

Abstract
Introduction
Sickle cell anemia (SCA) is a severe, inherited hemoglobin disorder affecting 100,000 persons in the US and millions worldwide. Hydroxyurea, a once daily oral medication, has emerged as the primary disease-modifying therapy for SCA. The accumulated body of evidence over 30 years demonstrates that hydroxyurea is a safe and effective therapy for SCA, but hydroxyurea remains underutilized for a variety of reasons.
Areas covered
In this review, we summarize the available evidence regarding the pharmacology, clinical, and laboratory benefits, and safety of hydroxy-urea therapy for the treatment of SCA. The purpose of this review is to provide the reader a comprehensive understanding of hydroxyurea and to reinforce the fact that hydroxyurea is a safe and effective medication for the treatment of SCA.
Expert opinion
In our opinion, hydroxyurea therapy should be considered standard-of-care for SCA, representing an essential component of patient management. Early initiation and broader use of hydroxyurea will alter the natural history of SCA, so affected children can live longer and healthier lives. In addition, hydroxyurea use should be extended to low-resource settings such as sub-Saharan Africa, where the burden of SCA and the need for hydroxyurea is arguably the greatest.
Keywords: fetal hemoglobin, hemoglobinopathies, hydroxyurea, sickle cell anemia
1. Introduction
1.1 Genetics and pathophysiology of sickle cell anemia (SCA)
Sickle cell disease (SCD) is a serious, inherited blood disorder associated with significant morbidity and early mortality. SCD affects approximately 100,000 persons in the US and millions of persons worldwide [1,2]. SCD includes a group of congenital hemolytic anemias all characterized by the predominance of sickle hemoglobin (HbS), which results from a single β-globin gene mutation, leading to an amino-acid substitution (Glu→Val, βA→βS) and formation of the abnormal HbS tetramer. Under deoxygenated states, HbS undergoes rapid intracellular polymerization, which damages the erythrocyte membrane and significantly alters both its shape and its flexibility while carrying oxygen to the body. These stiff and ‘sickled’ red blood cells have a decreased lifespan, and result in both acute and chronic hemolysis. They also undergo a complex process known as vaso-occlusion, in which sickled erythrocytes and other circulating cells adhere to the vascular endothelium, aggregate together to disrupt blood flow especially in the small blood vessels, and cause hypoxia to tissues and vital organs. The pathophysiology of vaso-occlusion is complex and involves more than overt occlusion by sickled erythrocytes, including the detrimental effects of chronic inflammation and abnormalities in the function of the vascular endothelium. The most common and most serious form of SCD is homozygous HbSS, also called sickle cell anemia (SCA), in which HbS is inherited from both parents. Other common and clinically significant forms of SCD include HbS inherited with HbC (known as HbSC) or with beta-thalassemia (known as HbS-β thalassemia). Serious clinical manifestations of SCA include hemolytic anemia, splenic dysfunction with increased susceptibility to infection, painful acute vaso-occlusive events, stroke, chronic organ dysfunction, and early mortality. In the US and Europe, newborn hemoglobinopathy screening allows early preventive treatments such as penicillin prophylaxis and provision of important immunizations, which together have significantly reduced the early mortality [3,4]. But in many resource poor settings where the sickle cell burden is greatest, early diagnostic capacities are lacking and childhood mortality is high [5,6].
1.2 Clinical manifestations of SCA
The clinical sequelae of SCA begin within the first few years of life, when the amount of fetal hemoglobin (HbF) decreases within the erythrocytes and HbS begins to predominate. HbF serves as a potent inhibitor of intracellular HbS polymerization, but its post-natal physiological decline reduces these protective effects. The spleen is affected earliest, typically in the first 1–2 years of life, but the kidney, brain, and other organs also begin to reflect sickle-related injury in early childhood from chronic and repeated sickling events. Although disease manifestations are not always clinically evident, the abnormal sickled erythrocytes circulate continuously and cause repeated injury due to hypoxia/reperfusion events related to sickling. For these reasons, disease-modifying treatments are warranted in children with SCA, even in the absence of obvious clinical signs and symptoms. Therapies designed to increase HbF levels are particularly attractive, since they offer direct intracellular protection against in vivo HbS polymerization and sickling. Theoretically, HbF induction could help prevent acute clinical complications of SCA such as pain and stroke, while also ameliorating the long-term complications related to chronic organ injury.
1.3 Disease-modifying therapy for SCA
As recently as the 1980s, the only available disease-modifying therapy for SCA was intermittent red blood cell transfusions, which were commonly reserved for acute, life-threatening complications. In the 1990s, hydroxyurea emerged as a promising pharmacologic therapy for SCA, due primarily to its ability to induce HbF [7-9], and over the past 30 years has become the primary disease modifying treatment modality [10]. In multiple prospective research studies, hydroxyurea has consistently demonstrated improvement in the abnormal laboratory parameters of SCA, while also reducing the acute clinical complications such as painful vaso-occlusive events and acute chest syndrome, as well as the need for blood transfusions and hospitalization [11-19]. More recently, hydroxyurea has been associated with improved survival for both adults and children with SCA [20-23].
1.4 Introduction to hydroxyurea therapy for SCA
Although there is recent progress in the development of new therapeutic agents that target specific parts of the pathophysiology of sickling, such as erythrocyte-endothelium adhesion or hypercoagulability, hydroxyurea remains the only carefully studied, widely available, and clinically effective pharmacologic therapy for SCA. For a variety of reasons, hydroxyurea was initially reserved only for adult patients with frequent and severe clinical complications; however, recent guidelines from the National Heart, Lung, and Blood Institute (NHLBI) now recommend that hydroxyurea be utilized much more broadly, including offering hydroxyurea to all infants with SCA beginning at 9 months of age, regardless of clinical severity [10]. Despite the substantial body of evidence demonstrating the benefits of hydroxyurea with an acceptable short- and long-term toxicity profile, there remain lingering concerns by some providers and patients regarding its safety. In this review, we summarize the evidence regarding the safe and effective use of hydroxyurea treatment for SCA.
