

Graphical abstract

Keywords: Ubiquitin Specific Protease 17, DUB3, Ubiquitin, Protein Refolding, USP17, Deubiquitinating
Introduction
Deubiquitinating enzymes (DUBs) reverse the process of ubiquitination by hydrolyzing ubiquitin from the protein substrate to which it is conjugated [1,2]. Thus far, approximately 100 DUBs have been identified in the human genome and they are involved in regulating a number of cellular processes and disease states [1,2]. DUBs are classified into six families, of which the largest family is the Ubiquitin Specific Protease (USP) family with roughly 60 members [1,2]. The USPs are multi-domain enzymes that can range in size from ∼40 kDa, in the case of USP12, up to ∼400 kDa, in the case of USP34 [1]. Each USP contains a catalytic domain, which shares the highly conserved papain-like fold composed of a catalytic triad of cysteine, histidine, and aspartate residues [1,2]. Other domains within the USPs are often important for protein-protein interactions and/or substrate recognition. A detailed review of the domains and their functions has been described elsewhere [2,3].
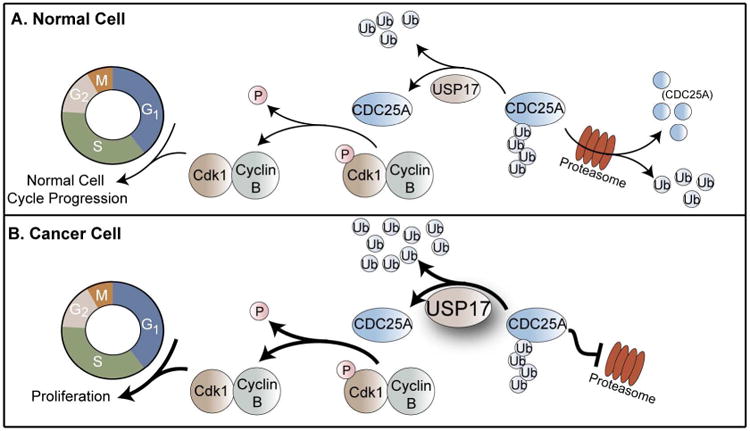
USP17 was originally identified as a member of the murine hematopoietic specific genes as DUB3 [4]. Expression of USP17 within the cell is cytokine-inducible and is required for cell-cycle progression through the G1-S and G2-M checkpoints. A well-characterized substrate of USP17 is the cell division cycle 25A (CDC25A) phosphatase. In a normal cell, CDC25A is responsible for dephosphorylation of Cdk1 and activation of Cdk1-CyclinB complex for the progression of the cell-cycle (Figure 1A) [5]. Cellular levels of CDC25A are regulated by the ubiquitin-proteasome system through Lys48 ubiquitin chains. During the G1-S and G2-M checkpoints, USP17 is expressed to deubiquitinate and stabilize CDC25A in order to promote cell-cycle progression through these checkpoints [5]. In numerous cancer phenotypes, expression of USP17 is upregulated, resulting in its expression outside of these checkpoints which causes an excess of stabilized CDC25A that applies stress to the cell-cycle resulting in unregulated cancer cell proliferation (Figure 1B) [5–7]. Other identified substrates of USP17 include the Ras converting enzyme 1 (RCE1) and the histone deacetylase dependent Sin3A co-repressor complex component, SDS3, both of which also play a role in cell-cycle regulation [8–10].
Figure 1. Role of USP17 in cell-cycle regulation and cancer.
CDC25A is a cell-cycle phosphatase that promotes cell-cycle progression by dephosphorylating Cdk1. CDC25A cellular levels are regulated through ubiquitination that signals for CDC25A degradation. (A) In a normal cell, USP17 deubiquitinates and stabilizes CDC25A to promote cell-cycle progression [5]. (B) In cancer cells, USP17 levels are upregulated which results in increased deubiquitination and stabilization of CDC25A. The increased levels of CDC25A therefore result in cancer cell proliferation [5].
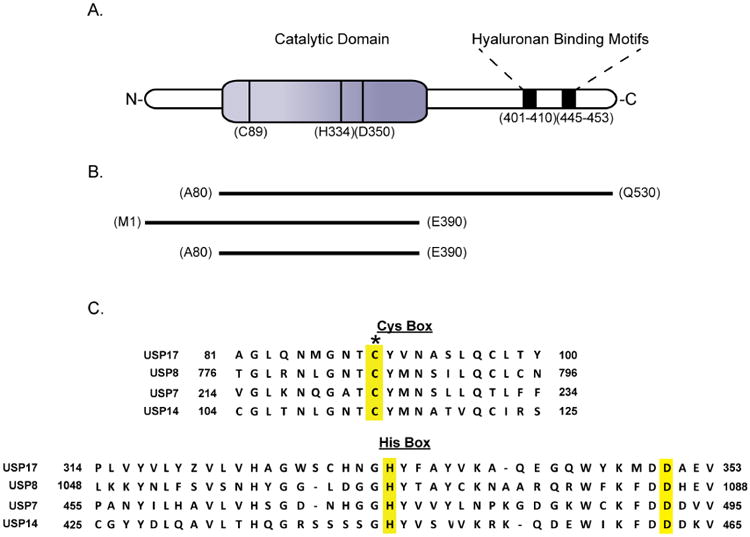
USP17 is a 58 kDa protein that has its catalytic domain located near the N-terminus as well as two hyaluronan binding motifs predicted to reside within the C-terminal region (Figure 2A) [4,11]. A sequence alignment of the catalytic regions of USP17 against three well-studied USPs is shown in Figure 2C [12]. As with other USPs, there are three highly conserved residues (Cys89, His334 and Asp350) that form the catalytic triad. Experimental binding studies by Ramakrishna and coworkers have shown the two predicted hyaluronan binding motifs bind to hyaluronan, a polysaccharide that is responsible for numerous cellular processes, including the regulation of cell division [11]. Many USPs are predicted to contain hyaluronan binding motifs, including the well-studied USP7, however the interactions between the USPs and hyaluronon itself is poorly understood [13]. In the case of USP17, the hyaluronan binding motifs have been shown to be important for the interaction of USP17 with its substrate, SDS3 [14].
Figure 2. Domain organization, constructs and primary sequences of USP17 and related DUBs.
(A) Illustration of the predicted domains of USP17 and the amino acids that make up the catalytic triad [4,11]. (B) Expression constructs generated in this study that were used in attempts to achieve soluble expression of USP17. (C) Clustal Omega sequence alignment of USP17 (NP_ 391627) with USP8 (NP_001122082), USP7 (NP_003461), and USP14 (NP_005142.1) [12]. The conserved catalytic triad residues are highlighted in yellow. The asterisk denotes the catalytic cysteine that was mutated to serine in the catalytic mutant.
McFarlane and coworkers have shown that the persistent overexpression of USP17 applies stress to the cell-cycle which results in proliferation of both breast and prostate cancers [4,6]. Combining the results of this study with the role of USP17 in CDC25A-driven cellular proliferation elucidated by Pereg and coworkers, strongly suggests that inhibitors of USP17 may serve as anti-cancer drugs. While previous work has been able to define the role of USP17 in celluo, without the ability to express and purify USP17, further characterization of USP17, as well as the identification of USP17 inhibitors, is severely limited. We describe here a reliable and robust method for the expression and purification of recombinant USP17 which enables the production of highly pure and active USP17 that is amenable to enzymatic and structural characterization, as well as inhibitor identification.
Materials and methods
Design of Expression Constructs
The human gene of usp17 (GenBank: Q0WX57.2) was codon optimized for E. coli expression, synthesized, and inserted into the pET11a expression vector by BioBasic Inc. The coding region for USP17 was subcloned into the pEV-L8 expression vector by methods described by Báez-Santos et al [15].
For baculovirus expression, BioBasic Inc. codon optimized usp17 (GenBank: Q0WX57.2) for Spodoptera frugiperda 9 (Sf9) expression, synthesized the gene, and inserted the gene into a modified pVL-1932 vector which includes a dual N-terminal 10His and a Green Fluorescent Protein (GFP) tag.
Site-Directed Mutagenesis
The catalytic mutant USP17-Cys89Ser pEV-L8 vector was prepared following the QuikChange site-directed mutagenesis protocol (Stratagene) and transformed into E. coli BL21 (DE3) cells for protein expression.
