

Abstract
Fluid movement within the heart generates substantial shear forces, but the effect of this mechanical stress on the electrical activity of the human heart has not been examined. The fast component of the delayed rectifier potassium currents responsible for repolarization of the cardiac action potential, Ikr, is encoded by the human ether-a-go-go related gene (hERG) channel. Here, we exposed hERG1a channel–expressing HEK293T cells to laminar shear stress (LSS) and observed that this mechanical stress increased the whole-cell current by 30–40%. LSS shifted the voltage dependence of steady-state activation of the hERG channel to the hyperpolarizing direction, accelerated the time course of activation and recovery from inactivation, slowed down deactivation, and shifted the steady-state inactivation to the positive direction, all of which favored the hERG open state. In contrast, the time course of inactivation was faster, favoring the closed state. Using specific inhibitors of focal adhesion kinase, a regulator of mechano-transduction via the integrin pathway, we also found that the LSS-induced modulation of the whole-cell current depended on the integrin pathway. The hERG1b channel variant, which lacks the Per-Arnt-Sim (PAS) domain, and long QT syndrome–associated variants having point mutations in the PAS domain were unaffected by LSS, suggesting that the PAS domain in hERG1a channel may be involved in sensing mechanical shear stress. We conclude that a mechano-electric feedback pathway modulates hERG channel activity through the integrin pathway, indicating that mechanical forces in the heart influence its electrical activity.
Keywords: hERG, patch clamp, membrane biophysics, electrophysiology, mechanotransduction, shear stress, heart, integrin, focal adhesions, cytoskeleton, LSS, laminar shear stress, focal adhesion kinase (FAK), Per-Arnt-Sim (PAS), action potential duration (APD), cardiac fluid movement, potassium channel, mechanical stress
Introduction
The beating of the heart is initiated by an elaborate and well-tuned electrical system that generates the cardiac action potential (AP).2 The shape and duration of the cardiac AP is held within close tolerances as small variations can lead to cardiac arrhythmias and even catastrophic heart failure. The firing of the AP, in turn, results in contractions of the musculature of the heart and hence blood flow through the body. The effect of mechanical forces at work during the heart beat on the electrical system of the heart have not, surprisingly, been investigated in detail so far. Regulation of ion channel activity plays a central role in controlling heart rate, rhythm, and contractility responses to cardiovascular demands. Dynamic beat to beat regulation of ion channels in the context of hemodynamic shear forces is still a gray area. Here, we investigate the influence of mechanical shear on a critical component of the electrical system a voltage-gated ion channel that plays a major role in the repolarization of the cardiac AP.
Voltage-gated ion channels are abundant in the heart, playing pivotal roles in the proper shaping of the cardiac action potential. Several neuronal voltage-gated potassium channels have recently been shown to be mechanosensitive (1). Voltage-gated cardiac channels (cardiac VGCs) share the same architecture as neuronal VGCs, raising the possibility that they are also mechanosensitive (2). The ventricles lack specialist stretch-activated channels, i.e. channels that are relatively insensitive to other stimuli but mediate cation currents in response to stretch (3, 4). The myocardium is continuously exposed to a variety of forces over each contraction and must therefore adapt to the associated mechanical stresses. One such major force is the shear stress that arises from blood flow and the sliding of myocardial layers against each other with each heart beat (5). If cardiac VGCs respond to shear forces (6, 7), they could contribute to both physiological and pathological mechano-electric feedback in the heart. However, it is unclear as to whether mechanosensitivity is an integral part of cardiac voltage-gated channel function and, if so, how gating transitions are modified under shear stress.
The human ether-a-go-go related gene (hERG) encodes a K+ channel that is responsible for the IKr current that contributes to the falling phase of the cardiac action potential. hERG channels, unlike other voltage-gated potassium channels, have a unique inactivation mechanism that limits potassium efflux during depolarization but rebounds during repolarization, thereby facilitating a significant outward K+ tail current (8, 9). Perturbation of these repolarizing currents could result in alteration of the cardiac action potential duration (APD) (10, 11). Prolonged APD is seen in disease states like long QT syndrome (12), whereas the reduced APD results in short QT syndromes (13–15). Mutations in hERG have been associated with both long and short QT syndromes as well as life-threatening arrhythmias. Chronic heart failure could follow perturbation of hERG channel function (16).
The transmission of external forces acting on cells to stretch-sensitive channels in the plasma membrane requires the intermediacy of physical structures. A collection of transmembrane proteins that transmits force and initiates transduction pathways within cells is the integrin family. Integrins are transmembrane glycoproteins forming heterodimers that act as extracellular matrix receptors and physically link the extracellular matrix to the cytoskeleton (17). The β1D splice variant is the major β isoform expressed in adult heart (18, 19). In myocytes, integrins are a component of costameres, which are macromolecular complexes with Z lines specialized for force transmission (19–21) and are activated by clustering of integrin receptors. Focal adhesion kinase binds to the cytoplasmic domain of β1 integrin, resulting in autophosphorylation of FAK at Tyr-397. Previous studies using heterologous expression of hERG channels in HEK293T cells to study its interaction with β1 integrins (22) have reported effects on current activation and cell spreading upon interaction of β1 integrins with the extracellular matrix. This, in turn, activates downstream effectors FAK (autophosphorylation at Tyr-397) and Rac1 in HEK293T cells. β1 integrins are highly expressed in cardiomyocytes and shown to vary with age of the myocytes (23). Blockage of hERG channels resulted in down-regulation of FAK phosphorylation (24) indicating a two-way communication between FAK and hERG. The FAK–integrin system could thus constitute a pathway to transmit forces that may arise both from intracellular (contractile) forces and forces from outside the cell (fluid shear stress) in the heart (25) to the hERG channel (26, 27).
We have expressed hERG channels in HEK293T cells and investigated the response of whole-cell currents to shear stress, reproduced in vitro by laminar flow in a patch-clamp recording chamber. Laminar shear stress is correlated with an increase in whole-cell currents together with shifts of the V1/2 of activation in the hyperpolarizing direction, the V1/2 of inactivation toward depolarizing potentials, acceleration of the activation kinetics, decrease in deactivation rates, and increase in the rate of recovery from inactivation. All of these changes favor the open state. Conversely, shear stress also increased the rate of inactivation favoring a non-conducting state. Shear effects were saturable and reversible. β1 integrins are highly expressed in HEK293T cells (22), and our data suggest that mechano-transduction occurs via the integrin pathway. The cytoskeleton participates in the shear response. hERG1b, a splice variant lacking the PAS domain, as well as long QT mutants harboring point mutations in the PAS domain were unresponsive to shear stress suggesting a possible role of the PAS domain in mechanosensation.
Results
Laminar shear stress increases hERG currents
The effect of laminar shear stress (LSS) on hERG-transfected HEK293T cells was investigated using the whole-cell patch-clamp configuration in the voltage-clamp mode. Voltage was stepped to a series of potentials from a holding potential of −80 mV and then taken to a test potential of −60 mV. The current just prior to the −60-mV step is taken as the step current, whereas that immediately after the step is the tail current. As shown in Fig. 1B, increased flow rate caused enhancement in tail current recorded at −60 mV from a step potential of 0 mV. Data are presented as percentage change from the value of the parameter at a baseline flow rate of 1 ml/min, which was taken as pre-shear. We present all data as mean ± S.D.
Figure 1.

Laminar shear stress mediated effects on hERG1a currents. A and B, representative current trace in response to a step potential to 0 mV (2 s) to activate hERG currents followed by a tail current elicited at −60 mV (3 s) from a holding potential of −80 mV. Pre-shear and shear stress response are indicated by arrows. B, scatter plot shows current enhancement during shear stress at both step and tail current (mean ± S.D.; p < 0.001, n = 20, paired t test). Scale bars, 2 nA and 500 ms. C, dose-dependent effect of fluid shear stress (flow rate in ml/min) on hERG1a tail currents. x axis denotes different flow rate, and y axis is the percentage increase during shear stress application (mean ± S.E., n = 5). D, time course of the effect of shear stress on hERG1a tail currents. Responses of hERG1a currents between pre-shear and laminar shear stress were reversible (mean ± S.E., n = 6). Individual data points at each given voltage are represented as mean ± S.E.