2. Body of review
2.1 Historical perspective
Hydroxyurea is a simple chemical compound (chemical formula CH4N2O2) first synthesized in 1869 by Dresler and Stein in a series of experiments attempting to extract derivatives of urea [24,25]. A century later, hydroxyurea was first introduced as a clinical agent, primarily as chemotherapy to treat various solid tumors and leukemias [26,27]. Due to early efficacy as an antineoplastic agent, hydroxyurea was first approved by the US FDA in 1967. Over the next several decades, the use of hydroxyurea expanded with efficacy for the treatment of chronic myelogenous leukemia, psoriasis, melanoma, ovarian cancer, polycythemia vera, and even HIV [28-33]. In the mid-1980s, hydroxyurea was shown in proof-of-principle studies to induce HbF in SCA [7,8], which was followed by a key adult Phase I/II study [9] and a pivotal Phase III trial a decade later [12]. Hydroxyurea received FDA approval for the treatment of adults with severe SCA in 1998 [34].
2.2 Mechanisms of action
Despite 30 years of research documenting its recognized laboratory and clinical benefits for SCA, the exact mechanisms by which hydroxyurea induces HbF and ameliorates the pathophysiology of SCA remain incompletely understood. The primary benefits of hydroxyurea for SCA relate to its ability to increase HbF levels [35], which inhibits intracellular HbS polymerization and prevents the sickling process within erythrocytes. However, the full benefits of hydroxyurea therapy in SCA are multifactorial, extending beyond HbF induction (Figure 1).
Figure 1.
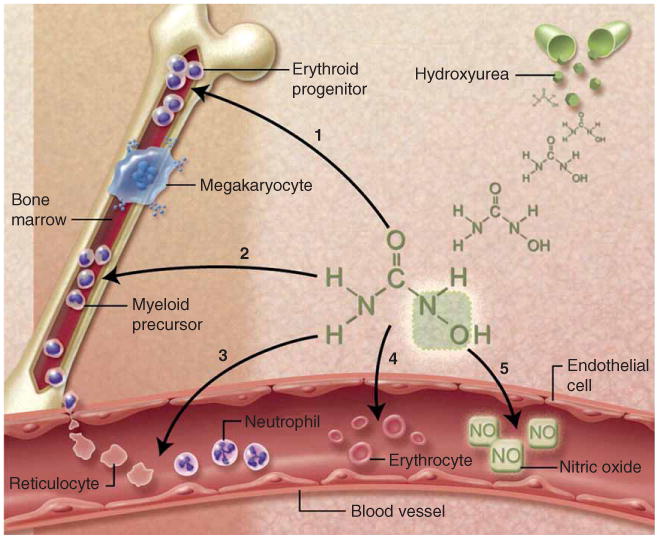
Multiple mechanisms of action byhydroxyurea for SCA. (1) Fetal hemoglobin induction through soluble guanylyl cyclase activation and altered erythroid kinetics; (2) lower neutrophil and reticulocyte counts from ribonucleotide reductase inhibition and marrow cytotoxicity; (3) decreased adhesiveness and improved rheology of circulating neutrophils and reticulocytes; (4) reduced hemolysis through improved erythrocyte hydration, macrocytosis, and reduced intracellular sickling; and (5) nitric oxide (NO) release with potential local vasodilatation and improved vascular response. Illustration courtesy of Alice Y. Chen.
Reproduced from [47] © the American Society of Hematology.
The primary and most well understood mechanism of action of hydroxyurea in vivo is the reversible inhibition of ribonucleotide reductase (RR), a critical enzyme that converts ribonucleosides into deoxyribonucleosides, which are required for the synthesis and repair of DNA [36]. Potent inhibition of RR leads to decreased intracellular pools of deoxyribonucleotide triphosphates and impedes progression of cellular division through S phase [37,38]. Temporary arrest of hematopoiesis through once-daily hydroxyurea results in altered erythroid kinetics upon recovery; such ‘stress erythropoiesis’ features higher HbF through recruitment of early erythroid progenitors that maintain their HbF-producing capacity [39,40]. Erythrocytes with more HbF are larger (higher MCV) and more deformable (better rheology). Figure 2 demonstrates the marked improvements between a peripheral blood smear of a child with SCA before and after hydroxyurea therapy. Myelopoiesis is also affected, leading to dose-dependent and reversible neutropenia. Other benefits include reduced expression of surface molecules that adhere to the endothelium [41], decreased degree of chronic inflammation as seen by decrease in WBC and platelet counts, increased levels of nitric oxide and cyclic nucleotides that may facilitate vascular dilatation and also induce HbF [42], and even direct salutary effects on the vascular endothelium [43].
Figure 2.
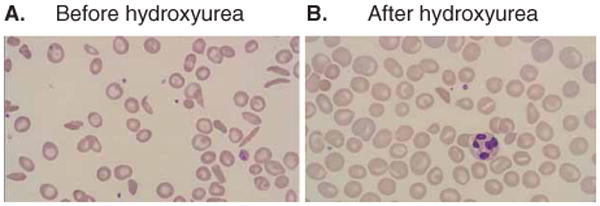
Peripheral blood smear changes with hydroxyurea therapy. Panel A illustrates the severe anemia and frequent irreversibly sickled erythrocytes commonly observed in untreated patients with SCA. Panel B demonstrates the improvement in anemia and increases in both the size and hemoglobin content of erythrocytes while on hydroxyurea therapy.
2.3 Clinical indications
In the 1980s, hydroxyurea was demonstrated to improve levels of HbF in anemic primates, which led to the first clinical application of hydroxyurea as treatment for SCA [7,8]. A critical Phase I/II trial confirmed that hydroxyurea increases HbF in adults with SCA without excessive bone marrow toxicity, leading to the landmark MultiCenter Study of Hydroxyurea in Sickle Cell Anemia (MSH). MSH was a Phase III double-blinded, placebo-controlled randomized trial of adults with severe SCA, and was halted early by the study sponsor, the National Heart, Lung, and Blood Institute, due to a highly significant reduction in the time to first painful crisis for the hydroxyurea-treated arm [12]. The study also demonstrated that hydroxyurea therapy reduced the frequency of acute chest syndrome, as well as the number of blood transfusions and hospitalizations; follow-up studies have demonstrated an association between hydroxyurea therapy and improved survival [20-23]. The clinical benefits of hydroxyurea were also associated with predictable laboratory benefits, including improvements in hemoglobin concentration, MCV, and HbF levels, along with reductions in WBC count (especially neutrophils), absolute reticulocyte count, and measures of hemolysis.