Expression, Solubilization, Refolding and Purification of USP17
Five liters of E. coli BL21 (DE3) cells harboring the USP17 pEV-L8 expression vector were grown from five separate one liter cultures of LB medium, each supplemented with 50 μg/mL Carbenicillin in 2 L Fernbach flasks. Shaking was performed at 37 °C and at 200 rpm in an Infors Multitron shaker until an A600 of 0.6 is reached. Cultures were then cooled for 30 minutes at 4 °C and USP17 expression was induced with a final concentration of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cultures were incubated at 25 °C with shaking at 200 rpms for an additional 18 hours.
E. coli cells were harvested by centrifugation at 3,000 × g at 4 °C for 20 minutes. The harvested cells were resuspended in lysis buffer (50 mM Tris pH 7.5, 10 mM β – mercaptoethanol (βME), and 150 mM NaCl) that was supplemented with 50 μg/mL DNase and 200 μg/mL Lysozyme, by adding 5 mL of lysis buffer for each gram of cells. The cells were sonicated on ice with a Branson Digital Sonifier at 60%, with cycles of 5 second pulses and 9 seconds of rest for a total of 12 minutes. The soluble lysate was separated from the inclusion body fraction by centrifugation at 28,880 × g at 4 °C for 30 minutes. The supernatant was decanted from the inclusion body pellet and the resulting inclusion body was washed 3 times by homogenization with 50 mL of lysis buffer containing 1% Triton™ X-100. Between each wash, the sample was centrifuged at 28,800 × g for 10 minutes. A final wash was performed identically with unsupplemented lysis buffer.
The washed inclusion body was then re-solubilized in 250 mL of Solubilization Buffer (6 M GuHCl, 50 mM CHES pH 9.5, and 10 mM βME) in a sealed Nalgene bottle. The bottle was placed on a stir plate at room temperature overnight with a magnetic stir bar set to stir the solution at 600 rpms. The 250 mL of solubilized inclusion body was refolded, dropwise, at room temperature into 5 L of Refolding Buffer (50 mM CHES pH 9.5, 10 mM βME, 0.7 M GuHCl, and 5% Glycerol) in a Nalgene bottle stirring at 600 rpms. Once all the protein had been refolded, the bottle was capped and the sample was incubated for 30 minutes with continuous stirring at 600 rpms. The refolded protein was then concentrated to 1 mg/mL in a volume of ∼500 mL using the Millipore Prep/Scale-Tangential Flow Filter concentrating system with a 1.5 L, 10,000 MWCO cartridge. The concentrated protein was centrifuged at 28,800 × g at 4°C for 30 minutes and filtered through a 0.45 μm filter to remove precipitant.
A 5 mL HisTrap Ni-NTA (GE Healthcare) chromatography column was equilibrated with Buffer A (50 mM CHES pH 9.5, 10 mM βME, 0.7 M GuHCl, 20 mM Imidizole, and 5% Glycerol). The filtered-refolded protein was then loaded onto the equilibrated HisTrap Ni-NTA column overnight at 4 °C with a flow rate of 0.5 mL/min. Unbound proteins were washed from the column using 15 column volumes of Buffer A that was applied at a flow rate of 2 mL/min. Bound proteins were eluted from the column using a 20 column volume gradient of 0-100% Buffer B (50 mM CHES pH 9.5, 10 mM βME, 0.7 M GuHCl, 450 mM Imidizole, and 5% Glycerol) that was applied at a flow rate of 2 mL/min. Fractions of 5 mL were collected throughout the gradient. The concentration of protein in the fractions was measured using the Microassay Bio-Rad Bradford Protein Assay. Peak fractions containing USP17, as determined by SDS-PAGE and specific activity (described below), were pooled. The pool was concentrated to 7 mg/mL by Ultrafiltration with a 30,000 MWCO membrane (Millipore) and injected onto a 300 mL Superdex 75 (GE Healthcare) size-exclusion chromatography (SEC) column equilibrated with SEC Buffer (50 mM CHES pH 9.5, 10 mM βME, 0.7 M GuHCl, and 5% Glycerol). The column was run at 1 mL/min and 5 mL fractions were collected continuously throughout. Peak fractions containing USP17, as confirmed by SDS-PAGE and specific activity, were pooled, flash-frozen in liquid nitrogen, and stored at -80 °C. The catalytic mutant, USP17-Cys89Ser, was purified identically except the activity assays were omitted since the enzyme was inactive.
USP17 was also expressed using the BD BaculoGold system and Sf9 cells according to the manufactures instructions (BD Biosciences). After 3 days of expression, the infected Sf9 cells were harvested by centrifugation at 3000 × g for 20 minutes at 4 °C. The cells were resuspended in 500 mM NaCl, 10 mM Imidizole, 10 mM βME, and 20 mM Tris at pH 7.5. The resuspended cells were then lysed by sonication with a Branson Digital Sonifer at 50% with cycles of 5 second pulses and 5 seconds of rest for a total of 4 minutes. The lysate was clarified by centrifugation at 80,000 × g for 1 hour at 4 °C. The clarified lysate was initially an alyzed for USP17 expression via SDS-PAGE and fluorescence imaging. Prior to staining, the resulting SDS-PAGE gel was placed on an UV lightbox to observe the GFP tag. The gel was then stained with Coomassie Brilliant blue for visualization of all the protein bands.
Extinction Coefficient Determination
The concentration of pure USP17 or USP17-Cys89Ser was determined using a Take3 Micro-Volume plate with a Biotek Synergy H1 plate reader. The extinction coefficient of purified USP17 was experimentally determined to be 58,104 M-1cm-1 for wild-type, and 58,338 M-1cm-1 for USP17-Cys89Ser as described by von Hippel et al [16].
Steady-state Kinetic Studies
A fluorogenic activity assay for USP17 was developed for a 96-well, black half-volume plate (Corning Costar) to evaluate the kinetics of the deubiquitinating activity of USP17. Two substrates were used, ubiquitin 7-amino-4-methylcoumarin (Ub-AMC, LifeSensors) and ubiquitin-rhodamine110 (Ub-Rho110, Boston Biochem). Assays were performed in 30 μL reaction volumes and in triplicate. Enzymatic reactions were initiated by the addition of 15 μL of enzyme followed by brief shaking (10 seconds) using the plate reader shaker. The rate of hydrolysis was analyzed at 10 second intervals over a time period of 30 minutes with a BioTeK Synergy H1 plate reader equilibrated at 25 °C. Both substrates were measured as a filter based assay with an excitation wavelength of 360 nm and an emission wavelength of 460 nm for Ub-AMC and an excitation wavelength of 485 and an emission wavelength of 528 for Ub-Rho110. The initial slope of each reaction was measured as Arbitrary Fluorescence Units (AFU) per unit time (AFU/sec). These values were then converted to the amount of product produced per unit time (μM/sec) using the ‘fluorescence extinction coefficient’ of the product (either released AMC or Rho110). To determine of the fluorescence extinction coefficient of the product for each substrate, the reactions were allowed to plateau and the maximum AFUs were measured and potted in terms of substrate concentration.
To determine the Km and kcat values of USP17, the enzyme concentration was held constant at 3.125 nM and the substrate concentrations (Ub-AMC or Ub-Rho110) were varied from 0.01 μM to 10 μM. The enzyme was diluted in Assay Buffer (50 mM Tris pH 7.5, 5 mM Dithiothreitol (DTT), 0.1 mg/mL BSA) and the substrates were diluted in Substrate Buffer (50 mM Tris pH 7.5, 1 mM EDTA, 100 mM NaCl, and 0.05% CHAPS). The resulting rate values in μM/min were then converted to turnover number in units of sec-1 by dividing the rates by the enzyme concentration (V/[E]). The turnover values were then plotted as a function of substrate concentration and the data were fit to the Michaelis-Menten equation using non-linear regression and the Enzyme Kinetics Module in the program SigmaPlot (v13: Systat Software Inc.).
Enzyme Specific Activity
The specific activity at each step of the purification was determined to evaluate enzyme purity and yields with 0.5 μM Ub-Rho110 in Assay Buffer at 25 °C. A unit of USP17 activity is defined as the number of micromoles of product produced per minute. Specific activity is reported as units per mg of protein.