Flow rates between 1 and 15 ml/min were tested, and tail current at −60 mV was found to be significantly greater under shear than pre-shear at all flow rates above 1 ml/min. The data suggest two states for the whole-cell current and fit well to a Boltzmann function with a midpoint at 5.2 ± 0.10 ml/min (n = 5). Currents at flow rates of 7, 10, and 15 ml/min were essentially indistinguishable, indicating that shear-induced changes in hERG physiology had saturated by 7 ml/min (Fig. 1C). All further studies compared recordings at flow rates of 1 and 10 ml/min. The corresponding shear stress to which the cells in the recording chamber were exposed is not precisely known. However, assuming a linear velocity variation in the direction orthogonal to flow, the shear stress is estimated to be 11.3 dyn/cm2 at 10 ml/min (28). In addition to enhancement of tail current, the step current also increased on application of shear stress (Fig. 1, A and C). An increase of around 27% from 42.5 ± 10.02 to 54 ± 16.48 pA/pF (n = 16, p < 0.001, paired t test) was observed for a step to 0 mV. Tail currents in the same experiment increased from 51.2 ± 13.80 pA/pF at 1 ml/min flow to 66.8 ± 16.56 pA/pF (n = 20, p < 0.001) at 10 ml/min, an increase of 30.4%. Around 88% of the cells recorded showed an increase in current in both step and tail current on being subjected to shear stress. However, some cells displayed an increase in either step or tail current but not both.
The reversibility of the phenomenon was tested at −60 mV by switching flow rates between 1 and 10 ml/min (Fig. 1D). After acquiring a baseline at 1 ml/min, flow was increased to 10 ml/min for 390 s, then allowed to recover at 1 ml/min for 360 s before returning to 10 ml/min. Tail currents increased by the first measured time point, which was at 20 s after switching to 10 ml/min on both occasions, and they continued to increase with a time constant of 91.4 s (fit is shown with a smooth black line, Fig. 1D). Saturation of the response was seen in about 300 s. Decay to baseline was slower, with a time constant of 154.8 s (fit is shown with a smooth black line, Fig. 1D). Recovery saturated at a value that was 13% higher than the original baseline. The second episode of high flow resulted in an increase of current to a value that was 95% of the first cycle (n = 6).
Another mechanical stress is membrane stretching by cell swelling. We recorded hERG1a potassium currents in whole-cell mode first under iso-osmotic conditions and then under hypo-osmotic conditions for a period of 2.5 min to induce cell swelling at a constant flow rate of 1 ml/min (Fig. S4A). Osmotic strength was modified using sorbitol to maintain the ionic composition. On switching from iso-osmotic to hypo-osmotic solutions, the step currents increased from 56.22 ± 12.40 to 69.16 ± 13.14 pA/pF at 0 mV (an increase of 23.2%), and tail currents increased from 80.66 ± 20.33 to 97.97 ± 17.17 pA/pF at −60 mV (an increase of 21.4%). Statistical comparison was done using paired t test, n = 6, p = 0.004, and p = 0.03 for step and tail currents, respectively. Cell capacitance did not change significantly (p > 0.05, n = 6, paired t test) between iso-osmotic (15.18 ± 4.87 pF) and hypo-osmotic conditions (14.54 ± 4.48 pF), which renders the fusion of channel containing vesicles with the plasma membrane an unlikely source of the increased current (Fig. S4B).
The initial survey of sensitivity to shear stress was restricted to a 0-mV step (Fig. 2, A–C). Analysis of the full range of step potentials at 10 ml/min reveals that the step current is enhanced in the presence of shear at all potentials below +10 mV (Fig. 2C). Additionally, when the I–V plots are normalized, a shift of 10 mV to hyperpolarizing potentials can be observed in the presence of shear (Fig. 2C, inset). This means that normalized current is higher under shear than in its absence at any potential below the peak potential of +10 mV. The combination of voltage shift and increased amplitude result in the two curves being indistinguishable above +10 mV (Fig. 2C).
Figure 2.

Laminar shear stress effect on hERG1a channel activation. A and B, representative current traces in response to a series of membrane depolarizations in a HEK293T-expressing hERG1a. Currents were elicited by a series of 2-s depolarization steps (between −70 and +70 mV) followed by a repolarization step to −60 mV. Scale bar, 2 nA and 1 s. C, current-voltage (I–V) relation curve plotted from current measured at the end of depolarizing step potentials (relative current amplitude, I/Imax, during depolarizing step potentials plotted against the step potential, Imax = maximal current during application of shear stress). D, voltage-dependent activation curves plotted from peak tail currents during a repolarization step to −60 mV after depolarizarion to various potentials. Data were normalized to the maximum current in the same curve. The solid lines and dashed lines correspond to the fitted Boltzmann functions. The V1/2 and the slope factor for pre-shear state is 0.6 ± 0.23 and 8.26 ± 0.0.38 mV, respectively, and during laminar shear stress was −8.8 ± 0.39 and 9.165 ± 0.0.34 mV, respectively. Post-shear switch to pre-shear flow rate yielded a V1/2 of −2.4 ± 0.46 and a slope factor of 5.57 ± 0.040 mV (mean ± S.D., n = 7). Individual data points at each given voltage are represented as mean ± S.E.
The tail current seen immediately after stepping to the test potential of −60 mV is proportional to the total number of channels that had opened during the step pulse. The voltage dependence of channel opening could be fit with a Boltzmann function with a mid-point (V1/2) at 0.6 ± 0.23 mV at a flow rate of 1 ml/min and at −8.8 ± 0.39 mV at 10 ml/min, a hyperpolarizing shift of −9.4 mV (Fig. 2D, n = 7). A mid-point (V1/2) of −2.4 ± 0.46 was observed after switching back to 1 ml/min flow rate, which is close to the pre-shear value (Fig. 2D, dashed lines, n = 7). No change in reversal potential was seen (Fig. 3A).
Figure 3.

Effect of laminar shear stress on deactivation properties of hERG1a. A. reversal potential measurement from a fully activated I–V. Currents were elicited by a 2-s depolarizing step potential to 0 mV followed by a 3-s repolarization step (between −120 and +20 mV). Scale bar, 2 nA and 1 s (n = 4). B and C, tail currents measured between −120 and −40 mV with an interval of 10 mV were fitted with a double-exponential function. Time constants of fast and slow components at −60 and −100 mV are plotted for comparison between pre-shear and shear stress (p < 0.05, n = 5, paired t test). Individual data points at each given voltage are represented as mean ± S.E.
LSS affects hERG channel activation and deactivation rates
The tail current is proportional to the number of channels open at the end of the step pulse. Decay of the tail current is due to the closing of these channels, i.e. deactivation. The decay of the tail current could be fit to two exponentials. At −60 and −100 mV, both the deactivation time constants (τdeact) (Fig. 3, B and C) increased with shear 161.3 ± 38.2 to 173 ± 38.07 ms (p < 0.0001, n = 5, paired t test) and 63 ± 18.24 to 92 ± 39.23 ms (p = 0.01, n = 5, paired t test), respectively, for the fast component, which includes 90% of the deactivation, and 1040 ± 161 to 1137 ± 57.92 ms (p = 0.01, n = 5, paired t test) and 343.8 ± 93.39 to 558.9 ± 168.1 ms (p = 0.02, n = 5, paired t test), respectively, for the slow component. Hence the outward current through hERG lasts longer under shear. Examination of kinetic transitions provides more information regarding the influence of shear stress on hERG current activation. Fig. 4A illustrates the envelope of tails protocol used to measure the activation time constant uncontaminated by the faster inactivation process (29). Because the inactivation of HERG channels is released at a faster rate than the channels deactivate (30), this is a more specific measure of activation. The tail current at −60 mV, which is proportional to the number of channels having transitioned from closed to open during the preceding step to 0 mV, is plotted as a function of time in Fig. 4B. Peak tail current values at different time points were fit with a single exponential function as shown. The analysis reveals a t½ of 279.9 ± 5.9 ms at a flow rate of 1 ml/min and t½ of 155.5 ± 17 ms under LSS (Fig. 4C), indicating that transitions from the closed to the open state almost double under shear (p < 0.0001, n = 8, paired t test).
Figure 4.

Time course of activation is faster after application laminar shear stress. A, currents evoked using an envelope of tails protocol to determine the time course of activation. Peak tail current was evoked by a step to −60 mV following a pre-pulse of increasing duration to 0 mV. Holding was −80 mV. B, apparent activation is faster during shear stress application. Individual data points at each given voltage are represented as mean ± S.E. The peak amplitudes of the tail currents (A) were plotted against test pulse duration and fitted to a single exponential function. C, time constant of activation (t½) was 279.9 ± 58.2 ms at a flow rate of 1 ml/min and 155.5 ± 49.2 ms under LSS (mean ± S.D., p < 0.0001, n = 8 paired t test).