Because hydroxyurea was historically a ‘cancer drug’ and was tested primarily in patients with severe SCA symptoms, its clinical indication was initially reserved for older and sicker patients. Even today, the US FDA label is highly restrictive, stating that hydroxyurea is indicated ‘to reduce the frequency of painful crises and to reduce the need for blood transfusions in adult patients with sickle cell anemia with recurrent moderate to severe painful crises (generally at least 3 during the preceding 12 months)’. However, the compelling results of the adult SCA trials led to prospective testing in children, and the use of hydroxyurea has steadily expanded within the pediatric population. As with adults, clinical trials and observations demonstrated that the salutary laboratory and clinical benefits of hydroxyurea were also seen in children without significant toxicity [14-17].
Due to its numerous benefits and safe short-term toxicity profile, early initiation of hydroxyurea might reduce or prevent the chronic organ damage known to begin in early childhood, particularly sickle-related injury to the brain, kidney, and spleen. Pilot studies demonstrated that hydroxyurea treatment could preserve splenic and brain function, and did not result in delayed growth or other serious toxicities [14-16]. These pilot studies led to the multicenter BABY HUG study, a Phase III double-blinded, placebo-controlled randomized trial of infants 9–17 months of age with SCA; enrollment intentionally did not require clinical severity. Although the primary study endpoints of preventing organ damage were not met, BABY HUG demonstrated positive results in almost every secondary study endpoint, whether related to acute clinical complications or measures of organ function. Infants taking hydroxyurea had fewer episodes of dactylitis and other painful vaso-occlusive events, fewer transfusions, and reduced numbers of hospitalizations [11]. These results were especially impressive given that the study used a relatively low, fixed-dose of hydroxyurea (20 mg/kg/day) without escalation to maximum tolerated dose (MTD), since other studies have demonstrated improved clinical and laboratory benefits at MTD [17-19]. The role of hydroxyurea has also been studied prospectively in the setting of cerebrovascular disease, both in children with overt stroke and more recently in children with abnormal transcranial Doppler velocities [44,45]. Preliminary results suggest that hydroxyurea may be beneficial in reducing the severity of cerebrovascular disease for children with SCA, and a large multicenter trial was recently stopped early by NIH for reaching its primary study endpoint (TWiTCH, ClinicalTrials.gov NCT01425307).
As the benefits of hydroxyurea become more publicized and accepted, and as providers and patients become more comfortable with drug dosing and safety, its use in SCA is expanding. There are now many adults and children who have been receiving hydroxyurea for 10–20 years with sustained laboratory and clinical benefits [13,19,21-23,46]. With long-term use, there is a growing body of evidence suggesting that hydroxyurea is associated with improved survival [20-23]. In 2014, the NHLBI released new evidence-based clinical guidelines for the management of patients with SCA and now recommends that adults be treated for many clinical indications; further, all children with SCA should be offered hydroxyurea therapy beginning at 9 months of age, regardless of clinical severity [10].
2.4 Variable response
The effects of hydroxyurea are dose dependent, and many clinical trials have demonstrated that the laboratory and clinical benefits of hydroxyurea are optimized when escalated to MTD [17,18,47]. The clinical effectiveness of dose escalation to MTD outside the setting of clinical trials, including provider comfort with dose escalation and patient/family satisfaction with the escalation process, has yet to be clearly demonstrated and remains an active area of research. MTD is defined as a stable and tolerated dose (mg/kg/day) that achieves a target range of mild marrow suppression, most commonly determined by the absolute neutrophil count but also by the absolute reticulocyte count. At MTD, almost all patients with SCA have a significant increase in the total hemoglobin concentration and the MCV, but the laboratory benefits of hydroxyurea are most often defined by the amount of HbF achieved. All children with good adherence will respond to hydroxyurea, but there is substantial interpatient variability in both the MTD itself and the %HbF levels achieved [39,46-48]. For example, some patients tolerate an MTD as high as 35 mg/kg/day before reaching appropriate myelosuppression, while others can only tolerate a dose of 15 mg/kg/day. Similarly, some patients achieve HbF levels greater than 40%, while others are never able to reach 20%. These observations suggest important individual pharmacokinetics (PK), pharmacodynamics (PD), and pharmacogenomics (PGx) differences that contribute to the phenotypic variability in both the dosing and response to hydroxyurea therapy.
Hydroxyurea has excellent bioavailability in its oral form and is rapidly cleared from the circulation with a half-life of 2–3 h in most adults and children [9,48-50]. Peak serum concentrations for children with SCA are reached quickly, and average 20–30 μg/ml (∼ 250–400 μM) with undetectable concentrations approximately 6–8 h after the initial dose [48]. One large analysis of PK data for children with SCA demonstrated that there appear to be two distinct oral absorption profiles (Figure 3)–one with hydroxyurea absorbed rapidly and peak concentrations within 15–30 min and another with slower absorption and peak concentrations at 60–120 min [48]. The fast profile was associated with a higher MTD, suggesting that rapid absorption and clearance allow higher hydroxyurea doses. Despite their variable PK parameters, patients at hydroxyurea MTD trended toward an average area under the curve of 115 μg*h/ml [51].
Figure 3.
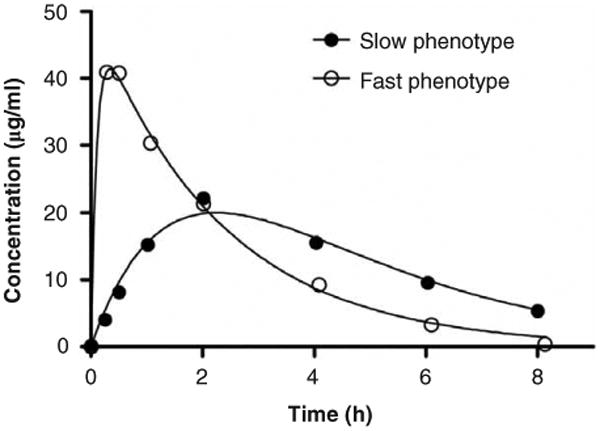
Concentration-time profiles of hydroxyurea in young patients with SCA. Shown are representative examples of ‘Fast’ phenotype with peak concentration at 15–30 min (59% of patients) and the ‘Slow’ phenotype with Cmax at either 60 or 120 min (41% of patients).