Enzyme Stability Studies
Aliquots of purified enzyme at concentrations of 1.26 mg/mL were incubated at 4 °C, 25 °C, or 37 °C for 1, 2, or 3 days. At each time point, the sample was centrifuged at 13,500 × g for five minutes and the protein concentration was measured by absorbance at 280 nm using the determined extinction coefficient of USP17 (see above). The specific activity of the samples was determined with 6.25 nM of the enzyme and 0.5 μM Ub-Rho110, then normalized to the specific activity measured for day 0.
Analytical Size-Exclusion Chromatography
The proteins ferritin, aldolase, conalbumin, and ovalbumin from the GE Healthcare High Molecular Weight calibration kit were diluted to 3 mg/mL in Analytical Buffer (50 mM HEPES pH 7.5, 0.7 M GuHCl, and 2 mM Tris(2-carboxyethyl)phosphine) (TCEP)), with the exception of ferritin, which was diluted to 0.3 mg/mL. Bovine serium albumin (BioRad) was diluted to 1.23 mg/mL. The proteins were combined into three separate injections for resolution: aldolase and ovalbumin, conalbumin and ferritin, and BSA. Each set of proteins was injected onto a 24 mL (Vt) Superdex 200 Increase 10/300 analytical SEC column, and run using an isocratic gradient at 4 °C and a flow rate of 0.5 mL per minute. For each protein the elution volume (Ve) was measured. Dextran (GE Healthcare) was injected at 1 mg/mL to determine the void volume (Vo) of the column under these conditions. The average distribution constant, (Kaverage), was determined by Equation 1.
| (Equation 1) |
The Kaverage was determined for each protein and was plotted against the log Mr to build a standard curve. The standard curve was determined using linear regression which resulted in Equation 2.
| (Equation 2) |
An aliquot of 500 μL of USP17 at a concentration of 1.26 mg/mL in Analytical Buffer was injected onto the same column under identical conditions as the standard proteins. The Kaverage of USP17 was determined and the molecular weight was calculated using Equation 2.
Analytical Ultracentrifugation
Analytical Ultracentrifugation was performed with the assistance of the Purdue Biophysical Analysis Laboratory. To determine the oligomeric state of USP17, sedimentation velocity experiments were performed at 25 °C with a Beckman-Coulter XLA ultracentrifuge at a concentration of 5.6 μM (0.4 mg/mL) of USP17 in Analytical Buffer. The sample was run in an AN-60 Ti rotor at 50,000 rpm. Absorbance optics at 280 nm was utilized for protein detection. The data was analyzed as described by Tomaret al [17].
Dynamic Light Scattering
Three milligrams of thawed USP17 was concentrated to ≤ 500 μL (5 mg/mL) using Ultrafiltration with a 10,000 MWCO membrane (Millipore) and was then injected onto a 24 mL Superose 6 Increase (GE Healthcare) SEC column equilibrated with Analytical Buffer. The column was run at a flow rate of 1 mL/min throughout. The peak of USP17 was pooled and analyzed by SDS-PAGE, and the specific activity was determined. USP17 was then in Analytical Buffer to three concentrations: 0.5, 0.75, and 1 mg/mL. Dynamic light scattering (DLS) was performed for each concentration using a Malvern Zetasizer Nano S with a HeNe laser at 633 nm. The three concentrations of USP17 were measured at a fixed scattering angle of 173° (back scatter) in 100 μL aliquots at a constant temperature of either 4 °C or 25 °C. Size analysis was performed by number distribution curves and were utilized to determine the molecular weight and oligomeric state of USP17 using the Malvern DTS Software. The number size distribution (%) represents the number of particles in the different size bins as determined by the Malvern DTS Software.
pH Evaluation on Kinetic Activity
The effect of pH on the activity of USP17 was tested from pH 6.0 to 10.5 in half-step increments using a wide pH range buffer system (75 mM Tris, 25 mM Acetic Acid, 25 mM MES, 25 mM Glycine, 0.1 mg/mL BSA, 0.01% Triton™ X-100, and 5 mM DTT) at each pH tested [18]. USP17 was diluted to a final concentration of 6.25 nM. Ub-Rho110 was diluted in the same buffer at each pH to two concentrations (0.5 μM and 5 μM) that are below and above the Km value. Enzymatic activity was measured in triplicate as described above. The resulting data were fit to a bell-shaped profile representing two ionizations, pKa and pKb, with the Enzyme Kinetics Module of SigmaPlot (v13: Systat Software Inc.).
Ubiquitin Chain Processing Assays
The substrate specificity of USP17 was tested against the di-ubiquitin chains of the eight linkage types (Lys6, Lys11, Lys29, Lys33, Lys48, or Lys63 (Boston Biochem) and linear (LifeSenors)), as well as Lys11(Boston Biochem), Lys48, or Lys63 (LifeSensors) tetra-ubiquitin chains. For reactions containing USP17 a concentration of 40 nM was used. Each di-ubiquitin reaction contained 4 μM of the ubiquitin chain and was incubated with or without enzyme at 25 °C for 120 minutes. The tetra-ubiquitin reactions contained 2 μM of the ubiquitin chains and were incubated with or without enzyme for a range of times from 10 to 120 minutes at 25 °C. Reactions were quenched with NuPAGE LDS sample buffer (Invitrogen) and analyzed by NuPAGE Novex 4-12% Bis-Tris mini gels (Invitrogen) stained with Coomassie Brilliant Blue.
Results
The methodology for the expression and purification of recombinant USPs is rather diverse. A number of expression constructs and methods have been utilized to produce recombinant USPs, a summary of which is provided in Table 1. While there are a few USPs (i.e. USP11 and USP16) that can be expressed and purified from E. coli as their full-length constructs, many USPs are insoluble when expressed from E. coli [19,20]. An alternative approach for the expression of full-length USPs has been to utilize the labor-intensive baculovirus expression systems which has successfully produced full-length USP1, USP7, and USP12 enzymes [3,18,20]. However, the most common approach for producing recombinant USPs is to truncate the USP of interest into its individual domains in order to obtain high yields of soluble protein from E. coli. With this approach, high priority is placed on the catalytic domain, as demonstrated for the well-characterized USP7, USP8, and USP14 catalytic domain constructs [3,19–23]. However, it is often found that the enzymatic activity of the catalytic domain is significantly lower than that for the full-length enzyme, making production of full-length USPs a necessity for advanced studies. We therefore pursued approaches for the production of full-length USP17.
Table 1. USP Expression Systems Utilized to Date.
|
|
|||
|---|---|---|---|
| Construct | Expression System | Reference | |
| USP1 | Full-Length | BES | (17) |
| USP7 | Full-Length | BES | (3) |
| USP7CD | Catalytic Domain | E. coli | (3) |
| USP8CD | Catalytic Domain | E. coli | (22) |
| USP11 | Full-Length | E. coli | (18) |
| USP12 | Full-Length | BES | (19) |
| USP14CD | Catalytic Domain | E.coli | (21) |
| USP16 | Full-Length | E.coli | (19) |
| USP17 | Full-Length | Refolded | This study |
High yields of USP17 can be obtained by refolding from bacterial inclusion bodies
USP17 with a N-terminal 6His tag was expressed and purified from E. coli inclusion bodies. The expression and refolding procedure involved washing the resulting inclusion bodies with 1% Triton™ X-100 by homogenization and then solubilizing the protein using 6 M GuHCl. Refolding of active USP17 was achieved by rapid dilution of the protein into a refolding buffer containing only 0.7 M GuHCl. Purification of the active, refolded USP17 was achieved in two steps, nickel-metal-chelate affinity chromatography followed by SEC. From 5 L of E. coli culture, approximately 20 mg of USP17 can be obtained, and the enzyme is >95% pure as judged by SDS-PAGE (Figure 3A). From SDS-PAGE analysis, USP17 has an estimated weight of 60 kDa, which is close to the expected size of USP17 with a 6His tag (∼62 kDa). The specific activity and yield of USP17 was measured at each step during the purification, and the results of which are summarized in Table 2. The activity measurements are normalized to the refolding step, the first step where active enzyme is present. The resulting calculated yield of USP17 was approximately 10% and the overall improvement in purification was 5-fold. In contrast, no activity could be measured for the catalytic mutant USP17-Cys89Ser suggesting the activity measured for wild-type USP17 was specific to USP17, and not to a contaminating enzyme that co-purified with USP17.
Figure 3. SDS-PAGE analysis of USP17 purifications.