Homomeric hERG1b channels do not respond to laminar shear stress
hERG1b is a splice variant that lacks the PAS domain found in the N-terminal cytoplasmic tail of hERG1a. hERG1b also has a unique N-terminal cap comprising 20 amino acid residues (31, 32). It has been reported earlier that hERG1b fails to form functional channels in HEK293T (29). However, with 2 μg of DNA we were able to get measurable currents (step current density, 36.2 ± 11.59 pA/pF, and tail current density: 11.4 ± 4.39 6 pA/pF). As shown in Fig. 5A and Fig. S1A, laminar shear stress had no effect on hERG1b current activation (p > 0.05, n = 7, paired t test).
Figure 5.

Contribution of PAS domain on hERG mechanosensitivity. A, hERG1b currents show no response to laminar shear stress (mean ± S.D., p > 0.05, n = 7, paired t test). B, hERG1b co-transfected with 1–131 amino acids (PAS domain). Tail currents responded to shear stress (mean ± S.D., p < 0.05, n = 5, paired t test). C, response of heterotetrameric hERG1a/1b channels to laminar shear stress (mean ± S.D., p < 0.01, n = 7–8, paired t test). ns, non-significant.
Because hERG1b lacks the PAS domain and is resistant to shear stress, we tested the role of the PAS domain in the shear response. The PAS domain alone, expressed as an independent polypeptide, interacts directly with hERG1a/1b heteromeric channels, and it has been suggested that it occupies sites left vacant by hERG1b subunits (33–35). Expressing the PAS domain together with hERG1b resulted in a large increase in currents (Fig. S1B). As shown in Fig. 5B, increasing flow rates to 10 ml/min had little effect in step currents in this system but did increase the tail currents by around 16% (73.1 ± 26.04 to 87.2 ± 29.43 pA/pF, p = 0.01, n = 5, paired t test). The PAS domain thus restored some degree of mechanosensitivity.
Both hERG1a and hERG1b are co-expressed in cardiac tissue, and the heteromultimer has been well characterized in heterologous expression systems (29, 34, 36). Cells transfected with 1 μg of DNA of each construct exhibited substantial currents resembling native Ikr consisting of a large outward current and a fast deactivating tail current that is much larger than in the cells transfected with hERG1a alone (Fig. S1C). As shown in Fig. 5C, increasing the flow rate to 10 ml/min increased current still further from 116.8 ± 11.15 to 128.4 ± 12.11 pA/pF (p = 0.01, n = 7) for step currents and from 63.52 ± 21.46 to 77.10 ± 20.48 pA/pF (p = 0.00, n = 8, paired t test) for tail currents. The fractional increases observed were significantly smaller than those observed for hERG1a alone (10% versus 27% for step currents and 21.4% versus 30.4% for tail currents).
Laminar shear stress effects on inactivation and recovery from inactivation
We investigated the inactivation properties of the hERG1a channel (Fig. 6). A first three-step protocol was applied to investigate the voltage-dependent steady-state inactivation. After reaching a steady state at +40 mV, the channels were subjected to brief (10 ms) hyperpolarizing pulses ranging from +30 to −140 mV. The peak current amplitude after a return to +40 mV was normalized and plotted against the potential of the hyperpolarizing step (Fig. 6A). The data were fit to a Boltzmann function with a mid-point V1/2 at −54.6 ± 3.3 mV at a flow rate of 1 ml/min and −51.5 ± 3.63 mV at 10 ml/min, which is not significantly different (p > 0.05, n = 7, paired t test).
Figure 6.

Effect of LSS on inactivation properties of hERG1a. A, currents were measured at +40 mV following a series of 10-ms steps to a range of voltages from +30 to −140 mV in 10-mV decrements. The data were fitted to a Boltzmann function. Steady-state inactivation plot showing shift of V1/2 and slope factor from −54.64 ± 3.3 and −21.01 ± 3.38 mV, respectively (pre-shear) to −51.45 ± 3.63 and −20.13 ± 3.66 mV, respectively (shear stress). A right shift V1/2 of 3.1 mV (n = 7). Scale bar, 5 nA and 1 s. B, 200-ms pulse to +40 mV to activate and then inactivate hERG followed by a brief hyperpolarizing step (10 ms) to −120 mV to allow the channels to recover from inactivation. In the third pulse, carrying the potential between 0 and +40 mV allowed the inactivation time course to be measured as a function of voltage. The time constant of onset of inactivation was estimated by fitting the decay of the currents in the third pulse to a single exponential function and plotted as a function of test potential (p < 0.01 at 0 mV, n = 9, paired t test). Scale bar, 4 nA and 50 ms. C, tail currents were evoked at different potentials from −60 to +10 mV following a 4-s, +20-mV pulse. Plot quantification showing data from recovery from inactivation at −50 mV is faster during shear stress. Time constants were measured as the single exponential fit to the rising phase of the tail current (p < 0.001 at −50 mV, n = 5, paired t test). Scale bar, 2 nA and 1 s. Individual data points at each given voltage are represented as mean ± S.E.
The kinetics of inactivation were studied by activating the channels with a 200-ms depolarizing step to +40 mV followed by a brief hyperpolarizing step (10 ms) to −120 mV to allow the channels to recover from inactivation. The hyperpolarizing step was followed by steps to various potentials from 0 to +40 mV. The onset of inactivation is seen in the decline of the observed current to the steady state (Fig. 6B). Fitting the current traces to a single exponential function yields the time constant of inactivation (τinact). LSS decreased the time constant for inactivation (Fig. 6B) (13.3 ± 4.2 to 9.3 ± 2.2 ms at 0 mV, p = 0.005, n = 9, paired t test). Shear thus decreases the dwell time in the open state and effectively suppresses outward current during positive voltage commands (29, 36).
We further determined the time constants for the recovery from inactivation (τrecovery). After reaching a steady state at +20 mV, tail currents were evoked at different potentials from −60 to +10 mV. Single exponential fits to the rising phase of the tail current yielded time constants for recovery at each test potential (Fig. 6C). Recovery was found to be faster during LSS as compared with pre-shear state at all potentials tested. At −50 mV, τrecovery was found to be 9 ± 2.15 and 7.7 ± 1.65 ms in pre-shear and post-shear conditions, respectively (p = 0.001, n = 5, paired t test), consistent with the resurgent current and larger tail current amplitudes seen in Fig. 1, A and B.
Laminar shear stress requires an intact cytoskeleton
10 μm cytochalasin D was used to depolymerize actin filaments (Fig. 7, A and A(i), and Fig. S2, A and C). After a 40-min treatment of hERG1a-transfected HEK293T cells, step currents were unaffected by shear (74.5 ± 42.38 pA/pF at 1 ml/min and 77.1 ± 47.84 pA/pF at 10 ml/min). A 14.5% increase in tail current, from 59.9 ± 17.71 to 68.5 ± 18.91 pA/pF (p = 0.003, n = 5, paired t test), is smaller than the 30.4% increase in tail currents observed with untreated cells, but the response was not completely eliminated.
Figure 7.

Shear stress and cytoskeleton. Representative current traces from hERG1a channel before (pre-shear) (A) and during laminar shear stress (shear stress) (B) are indicated by arrows. Currents were elicited by a depolarizing step to 0 mV followed by a repolarization to −60 mV from a holding potential of −80 mV. A(i), 10 μm cytochalasin D was incubated for 40 min to disrupt actin filaments. Scatter plot shows partial mechanosensitivity as seen with increase in the tail current after LSS application (mean ± S.D., p < 0.05, n = 5, paired t test). Scale bar, 1 nA and 500 ms. B(i), 10 μm nocodazole incubated for 2 h to destroy the microtubule network. Scatter plot shows increase in hERG currents during application of shear stress (mean ± S.D., p < 0.01, n = 5, paired t test). Scale bar, 1 nA and 500 ms. C (untreated versus cytochalasin (Cyto)/nocodazole), 10 μm cytochalasin D was incubated for 40 min to disrupt actin filaments (mean ± S.D., p < 0.05, n = 5, Mann-Whitney test). 10 μm nocodazole was incubated for 2 h to destroy the microtubule network (mean ± S.D., p > 0.05, n = 5, Mann-Whitney test). ns, non-significant.