Reproduced with permission from [48] © the American Society of Hematology.
Using historical PK and PD data for children with SCA who achieved hydroxyurea MTD, we have recently developed a population PK-model and individualized dosing strategy that is under investigation in the prospective Therapeutic Response Evaluation and Adherence Trial (TREAT, ClinicalTrials.gov NCT02286154). This Bayesian approach has the potential to optimize the dose titration of hydroxyurea therapy for children with SCA, such that the clinical benefits at MTD are achieved more quickly than using standard dose escalation [52].
2.5 Safety evaluation
Hydroxyurea is a well-tolerated medication with very few short- or long-term toxicities. The most commonly encountered short-term toxicity includes mild and reversible cytopenias, which are an expected result and actually used to determine the MTD. Although all cell lines can be affected, the most commonly encountered hematological toxicity includes mild to moderate neutropenia, followed by reticulocytopenia and then thrombocytopenia. In clinical trials, about 20% of participants develop mild cytopenias, which are always reversible by either temporarily suspending therapy or, if persistent, slightly decreasing the hydroxyurea dose [11,16,47]. Because of the predicted myelosuppression, hydroxyurea initiation and dose escalation requires complete blood counts on a monthly basis. Once MTD is reached, laboratory monitoring can be less frequent. Table 1 illustrates the common hematological criteria used to define hydroxyurea toxicities and to assist the dose escalation process to MTD. If any toxicity criteria are present, hydroxyurea should be temporarily suspended for 1 week, to allow blood count recovery. However, if there is no toxicity and all of the escalation criteria are met, then the hydroxyurea dose can be escalated in increments of ∼5 mg/kg/day every 8 weeks until MTD is achieved. MTD is most commonly defined an absolute neutrophil count of 1.5 – 3.0 × 109/l , which represents a mild degree of neutropenia that is not associated with an increased risk of infection.
Table 1.
Hematological parameters used to define the hydroxyurea maximum tolerated dose (MTD) with mild marrow suppression.
| Parameter | Toxicity criteria | Escalation criteria | Target MTD value |
|---|---|---|---|
| Absolute neutrophil count (ANC, × 109/l) | < 1.0 | > 3.0 | 1.5–3.0 |
| Absolute reticulocyte count (ARC, × 109/l) | < 80 (unless Hb ≥ 9.0) | > 200 | 100–200 |
| Platelets (× 109/l) | < 80 | > 150 | > 80 |
Although early clinical trials monitored renal and liver function closely during hydroxyurea therapy, there is no evidence to suggest that hydroxyurea has adverse effects upon renal or hepatic function [14]. However, renal function should be measured at treatment initiation, since hydroxyurea is primarily cleared through renal excretion. The most common reported clinical side effects of hydroxyurea therapy include mild gastrointestinal upset, darkening of the skin or nail beds, and rarely mild thinning of the hair. Table 2 provides a summary of hydroxyurea toxicities ranging from very rare to more common.
Table 2.
Recognized and potential toxicities of hydroxyurea therapy in SCA.
| Very rare (≪ 1%) | Rare (< 1%) | Occasional (1–10%) | Common (> 10%) |
|---|---|---|---|
| Allergic reaction | Diarrhea | Anemia (mild, dose-dependent) | Leukopenia (mild, dose-dependent) |
| Increased ALT | Gastritis | Anorexia | Nail/Skin hyperpigmentation |
| Increased Creatinine | Hypersplenism | Hair thinning | Neutropenia (mild, dose-dependent) |
| Malignancy | Pancytopenia | Nausea | Reticulocytopenia (mild, dose-dependent) |
| Skin ulcers | Rash Vomiting | Thrombocytopenia |
Before the advent of early identification of SCA by newborn screening and aggressive interventions with penicillin prophylaxis and pneumococcal vaccination, the possible long-term side-effects of any disease-modifying therapy such as hydroxyurea were not an important consideration, since survival to adulthood was rare. Now, in high-resource settings with comprehensive sickle cell care including increased use of hydroxyurea, most children with SCA will reach adulthood. Concerns regarding potential long-term effects of hydroxyurea are now highly relevant and include potential effects on fertility, pregnancy risks including teratogenicity, and concern for increased risk of malignancy.
Some early reports suggested that hydroxyurea was associated with an increased risk of leukemic transformation among patients with myeloproliferative neoplasms, which was extrapolated by both patients and healthcare providers to suggest hydroxyurea might also confer an increased risk of leukemia in SCA [53]. However, subsequent studies demonstrated that hydroxyurea does not lead to an increased risk of leukemia for patients with myeloproliferative neoplasms, despite those being intrinsically pre-leukemic conditions [54]. Importantly, SCA is unlike myeloproliferative neoplasms such as polycythemia vera or essential thrombocytosis, and is not considered a preleukemic condition. Many patients have taken hydroxyurea for over 10–20 years, with no evidence to suggest an increased risk of malignancy. Of course, patients with SCA carry the same risk of malignancy as the general population and numerous cases in the pre-hydroxyurea era have been reported [55]. With increased survival due to hydroxyurea therapy, there will inevitably be some patients with SCA who develop leukemia or other malignancies, but this should not automatically indict hydroxyurea as the causative agent. Close continued longitudinal monitoring of patients with long-term exposure to hydroxyurea therapy will be essential to elucidate any increased risk of malignancy associated with treatment.
