhodopsin. Biochemistry 36, 9253–9260 10.1021/bi970772g [DOI] [PubMed] [Google Scholar]
- 10. Scheerer P., and Sommer M. E. (2017) Structural mechanism of arrestin activation. Curr. Opin Struct. Biol. 45, 160–169 10.1016/j.sbi.2017.05.001 [DOI] [PubMed] [Google Scholar]
- 11. Rasmussen S. G. F., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T. A., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., and Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex., Nature 477, 549–555 10.1038/nature10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H. E., Zhang C., Moeller A., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han M., Smith S. O., and Sakmar T. P. (1998) Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry 37, 8253–8261 10.1021/bi980147r [DOI] [PubMed] [Google Scholar]
- 14. Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., and Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. U.S.A. 109, 119–124 10.1073/pnas.1114089108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goncalves J. A., South K., Ahuja S., Zaitseva E., Opefi C. A., Eilers M., Vogel R., Reeves P. J., and Smith S. O. (2010) Highly conserved tyrosine stabilizes the active state of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 107, 19861–19866 10.1073/pnas.1009405107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elgeti M., Kazmin R., Heck M., Morizumi T., Ritter E., Scheerer P., Ernst O. P., Siebert F., Hofmann K. P., and Bartl F. J. (2011) Conserved Tyr223(5.58) plays different roles in the activation and G-protein interaction of rhodopsin. J. Am. Chem. Soc. 133, 7159–7165 10.1021/ja200545n [DOI] [PubMed] [Google Scholar]
- 17. Szczepek M., Beyrière F., Hofmann K. P., Elgeti M., Kazmin R., Rose A., Bartl F. J., von Stetten D., Heck M., Sommer M. E., Hildebrand P. W., and Scheerer P. (2014) Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat. Commun. 5, 4801 10.1038/ncomms5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manglik A., and Kobilka B. (2014) The role of protein dynamics in GPCR function: Insights from the β2AR and rhodopsin. Curr. Opin. Cell Biol. 27, 136–143 10.1016/j.ceb.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim T. H., Chung K. Y., Manglik A., Hansen A. L., Dror R. O., Mildorf T. J., Shaw D. E., Kobilka B. K., and Prosser R. S. (2013) The role of ligands on the equilibria between functional states of a G protein–coupled receptor. J. Am. Chem. Soc. 135, 9465–9474 10.1021/ja404305k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., and Kobilka B. K. (2013) The dynamic process of β(2)-adrenergic receptor activation. Cell 152, 532–542 10.1016/j.cell.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fahmy K., Siebert F., and Sakmar T. P. (1995) Photoactivated state of rhodopsin and how it can form. Biophys. Chem. 56, 171–181 10.1016/0301-4622(95)00030-2 [DOI] [PubMed] [Google Scholar]
- 22. Arnis S., Fahmy K., Hofmann K. P., and Sakmar T. P. (1994) A conserved carboxylic acid group mediates light-dependent proton uptake and signaling by rhodopsin. J. Biol. Chem. 269, 23879–23881 [PubMed] [Google Scholar]
- 23. Vogel R., Mahalingam M., Lüdeke S., Huber T., Siebert F., and Sakmar T. P. (2008) Functional role of the “ionic lock”—an interhelical hydrogen-bond network in family A heptahelical receptors. J. Mol. Biol. 380, 648–655 10.1016/j.jmb.2008.05.022 [DOI] [PubMed] [Google Scholar]