2 h after cell plating, we used nocodazole to disrupt the microtubule network, and we examined the effect of shear stress. hERG1a-transfected HEK293T cells responded to shear stress after a 2-h treatment with 10 μm nocodazole (Fig. 7, B and B(i)). The treated cells did not change morphology. Interestingly, tail current amplitudes were lower than the step current amplitudes in these cells. Shear stress increased the step current by 22.6%, i.e. from 54.1 ± 13.91 to 64.4 ± 16.10 pA/pF (p = 0.01, n = 5, paired t test) and the tail current by 33.6%, i.e. from 35.3 ± 13.3 to 47.2 ± 16.6 pA/pF (p = 0.03, n = 5, paired t test). The increases seen are similar to those observed with untreated cells.
The extent to which disruption of the cytoskeleton affected the stress response was evaluated in terms of the attenuation of the stress response from that seen in untreated cells. Cytochalasin D essentially eliminated the effect of shear on step currents, while halving that on tail currents, as compared with untreated cells (mean ± S.D., p < 0.01 and p < 0.05 for step and tail current, respectively, n = 5, Mann-Whitney test). In contrast, nocodazole treatment had no effect on tail current sensitivity to shear, while marginally attenuating the step current response (mean ± S.D., p > 0.05, n = 5, Mann-Whitney test) (Fig. 7C).
Involvement of the integrin/FAK mechano-transduction pathway
Integrins are central to the mechano-transduction process, conveying mechanical forces into the cell. β1 integrins and their downstream effector FAK are associated with hERG1a in HEK293 cells (22). Fluid shear forces induce rapid remodeling of focal adhesions formed by focal adhesion kinases (37). The FAK is tightly coupled to integrins and responds to integrin clustering during fluid shear stresses by autophosphorylation and activation (27). RGD peptides inhibit the binding of integrins to fibronectin (38). 0.1 mm RGD peptide incubated for 30 min changed the morphology of the cells plated on fibronectin (Fig. S2, A and B) indicating that RGD peptides were able to inhibit the binding of integrins to the ECM. The treated cells responded to fluid shear stress by a small decrease in step and tail currents as shown in Fig. 8A. The step currents declined from 38.98 ± 21.95 to 31.26 ± 18.05 pA/pF (p = 0.003, n = 8, paired t test) during shear stress and the tail current from 63.11 ± 29.95 to 53.45 ± 28.25 pA/pF (p = 0.004, n = 8, paired t test), which are decreases of 19 and 15.33% in hERG1a step and tail current, respectively.
Figure 8.

Shear stress and integrin pathway. Representative current traces from hERG1a channel before (pre-shear) and during laminar shear stress (shear stress) are indicated by arrows. Currents were elicited by a depolarizing step to 0 mV followed by a repolarization to −60 mV from a holding potential of −80 mV. A, 0.1 mm RGD peptide incubated for 30 min to block integrin binding to fibronectin. Scatter plot shows complete loss of mechanosensitivity as seen in both step and tail current (mean ± S.D., p < 0.01, n = 8, paired t test) during LSS application. Scale bar, 1 nA and 500 ms. B, 50 μm FAK inhibitor 14 dialyzed in the cell through a patch pipette for 15 min. Scatter plot shows attenuation of mechanosensitivity of hERG currents in step current (mean ± S.D., p < 0.05, n = 8, paired t test) and tail current (mean ± S.D., p < 0.01, n = 8, paired t test) during application of shear stress. Scale bar, 1 nA and 500 ms. ns, non-significant.
FAK is an early downstream effector of integrins and has been shown to be activated in stretched cardiomyocytes (39). FAK involvement in the response to LSS was tested by preventing the activation of FAK by inhibiting the autophosphorylation at tyrosine 397. When dialyzed intracellularly with 50 μm FAK inhibitor 14 (1,2,4,5-benzenetetramine tetrahydrochloride) for 15 min, the enhancement of hERG1a currents was abrogated. Indeed, the currents declined under shear to an even greater degree than on inhibiting integrin. The step current density declined from 57.77 ± 27.55 to 45.67 ± 15.86 pA/pF (p < 0.05, n = 8, paired t test) under LSS and the tail current from 71.14 ± 28.39 to 53.58 ± 21.52 pA/pF (p = −0.007, n = 8, paired t test), i.e. 20.9 and 25% decreases in hERG1a step and tail currents, respectively (Fig. 8B). Together, these results indicate that integrins play a critical role in the mechanosensitivity of hERG channels and form the mechanosensor. The mechanical signal is then conveyed via the activation of FAK to hERG1a channels.
Long and short QT mutants show differential shear sensitivity
Mutations in the human ether-a-go-go-related gene (HERG) cause LQT, an inherited disorder of cardiac repolarization (12, 40, 41, 43–46). Reduction of IKr causes delayed myocyte repolarization (47) and an increased risk of life-threatening ventricular arrhythmia. As seen in Fig. 5C, heterotetrameric hERG1a/1b channels have low mechanosensitivity. This could be due to the presence of hERG1b, which is neither sensitive to LSS nor possesses a PAS domain (Fig. 5A). The add-back of the PAS domain restores some mechanosensitivity (Fig. 5B). This suggests that interactions involving the PAS domain could be crucial for the electrical response of hERG1a to mechanical stress.
Responses of two long QT type 2 mutants, with mutations in the PAS domain, were analyzed under laminar shear stress. Both N33T and R56Q mutations in the PAS domain of the hERG1a channel have accelerated channel deactivation and altered inactivation gating (48) due to disruption of the interaction of the N-terminal region with the rest of the channel. Laminar shear stress had minimal effect on the N33T mutant (Fig. 9A and Fig. S3A). There was a small but significant increase in step current density (43.49 ± 18.38 pA/pF pre-shear versus 47.05 ± 20.61 pA/PF) and tail current (40.1 ± 17.21 pA/pF versus 43.29 ± 18.1 pA/pF) observed during LSS (p = 0.03, n = 7, paired t test). As shown in Fig. 9B and Fig. S3B, the R56Q mutant showed a non-significant response to LSS (step current density, 152.6 ± 38.03 pA/pF pre-shear versus 156.4 ± 38.13 pA/pF under shear stress, and tail current density, 139.2 ± 36.69 pA/pF pre-shear versus 150.3 ± 46.63 pA/pF under shear stress, p > 0.05, n = 7, paired t test). Thus, both N33T and R56Q mutations in the PAS domain display reduced shear sensitivity.
Figure 9.

Shear sensitivity of hERG mutants. A, scatter plot shows response of N33T mutant to shear stress (mean ± S.D., p < 0.05, n = 7, paired t test). B, scatter plot shows response of R56Q mutant to shear stress (mean ± S.D., p > 0.05, n = 7, paired t test). C, scatter plot shows response of N588K mutant to shear stress. Both step (mean ± S.D., p < 0.001, n = 7, paired t test) and tail current (mean ± S.D., p < 0.001, n = 7, paired t test) showed significant increase in current during shear stress. ns, non-significant.
The short QT mutant, N588K, is a gain-of-function mutation that abolishes hERG rectification and passes more current during the cardiac AP (13–15, 49). N588K showed significant mechanosensitivity to LSS. As shown in Fig. 9C and Fig. S3C, the step current density increased by 22.8% from 160.45 ± 37.77 to 197 ± 35.4 pA/pF under shear stress (p = 0.003, n = 7), and tail current increased by 31.5% from 39.25 ± 9.3 to 51.62 ± 8.4 pA/pF (p = 0.003, n = 7, paired t test). Hence, the N588K mutation does not affect mechanosensitivity.
Shear stress increases hERG1a currents during cardiac action potential
Our data suggest that hERG channels would open earlier in the course of the cardiac AP under shear stress and also pass more current. To test this prediction, we subjected cells expressing hERG1a to a voltage protocol derived from a pre-recorded action potential from a rabbit ventricular myocyte (Fig. 10) (29). Current traces before and during shear stress are shown. hERG channels passed little current during phase 0, phase 1, and phase 2 of the action potential and peaked during phase 3, consistent with their role in the repolarizing phase of the cardiac AP. hERG currents increased even more at this phase under the influence of shear stress. Analysis of current traces before and during shear stress indicated that there was a 30.2 ± 1.31% increase in peak current during the action potential voltage command (n = 7).
Figure 10.