Effects of hydroxyurea upon fertility and pregnancy represent another common concern for both patients and providers. Similar to the aforementioned cancer risk, there is no clearly defined or observed negative impact of hydroxyurea therapy upon fertility, and no clinical evidence of teratogenicity, but further investigation is certainly warranted as more treated patients reach adulthood. Men with SCA, regardless of hydroxyurea exposure, have numerous sperm and reproductive abnormalities that could decrease fertility [56-62]. Animal models documented adverse effects of hydroxyurea upon sperm function and morphology [63], but very few data in humans confirm or even suggest that hydroxyurea therapy adversely affects fertility [64]. In the MSH trial and follow-up study, despite the recommendation that participants use contraception to prevent pregnancy, 27 male MSH participants reported 40 pregnancies with good outcomes [65]. There are even fewer data evaluating the effects of hydroxyurea on fertility in women, although pregnancy is recognized as high risk in SCA with greatly increased risk of miscarriage and maternal death [66]. Similar to men, many successful pregnancies occurred in women taking hydroxyurea on and after the MSH study [65]. As more men and women with SCA live healthier lives into adulthood, it will be critical to carefully evaluate the effects of hydroxyurea exposure on fertility, but current data do not suggest that therapy leads to infertility. Data regarding the potential effects of hydroxyurea on women during pregnancy and lactation are lacking, and prospective studies are sorely needed, since the risks of discontinuing or withholding effective hydroxyurea treatment are substantial to both the mother with SCA and her baby.
2.6 International opportunities
Despite the proven benefits of hydroxyurea therapy for patients with SCA, treatment to date has been limited almost entirely to high-resource settings. However, as noted earlier, SCA is a global disease that affects over 300,000 new infants each year with the highest burden in the low-resource countries of sub-Saharan Africa. New efforts by the World Health Organization (WHO) have challenged countries in the African Region affected by SCA to promote sickle cell screening and to develop national sickle cell strategies. Their recommendations also describe the importance of hydroxyurea as a critical and affordable disease-modifying treatment, and the WHO list of Essential Medicines for Children now includes hydroxyurea for the treatment of hemoglobin disorders [67,68].
It is imperative that the safety, feasibility, and benefits of hydroxyurea be demonstrated in these low-resource settings, which are associated with unique co-morbidities such as malaria, HIV, and severe malnutrition. Several ongoing clinical research trials will obtain critical pilot data regarding the use of hydroxyurea in Africa, including the Realizing Effectiveness Across Continents with Hydroxyurea (REACH, ClinicalTrials.gov NCT01966731) [69] and Novel use Of Hydroxyurea in an African Region with Malaria (NOHARM, ClinicalTrials.gov NCT01976416). These data will assist the development of local treatment guidelines for hydroxyurea use, as part of a national strategy for the treatment of SCA in these low-resource settings. As the safety and benefits of hydroxyurea are demonstrated, local Ministries of Health and international philanthropic organizations must work together with the pharmaceutical industry to increase the availability of hydroxyurea and decrease its price, with the goal of wide availability to the many suffering patients who would benefit greatly from this important therapy.
3. Conclusions
Hydroxyurea is the only available oral medication with proven disease-modifying benefits for the treatment of SCA. There is a large and overwhelming body of evidence consistently documenting the safety and efficacy of hydroxyurea for both adults and children with SCA. Hydroxyurea provides many benefits for SCA including reduction of both acute and chronic complications of this serious and life-threatening disease. Although there were and still remain concerns regarding the long-term safety of hydroxyurea, thousands of patients have now been treated effectively for many years, without any signal to suggest that hydroxyurea therapy results in significant toxicity. Despite the consistent evidence documenting its safety and efficacy, hydroxyurea remains underutilized in SCA, primarily due to provider inexperience and unfounded safety concerns expressed by both providers and patients [69].
4. Expert opinion
4.1 Expansion of hydroxyurea use
We advocate that hydroxyurea now be an integral part of the standard care package for all persons with SCA, just as penicillin prophylaxis is universally recommended for all children through the age of 5 years. There are no cardiologists who would withhold therapy for severe hypertension, or endocrinologists who would withhold insulin from a patient with diabetes mellitus; similarly, we advocate that hydroxy-urea should not be withheld from patients with SCA. The NHLBI guidelines recommend the widespread use of hydroxyurea [10] and we are strongly urging clinical providers not only in the US and Europe, but also across sub-Saharan Africa, the Middle East, and India, to follow these guidelines. There is now ample and incontrovertible evidence to suggest that hydroxyurea is effective in SCA for reducing the acute complications while increasing the quality of life, and a growing body of evidence documenting improved organ function and prolonged survival. Although some questions remain about its long-term risks, hydroxyurea therapy should not be limited on theoretical grounds. While recognizing that hydroxyurea is not a true cure for SCA, many treatments are prescribed for efficacy despite not being curative. Although other effective disease-modifying therapies will likely be developed over the next several decades, it must be emphasized that hydroxyurea is highly effective, widely available now, and very inexpensive. We strongly believe that all patients with SCA, from infancy to adulthood, deserve and will undoubtedly benefit from hydroxyurea therapy.
If hydroxyurea is so effective, why are not more patients taking it [70]? Part of the problem relates to an unexplained reluctance to consider SCA as a life-threatening disease warranting aggressive preventive therapy. Historically, SCA has been managed symptomatically, meaning that many persons affected by SCA did not receive any treatment at all until they presented with an acute complication or until serious organ damage had already occurred. Until the past decade, when chronic transfusion therapy and hydroxyurea have become increasingly used in children, there were no routine therapies available for children affected by SCA. If a child had fever, they received antibiotics; if there was severe anemia, they might receive a blood transfusion; and if there was pain, they would receive analgesics. Severe, painful vaso-occlusive crises are the most overt and recognizable symptom of SCA, but SCA is a chronic condition characterized by ongoing severe hemolytic anemia from damaged red blood cells and inexorable in vivo sickling, resulting in poor tissue oxygenation and slow but steady damage to nearly every organ system of the body. Fortunately, organ systems in patients with SCA are resilient and can function fairly effectively without any notable symptoms, until the damage is substantial and often irreversible. Chronic kidney disease, severe cerebrovascular disease, and multi-organ failure including pulmonary hypertension lead to premature death in SCA. Each of these chronic conditions is the result of years of damage by poor tissue oxygenation and vaso-occlusion secondary to circulating sickled cells. For these reasons, it is important to implement disease-modifying therapy to prevent these complications, rather than providing treatments only after the damage has occurred.