(A) Purification of full-length USP17 from the E. coli expression system. (B) Lysate of Sf9 cells expressing GFP-tagged USP17 stained with Coomassie Brilliant Blue (Lane 1) and UV visualization of GFP tag (Lane 2). (C) The catalytic mutant, USP17-Cys89Ser, catalytic mutant purification from E. coli expression system. MW = molecular weight standards
Table 2. Summary of USP17 Purification Steps and Resulting Yields.
| Sample | Total Protein (mg) | Specific Activity* (Unit/mg) | Total Units | % Yield | Fold Purification |
|---|---|---|---|---|---|
| Soluble Lysate | 870 | 0.006 | 5.37 | N/A | N/A |
| Refolded Protein | 494 | 0.028 | 14.01 | 100 | 1 |
| Nickel Pool | 72 | 0.123 | 3.84 | 13 | 4 |
| SEC Pool | 21 | 0.147 | 3.12 | 10.5 | 5 |
Unit = μmoles product produced per minute. Specific Activity of refolded USP17 from inclusion body with fluorogenic substrate Ub-Rho110
During the development of the USP17 purification procedure, it was evident from SDS-PAGE analyses and confirmed by western blot with an antibody towards the 6His tag (data not shown) that USP17 is prone to some truncation. These truncations are either the result of self-degradation of USP17 or the susceptibility of USP17 to protease degradation during expression. While a high yield of the full-length enzyme can be purified away from the truncations by the refolding method described above (Figure 3A), three different expression methods were attempted to identify the cause in attempt to reduce the amount of degradation observed. First, USP17 was expressed with a N-terminal 10His-GFP tag by baculovirus infection of Sf9 cells. The yield of USP17 from Sf9 cells was significantly less than what can be obtained by refolding and degradation was still apparent, as observed with a GFP-tagged version of USP17 (Figure 3B). Second, we expressed, refolded from inclusion body, and purified a catalytic mutant of USP17, USP17-Cys89Ser (Figure 3C), to evaluate if the degradation is autocatalytic or the result of proteases within the cell. During the purification of USP17-Cys89Ser, the degradation pattern of USP17 was again observed (Figure 3C) indicating that USP17 is susceptible to protease degradation during expression. This conclusion is further supported by the absence of truncations appearing after the purification is complete, suggesting purified full-length USP17 is stable and not prone to self-degradation. Third, attempts were made to truncate USP17 to improve solubility and prevent degradation (Figure 2B), a technique utilized by many other USPs as summarized in Table 1. However, none of the USP17 truncations expressed solubly or were able to refold in a stable form from E. coli inclusion bodies. In summary, the refolding and purification methodology described above is the best procedure for obtaining high yields of recombinant full-length USP17.
Three significant observations were made regarding the stability of USP17 during the optimization of the refolding and purification of USP17 from inclusion body. First, during the optimization of the refolding buffer, it was found that if the GuHCl concentration was reduced to < 0.7 M, USP17 precipitated immediately upon rapid dilution. Second, while USP17 can actively refold at a pH of 7.5, significantly more precipitant is observed than when USP17 is refolded at pH 9.5. Finally, to obtain a high yield of pure full-length USP17, 5% glycerol is required in the refolding buffer and all subsequent purification buffers to aid in stabilization. Without the glycerol present, USP17 would precipitate throughout the purification, specifically when concentrated for SEC. Furthermore, the glycerol acts as a cryo-protectant when USP17 is flash frozen in liquid nitrogen and stored at -80 °C.
Thermal stability of USP17
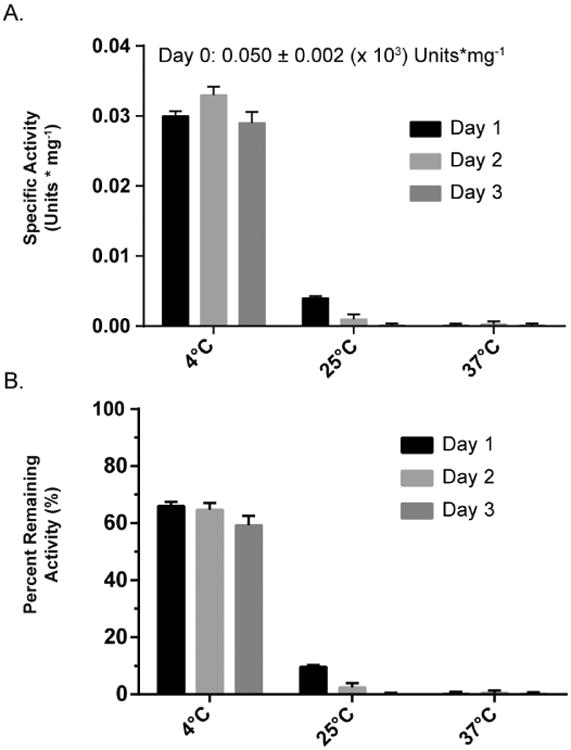
The stability of USP17 at different temperatures was tested by incubating USP17 at 4 °C, 25 °C, or 37 °C for 1, 2, or 3 days. After incubation, the samples were centrifuged and the remaining USP17 concentration and specific activity was measured to evaluate the effect temperature and incubation time had on the activity of the enzyme (Figure 4A). The specific activity was normalized to the measured activity at the beginning of day one for comparison (Figure 4B). A large amount of precipitation was observed in the samples incubated at 37 °C for each day. Almost a complete loss in specific activity was observed after day 1 and each day after, suggesting the remaining soluble protein was inactive. Similar to the samples incubated at 37 °C, large amounts of precipitation was observed for each sample stored at 25 °C. After day 1, the 25 °C samples only retained ∼10% activity. After days 2 and 3 the samples 25 °C retained minimal activity. The samples stored at 4 °C had minimal precipitation and only lost ∼35% of the total activity after day 1. Unlike the other temperatures tested, no further loss in total activity was observed after days 2 and 3 for the 4 °C samples. This suggests that any aggregation or inactivation that occurred during the incubation period happened within the first 24 hours and not thereafter. The remaining specific activity and temperature are summarized in Figure 4.
Figure 4. Retention of USP17 specific activity over time and at different temperatures.
Aliquots of USP17 were stored at 4 °C, 25 °C, or 37 °C for 1, 2, or 3 days. At each time point the sample was centrifuged, and the remaining enzyme concentration was measured by A280 and adjusted with the determined extinction coefficient to 6.25 nM. (A) Remaining activity was measured with 0.5 μM Ub-Rho110. (B) Specific activity was calculated as described in the Materials and Methods with the same concentrations of substrate and enzyme as (A). All assays were done in triplicate. Error, Standard Deviation.
USP17 is a monomer in solution
To analyze the homogeneity and oligomeric state of refolded USP17, we performed analytical-SEC, Analytical Ultracentrifugation (AUC), and dynamic light scattering (DLS). For each method, USP17 was diluted in the same buffer for consistency as described in the Materials and Methods. For the analytical-SEC analysis, a 500 μL sample of USP17 at a concentration of 1.3 mg/mL was passed over a 24 mL Superdex 200 (GE Healthcare) SEC column at 4 °C. The elution profile (Figure 5A), displayed a broad peak and the average distribution constant, Kaverage, of the maximal height, as well as the half maximal heights, was calculated. Using the standard curve constructed under the same conditions (Figure 5B), the molecular weight of the USP17 peak corresponded to 122 kDa and ranged from 86 to 153 kDa.
Figure 5. Determination of the oligomeric state of USP17.