- 24. Mahalingam M., Martínez-Mayorga K., Brown M. F., and Vogel R. (2008) Two protonation switches control rhodopsin activation in membranes. Proc. Natl. Acad. Sci. U.S.A. 105, 17795–17800 10.1073/pnas.0804541105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomsen A. R. B., Plouffe B., Cahill T. J. 3rd, Shukla A. K., Tarrasch J. T., Dosey A. M., Kahsai A. W., Strachan R. T., Pani B., Mahoney J. P., Huang L., Breton B., Heydenreich F. M., Sunahara R. K., Skiniotis G., Bouvier M., and Lefkowitz R. J. (2016) GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 10.1016/j.cell.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Min K. C., Zvyaga T. A., Cypess A. M., and Sakmar T. P. (1993) Characterization of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Mutations on the cytoplasmic surface affect transducin activation. J. Biol. Chem. 268, 9400–9404 [PubMed] [Google Scholar]
- 27. Sung C. H., Davenport C. M., and Nathans J. (1993) Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J. Biol. Chem. 268, 26645–26649 [PubMed] [Google Scholar]
- 28. Sommer M. E., Elgeti M., Hildebrand P. W., Szczepek M., Hofmann K. P., and Scheerer P. (2015) Structure-based biophysical analysis of the interaction of rhodopsin with G protein and arrestin. Meth. Enzymol. 556, 563–608 10.1016/bs.mie.2014.12.014 [DOI] [PubMed] [Google Scholar]
- 29. Kim M., Vishnivetskiy S. A., Van Eps N., Alexander N. S., Cleghorn W. M., Zhan X., Hanson S. M., Morizumi T., Ernst O. P., Meiler J., Gurevich V. V., and Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412 10.1073/pnas.1216304109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beyrière F., Sommer M. E., Szczepek M., Bartl F. J., Hofmann K. P., Heck M., and Ritter E. (2015) Formation and decay of the arrestin·rhodopsin complex in native disc membranes. J. Biol. Chem. 290, 12919–12928 10.1074/jbc.M114.620898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feuerstein S. E., Pulvermüller A., Hartmann R., Granzin J., Stoldt M., Henklein P., Ernst O. P., Heck M., Willbold D., and Koenig B. W. (2009) Helix formation in arrestin accompanies recognition of photoactivated rhodopsin. Biochemistry 48, 10733–10742 10.1021/bi900544p [DOI] [PubMed] [Google Scholar]
- 32. Pulvermüller A., Schroder K., Fischer T., and Hofmann K. P. (2000) Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J. Biol. Chem. 275, 37679–37685 10.1074/jbc.M006776200 [DOI] [PubMed] [Google Scholar]
- 33. Siebert F. (1995) Application of FTIR spectroscopy to the investigation of dark structures and photoreactions of visual pigments. Isr. J. Chem. 35, 309–323 10.1002/ijch.199500033 [DOI] [Google Scholar]
- 34. Beck M., Sakmar T. P., and Siebert F. (1998) Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry 37, 7630–7639 10.1021/bi9801560 [DOI] [PubMed] [Google Scholar]
- 35. Knierim B., Hofmann K. P., Ernst O. P., and Hubbell W. L. (2007) Sequence of late molecular events in the activation of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 104, 20290–20295 10.1073/pnas.0710393104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoersch D., Otto H., Wallat I., and Heyn M. P. (2008) Monitoring the conformational changes of photoactivated rhodopsin from microseconds to seconds by transient fluorescence spectroscopy. Biochemistry 47, 11518–11527 10.1021/bi801397e [DOI] [PubMed] [Google Scholar]
- 37. Fahmy K., and Sakmar T. P. (1993) Regulation of the rhodopsin-transducin interaction by a highly conserved carboxylic acid group. Biochemistry 32, 7229–7236 10.1021/bi00079a020 [DOI] [PubMed] [Google Scholar]
- 38. Elgeti M., Rose A. S., Bartl F. J., Hildebrand P. W., Hofmann K. P., and Heck M. (2013) Precision vs flexibility in GPCR signaling. J. Am. Chem. Soc. 135, 12305–12312 10.1021/ja405133k [DOI] [PubMed] [Google Scholar]
- 39. Vogel R., Martell S., Mahalingam M., Engelhard M., and Siebert F. (2007) Interaction of a G protein-coupled receptor with a G protein-derived peptide induces structural changes in both peptide and receptor: A Fourier-transform infrared study using isotopically labeled peptides. J. Mol. Biol. 366, 1580–1588 10.1016/j.jmb.2006.12.019 [DOI] [PubMed] [Google Scholar]
