

 Peptides with various C-terminal functionalization, including peptides and dendrimers, were prepared via SPPS and an efficient on-resin modification.
Peptides with various C-terminal functionalization, including peptides and dendrimers, were prepared via SPPS and an efficient on-resin modification.
Abstract
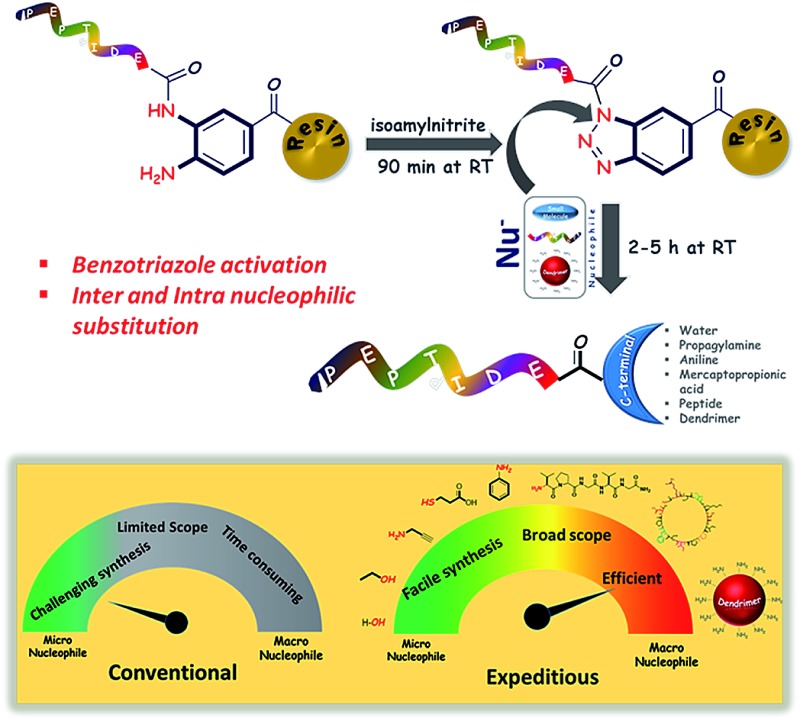
A convenient and efficient chemical toolbox was developed for the on-resin C-terminal functionalization of various peptides. By transforming resin-bound 3,4-diaminobenzoic acid species with isoamyl nitrite, the resulting resin-bound benzotriazole entity can be efficiently displaced by nucleophiles during cleavage of the peptide–resin connection in a short reaction time. The resin cleavage step allowed for the use of various nucleophiles including water, EtOH, amines, thiol, and G5 poly(amidoamino) dendrimers with yields ranging from 66% to 82% within 5 h. This method was successfully applied to prepare the elastin sequence (VPGVG)4 through on-resin ligation in 77% yield in one day and a head-to-tail cyclic peptide, sunflower trypsin inhibitor-1, in 42% yield.
Introduction
Peptides with various functional groups at the C-terminal are in high demand for numerous purposes such as the introduction of a thioester for native chemical ligation (NCL),1 preparation of therapeutic agents,2,3 and peripheral functionalization of macromolecules as imaging agents.4 Solid-phase peptide synthesis (SPPS) is considered to be the most important peptide synthesis technique as it provides precise control over the structure and allows exact post-synthetic modifications.5 However, the use of SPPS to functionalize the C-terminus of a peptide is problematic since SPPS-produced peptides are generally synthesized from the C-terminus to the N-terminus. One efficient method to functionalize the C-terminus of a synthetic peptide is the introduction of a labile group between the C-terminus and a solid-support,6 however, the stability of the labile group limits the available reactions. Alternatively, a stable moiety that is activated at the cleavage step provides a more reliable and feasible approach for modifying the C-termini of peptides and both on-resin and off-resin activation approaches have been reported.