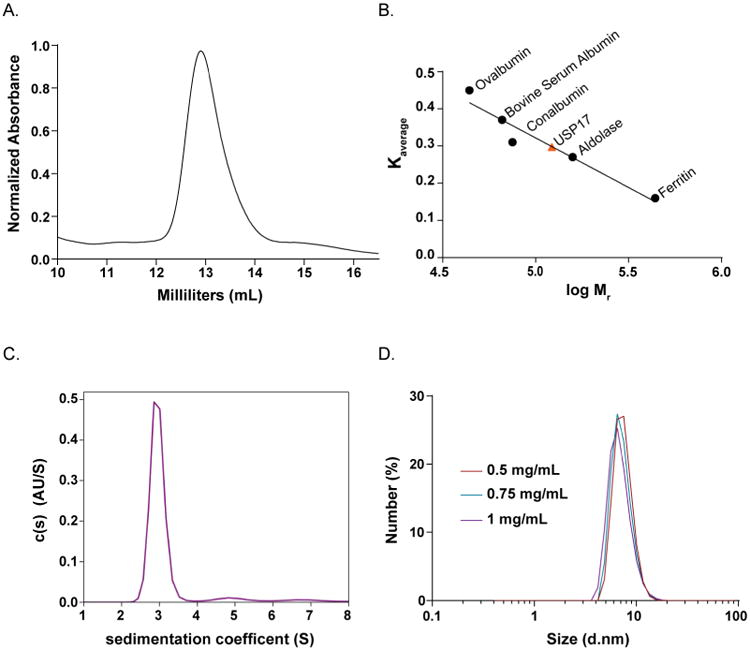
(A) Elution profile of USP17 from a 24 mL Superdex 200 (GE Healthcare) SEC column. (B) Standard curve built from the average distribution coefficient (Kaverage) vs log (Mr) of well characterized proteins, black circles. The Kaverage of USP17 was determined from the elution profile in (A) and fit to the standard curve (Equation 2) to determine the molecular weight of USP17, orange triangle. (C) AUC sedimentation velocity (AUC-SV) analysis at a concentration of 5.6 μM. Plot of the distribution of sedimentation coefficients (C (s)) versus s, where s is plotted in Svedberg units, (S) calculated from AUC sedimentation velocity experiments. (D) DLS curves at 4 °C of USP17 at three concentrations 0.5 mg/mL (deep red), 0.75 mg/mL (teal), and 1 mg/mL (purple).
To further analyze of the oligomeric state of USP17, we performed analytical ultracentrifugation (AUC). For this method, USP17 was analyzed at a single concentration of 5.6 μM (0.4 mg/mL) at 50,000 rpm and 25 °C. Under these conditions, the resulting sedimentation coefficient was determined to be 2.955 ± 0.002 which corresponds to a molecular weight of 65.2 kDa (Figure 5C).
Due to the discrepancies between the analytical-SEC and AUC results, we utilized the versatility of DLS to evaluate the oligomeric state of USP17 at both 4 °C and 25 °C degrees and at varying concentrations. USP17 was passed over a Superose 6 Increase (GE Healthcare) SEC column and the peak fraction of pure USP17 was concentrated to 1 mg/mL. USP17 was evaluated with DLS at 1, 0.75 and 0.5 mg/mL and at both 4°C and 25 °C. The hydrodynamic radius from the measurements at 4 °C (Figure 5D) for each concentration measured was between 3.5 ± 0.8 nm for 0.75 mg/mL and 3.75 ± 0.8 nm for 0.5 mg/mL. These numbers equate to a molecular weight range of 64 ± 2 kDa to 73 ± 2 kDa. The hydrodynamic radii broadened slightly for each concentration when measured at 25 °C (Supplemental Figure 1). For example, at 0.75 mg/mL of USP17 the hydrodynamic radius was 3.5 nm at 4 °C and 4 nm at 25 °C, which increased the calculated molecular weight from 64 kDa to 93 kDa respectively. The hydrodynamic radius and the resulting molecular weights from all conditions tested are summarized in Supplemental Table 1.
Steady-State Kinetic Characterization of USP17
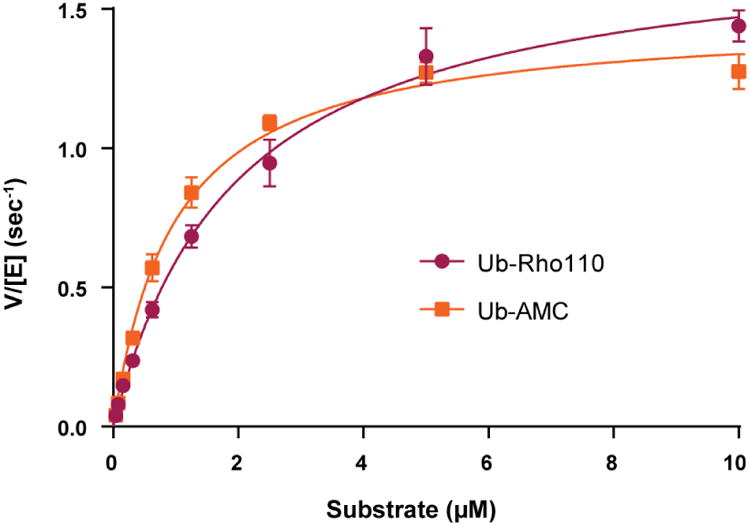
The steady-state kinetic parameters were determined for USP17 by measuring the kinetic response of USP17 to increasing concentrations of two fluorogenic substrates, Ub-AMC and Ub-Rho110. As shown in Figure 6, USP17 can be readily saturated by both substrates. The data in Figure 6 were fit to the Michaelis-Menten equation, and the resulting kinetic parameters from those fits are summarized in Table 3. The kinetic parameters suggest that USP17 recognizes and hydrolyzes both Ub-AMC and Ub-Rho110 with somewhat similar catalytic efficiencies (kcat/KM = 1,500 (×103) * M-1 * sec-1 for Ub-AMC and 880 (×103) * M-1 * sec-1 for Ub-Rho110). Ub-AMC is the more commonly utilized fluorogenic substrate within the deubiquitinating enzyme literature. As such, Table 4 compares the kinetic parameters of USP17 with Ub-AMC, to those of other human USPs characterized to date [19,24]. Interestingly, USP17 catalyzes the hydrolysis of Ub-AMC 3-fold more efficiently than USP7 as a result of the lower Km value associated with USP17. Similar to other USPs, USP17 poorly catalyzes the hydrolysis of the fluorogenic peptide substrate RLRGG-AMC and the ubiquitin-like modifier substrate ISG15-AMC, that is involved in the innate immune response (data not shown).
Figure 6. Kinetic response of USP17 to increasing concentrations of two fluorogenic substrates, Ub-Rho110 (purple, circles) and Ub-AMC (orange, squares).
The activity of USP17 was monitored with substrate concentrations from 0.04 to 10 μM with 3.125 nM USP17. Assays were performed in triplicate. Curves were fit to the Michaelis-Menten equation and the kinetic parameters, Km and kcat, were determined for each substrate and presented in Table 3. Error, Standard Deviation.
Table 3. Kinetic Parameters of USP17 with Ub-AMC and Ub-Rho110.
|
|
||
|---|---|---|
| Ub-AMC | Ub-Rho110 | |
| Km (μM) | 0.98 ± 0.06 | 2.0 ± 0.1 |
| kcat (s-1) | 1.47 ± 0.02 | 1.76 ± 0.05 |
| kcat/Km (×103 * s-1 * M-1) | 1500 ± 90 | 880 ± 50 |
Table 4. Kinetic Parameters of Well Characterized USPs with Ub-AMC.
|
|
|||
|---|---|---|---|
| Km (μM) | kcat (s-1) | kcat/Km (×103 s-1 * M-1) | |
| aUSP1 (21-785) | 9.71 ± 0.85 | 0.079 ± 0.003 | 9 |
| bUSP2CD | 2.4 ± 0.2 | 0.35 ± 0.03 | 146 |
| aUSP7 | 2.89 ± 0.1 | 1.37 ± 0.01 | 482 |
| aUSP8CD | 17.30 ± 2.5 | 7.90 ± 0.46 | 464 |
| aUSP11 | 0.77 ± 0.13 | 0.074 ± 0.003 | 96 |
| aUSP12 | 12.01 ± 4.2 | 0.0023 ± 0.002 | 0.2 |
| aUSP16 | 1.42 ± 0.25 | 0.069 ± 0.003 | 49.3 |
| aUSP17 | 0.98 ± 0.06 | 1.47 ± 0.02 | 1,500 |
Values obtained from Faesen et al (18).
values obtained from Bozza et al (23).