- 40. Elgeti M., Ritter E., and Bartl F. J. (2008) New insights into light-induced deactivation of active rhodopsin by SVD and global analysis of time-resolved UV/vis- and FTIR-data. Z. Phys. Chem. 222, 1117–1129 10.1524/zpch.2008.5392 [DOI] [Google Scholar]
- 41. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 10.1016/j.bbabio.2007.06.004 [DOI] [PubMed] [Google Scholar]
- 42. Park J. H., Scheerer P., Hofmann K. P., Choe H.-W., and Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin., Nature 454, 183–187 10.1038/nature07063 [DOI] [PubMed] [Google Scholar]
- 43. Hauser K., Engelhard M., Friedman N., Sheves M., and Siebert F. (2002) Interpretation of amide I difference bands observed during protein reactions using site-directed isotopically labeled bacteriorhodopsin as a model system. J. Phys. Chem. A 106, 3553–3559 10.1021/jp012926e [DOI] [Google Scholar]
- 44. Van Eps N., Caro L. N., Morizumi T., Kusnetzow A. K., Szczepek M., Hofmann K. P., Bayburt T. H., Sligar S. G., Ernst O. P., and Hubbell W. L. (2017) Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. U.S.A. 201620405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ostermaier M. K., Peterhans C., Jaussi R., Deupi X., and Standfuss J. (2014) Functional map of arrestin-1 at single amino acid resolution. Proc. Natl. Acad. Sci. U.S.A. 111, 1825–1830 10.1073/pnas.1319402111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi W., Sports C. D., Raman D., Shirakawa S., Osawa S., and Weiss E. R. (1998) Rhodopsin arginine-135 mutants are phosphorylated by rhodopsin kinase and bind arrestin in the absence of 11-cis-retinal. Biochemistry 37, 4869–4874 10.1021/bi9731100 [DOI] [PubMed] [Google Scholar]
- 47. Arnis S., and Hofmann K. P. (1993) Two different forms of metarhodopsin II: Schiff base deprotonation precedes proton uptake and signaling state. Proc. Natl. Acad. Sci. U.S.A. 90, 7849–7853 10.1073/pnas.90.16.7849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghanouni P., Schambye H., Seifert R., Lee T. W., Rasmussen S. G., Gether U., and Kobilka B. K. (2000) The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J. Biol. Chem. 275, 3121–3127 10.1074/jbc.275.5.3121 [DOI] [PubMed] [Google Scholar]
- 49. Strachan R. T., Sun J. P., Rominger D. H., Violin J. D., Ahn S., Rojas Bie Thomsen A., Zhu X., Kleist A., Costa T., and Lefkowitz R. J. (2014) Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 289, 14211–14224 10.1074/jbc.M114.548131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schafer C. T., Fay J. F., Janz J. M., and Farrens D. L. (2016) Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state., Proc. Natl. Acad. Sci. U.S.A. 113, 11961–11966 10.1073/pnas.1606347113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meyer C. K., Böhme M., Ockenfels A., Gartner W., Hofmann K. P., Ernst O. P., and Gärtner W. (2000) Signaling states of rhodopsin. Retinal provides a scaffold for activating proton transfer switches. J. Biol. Chem. 275, 19713–19718 10.1074/jbc.M000603200 [DOI] [PubMed] [Google Scholar]
- 52. Vogel R., Fan G. B., Sheves M., and Siebert F. (2000) The molecular origin of the inhibition of transducin activation in rhodopsin lacking the 9-methyl group of the retinal chromophore: A UV-vis and FTIR spectroscopic study. Biochemistry 39, 8895–8908 10.1021/bi000852b [DOI] [PubMed] [Google Scholar]
- 53. Vogel R., Lüdeke S., Siebert F., Sakmar T. P., Hirshfeld A., and Sheves M. (2006) Agonists and partial agonists of rhodopsin: retinal polyene methylation affects receptor activation. Biochemistry 45, 1640–1652 10.1021/bi052196r [DOI] [PubMed] [Google Scholar]
- 54. Knierim B., Hofmann K. P., Gärtner W., Hubbell W. L., and Ernst O. P. (2008) Rhodopsin and 9-demethyl-retinal analog: Effect of a partial agonist on displacement of transmembrane helix 6 in class A G protein-coupled receptors. J. Biol. Chem. 283, 4967–4974 10.1074/jbc.M703059200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.