The off-resin activation approaches7–9 have usually involved complicated preparation procedures resulting in side-product formation that has undermined the advantages of SPPS. One representative example by Dawson and coworkers is the use of 3,4-diaminobenzoic acid (Dbz) as an efficient linker for the preparation of peptide thioesters.8 However, the diacylation resulted in the need to carry out a purification step prior to the cyclization to N-acyl-benzimidazolinone (Nbz). Although this issue has been addressed with a newly developed substituted Dbz,10,11 the process still required off-resin cyclization. Moreover, Nbz, being a poor leaving group, is often associated with slow reactions and poor yields.12 Liu and coworkers have developed a strategy using a benzotriazole (Bt) moiety for producing peptide thioesters.13 However, this approach required off-resin cyclization of Dbz and importantly, the thioesterification was limited to aqueous buffer conditions incompatible with protected peptides. Therefore, it remains a demanding challenge to develop a straightforward method for diverse C-terminal modifications of peptides off-resin.
On-resin modification at the C-terminal of peptides is rare but has been shown to be a convenient approach, in which the cleavage occurs simultaneously with the incorporation of the functional group at the C-terminus. Several linkers, such as a pyroglutamyl imide,14 aryl sulfonamide,15 and aryl hydrazides16 have been reported. However, each of these methods suffers from disadvantages such as poor leaving groups and being specific for the introduction of a single type of functional group. Recently, a cyclic urethane technique was reported as a universal method for on-resin C-terminal modification.17 However, selective deprotection of the first serine residue during the activation process limits the amino acid residues available for this SPPS. Therefore, developing a straightforward on-resin method to activate the C-termini of peptides for introducing various moieties is in high demand in peptide chemistry and its related applications.
The benzotriazole (Bt) group is considered to be an efficient linker for the traceless synthesis of large compounds and is compatible with various types of synthetic transformations in solution and solid-phase syntheses.18 We envisioned Bt to be an efficient linkage moiety in SPPS for the purpose of on-resin C-terminal modifications. Herein, we report a new strategy of on-resin modification of the C-terminus of peptides using nucleophilic treatment of resin-bound Bt (RB-Bt) obtained via isoamyl nitrite-promoted on-resin cyclization of o-aminoanilide. This robust activation technique provided a new and efficient means to access large arrays of C-terminal peptide modifications including long peptides, cyclic peptides, and peptide-decorated polymers through SPPS chemistry. This approach is shown in Fig. 1.
Fig. 1. Representative scheme for the on-resin C-terminal modification of peptide analogues. A linear analogue has the Dbz moiety on one peptide. Branched analogues have two peptides attached to one Dbz moiety.

Result and discussion
Preparation of Bt resin and the on-resin cleavage method
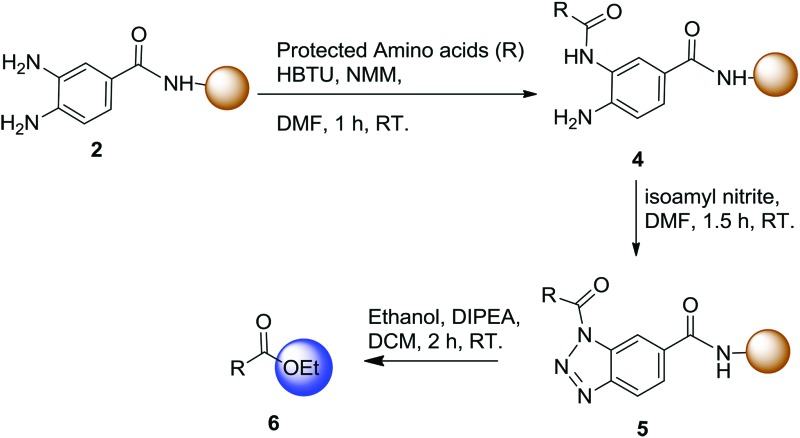
The synthesis started from the preparation of the Dbz-derived linker on a solid support. We coupled o-aminonitrobenzoic acid (1) onto the Rink amide resin followed with the reduction of the nitro group of the resin-bound scaffold using SnCl2 to afford the desired product 2. To confirm the structure of 2, we further cleaved the resin to obtain 3. We affirmed the structure of 3 using NMR and MS analysis, which indirectly confirmed the successful preparation of 2 (Scheme 1).
Scheme 1. Preparation of the Dbz linker.