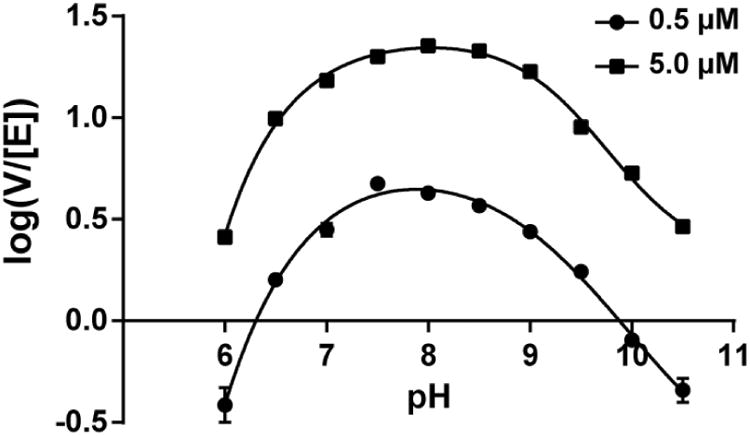
Effect of pH on USP17 Activity
The kinetic response of USP17 to variable pH values over the pH range of 6 to 10.5 was measured using Ub-Rho110 as a substrate. The results are shown in Figure 7. The rates of hydrolysis of Ub-Rho110 were evaluated at subsaturating (0.5 μM) and near saturating (5 μM) concentrations of Ub-Rho110 to look for an observed shift in pKa values. Ten pH values spaced 0.5 pH units apart were chosen and the rates were measured in triplicate. The data in Figure 7 assumes typical bell-shaped curves and therefore the data were fit to the equation that describes the kinetic model of an enzyme undergoing two ionizations with two pKa values (pKa and pKb) using SigmaPlot (v13: Systat Software Inc.). The resulting fits are shown in Figure 7 for both subsaturating and saturating substrate concentrations and the resulting pKa for 0.5 μM Ub-Rho is 6.83 ± 0.07, which is similar to the pKa value at 5 μM Ub-Rho110 which is 6.75 ± 0.04. The pKb value at 0.5 μM Ub-Rho110 is 9.13 ± 0.07 which is similar to the pKb value of 9.35 ± 0.04 at 5 μM Ub-Rho110. The lack of any significant differences in the pKa and pKb values at subsaturating and saturating concentrations suggests that ionizations in the free enzyme (E) and substrate-bound enzyme complex (ES) are the same and that substrate binding does not influence the pKa values.
Figure 7. Kinetic response of USP17 at varying pH.
The activity of USP17 was measured with Ub-Rho110 from pH 6 to 10.5 with 6.25 μM USP17. Two concentrations of Ub-Rho110 surrounding the Km value were tested, 0.5 μM and 5 μM. Assays were performed in triplicate. Curves were fit to a bell-shaped rate profile as described in the Material and Methods.
Substrate Specificity of USP17 for Different Ubiquitin Chain Linkages
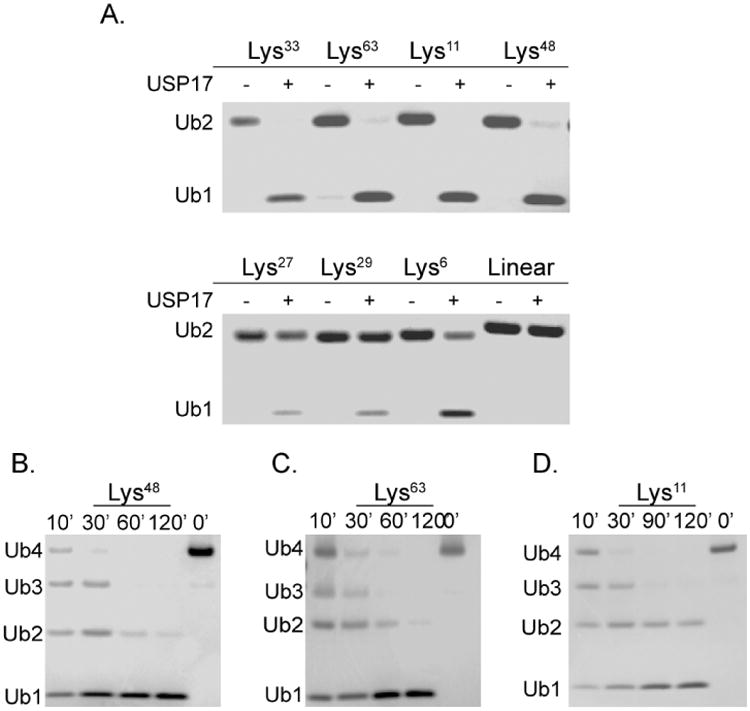
Ubiquitination of protein substrates involves one of the seven lysine residues of ubiquitin. The topology of the ubiquitin chain is directly dependent on the specific lysine residue that is used to conjugate one ubiquitin to the next. These differences can confer an additional layer of specificity for a particular USP beyond recognition of the protein substrate alone. Previous cell-based assays have demonstrated that USP17 can process Lys63 chains conjugated to RCE1 and SDS3 and Lys48 chains conjugated to CDC25A [5,9,10]. To further explore the ability of USP17 to recognize and cleave other ubiquitin linkages, we investigated the ability of USP17 to hydrolyze all seven lysine linkages (Lys6, Lys11, Lys27,Lys29, Lys33, Lys48, or Lys63) using di-ubiquitin as well as its ability to cleave a linear peptide linkage between two ubiquitins (linear). The di-ubiquitin chains were incubated with USP17 and the reaction products were analyzed by SDS-PAGE (Figure 8A). USP17 was able to process all linkages, to a certain extent, with the exception of linear di-ubiquitin. As was expected, USP17 was able to efficiently process Lys48 and Lys63 almost fully to mono-ubiquitin, as well as Lys11 and Lys33 (Figure 8A, Top Panel). A substantial amount of uncleaved di-ubiquitin remained for Lys27, Lys29, and Lys6 (Figure 8A, Bottom Panel). A comparison of these cleavage patterns to other characterized USPs is displayed in Table 5. We then evaluated how USP17 processed longer ubiquitin chains by incubating USP17 with tetra-ubiquitin chains of Lys48 (Figure 8B), Lys63 (Figure 8C), or Lys11 (Figure 8D) by evaluating the cleavage pattern over time. The data suggests that all three chain types are processed by exo-trimming to a similar extent within the time range tested.
Figure 8. Processing of di-Ubiqutin and tetra-Ubiquitin substrates by USP17.
For all reactions, 40 nM USP17 was combined with ubiquitin substrate and allowed to incubate at 25 °C. (A) Survey of the hydrolysis of di-Ubiquitin by USP17. Each assay contained 4 μM of the di-Ubiquitin linkage (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63, and linear) and was allowed to incubate for 2 hr. Di-Ubiquitin was incubated without enzyme for comparison. Time-dependent hydrolysis of Lys48 (B), Lys63 (C), or Lys11 (D) tetra-Ubiquitin by USP17. Aliquots were removed at four time points and quenched with sample buffer, then analyzed by SDS-PAGE. Polyubiquitin chains incubated without USP17 serve as negative control, 0′.
Table 5. Comparison of Di-Ubiquitin Linkage Preferences of Various USPs.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Lys6 | Lys11 | Lys27 | Lys29 | Lys33 | Lys48 | Lys63 | Linear | |
| USP1 (21-785) | +++ | ++ | + | + | ++ | +++ | +++ | - |
| USP7 | +++ | +++ | ++ | + | ++ | +++ | +++ | - |
| USP8CD | +++ | +++ | +++ | +++ | +++ | +++ | +++ | - |
| USP11 | +++ | +++ | +++ | ++ | +++ | +++ | +++ | - |
| USP16 | ++ | +++ | +++ | +++ | +++ | +++ | +++ | + |
| aUSP17 | ++ | +++ | + | + | +++ | +++ | +++ | - |
Evaluation of chain linkage preferences was obtained from Faesen et al (18).
values are reported from this paper
Discussion
The USP family of DUBs has a wide functional diversity in maintaining homeostasis within the cell through their regulation of many signaling pathways, including the ubiquitin-proteasome, DNA repair, and the cell-cycle. As a result, the mis-regulation of the USPs themselves frequently results in the development of cancer. With this realization, there is a great need to expand upon the number of USPs that have been expressed and purified, allowing for more in depth kinetic, biochemical, biophysical and structural studies of the USP family. Furthermore, these advanced approaches can be used in conjunction with the development of small molecule inhibitors to further probe the functional roles of the USPs in cells, and ultimately the development of therapeutic compounds. Of the USPs that have been expressed via recombinant systems, few of them are of the full-length construct due to their inherent insolubility when expressed in E. coli. Therefore, it is not uncommon to see full-length USPs expressed from the more labor-intensive, often low-yielding baculovirus expression systems. To obtain higher yields of soluble USPs, the individual catalytic domains or other domains of USPs have more often been produced.