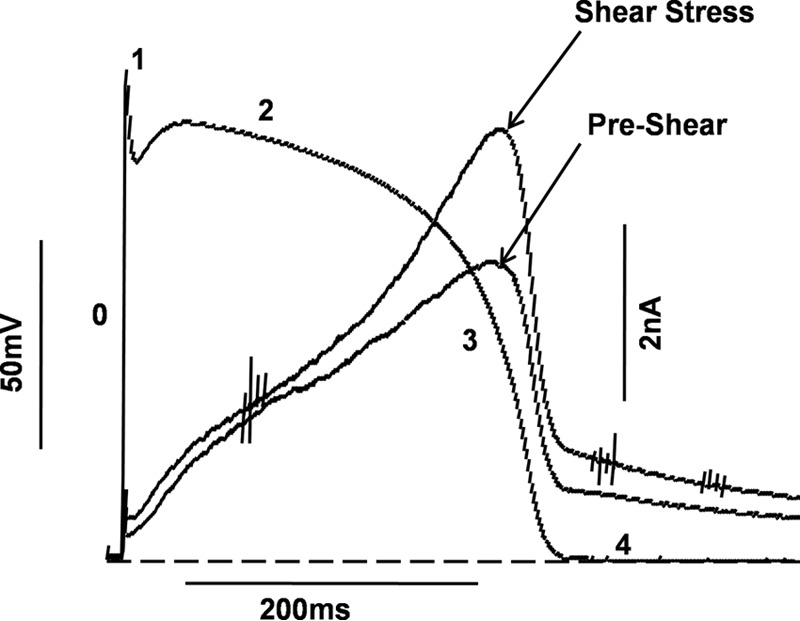
Enhancement of hERG1a currents during rabbit action potential stimulus. Different phases of the action potential are labeled as 0–4. LSS increases total potassium ions conducted by hERG1a channels during an action potential stimulus (scale bar, 50 mV and 200 ms). Typical current traces before and during shear stress are shown (Scale bar, 2 nA and 200 ms). Dashed line represents the zero current level (n = 7).
Discussion
The issue of the mechanosensitivity of the cardiac electrical system is a crucial one and also one that is difficult to address. The magnitude of shear stress experienced by a single cardiomyocyte in vivo in a working myocardium is difficult to determine (5). Myocytes are cyclically deformed by variable diastolic shear and squeeze forces associated with interlaminar fluid flow and myocardial layers sliding against each other (50–52). Recourse then has to be taken to examine components of the electrical system in heterologous systems subjected to mechanical stress. The impact of laminar shear stress on ionic currents by bath perfusion has previously been studied in HEK293 cells heterologously expressing cardiac voltage-gated sodium and calcium channels by whole-cell patch clamp (6, 7, 53). The crucial role played by potassium channels in the control of cell excitability has gained considerable attention after discovering that among the causative factors of the long Q-T syndrome, a human genetic abnormality of the cardiac action potential repolarization, were mutations in HERG. These mutations result in alterations in the electrophysiology of heterologously expressed hERG channels (43–46).
Membrane stretch induced either by pressure using the patch pipette or variations in osmolarity has been reported to affect voltage-gated K+ currents (1). The kinetics reported are unlikely to be compatible with beat-to-beat modulation of cardiac currents (50, 53, 54). Our experiments demonstrate that hERG channels respond to both osmotically induced stretch (Fig. S4) and shear stress. We have chosen to study shear because it is most likely to mimic the effects of the hemodynamic shear stress and because of the marked repeatability of its effects (55).
In this study, we have investigated mechano-electrical coupling in hERG channels using whole-cell voltage clamp of hERG-transfected HEK293T cells under fluid shear stress. Shear increases the observed hERG conductance in both the step and tail currents. The variation of conductance with shear could be well fit by a Boltzmann function suggesting that the channel exists in two states and that shear facilitates switching between the states: a “low-shear” state and a “high-shear” state. The two states differ in a number of parameters: voltage dependence of activation and inactivation; deactivation; time constant of channel opening; inactivation; and recovery from inactivation.
Effect on activation properties
An analysis of the voltage dependence of the tail currents shows that laminar shear stress induces a hyperpolarizing shift of 9 mV in the I–V curve for the step current (Fig. 2). These shifts are reversible over two cycles of shear and relaxation spread over more than 10 min (Figs. 1D and 2D). They are thus unlikely to arise from a time-dependent drift of channel properties as previously reported for Shaker K+ channels (56). The rate of channel opening was faster under shear than in control cells. In addition, deactivation is slowed significantly under shear (Fig. 3, B and C). These changes in activation properties favor the open state and could result in a significant increase in repolarization current.
Effect on inactivation properties
Both onset of and recovery from inactivation are influenced by fluid shear. The shift toward depolarizing potentials in the fractional availability curve seen under LSS is qualitatively consistent with faster recovery from inactivation. Enhanced rate of onset of inactivation would reduce outward K+ currents and thereby aid in faster upstroke velocity due to Na+ influx through sodium channels. However, rapid recovery from inactivation will result in earlier K+ current rebound during repolarization, thus shortening the APD. Because of faster inactivation upon application of LSS, there is a change in rectification voltage that shifts toward hyperpolarized potentials. This property of hERG channel could avert premature beats due to pressure overload in the heart (57–59), thereby serving a protective role during increased fluid shear stress.
hERG1b channels are insensitive to shear
Neither step currents nor tail currents observed in hERG1b-expressing HEK cells are affected by shear to a significant extent (Fig. 5A). hERG1a and hERG1b co-assemble in cardiac cells, although information regarding native stoichiometry is unavailable (29, 34, 60). Co-expression of the two constructs led to significantly larger currents than observed in cells expressing either construct alone (Fig. 5C). However, these currents were less sensitive to shear than was hERG1a alone. We ascribe this decreased sensitivity to the fact that hERG1b channels are insensitive to shear and hERG1b subunits probably contribute to a significant fraction of the channels under study. hERG1b is shorter than hERG1a primarily due to an N-terminal deletion that removes the PAS domain. Expression of the PAS domain alone together with hERG1b partially restores sensitivity but only to tail currents (Fig. 5B). Heteromultimeric hERG1a/1b channels display sensitivity intermediate between hERG1a alone and hERG1b + PAS.
N-terminal region is responsible for shear sensitivity
Genetic alterations in the PAS domain of hERG channels have been linked to LQT syndromes. Homomeric hERG1a channels have relatively slow channel activation and recovery from inactivation, the two major components that determine K+ current amplitude during repolarization. Heteromeric hERG1a/1b channels have fewer PAS domains and show a significant increase in current amplitude due to faster activation and recovery from inactivation (29). Antibodies directed against the PAS domain also increase current amplitude by modulating hERG channel kinetics. Interaction of the PAS domain with the cyclic nucleotide-binding homology domain (CNBh domain) present in the C-terminal region (61) of the hERG channel has been implicated in these changes in current amplitude (62). It is conceivable that shear stress might also modulate the interaction of the PAS domain with the CNBh domain thereby affecting the gating kinetics.
The N33T and R56Q mutations in the PAS domain extend the cardiac AP resulting in a long QT interval. Both the hERG channel mutants, N33T and R56Q, are less sensitive to LSS (Fig. 9, A and B, and Fig. S3).
Loss-of-function mutations in hERG result in an increase in the QT interval. Shear tends to shorten the QT interval and could have played a protective role. However, long QT mutants do not respond to shear (Fig. 9 and Fig. S3), and thus the protective role played by hERG during increased shear stress is lost in patients carrying loss-of-function mutations. The gain-of-function mutant N588K carries more charge during action potential clamp experiments (15), consequently shortening the cardiac action potential. Shear stress leads to even more current through an already overactivated channel thus predisposing the mutation carriers toward life-threatening conditions.
Our data on deletion of the PAS domain (hERG1b), mutations within the PAS domain (N33T and R56Q), and a mutation outside the PAS domain (N588K, which retains mechanosensitivity) implicate the PAS domain in transducing mechanical shear to the modulation of hERG electrical activity.
Integrin pathway and cytoskeletal elements involvement in the mechanosensation of hERG channels
If the PAS domain is a major contributor to mechanosensitivity, then it is likely that it interacts with elements that transduce mechanical stress at the cell membrane to internal structures. hERG channels are physically associated with β1 integrins and undergo current activation after adhesion of integrins to fibronectin followed by association of focal adhesion kinase that undergoes tyrosine phosphorylation (22). Integrins are the main receptors that connect the cytoskeleton to the ECM (27, 63, 64). There is bi-directional transmission of force: the tractional force from the cytoskeleton is passed onto the ECM via integrins, and stresses applied by the ECM to the cell surface are transmitted and transduced by integrins. The transduction of physical forces into chemical forces by integrins involves the triggering of multiple signaling pathways (42). Autophosphorylation of focal adhesion kinases on tyrosine 397 is sensitive to the tethering of integrin to a rigid substratum, and it is responsible for cell movement in durotaxis (27). Thus, FAK appears to be an important component of the integrin-mediated mechano-transduction apparatus.