4.2 Comparison of hydroxyurea to other available treatments
Currently, available disease-modifying therapies for SCA include hydroxyurea, intermittent or regular blood transfusions, or stem cell transplantation. Blood transfusions are helpful in SCA and can be life-saving in certain acute settings, but have many short- and long-term complications, including acute transfusion reactions, transmission of infection, the development of allo- and auto-antibodies to erythrocyte antigens, and severe transfusion-acquired iron overload leading to further damage to organs such as the heart, pancreas, and liver. Stem cell transplantation can be curative for SCA, but is extremely limited as a practical therapy even in high-resource settings due to lack of matched donors, high cost, and concern about long-term effects including death and sterility. Hydroxyurea, as discussed here, is available as a once daily oral medication and is associated with few short- or long-term toxicities. There are risks and benefits of each of these three therapies, but it is our opinion that hydroxyurea is by far the most logical starting point when selecting a disease-modifying therapy for SCA. Over the next few years, we anticipate that hydroxyurea treatment will become even more effective, as personalized treatment strategies including gene-based drug dosing and PK-based dosing regimens become available.
4.3 Benefits of improved hydroxyurea use
With the early initiation of hydroxyurea, we anticipate that the natural history, so eloquently described through the Cooperative Study for SCD over 30 years ago, will change [71]. As hydroxyurea becomes more widely accepted as the standard of care to prevent both acute and chronic complications of SCA, infants who initiate hydroxyurea therapy at a very young age will represent a ‘new generation’ of SCA patients who live healthier and longer lives. These children may not ever know the frequent debilitating pain suffered by many patients with SCA, and will instead become highly functional persons with the ability to attend school and maintain employment without significant disabilities. While some questions remain about the long-term risks and benefits of hydroxyurea therapy, there should be no question that infants born today with SCA have a much greater chance at a healthy and long life than their counterparts just a few decades ago.
4.4 Hydroxyurea use in limited-resource settings
It is unjust to describe dramatic improvements in the management and outcomes for persons affected by SCA, without recognizing and acting upon the tremendous disparities that occur between high- and low-resource settings. Only a very small percentage of global SCA births occur among people with local medical and financial resources that allow early diagnosis of SCA and early access to hydroxyurea therapy. It is imperative for the global sickle cell community to work together to level the playing field, in order to improve both the length and quality of life for hundreds of thousands of infants born with SCA each year in low-resource settings, primarily sub-Saharan Africa and India. Upon obtaining critical pilot data that confirm safety, feasibility, and benefits of hydroxyurea for affected patients living in low-resource settings, the wide scale introduction of hydroxyurea therapy has the potential to significantly improve the otherwise dismal natural history of SCA around the world.
Article highlights.
Hydroxyurea is the only approved medication with proven benefits for the treatment of SCA.
Multiple clinical trials in adults and children have demonstrated that hydroxyurea decreases nearly all clinical complications of SCA and evidence suggests that hydroxyurea use is also associated with decreased mortality.
Over 30 years of evidence has demonstrated that hydroxyurea is a safe medication with no significant long-term side effects identified to date.
The use of hydroxyurea is mostly limited to the high-resource settings of the US and Europe with limited use in sub-Saharan Africa, where 75% of the global burden of SCA exists.
We strongly advocate that hydroxyurea be considered for all persons affected by SCA, including those living in limited-resource settings, in order to improve both the quality and length of life.
This box summarizes key points contained in the article.
Footnotes
Declaration of interests: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Hassell KL. Population Estimates of Sickle Cell Disease in the U.S. Am J Prev Med. 2010;38(4):S512–21. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Piel FB, Hay SI, Gupta S, et al. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013;10(7):e1001484. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447–52. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92(7):905–12. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]
- 5.McGann PT. Sickle cell anemia: an underappreciated and unaddressed contributor to global childhood mortality. J Pediatr. 2014;165(1):18–22. doi: 10.1016/j.jpeds.2014.01.070. [DOI] [PubMed] [Google Scholar]
- 6.Grosse SD, Odame I, Atrash HK, et al. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6 Suppl 4):S398–405. doi: 10.1016/j.amepre.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Letvin NL, Linch DC, Beardsley GP, et al. Augmentation of Fetal-Hemoglobin Production in Anemic Monkeys by Hydroxyurea. N Engl J Med. 1984;310(14):869–73. doi: 10.1056/NEJM198404053101401. [DOI] [PubMed] [Google Scholar]
- 8.Platt OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74:652–6. doi: 10.1172/JCI111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79(10):2255–65. [PubMed] [Google Scholar]
- 10••.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of Sickle Cell Disease: Summary of the 2014 Evidence-Based Report by Expert Panel Members. JAMA. 2014;312(10):1033–48. doi: 10.1001/jama.2014.10517. The 2014 NHLBI Sickle Cell guidelines suggest that hydroxyurea be offered to all children with SCA beginning at 9 months of age, regardless of clinical severity. [DOI] [PubMed] [Google Scholar]