One challenge of this work was preparing the Bt linkages on the solid support without damaging the synthetic peptide. Although the Bt moiety has been used as an auxiliary agent in many solid-phase syntheses, the conditions reported for its preparation were harsh with regard to maintaining the side chain-protected amino acid residues, leading to undesired isomerization.19 To overcome this issue, a revised Katritzky method19 was used for cyclization of 2 to form the RB-Bt. To optimize the cyclization at the o-aminoanilide, we tried both sodium nitrite and isoamyl nitrite for the formation of the Bt moiety. The transformation required a longer reaction time with only a 30–40% conversion when using sodium nitrite, whereas the reaction was accomplished within 90 min to 3 h with 85–99% conversion when employing isoamyl nitrite. The reaction with isoamyl nitrile (10 eq.) in both DMF and dichloromethane (DCM) gave the highest yield (99%) (Table S1†). Therefore, we selected isoamyl nitrile in DMF for the following investigation so that it would be compatible with most SPPS preparations.
To evaluate the compatibility of individual amino acids with activation and nucleophilic acyl substitution, a leucine analogue was first synthesized using this established procedure with ethanol as the nucleophile to afford 6a in 91% isolated yield (Table 1). Then, we also investigated the suitability of other amino acids in the first position next to the Dbz linker to determine the scope of this method (Table 1). Using the described procedure, the desired ester products 6b–j were obtained with high purities and yields (78–90%). One critical issue in C-terminal activation is the possible racemization of the introduced amino acid residue. Remarkably, no racemization of any amino acid was observed (Fig. S43–S52†). Thus, this method is compatible with various amino acids and protecting groups commonly used in solid-phase peptide synthesis.
Table 1. Results from an investigation of the scope of the activation/cleavage procedure.
| |||
| Entry | R | Product | Yield a (%) |
| 1 | Fmoc-l-Leu– | 6a | 91 |
| 2 | Fmoc-l-Phe– | 6b | 90 |
| 3 | Fmoc-l-Asp(OtBu)– | 6c | 87 |
| 4 | Fmoc-l-Gln(Trt)– | 6d | 86 |
| 5 | Fmoc-l-His(Trt)– | 6e | 82 |
| 6 | Fmoc-l-Cys(Trt)– | 6f | 84 |
| 7 | Fmoc-l-Lys(Boc)– | 6g | 88 |
| 8 | Fmoc-l-Trp(Boc)– | 6h | 86 |
| 9 | Fmoc-l-Arg(pbf)– | 6i | 78 |
| 10 | Fmoc-l-Met– | 6j | 90 |
aBased on resin loading amount.
Preparation of C-functionalized peptides
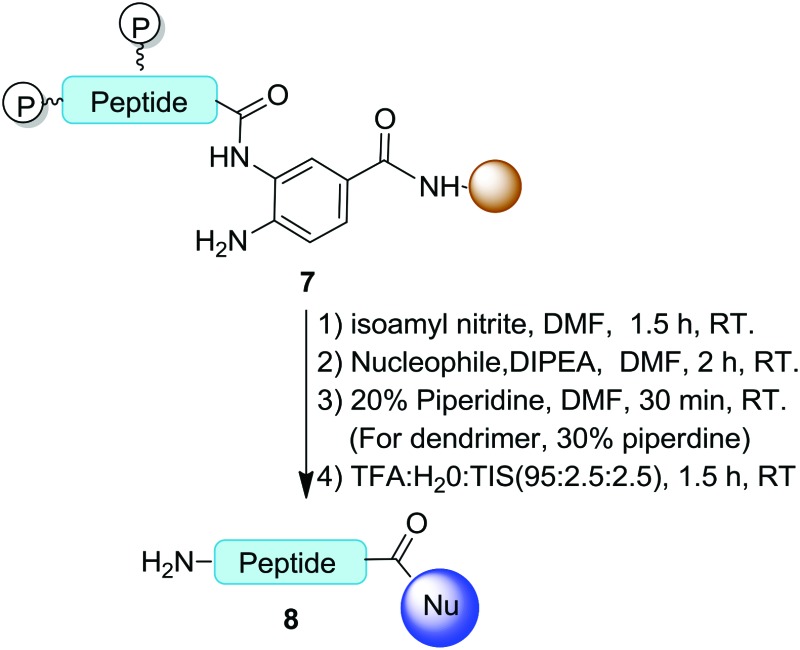
To study the scope of available nucleophiles, the resulting on-resin products 7 were reacted with various nucleophiles including water, aliphatics, aromatic amines, and a thiol (Table 2). After the removal of Fmoc, the crude products were treated with a TFA cocktail for deprotection to afford the final desired products 8. From this approach, a simple hydrolysis gave the corresponding acid 8a in 82% yield. Peptide 8b resulted from the treatment of propargylamine for 2 h. The cleavage reaction with aniline to produce 8c required a longer reaction time of 5 h. Note that thioesters are versatile intermediates with great potential in chemical biology by means of NCL.20 Our method provided an easy access to synthetic C-terminal peptide thioester 8d in 80% yield as a result of a 2 h treatment with mercaptopropionic acid. Overall, the cyclization and cleavage gave peptide products with 66–82% isolated yields and 90–95% purities as characterized by HPLC.