As shown in Fig. 8, preventing the binding of integrins to fibronectin or blocking of FAK autophosphorylation at Tyr-397 completely altered the effect of fluid shear stress on hERG1a currents, implicating integrins and their downstream effector FAK in the mechanosensitivity of the hERG1a channels. Disrupting cytoskeleton elements attenuated shear responses of hERG channels, but the effect was less pronounced than on disrupting integrin signaling. A fall-out of our findings is that drugs that affect integrin function could in turn affect mechanosensitivity of hERG channels. This would pose a cardiac liability that normally goes unnoticed during regular preclinical screening.
hERG currents during a cardiac action potential
Our data indicate that shear shifts hERG channels from a “low-shear” state to a “high-shear” state that opens more rapidly and at less depolarizing potentials, which inactivates more rapidly and also recovers more rapidly from inactivation. The implication would be that more current would be passed by the “ high-shear state” of the channel in the course of the cardiac action potential. Our experiment with the dynamic clamp (Fig. 10) simulates the voltage changes over the course of an actual cardiac AP. hERG currents are, in fact, over 30% larger under shear than in its absence. The caveat here is that the shear was applied continuously and not in a pulsatile manner as in the heart. It is not clear how myocytes, and in particular the integrin signal transduction pathway, integrate the mechanical stresses of a beating heart. Our data demonstrate that hERG currents are sensitive to continuous shear, although the behavior under pulsatile shear remains to be established.
Experimental procedures
Cell culture and transfection
HEK293T cells were routinely cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum, 0.5% penicillin/streptomycin (Sigma) and incubated at 37 °C in 5% CO2. hERG1a and hERG1b in pXOOM vector were gifts from Dr. Nicole Schmitt, University of Copenhagen, Denmark. Long QT mutants, R56Q and N33T, and the PAS domain (1–131 amino acids) in pCDNA3.1 were kind gifts from Mathew Trudeau, University of Maryland, and N588K in pCDNA3.1 was a gift from Jules C. Hancox, University of Bristol, UK. Plasmids were transfected using Effectene reagent (Qiagen). 48 h after transfection, cells were dissociated by treating them with 0.05% trypsin plus 0.02% EDTA and seeded on fibronectin (Sigma)-coated coverslips (20). Patch-clamp recordings were performed 5–6 h after plating.
Generation of shear stress
Shear stress was generated by a gravity-fed perfusion system placed 40 cm above the experimental chamber and attached to a flow regulator. The bath chamber incorporated diamond-shaped fluidics yielding laminar flow throughout the bath (Warner Instruments). A schematic of this chamber is illustrated in Fig. S5. The liquid surface of the perfusion chamber during experiments was kept flat (neither concave nor convex). Shear stress levels were calculated from the flow rates and tube geometry on the basis of the fully developed Poiseuille flow solution in a tube of circular cross-section as described previously (28).
The magnitude of the shear stress at the outlet was calculated by using Equation 1,
| (1) |
where τ is the shear stress in dyn·cm−2; μ is the viscosity of the fluid (dyn·s/cm−2); Q is the volumetric flow rate (cm3·s−1); and r is the internal diameter of the inlet tube. Most measurements were made at a flow rate of 10 ml/min, which provided maximum activation of the hERG current.
Electrophysiology
Voltage-clamp recordings were performed on hERG-transfected HEK293T cells in the whole-cell patch configuration using a patch-clamp amplifier (EPC10, HEKA). Patch pipettes had resistances between 1 and 3 megohms. Series resistance compensation was typically 60–70%, such that voltage errors were less than 5 mV. No leak subtraction was applied; cells exhibiting leak conductance of >10% maximal conductance were excluded from the study. Data were acquired using Patchmaster (HEKA) acquisition software. To study the current-voltage relationship, cells were held at −80 mV and stepped to a range of potentials for 3 s at 10-mV increments, followed by a step repolarization to −60 mV for 2 s to measure the outward tail current. The tail current amplitudes were used for plotting the steady-state activation. To study the time constants of activation, deactivation of HERG, and inactivation properties, specific pulse protocols were used as described in the figure legends. Changes in the hERG K+ channel current amplitude in response to step changes in flow rates were monitored using a single step depolarization to 0 mV for 2 s from a holding potential of −80 mV, followed by a hyperpolarizing step to −60 mV for 3 s to elicit the tail current. The pipette solution contained (mmol/liter) KCl 130, MgCl2 2, CaCl2 0.5, EGTA 5, Mg-ATP 4, and HEPES 10, adjusted to pH 7.2 with KOH, and osmolarity was maintained at 307–310 mosm/kg. External bath solution (in mmol/liter) contained NaCl 150, CaCl2 1.8, KCl 4, MgCl2 1, glucose 10, and HEPES 10, adjusted to pH 7.4 with NaOH, and osmolarity was maintained at 321–327 mosm/kg. For cell swelling experiments, the iso-osmotic extracellular solution (in mmol/liter) contained 100 mm NaCl, CaCl2 1.8, KCl 4, MgCl2 1, glucose 10, HEPES 10, and sorbitol 80 adjusted to pH 7.4 with NaOH. The hypo-osmotic solution lacked sorbitol. Experiments were performed at room temperature. Electrical signals were filtered at 2 kHz and digitized at 5 kHz.
Materials
Colchicine (10 μm) and nocodazole (μm) were obtained from Sigma. RGD peptide and FAK14 inhibitor (50 μm) were obtained from Santa Cruz Biotechnology.
Statistics
Data were analyzed with Fitmaster (HEKA), Clampfit (Axon), and Origin (OriginLab, Northampton, UK). Conductance and voltage data were fitted to a single Boltzmann function: I/Imax = (Imin) + (Imax − Imin)/{1 + exp((V1/2 − V)/k)}, where V1/2 is the half-activation potential; V is the test voltage, and k is the slope factor. Current relaxations with repolarizing voltage steps (deactivation) and the rising phase of the repolarizing voltage step (recovery from inactivation) were fit with an exponential function (y = Ae(−t/τ)), where t is time, and τ is the time constant of deactivation or recovery from inactivation. The time course of hERG current activation was measured using single exponential fits of the peak tail current envelope recorded at −60 mV. The inactivation time constant was determined using a three-step protocol, as described in Fig. 6 (legend), to isolate inactivating currents, which were fit with a single-exponential function. Steady-state inactivation was measured using a separate three-step protocol, as described in Fig. 6 (legend). The instantaneous currents at 40 mV were normalized and fit with a Boltzmann function. Values presented are mean ± S.D. Paired t test and Mann-Whitney test were used to compare the difference between two groups. Significance level was set at a value of p < 0.05. p < 0.05, p < 0.01, and p < 0.001 are represented as *, **, and ***, respectively, in the figures. n represents the number of experiments.
Author contributions
S. R. and M. K. M. conceptualization; S. R. and M. K. M. data curation; S. R. and M. K. M. software; S. R. and M. K. M. formal analysis; S. R. and M. K. M. validation; S. R. and M. K. M. investigation; S. R. and M. K. M. visualization; S. R. and M. K. M. methodology; S. R. and M. K. M. writing-original draft; S. R. and M. K. M. writing-review and editing; M. K. M. resources; M. K. M. supervision M. K. M. funding acquisition; M. K. M. project administration.
Supplementary Material
Acknowledgments
We thank members of our laboratories for discussion. We thank Savita Bhagat, Sahil Lall, and Ankita Kishor for help in cell culture-related experiments. We thank Dr. Manjunath Ramarao and Dr. Harinath Sale on expert guidance and facilitating the research work.
This work was supported by internal funds of the Tata Institute of Fundamental Research. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
- AP
- action potential
- LSS
- laminar shear stress
- FAK
- focal adhesion kinase
- PAS
- Per-Arnt-Sim
- APD
- action potential duration
- pF
- picofarad
- VGC
- voltage-gated channel
- ECM
- extracellular matrix
- CNBh
- cyclic nucleotide-binding homology domain
- hERG
- human ether-a-go-go related gene.