- 11••.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG) Lancet. 2011;377(9778):1663–72. doi: 10.1016/S0140-6736(11)60355-3. The important results from the Phase III double-blinded, placebo-controlled randomized BABY HUG trial that demonstrated the benefits of hydroxyurea for very young children (9–18 months of age at enrollment) with SCA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12••.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317–22. doi: 10.1056/NEJM199505183322001. The important results from thePhase III double-blinded, placebo-controlled randomized MSH trial that first demonstrated the dramatic clinical benefits of hydroxyurea for adults with very severe SCA. [DOI] [PubMed] [Google Scholar]
- 13.Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 14.Kinney TR, Helms RW, O'Branski EE, et al. Safety of Hydroxyurea in Children With Sickle Cell Anemia: Results of the HUG-KIDS Study, a Phase I/II Trial. Blood. 1999;94(5):1550–4. [PubMed] [Google Scholar]
- 15.Hankins JS, Helton KJ, McCarville MB, et al. Preservation of spleen and brain function in children with sickle cell anemia treated with hydroxyurea. Pediatr Blood Cancer. 2008;50(2):293–7. doi: 10.1002/pbc.21271. [DOI] [PubMed] [Google Scholar]
- 16.Wang WC, Wynn LW, Rogers ZR, et al. A two-year pilot trial of hydroxyurea in very young children with sickle-cell anemia. J Pediatr. 2001;139(6):790–6. doi: 10.1067/mpd.2001.119590. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerman SA, Schultz WH, Davis JS, et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood. 2004;103(6):2039–45. doi: 10.1182/blood-2003-07-2475. [DOI] [PubMed] [Google Scholar]
- 18.Estepp JH, Smeltzer MP, Kang G, et al. Higher Fetal Hemoglobin Following Escalation of Hydroxyurea to Maximum Tolerated Dose Provides Clinical Benefit to Children with Sickle Cell Anemia. Blood. 2014;124(21) ASH Annual Meeting Abstract. [Google Scholar]
- 19.Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood. 2005;106(7):2269–75. doi: 10.1182/blood-2004-12-4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Lopes de Castro Lobo C, Pinto JFC, Nascimento EM, et al. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol. 2013;161(6):852–60. doi: 10.1111/bjh.12323. Important and novel report describing the survival benefits of hydroxyurea for Brazilian children with SCA. [DOI] [PubMed] [Google Scholar]
- 21•.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS) Blood. 2010;115(12):2354–63. doi: 10.1182/blood-2009-05-221333. This article describes the marked survival benefits for patients taking hydroxyurea compared to those who were not, despite the fact that those selected to take hydroxyurea were pre-identified as those with more severe forms of SCA. [DOI] [PubMed] [Google Scholar]
- 22•.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85(6):403–8. doi: 10.1002/ajh.21699. Long-term results from the initial MSH study that demonstrate the improved survival for adults with SCA taking hydroxyurea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23•.Lê Q, Gulbus B, Dedeken L, et al. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer. 2015 doi: 10.1002/pbc.25608. [Epub ahead of print] First report from the US or Europe describing the improved mortality associated with hydroxyurea therapy for children with SCA. [DOI] [PubMed] [Google Scholar]
- 24.Dresler W, Stein R. Ueber den hydroxylharnstoff. Justus Liebigs Annalen der Chemie. 1869 [Google Scholar]
- 25.Rees DC. The rationale for using hydroxycarbamide in the treatment of sickle cell disease. Haematologica. 2011;96(4):488–91. doi: 10.3324/haematol.2011.041988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole DR, Beckloff GL, Rousselot LM. Clinical results with hydroxyurea in cancer chemotherapy. NY State J Med. 1965;65:2132–6. [PubMed] [Google Scholar]
- 27.Bloedow CE. Phase II studies of hydroxyurea (NSC-32065) in adults: miscellaneous tumors. Cancer Chemother Rep. 1964;40:39–41. [PubMed] [Google Scholar]
- 28.Dahl M, Comaish JS. Long-term effects of hydroxyurea in psoriasis. BMJ. 1972;5840:585–7. doi: 10.1136/bmj.4.5840.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy BJ. Hydroxyurea therapy in chronic myelogenous leukemia. Cancer. 1972;29(4):1052–6. doi: 10.1002/1097-0142(197204)29:4<1052::aid-cncr2820290454>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Donovan PB, Kaplan ME, Goldberg JD, et al. Treatment of polycythemia vera with hydroxyurea. Am J Hematol. 1984;7(4):329–34. doi: 10.1002/ajh.2830170402. [DOI] [PubMed] [Google Scholar]
- 31.Lori F, Malykh A, Cara A, et al. Hydroxyurea as an inhibitor of human immunodeficiency virus-type 1 replication. Science. 1994;5186:801–5. doi: 10.1126/science.7973634. [DOI] [PubMed] [Google Scholar]
- 32.Ariel IM. Therapeutic effects of hydroxyurea. Experience with 118 patients with inoperable solid tumors. Cancer. 1970;25(3):705–14. doi: 10.1002/1097-0142(197003)25:3<705::aid-cncr2820250331>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 33.Cassileth PA, Hyman GA. Treatment of malignant melanoma with hydroxyurea. Cancer Res. 1967;27(10):1843–5. [PubMed] [Google Scholar]
- 34.Segal JB, Strouse JJ, Beach MC, et al. Hydroxyurea for the treatment of sickle cell disease. Evid Rep Technol Assess. 2008;165:1–95. [PMC free article] [PubMed] [Google Scholar]
- 35.Lebensburger JD, Pestina TI, Ware RE, et al. Hydroxyurea Therapy Requires HbF Induction for Clinical Benefit in a Sickle Cell Mouse Model. Haematalogica. 2010;95(9):1599–603. doi: 10.3324/haematol.2010.023325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elford HL. Effect of hydroxyurea on ribonucleotide reductase. Biochem Biophys Res Commun. 1968;33(1):129–35. doi: 10.1016/0006-291x(68)90266-0. [DOI] [PubMed] [Google Scholar]
- 37.Alvino GM, Collingwood D, Murphy JM, et al. Replication in Hydroxyurea: It's a Matter of Time. Mol Cell Biol. 2007;27(18):6396–406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koç A, Wheeler LJ, Mathews CK, Merrill GF. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem. 2004;279(1):223–30. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- 39.Baliga BS, Pace BS, Chen HH, et al. Mechanism for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. Am J Hematol. 2000;65(3):227–33. doi: 10.1002/1096-8652(200011)65:3<227::aid-ajh9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 40.Stamatoyannopoulos G, Veith R, Galanello R, et al. Production in Stressed Erythropoiesis: Observations and Kinetic Models. Ann NY Acad Sci. 1985;445:188–97. doi: 10.1111/j.1749-6632.1985.tb17188.x. [DOI] [PubMed] [Google Scholar]
- 41.Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88(12):4701–10. [PubMed] [Google Scholar]