Table 2. Results of on-resin nucleophilic cleavage.
| ||||
| Entry | Peptide | Nucleophile | Product | Yield a (%) |
| 1 | HSSKLQL (7a) | H2O | 8a | 82 |
| 2 | HSSKLQL (7a) | Propargylamine | 8b | 78 |
| 3 | LMYKA (7b) | Aniline | 8c | 66 |
| 4 | Ac-AYRGA (7c) | Mercaptopropionic acid | 8d | 80 |
| 5 | Leu (7d) | (G:5)-dendri-PAMAM–(NH2)128 | 8e b | 75 d |
| 6 | HSSKLQL (7e) | (G:5)-dendri-PAMAM–(NH2)128 | 8f c | 70 d |
aBased on resin loading amount.
bThe product is (G:5)-dendri-PAMAM–(Leu)60.
cThe product is (G:5)-dendri-PAMAM–(LQLKSSH)32.
dBased on the amount of dendrimer used.
Preparation of peptide-decorated dendrimers
To further explore available nucleophiles, amine-terminated poly(amidoamino) (PAMAM) dendrimers were used to synthesize amino acid/peptide-decorated dendrimers. PAMAM dendrimers are widely used in material sciences and biomedical investigations.21 Because of their dense distribution of peripheral amines, the peripheral modification of dendrimers remains a challenge. Therefore, they are good examples for testing the efficiency of the above approach for functionalizing macromolecules. Using hydrophobic leucine as the model, the cleavage of a leucine off the resin by the fifth-generation (G5) PAMAM dendrimer in DMF was carried out at rt for 2 h. Subsequent removal of the Fmoc groups afforded the desired corresponding leucine-decorated PAMAM dendrimer 8e in 75% yield (Table 2, entry 5). Integrating the characteristic leucine and PAMAM 1H NMR signals at 0.9 ppm and 2.6 ppm, respectively, indicated that the dendrimer-functionalized peptide product consisted of sixty leucine residues and was obtained in a yield of 75%.
Additionally, the peptide HSSKLQL was prepared with the Bt resin. The resin was activated and then treated with G5 PAMAM dendrimers to afford the corresponding HSSKLQL-decorated PAMAM dendrimer 8f in 70% yield and a functionalization number of 32 (Table 2, entry 6). This demonstrated that the Bt resin was highly effective for constructing peptide-modified dendrimers after a few hours of coupling time. This result also indicated that this approach allows bulky nucleophiles such as polymers to be used while the peptide remains on the resin.