Similar to other USPs, we found that USP17 is highly insoluble when expressed in E. coli. USP17 is a critical DUB involved in many pathways that dictate cell-cycle progression. Both in cellulo and in vitro based studies have elucidated the importance of USP17's activity on the stabilization of phosphatase CDC25A [5]. However, until our work reported here, recombinant expression and purification methods for USP17 had not been developed or reported, which greatly limited our ability to study this important enzyme. We were able to develop and optimize a reproducible procedure for the expression, refolding, and purification of USP17 from E. coli inclusion bodies. With large quantities of recombinant USP17 in hand, we were able to interrogate the biochemical, kinetic and oligomeric properties of USP17 and determine its substrate specificity towards ubiquitin and ubiquitin-like substrates. The results obtained build upon our knowledge of the USP family and have important implications for the development of anti-cancer therapeutics.
Our observations during the expression of USP17 from both baculovirus and E. coli revealed that USP17 is prone to proteolytic degradation. As is commonly observed with recombinant proteins, neither expression system may contain the required machinery to properly refold USP17, resulting in unfolded regions of USP17 being susceptible to proteolysis. It is not likely that USP17 itself is responsible for its degradation, as similar degradation patterns are observed for the refolded catalytic mutant. This evidence suggests that proteases native to Sf9 and E. coli are responsible for the observed degradation of USP17.
Three approaches were utilized to determine the oligomeric state of USP17: analytical-SEC, AUC, and DLS techniques. USP17 was determined to be a monomer in solution by AUC when evaluated at 25 °C and at a co ncentration of 0.4 mg/mL. It was also observed to be a monomer in solution at 4 °C and at concentrations ranging from 0.5 to 1 mg/mL by DLS. However, USP17 was determined to shift to a more dimeric state when evaluated by either analytical-SEC at 1.3 mg/mL and 4 °C, or by DLS at concentrations ranging from 0.75 to 1 mg/mL and 25 °C. Taken together, the results suggest USP17 primarily assumes a monomeric state at lower concentrations (i.e. under the conditions of our kinetic assays) but the equilibrium may shift towards a dimer at higher USP17 concentrations (i.e > 1 mg/ml), and this dimerization may have some temperature dependence.
The kinetic parameters of USP17 were determined for both Ub-Rho110 and Ub-AMC substrates, and only modest differences in the kinetic parameters were observed. Of greater interest are the comparisons of the kinetic parameters for USP17 with Ub-AMC and the reported kinetic parameters for other USPs (Table 4) [19]. The catalytic efficiency for USP17 (kcat/Km) is 3-fold higher than that of the very efficient and full-length USP7 [19] and is 7,500 times more efficient than the catalytic domain of USP2. The high level of USP17 catalytic activity provides further evidence of the significant role USP17 has in progressing the cell-cycle. However, we need to keep in perspective that all of the kinetic studies on USPs have been performed with an artificial substrate, Ub-AMC, and may not represent catalysis on authentic cellular substrates.
A number of USPs have so far been investigated for their ability to recognize and process ubiquitin chains, and each has been found to exhibit its own degree of chain linkage specificity or promiscuity (Table 5). This specificity is partially implied by the different topologies the specific ubiquitin chains assume and whether or not the USP is capable of accessing each iso-peptide bond. Faesen and coworkers performed a di-ubiquitin panel assessing the ability of a variety of USPs to process the eight different ubiquitin chain types over time [19]. While subtle differences could be observed between the USP's ability to process the chains, it was clear that most of the USPs tested were promiscuous and could process the majority of the chains with the exception of the linear chain [19]. However, USP17 appears to be slightly more specific about the topology of each linkage than the other members of the USP family. While USP17 could process each of the ubiquitin chain types, with the exception of linear ubiquitin, it did so to varying degrees. USP17 easily processed Lys48 and Lys63, supporting the previous cell-based findings [5,9,10]. Interestingly, USP17 processed the Lys33 chain relatively well, a chain type that is not easily processed by most USPs, and is related to a handful of pathways which use ubiquitination to regulate the function of the substrate rather than target it for degradation [25]. Recently Lys33 ubiquitin chains have been associated with the modulation of kinase activity [26]. USP17 also easily processed Lys11, which have been described as a strong degradation signal similar to the Lys48 chains [7,19,27]. The observed cleavage pattern by USP17 is most similar to the cell-cycle dependent USP1.
We also determined the efficiency by which USP17 processes longer ubiquitin chains including Lys48, Lys63 and Lys11 linked tetra-ubiquitin chains. The cleavage pattern for each chain type suggest USP17 efficiently deubiquitinates each of these substrates and utilizes an exo-trimming mechanism as no accumulation of a single intermediate ubiquitin species other than mono-ubiquitin was observed [28]. Furthermore, as these chains are suspected by structural studies to assume significantly different topologies, these findings suggest that the specificity of USP17 is more likely dependent on the protein substrate to which the ubiquitin chain is conjugated to, not the chain itself [28].
The effects of pH on the ability of USP17 to process substrate was evaluated by varying the pH and Ub-Rho110 concentration. From a global prospective, USP17 remains active within a wide range of pH values (6 to 10.5), however USP17 was most active between pH values of 7 and 9. The estimated pKa and pKb values suggest that the histidine and cysteine of USP17's catalytic triad assume the appropriate ionization states to perform catalysis within this range. Minimal differences were observed between the estimated pKa and pKb values at the two substrate concentrations, suggesting that the binding of the substrate is not influencing the pKa values on the free enzyme or substrate upon formation of the USP17- Ub-Rho110 complex.
USP17 plays an important role in cell-cycle progression and proliferation and regulation of USP17 activity in these processes requires a careful balance. When persistent USP17 expression occurs, it can apply stress to the cell-cycle resulting in proliferation of cancer cells [6]. The overexpression of USP17 is thought to be a contributing factor in the proliferation of prostate, breast, hematopoietic, and non-small cell lung cancers by perturbing the cell-cycle checkpoints [5–7]. Previous work has shown that in cancer, knock-down of overexpressed USP17 by shRNA leads to a reduction in the size of breast cancer xenographs [5]. These studies suggest that USP17 may be a novel target for anti-cancer drug treatment. Therefore, the development of this robust expression and purification system for USP17, and the biochemical, kinetic and oligomeric state characterization described here, will pave the way for the discovery and design of inhibitors targeting USP17. Furthermore, the enzyme has a high catalytic turnover of fluorescent ubiquitin substrates and is stable enough to allow for high-throughput screening of large compound libraries.
Supplementary Material
Supplemental Figure 1. DLS results at different temperatures. Representative DLS curves of USP17 at two temperatures: 4 °C (Orange) and 25 °C (Blue) at three concentrations of USP17 (A) 0.5 mg/mL, (B) 0.75 mg/mL, and (C) 1 mg/mL. All assays were performed in triplicate.
Highlights.
USP17 is highly upregulated in cancer and is a viable anti-cancer drug target
USP17 was purified from E.coli inclusion bodies and is highly active
USP17 appears to function as a monomer in solution
USP17 can recognize and cleave Lys11, Lys33, Lys48 and Lys63 ubiquitin linkages
Purified USP17 can be used for high-throughput screening to identify inhibitors
Acknowledgments
We wish to thank Dr. Jia Ma of the Bindley Biosciences Center Biophysical Analysis Laboratory for helping us with that AUC data collection and analysis.
Funding: This work was supported by a grant from the Walther Cancer foundation (to ADM) and a Purdue Research Foundation grant from the Purdue Center for Cancer Research (to NMH). DNA sequencing was partially supported by the Purdue Center for Cancer Research DNA Sequencing Shared Resource which is partially supported by a grant from the National Institutes of Health [P30 CA023168].