References
- 1. Schmidt D., del Mármol J., and MacKinnon R. (2012) Mechanistic basis for low threshold mechanosensitivity in voltage-dependent K+ channels. Proc. Natl. Acad. Sci. U.S.A. 109, 10352–10357 10.1073/pnas.1204700109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Isenberg G., Kazanski V., Kondratev D., Gallitelli M. F., Kiseleva I., and Kamkin A. (2003) Differential effects of stretch and compression on membrane currents and [Na+]c in ventricular myocytes. Prog. Biophys. Mol. Biol. 82, 43–56 10.1016/S0079-6107(03)00004-X [DOI] [PubMed] [Google Scholar]
- 3. Morris C. E. (2011) Voltage-gated channel mechanosensitivity: fact or friction? Front. Physiol. 2, 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yellen G. (1998) The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31, 239–295 10.1017/S0033583598003448 [DOI] [PubMed] [Google Scholar]
- 5. Boycott H. E., Barbier C. S., Eichel C. A., Costa K. D., Martins R. P., Louault F., Dilanian G., Coulombe A., Hatem S. N., and Balse E. (2013) Shear stress triggers insertion of voltage-gated potassium channels from intracellular compartments in atrial myocytes. Proc. Natl. Acad. Sci. U.S.A. 110, E3955–E3964 10.1073/pnas.1309896110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beyder A., Rae J. L., Bernard C., Strege P. R., Sachs F., and Farrugia G. (2010) Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 588, 4969–4985 10.1113/jphysiol.2010.199034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beyder A., Strege P. R., Reyes S., Bernard C. E., Terzic A., Makielski J., Ackerman M. J., and Farrugia G. (2012) Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel Na(v) 1.5: a novel mechanism of drug action. Circulation 125, 2698–2706 10.1161/CIRCULATIONAHA.112.094714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trudeau M. C., Warmke J. W., Ganetzky B., and Robertson G. A. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95 10.1126/science.7604285 [DOI] [PubMed] [Google Scholar]
- 9. Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., and Hill A. P. (2012) hERG K+ channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478 10.1152/physrev.00036.2011 [DOI] [PubMed] [Google Scholar]
- 10. Sanguinetti M. C., and Jurkiewicz N. K. (1990) Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215 10.1085/jgp.96.1.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanguinetti M. C., Jiang C., Curran M. E., and Keating M. T. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 10.1016/0092-8674(95)90340-2 [DOI] [PubMed] [Google Scholar]
- 12. Curran M. E., Splawski I., Timothy K. W., Vincent G. M., Green E. D., and Keating M. T. (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 10.1016/0092-8674(95)90358-5 [DOI] [PubMed] [Google Scholar]
- 13. McPate M. J., Duncan R. S., Hancox J. C., and Witchel H. J. (2008) Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br. J. Pharmacol. 155, 957–966 10.1038/bjp.2008.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McPate M. J., Zhang H., Adeniran I., Cordeiro J. M., Witchel H. J., and Hancox J. C. (2009) Comparative effects of the short QT N588K mutation at 37 degrees C on hERG K+ channel current during ventricular, Purkinje fibre and atrial action potentials: an action potential clamp study. J. Physiol. Pharmacol. 60, 23–41 [PubMed] [Google Scholar]
- 15. McPate M. J., Duncan R. S., Milnes J. T., Witchel H. J., and Hancox J. C. (2005) The N588K-HERG K+ channel mutation in the “short QT syndrome”: mechanism of gain-in-function determined at 37°C. Biochem. Biophys. Res. Commun. 334, 441–449 10.1016/j.bbrc.2005.06.112 [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Wang H., Zhang Y., Gao H., Nattel S., and Wang Z. (2004) Impairment of HERG K+ channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J. Biol. Chem. 279, 13289–13292 10.1074/jbc.C400025200 [DOI] [PubMed] [Google Scholar]
- 17. Wang N., Butler J. P., and Ingber D. E. (1993) Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- 18. Ross R. S. (2002) The extracellular connections: the role of integrins in myocardial remodeling. J. Card. Fail. 8, S326–S331 10.1054/jcaf.2002.129263 [DOI] [PubMed] [Google Scholar]
- 19. Zhidkova N. I., Belkin A. M., and Mayne R. (1995) Novel isoform of β1 integrin expressed in skeletal and cardiac muscle. Biochem. Biophys. Res. Commun. 214, 279–285 10.1006/bbrc.1995.2285 [DOI] [PubMed] [Google Scholar]
- 20. Borg T. K., Goldsmith E. C., Price R., Carver W., Terracio L., and Samarel A. M. (2000) Specialization at the Z line of cardiac myocytes. Cardiovasc. Res. 46, 277–285 10.1016/S0008-6363(99)00433-2 [DOI] [PubMed] [Google Scholar]
- 21. Danowski B. A., Imanaka-Yoshida K., Sanger J. M., and Sanger J. W. (1992) Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 118, 1411–1420 10.1083/jcb.118.6.1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cherubini A., Hofmann G., Pillozzi S., Guasti L., Crociani O., Cilia E., Di Stefano P., Degani S., Balzi M., Olivotto M., Wanke E., Becchetti A., Defilippi P., Wymore R., and Arcangeli A. (2005) Human ether-a-go-go-related gene 1 channels are physically linked to β1 integrins and modulate adhesion-dependent signaling. Mol. Biol. Cell 16, 2972–2983 10.1091/mbc.E04-10-0940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Browe D. M., and Baumgarten C. M. (2003) Stretch of β1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J. Gen. Physiol. 122, 689–702 10.1085/jgp.200308899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hofmann G., Bernabei P. A., Crociani O., Cherubini A., Guasti L., Pillozzi S., Lastraioli E., Polvani S., Bartolozzi B., Solazzo V., Gragnani L., Defilippi P., Rosati B., Wanke E., Olivotto M., and Arcangeli A. (2001) HERG K+ channels activation during β1 integrin-mediated adhesion to fibronectin induces an up-regulation of α(v)β(3) integrin in the preosteoclastic leukemia cell line FLG 29.1. J. Biol. Chem. 276, 4923–4931 10.1074/jbc.M005682200 [DOI] [PubMed] [Google Scholar]
- 25. Lorenzen-Schmidt I., Schmid-Schönbein G. W., Giles W. R., McCulloch A. D., Chien S., and Omens J. H. (2006) Chronotropic response of cultured neonatal rat ventricular myocytes to short-term fluid shear. Cell Biochem. Biophys. 46, 113–122 10.1385/CBB:46:2:113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwartz M. A. (2010) Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2, a005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Katsumi A., Orr A. W., Tzima E., and Schwartz M. A. (2004) Integrins in mechanotransduction. J. Biol. Chem. 279, 12001–12004 10.1074/jbc.R300038200 [DOI] [PubMed] [Google Scholar]
- 28. Barakat A. I., Leaver E. V., Pappone P. A., and Davies P. F. (1999) A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ. Res. 85, 820–828 10.1161/01.RES.85.9.820 [DOI] [PubMed] [Google Scholar]
- 29. Sale H., Wang J., O'Hara T. J., Tester D. J., Phartiyal P., He J.-Q., Rudy Y., Ackerman M. J., and Robertson G. A. (2008) Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with long-QT syndrome. Circ. Res. 103, e81–e95 10.1161/CIRCRESAHA.108.185249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith P. L., Baukrowitz T., and Yellen G. (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836 10.1038/379833a0 [DOI] [PubMed] [Google Scholar]
- 31. Jones D. K., Liu F., Vaidyanathan R., Eckhardt L. L., Trudeau M. C., and Robertson G. A. (2014) hERG 1b is critical for human cardiac repolarization. Proc. Natl. Acad. Sci. U.S.A. 111, 18073–18077 10.1073/pnas.1414945111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., and Mackinnon R. (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95, 649–655 10.1016/S0092-8674(00)81635-9 [DOI] [PubMed] [Google Scholar]