Preparation of a cyclic peptide and long peptide
With this encouraging result, we focused our effort on applying this method for the preparation of various peptide analogues including cyclic peptides and hydrophobic peptides. Synthesizing cyclic peptides is a challenging task and various methods have been reported for such syntheses.22 Cyclic peptides are important targets of peptide synthesis for their interesting biological properties and exciting potential in biomedical applications. Bt resin was used in the synthesis of sunflower trypsin inhibitor-1 (SFTI-1) (Scheme 2). SFTI-1 is a naturally occurring cyclic peptide of 14 residues with one disulfide bond and is a potent Bowman–Birk trypsin inhibitor.23 Recently, SFTI-1 analogues were successfully prepared by Chatterjee and coworkers.24 However, the cyclization required 24 h at 50 °C. An isolated yield of 35% was obtained after chromatographic purification. To explore the potential of the reported approach for preparing SFTI-1, the linear peptides were first synthesized on the Bt resin. After removal of the Fmoc group at the N-terminus, the resin-bound o-aminoanilide was activated to form the corresponding RB-Bt 10. Thereafter, the terminal amine served as the nucleophile to cyclize and simultaneously cleave the protected peptide from the resin in 5 h. The resulting cyclic peptide was deprotected, simultaneously triggering intramolecular disulfide bridge formation to give oxidized SFTI-1 with 42% yield and 94% purity. The results revealed that the intermolecular head-to-tail cyclization occurred successfully and, remarkably, isoamyl nitrite selectively cyclized the diamine into benzotriazole and did not affect the N-terminal amine.
Scheme 2. Preparation of cyclic peptide 11.

Regardless, preparation of hydrophobic peptide remains a challenge in SPPS. An elastin sequence, (VPGVG)n, was selected as the hydrophobic model because its synthesis and characterization are reported.25 The oligomers of this pentapeptide are difficult polypeptides to prepare through SPPS because of their hydrophobicity and rich network of hydrogen bonding as well as the fact that they are prone to aggregation.26 To demonstrate the usefulness of this procedure for preparing hydrophobic peptides, the preparation of the tetramer of VPGVG started from the preparation of the monomer (13), which was used as the nucleophile for the cleavage reaction of on-resin 12 to give 14 in 92% yield. The 10-mer peptide 14 was subjected to the same procedure resulting in a 93% yield of 15-mer 15, which was used to prepare the desired 20-mer (residue) peptide 16 in 90% yield (Scheme 3). Each reaction was completed in 2 h and the yields were calculated based on the amount of 12 used in the reactions. These results clearly demonstrated that various lengths of peptides can serve as nucleophiles for the Bt resin and suggests the preparation of long peptides can be achieved efficiently and conveniently using this method.
Scheme 3. Preparation of elastin peptide through ligation using Bt resin.

Conclusions
Herein, we describe the use of Bt-resin as a powerful synthetic tool for on-resin C-terminal modification of synthetic peptides. The most significant feature of this method is its compatibility with diverse nucleophiles for releasing C-terminal modified peptides from the solid support in a short time. In addition to small-molecule nucleophiles, the ability to use peptides as nucleophiles allows the preparation of polypeptides from a linear synthesis to on-resin ligation of small peptides resulting in acceleration of the preparation of long peptides. The on-resin cyclization was also used to efficiently prepare cyclic peptides. Moreover, the modular aspect of the method allowed for the attachment of peptides to dendrimers. The products are obtained in high purities and yields without the need for any chromatographic purification steps. This method should permit the preparation of various peptide analogues and peptide-decorated macromolecules for biomedical applications such as targeted drug delivery, molecular imaging, and immunology.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was support by the Ministry of Science and Technology of Taiwan (MOST 103-2628-M-037-001-MY3 and 105-2632-M-037-001-). The authors thanks Prof. Chi-Wi Ong (National Sun Yat-sen University, Taiwan) and Prof. Ling Peng (CINaM, CNRS UMR 7325, French) for discussions and proofreading.