Abbreviations
- DUB
deubiquitinating enzyme
- USP
Ubiquitin Specific Protease
- Ub
ubiquitin
- CDC25A
cell division cycle 25A
- RCE1
Ras converting enzyme 1
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- βME
β-mercaptoethanol
- AMC
7-amino-4-methylcoumarin
- Rho110
rhodamine110
- AFU
arbitrary fluorescence units
- DTT
dithiothreitol
- BSA
bovine serum albumin
- SEC
size-exclusion chromatography
- AUC
analytical ultracentrifugation
- DLS
dynamic light scattering
- Sf9
Spodoptera frugiperda 9
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fraile JM, Quesada V, Rodriguez D, Freije JMP, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–2388. doi: 10.1038/onc.2011.443. doi: http://www.nature.com/onc/journal/v31/n19/suppinfo/onc2011443s1.html. [DOI] [PubMed] [Google Scholar]
- 2.Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, Bernards R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. doi: http://dx.doi.org/10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Faesen AC, Dirac AMG, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP Activation by Its C-Terminal Ubiquitin-like Domain and Allosteric Regulation by GMP-Synthetase. Mol Cell. 2011;44:147–159. doi: 10.1016/j.molcel.2011.06.034. doi: http://dx.doi.org/10.1016/j.molcel.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 4.Burrows JF, McGrattan MJ, Rascle A, Humbert M, Baek KH, Johnston JA. DUB-3, a cytokine-inducible deubiquitinating enzyme that blocks proliferation. J Biol Chem. 2004;279:13993–14000. doi: 10.1074/jbc.M311291200. [DOI] [PubMed] [Google Scholar]
- 5.Pereg Y, Liu BY, O'Rourke KM, Sagolla M, Dey A, Komuves L, French DM, Dixit VM. Ubiquitin hydrolase Dub3 promotes oncogenic transformation by stabilizing Cdc25A. Nat Cell Biol. 2010;12:400–U226. doi: 10.1038/ncb2041. [DOI] [PubMed] [Google Scholar]
- 6.McFarlane C, Kelvin AA, de la Vega M, Govender U, Scott CJ, Burrows JF, Johnston JA. The Deubiquitinating Enzyme USP17 Is Highly Expressed in Tumor Biopsies, Is Cell Cycle Regulated, and Is Required for G(1)-S Progression. Cancer Res. 2010;70:3329–3339. doi: 10.1158/0008-5472.can-09-4152. [DOI] [PubMed] [Google Scholar]
- 7.McFarlane C, McFarlane S, Paul I, Arthur K, Scheaff M, Kerr K, Stevenson M, Fennell DA, Johnston JA. The deubiquitinating enzyme USP17 is associated with non-small cell lung cancer (NSCLC) recurrence and metastasis. Oncotarget. 2013;4:1836–1843. doi: 10.18632/oncotarget.1282. <Go to ISI>://WOS:000327401100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burrows JF, Kelvin AA, McFarlane C, Burden RE, McGrattan MJ, De la Vega M, Govender U, Quinn DJ, Dib K, Gadina M, Scott CJ, Johnston JA. USP17 Regulates Ras Activation and Cell Proliferation by Blocking RCE1 Activity. J Biol Chem. 2009;284:9587–9595. doi: 10.1074/jbc.M807216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaworski J, Govender U, McFarlane C, de la Vega M, Greene MK, Rawlings ND, Johnston JA, Scott CJ, Burrows JE. A novel RCE1 isoform is required for H-Ras plasma membrane localization and is regulated by USP17. Biochem J. 2014;457:289–300. doi: 10.1042/bj20131213. [DOI] [PubMed] [Google Scholar]
- 10.Ramakrishna S, Suresh B, Lee EJ, Lee HJ, Ahn WS, Baek KH. Lys-63-specific Deubiquitination of SDS3 by USP17 Regulates HDAC Activity. J Biol Chem. 2011;286:10505–10514. doi: 10.1074/jbc.M110.162321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin JM, Yoo KJ, Kim MS, Kim D, Baek KH. Hyaluronan- and RNA-binding deubiquitinating enzymes of USP17 family members associated with cell viability. BMC Genomics. 2006;7 doi: 10.1186/1471-2164-7-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG, Aniba M, Poch O, Thompson J, Blackshields G, Sievers F, Shi W, Wilm A, Higgins D, Bradley R, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L, Clamp M, Cuff J, Searle S, Barton G, Do C, Mahabhashyam M, Brudno M, Batzoglou S, Eddy S, Edgar R, Edgar R, Finn R, Mistry J, Tate J, Coggill P, Heger A, Pollington J, Gavin O, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer E, Eddy S, Bateman A, Gouy M, Guindon S, Gascuel O, Hogeweg P, Hesper B, Katoh K, Misawa K, Kuma K, Miyata T, Katoh K, Toh H, Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D, Lassmann T, Sonnhammer E, Liu Y, Schmidt B, Maskell D, Löytynoja A, Goldman N, Mizuguchi K, Deane C, Blundell T, Overington J, Morgenstern B, Frech K, Dress A, Werner T, Notredame C, Higgins D, Heringa J, Pirovano W, Feenstra K, Heringa J, Söding J, Thompson J, Koehl P, Ripp R, Poch O, Wilm A, Higgins D, Notredame C. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramakrishna S, Suresh B, Baek KH. Biological functions of hyaluronan and cytokine-inducible deubiquitinating enzymes. Biochim Biophys Acta-Reviews Cancer. 2015;1855:83–91. doi: 10.1016/j.bbcan.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Ramakrishna S, Suresh B, Bae SM, Ahn WS, Lim KH, Baek KH. Hyaluronan Binding Motifs of USP17 and SDS3 Exhibit Anti-Tumor Activity. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Báez-Santos YM, Mielech AM, Deng X, Baker S, Mesecar AD. Catalytic Function and Substrate Specificity of the Papain-Like Protease Domain of nsp3 from the Middle East Respiratory Syndrome Coronavirus. J Virol. 2014;88:12511–12527. doi: 10.1128/JVI.01294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. doi: http://dx.doi.org/10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 17.Tomar S, Johnston ML, St John SE, Osswald HL, Nyalapatla PR, Paul LN, Ghosh AK, Denison MR, Mesecar AD. Ligand-induced Dimerization of Middle East Respiratory Syndrome (MERS) Coronavirus nsp5 Protease (3CLpro): Implications for nsp5 Regulation and the Development of Antivirals. J Biol Chem. 2015;290:19403–22. doi: 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villamil MA, Chen J, Liang Q, Zhuang Z. A Noncanonical Cysteine Protease USP1 Is Activated through Active Site Modulation by USP1-Associated Factor 1. Biochemistry. 2012;51:2829–2839. doi: 10.1021/bi3000512. [DOI] [PubMed] [Google Scholar]
- 19.Faesen AC, Luna-Vargas MPA, Geurink PP, Clerici M, Merkx R, van Dijk WJ, Hameed DS, El Oualid F, Ovaa H, Sixma TK. The Differential Modulation of USP Activity by Internal Regulatory Domains, Interactors and Eight Ubiquitin Chain Types. Chem Biol. 2011;18:1550–1561. doi: 10.1016/j.chembiol.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Luna-Vargas MPA, Christodoulou E, Alfieri A, van Dijk WJ, Stadnik M, Hibbert RG, Sahtoe DD, Clerici M, De Marco V, Littler D, Celie PHN, Sixma TK, Perrakis A. Enabling high-throughput ligation-independent cloning and protein expression for the family of ubiquitin specific proteases. J Struct Biol. 2011;175:113–119. doi: 10.1016/j.jsb.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. Crystal Structure of a UBP-Family Deubiquitinating Enzyme in Isolation and in Complex with Ubiquitin Aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. doi: http://dx.doi.org/10.1016/S0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 22.Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, Cohen RE, Shi Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avvakumov GV, Walker JR, Xue S, Finerty PJ, Jr, Mackenzie F, Newman EM, Dhe-Paganon S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8) J Biol Chem. 2006;281:38061–38070. doi: 10.1074/jbc.M606704200. [DOI] [PubMed] [Google Scholar]
- 24.Bozza WP, Liang Q, Gong P, Zhuang Z. Transient Kinetic Analysis of USP2-Catalyzed Deubiquitination Reveals a Conformational Rearrangement in the K48-Linked Diubiquitin Substrate. Biochemistry. 2012;51:10075–10086. doi: 10.1021/bi3009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristariyanto YA, Choi SY, Rehman SAA, Ritorto MS, Campbell DG, Morrice NA, Toth R, Kulathu Y. Assembly and structure of Lys33-linked polyubiquitin reveals distinct conformations. Biochem J. 2015;467:345–52. doi: 10.1042/BJ20141502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys29/Lys33-linked polyubiquitin chains. Biochem J. 2008;411:249–260. doi: 10.1042/bj20080067. [DOI] [PubMed] [Google Scholar]
- 27.Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A. 2009;106:18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 2011;3:26. doi: 10.3410/b3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. DLS results at different temperatures. Representative DLS curves of USP17 at two temperatures: 4 °C (Orange) and 25 °C (Blue) at three concentrations of USP17 (A) 0.5 mg/mL, (B) 0.75 mg/mL, and (C) 1 mg/mL. All assays were performed in triplicate.