- 33. Gustina A. S., and Trudeau M. C. (2013) The eag domain regulates hERG channel inactivation gating via a direct interaction. J. Gen. Physiol. 141, 229–241 10.1085/jgp.201210870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phartiyal P., Sale H., Jones E. M., and Robertson G. A. (2008) Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J. Biol. Chem. 283, 3702–3707 10.1074/jbc.M708999200 [DOI] [PubMed] [Google Scholar]
- 35. Trudeau M. C., Leung L. M., Roti E. R., and Robertson G. A. (2011) hERG1a N-terminal eag domain-containing polypeptides regulate homomeric hERG1b and heteromeric hERG1a/hERG1b channels: a possible mechanism for long QT syndrome. J. Gen. Physiol. 138, 581–592 10.1085/jgp.201110683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larsen A. P., and Olesen S.-P. (2010) Differential expression of hERG1 channel isoforms reproduces properties of native IKr and modulates cardiac action potential characteristics. PLoS ONE 5, e9021 10.1371/journal.pone.0009021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlaepfer D. D., Hauck C. R., and Sieg D. J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71, 435–478 10.1016/S0079-6107(98)00052-2 [DOI] [PubMed] [Google Scholar]
- 38. Gehlsen K. R., Argraves W. S., Pierschbacher M. D., and Ruoslahti E. (1988) Inhibition of in vitro tumor cell invasion by Arg-Gly-Asp-containing synthetic peptides. J. Cell Biol. 106, 925–930 10.1083/jcb.106.3.925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burgess M. L., McCrea J. C., and Hedrick H. L. (2001) Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 122, 1739–1756 10.1016/S0047-6374(01)00296-2 [DOI] [PubMed] [Google Scholar]
- 40. Akimoto K., Furutani M., Imamura S., Furutani Y., Kasanuki H., Takao A., Momma K., and Matsuoka R. (1998) Novel missense mutation (G601S) of HERG in a Japanese long QT syndrome family. Hum. Mutat. 1998, Suppl. 1, S184–S186 [DOI] [PubMed] [Google Scholar]
- 41. Benson D. W., MacRae C. A., Vesely M. R., Walsh E. P., Seidman J. G., Seidman C. E., and Satler C. A. (1996) Missense mutation in the pore region of HERG causes familial long QT syndrome. Circulation 93, 1791–1795 10.1161/01.CIR.93.10.1791 [DOI] [PubMed] [Google Scholar]
- 42. Huang S., and Ingber D. E., (1999) The structural and mechanical complexity of cell-growth control. Nat. Cell Biol. 1, E131–E138 10.1038/13043 [DOI] [PubMed] [Google Scholar]
- 43. Satler C. A., Vesely M. R., Duggal P., Ginsburg G. S., and Beggs A. H. (1998) Multiple different missense mutations in the pore region of HERG in patients with long QT syndrome. Hum. Genet. 102, 265–272 10.1007/s004390050690 [DOI] [PubMed] [Google Scholar]
- 44. Satler C. A., Walsh E. P., Vesely M. R., Plummer M. H., Ginsburg G. S., and Jacob H. J. (1996) Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. Am. J. Med. Genet. 65, 27–35 10.1002/(SICI)1096-8628(19961002)65:1%3C27::AID-AJMG4%3E3.0.CO%3B2-V [DOI] [PubMed] [Google Scholar]
- 45. Tanaka T., Nagai R., Tomoike H., Takata S., Yano K., Yabuta K., Haneda N., Nakano O., Shibata A., Sawayama T., Kasai H., Yazaki Y., and Nakamura Y. (1997) Four novel KVLQT1 and four novel HERG mutations in familial long-QT syndrome. Circulation 95, 565–567 10.1161/01.CIR.95.3.565 [DOI] [PubMed] [Google Scholar]
- 46. Yoshida H., Horie M., Otani H., Takano M., Tsuji K., Kubota T., Fukunami M., and Sasayama S. (1999) Characterization of a novel missense mutation in the pore of HERG in a patient with long QT syndrome. J. Cardiovasc. Electrophysiol. 10, 1262–1270 10.1111/j.1540-8167.1999.tb00304.x [DOI] [PubMed] [Google Scholar]
- 47. Nakajima T., Furukawa T., Tanaka T., Katayama Y., Nagai R., Nakamura Y., and Hiraoka M. (1998) Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ. Res. 83, 415–422 10.1161/01.RES.83.4.415 [DOI] [PubMed] [Google Scholar]
- 48. Chen J., Zou A., Splawski I., Keating M. T., and Sanguinetti M. C. (1999) Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J. Biol. Chem. 274, 10113–10118 10.1074/jbc.274.15.10113 [DOI] [PubMed] [Google Scholar]
- 49. Cordeiro J. M., Brugada R., Wu Y. S., Hong K., and Dumaine R. (2005) Modulation of I(Kr) inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc. Res. 67, 498–509 10.1016/j.cardiores.2005.02.018 [DOI] [PubMed] [Google Scholar]
- 50. Costa K. D., Takayama Y., McCulloch A. D., and Covell J. W. (1999) Laminar fiber architecture and three-dimensional systolic mechanics in canine ventricular myocardium. Am. J. Physiol. 276, H595–H607 [DOI] [PubMed] [Google Scholar]
- 51. LeGrice I. J., Takayama Y., and Covell J. W. (1995) Transverse shear along myocardial cleavage planes provides a mechanism for normal systolic wall thickening. Circ. Res. 77, 182–193 10.1161/01.RES.77.1.182 [DOI] [PubMed] [Google Scholar]
- 52. Pasipoularides A. (2012) Diastolic filling vortex forces and cardiac adaptations: probing the epigenetic nexus. Hellenic J. Cardiol. 53, 458–469 [PMC free article] [PubMed] [Google Scholar]
- 53. Rosa A. O., Yamaguchi N., and Morad M. (2013) Mechanical regulation of native and the recombinant calcium channel. Cell Calcium 53, 264–274 10.1016/j.ceca.2012.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ou Y., Strege P., Miller S. M., Makielski J., Ackerman M., Gibbons S. J., and Farrugia G. (2003) Syntrophin γ2 regulates SCN5A gating by a PDZ domain-mediated interaction. J. Biol. Chem. 278, 1915–1923 10.1074/jbc.M209938200 [DOI] [PubMed] [Google Scholar]
- 55. Strege P. R., Holm A. N., Rich A., Miller S. M., Ou Y., Sarr M. G., and Farrugia G. (2003) Cytoskeletal modulation of sodium current in human jejunal circular smooth muscle cells. Am. J. Physiol. Cell Physiol. 284, C60–C66 10.1152/ajpcell.00532.2001 [DOI] [PubMed] [Google Scholar]
- 56. Schoppa N. E., and Sigworth F. J. (1998) Activation of shaker potassium channels I. Characterization of voltage-dependent transitions. J. Gen. Physiol. 111, 271–294 10.1085/jgp.111.2.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee S., Kim J.-C., Li Y., Son M.-J., and Woo S.-H. (2008) Fluid pressure modulates L-type Ca2+ channel via enhancement of Ca2+-induced Ca2+ release in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 294, C966–C976 10.1152/ajpcell.00381.2007 [DOI] [PubMed] [Google Scholar]
- 58. Lu Y., Mahaut-Smith M. P., Varghese A., Huang C. L., Kemp P. R., and Vandenberg J. I. (2001) Effects of premature stimulation on HERG K(+) channels. J. Physiol. 537, 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schillaci G., Verdecchia P., Borgioni C., Ciucci A., Zampi I., Battistelli M., Gattobigio R., Sacchi N., and Porcellati C. (1996) Association between persistent pressure overload and ventricular arrhythmias in essential hypertension. Hypertension 28, 284–289 10.1161/01.HYP.28.2.284 [DOI] [PubMed] [Google Scholar]
- 60. Larsen A. P., Olesen S.-P., Grunnet M., and Jespersen T. (2008) Characterization of hERG1a and hERG1b potassium channels—a possible role for hERG1b in the I Kr current. Pflügers Arch. 456, 1137–1148 [DOI] [PubMed] [Google Scholar]
- 61. Morais-Cabral J. H., and Robertson G. A. (2015) The enigmatic cytoplasmic regions of KCNH channels. J. Mol. Biol. 427, 67–76 10.1016/j.jmb.2014.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harley C. A., Starek G., Jones D. K., Fernandes A. S., Robertson G. A., and Morais-Cabral J. H. (2016) Enhancement of hERG channel activity by scFv antibody fragments targeted to the PAS domain. Proc. Natl. Acad. Sci. U.S.A. 113, 9916–9921 10.1073/pnas.1601116113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Taherian A., Li X., Liu Y., and Haas T. A. (2011) Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 11, 293 10.1186/1471-2407-11-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Luca M., Pellegrini G., Bondanza S., Cremona O., Savoia P., Cancedda R., and Marchisio P. C. (1992) The control of polarized integrin topography and the organization of adhesion-related cytoskeleton in normal human keratinocytes depend upon number of passages in culture and ionic environment. Exp. Cell Res. 202, 142–150 10.1016/0014-4827(92)90413-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.