- 42.Cokic VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide–dependent activation of soluble guanylyl cyclase. J Clin Invest. 2003;111(2):231–9. doi: 10.1172/JCI16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almeida CB, Scheiermann C, Jang JE, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood. 2012;120(14):2879–88. doi: 10.1182/blood-2012-02-409524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ware RE, Zimmerman SA, Sylvestre PB, et al. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. J Pediatr. 2004;145(3):346–52. doi: 10.1016/j.jpeds.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 45.Ware RE, Helms RW, SWiTCH Investigators Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) Blood. 2012;119(17):3925–32. doi: 10.1182/blood-2011-11-392340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hankins JS, Aygun B, Nottage K, et al. From infancy to adolescence: fifteen years of continuous treatment with hydroxyurea in sickle cell anemia. Medicine. 2014;93(28):e215. doi: 10.1097/MD.0000000000000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115(26):5300–11. doi: 10.1182/blood-2009-04-146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ware RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood. 2011;118(18):4985–91. doi: 10.1182/blood-2011-07-364190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Montalembert M, Bachir D, Hulin A, et al. Pharmacokinetics of hydroxyurea 1,000 mg coated breakable tablets and 500 mg capsules in pediatric and adult patients with sickle cell disease. Haematologica. 2006;91(12):1685–8. [PubMed] [Google Scholar]
- 50.Rodriguez GI, Kuhn JG, Weiss GR, et al. A bioavailability and pharmacokinetic study of oral and intravenous hydroxyurea. Blood. 1998;91(5):1533–41. [PubMed] [Google Scholar]
- 51.McGann PT, Sparreboom A, Mortier NA, et al. Hydroxyurea Pharmacokinetics for Predicting Maximum Tolerated Dose in Children with Sickle Cell Anemia. Blood. 2014;124(21):2707. (ASH Annual Meeting Abstract) [Google Scholar]
- 52.Dong M, McGann PT, Mizuno T, et al. A Prediction of Maximum Tolerated Dose (MTD) for Hydroxyurea Treatment using Bayesian Adaptive Control in Children with Sickle Cell Anaemia. 2015 (Manuscript under review) [Google Scholar]
- 53.Weinfeld A, Swolin B, Westin J. Acute leukaemia after hydroxyurea therapy in polycythaemia vera and allied disorders: prospective study of efficacy and leukaemogenicity with therapeutic implications. Eur J Haematol. 1994;52(3):134–9. doi: 10.1111/j.1600-0609.1994.tb01303.x. [DOI] [PubMed] [Google Scholar]
- 54.Björkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410–15. doi: 10.1200/JCO.2011.34.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulz WH, Ware RE. Malignancy in patients with sickle cell disease. Am J Hematol. 2003;7(4):249–53. doi: 10.1002/ajh.10427. [DOI] [PubMed] [Google Scholar]
- 56.Agbaraji VO, Scott RB, Leto S. Fertility studies in sickle cell disease: semen analysis in adult male patients. Int J Fertil. 1988;33(5):347–52. [PubMed] [Google Scholar]
- 57.Anele UA, Burnett AL. Erectile Dysfunction after Sickle Cell Disease–Associated Recurrent Ischemic Priapism: Profile and Risk Factors. J Sex Med. 2015;12(3):713–19. doi: 10.1111/jsm.12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrison BF, Anele UA, Reid ME, et al. Is testosterone deficiency a possible risk factor for priapism associated with sickle-cell disease? Int Urol Nephrol. 2014;47(1):47–52. doi: 10.1007/s11255-014-0864-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zemel BS, Kawchak DA, Ohene-Frempong K, et al. Effects of delayed pubertal development, nutritional status, and disease severity on longitudinal patterns of growth failure in children with sickle cell disease. Pediatr Res. 2007;61:607–13. doi: 10.1203/pdr.0b013e318045bdca. [DOI] [PubMed] [Google Scholar]
- 60.Abudu EK, Akanmu SA, Soriyan OO, et al. Serum testosterone levels of HbSS (sickle cell disease) male subjects in Lagos. Nigeria BMC Res Notes. 2011;4:298. doi: 10.1186/1756-0500-4-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osegbe DN, Akinyanju O, Amaku EO. Fertility in males with sickle cell disease. Lancet. 1981;2(8241):275–6. doi: 10.1016/s0140-6736(81)90525-0. [DOI] [PubMed] [Google Scholar]
- 62.DeBaun MR. Hydroxyurea therapy contributes to infertility in adult men with sickle cell disease: a review. Expert Rev Hematol. 2014;7(6):767–73. doi: 10.1586/17474086.2014.959922. [DOI] [PubMed] [Google Scholar]
- 63.Jones KM, Niaz MS, Brooks CM, et al. Adverse effects of a clinically relevant dose of hydroxyurea used for the treatment of sickle cell disease on male fertility endpoints. Int J Environ Res Public Helath. 2009;6(3):1124–44. doi: 10.3390/ijerph6031124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berthaut I, Guignedoux G, Kirsch-Noir F, et al. Influence of sickle cell disease and treatment with hydroxyurea on sperm parameters and fertility of human males. Haematologica. 2008;93(7):988–93. doi: 10.3324/haematol.11515. [DOI] [PubMed] [Google Scholar]
- 65.Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. J National Med Assoc. 2009;101(10):1046–51. doi: 10.1016/s0027-9684(15)31072-5. [DOI] [PubMed] [Google Scholar]
- 66.Oteng-Ntim E, Meeks D, Seet PT, et al. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: systematic review and meta-analysis. Blood. 2015;125(21):3316–25. doi: 10.1182/blood-2014-11-607317. [DOI] [PubMed] [Google Scholar]
- 67.World Health Organization: RegionalOffice for Africa. Sickle-cell disease: A strategy for the WHO African Region. 2010 Jun; [Google Scholar]
- 68.WHO Model List of Essential Medicines for Children, 4th List. Geneva, Switzerland: Apr, 2013. [Google Scholar]
- 69•.McGann PT, Tshilolo L, Santos B, et al. Hydroxyurea Therapy for Children with Sickle Cell Anemia: Rationale and Design of the REACH Trial. Pediatr Blood Cancer. 2015 doi: 10.1002/pbc.25705. in press. A description of the rationale for and design of the first multi-center trial of hydroxyurea for children with SCA in sub-Saharan Africa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stettler N, McKiernan CM, Melin CQ, et al. Proportion of adults with sickle cell anemia and pain crises receiving hydroxyurea. JAMA. 2015;313(16):1671–2. doi: 10.1001/jama.2015.3075. [DOI] [PubMed] [Google Scholar]
- 71.Gaston M, Rosse W. The cooperative study of sickle cell disease Review of study design and objectives. Am J Pediatr Hematol/Oncol. 1982;4(2):197–201. [PubMed] [Google Scholar]