Footnotes
†Electronic supplementary information (ESI) available: Experimental procedure and NMR spectra of synthetic peptides and relative derivatives. See DOI: 10.1039/c7sc03229c
References
- Bondalapati S., Jbara M., Brik A. Nat. Chem. 2016;8:407–418. doi: 10.1038/nchem.2476. [DOI] [PubMed] [Google Scholar]
- Songster M. F., Barany G. Methods Enzymol. 1997;289:126–174. doi: 10.1016/s0076-6879(97)89047-7. [DOI] [PubMed] [Google Scholar]
- Gao W., Liu W., Christensen T., Zalutsky M. R., Chilkoti A. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16432–16437. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J., Alewood P. F. Angew. Chem., Int. Ed. 2016;55:5124–5134. doi: 10.1002/anie.201508428. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. J. Am. Chem. Soc. 1963;85:2149–2154. [Google Scholar]
- Alsina J., Albericio F. Pept. Sci. 2003;71:454–477. doi: 10.1002/bip.10492. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. A., Simon M. D., Pentelute B. L. Org. Lett. 2016;18:1222–1225. doi: 10.1021/acs.orglett.5b03625. [DOI] [PubMed] [Google Scholar]
- Blanco-Canosa J. B., Dawson P. E. Angew. Chem. 2008;120:6957–6961. doi: 10.1002/anie.200705471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig T. G., Rohrbacher F., Pattabiraman V. R., Bode J. W. Angew. Chem., Int. Ed. 2014;53:12244–12247. doi: 10.1002/anie.201406097. [DOI] [PubMed] [Google Scholar]
- Weidmann J., Dimitrijević E., Hoheisel J. D., Dawson P. E. Org. Lett. 2016;18:164–167. doi: 10.1021/acs.orglett.5b03111. [DOI] [PubMed] [Google Scholar]
- Blanco-Canosa J. B., Nardone B., Albericio F., Dawson P. E. J. Am. Chem. Soc. 2015;137:7197–7209. doi: 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xu C., Lam H. Y., Lee C. L., Li X. Proc. Natl. Acad. Sci. U. S. A. 2013;110:6657–6662. doi: 10.1073/pnas.1221012110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-X., Fang G.-M., He Y., Qu D.-L., Yu M., Hong Z.-Y., Liu L. Angew. Chem., Int. Ed. 2015;54:2194–2198. doi: 10.1002/anie.201408078. [DOI] [PubMed] [Google Scholar]
- Tofteng A. P., Sørensen K. K., Conde-Frieboes K. W., Hoeg-Jensen T., Jensen K. J. Angew. Chem., Int. Ed. 2009;48:7411–7414. doi: 10.1002/anie.200903710. [DOI] [PubMed] [Google Scholar]
- Backes B. J., Ellman J. A. J. Am. Chem. Soc. 1994;116:11171–11172. [Google Scholar]
- Woo Y.-H., Mitchell A. R., Camarero J. A. Int. J. Pept. Res. Ther. 2007;13:181–190. [Google Scholar]
- Elashal H. E., Cohen R. D., Raj M. Chem. Commun. 2016;52:9699–9702. doi: 10.1039/c6cc04245g. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R., Lan X., Yang J. Z., Denisko O. V. Chem. Rev. 1998;98:409–548. doi: 10.1021/cr941170v. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R., Belyakov S. A., Tymoshenko D. O. J. Comb. Chem. 1999;1:173–176. [Google Scholar]
- Mende F., Seitz O. Angew. Chem., Int. Ed. 2011;50:1232–1240. doi: 10.1002/anie.201005180. [DOI] [PubMed] [Google Scholar]
- Yu M., Jie X., Xu L., Chen C., Shen W., Cao Y., Lian G., Qi R. Biomacromolecules. 2015;16:2588–2598. doi: 10.1021/acs.biomac.5b00979. [DOI] [PubMed] [Google Scholar]
- Tapeinou A., Matsoukas M.-T., Simal C., Tselios T. Pept. Sci. 2015;104:453–461. doi: 10.1002/bip.22669. [DOI] [PubMed] [Google Scholar]
- Mylne J. S., Colgrave M. L., Daly N. L., Chanson A. H., Elliott A. G., McCallum E. J., Jones A., Craik D. J. Nat. Chem. Biol. 2011;7:257–259. doi: 10.1038/nchembio.542. [DOI] [PubMed] [Google Scholar]
- Shelton P. M. M., Weller C. E., Chatterjee C. J. Am. Chem. Soc. 2017;139:3946–3949. doi: 10.1021/jacs.6b13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladini F., Araman C., Becker C. F. W. J. Pept. Sci. 2016;22:334–342. doi: 10.1002/psc.2871. [DOI] [PubMed] [Google Scholar]
- Dang B., Dhayalan B., Kent S. B. H. Org. Lett. 2015;17:3521–3523. doi: 10.1021/acs.orglett.5b01632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.